

Abstract
Beyond haemostasis, platelets have emerged as versatile effectors of the immune response. The contribution of platelets in inflammation, tissue integrity and defence against infections has considerably widened the spectrum of their role in health and disease. Here, we propose a narrative review that first describes these new platelet attributes. We then examine their relevance to microcirculatory alterations in multi-organ dysfunction, a major sepsis complication. Rapid progresses that are made on the knowledge of novel platelet functions should improve the understanding of thrombocytopenia, a common condition and a predictor of adverse outcome in sepsis, and may provide potential avenues for management and therapy.
Keywords: Platelets, Sepsis, Inflammation, Intensive care
Background
Sepsis is a syndrome based on a dysregulated immune response to infection also involving non-immunologic mechanisms, including neuroendocrine, cardiovascular and metabolic pathways [1–3]. Due to its prevalence and high mortality rate, sepsis is a major public health issue [4, 5]. The contribution of blood platelets to sepsis pathophysiology has been the subject of renewed attention. First, alterations of platelet count are commonly encountered in the intensive care unit (ICU). Using common platelet counts thresholds, thrombocytopenia accounts for 20–50% of patients for the whole part of intensive care settings [6–9]. Thrombocytopenia or the non-resolution of thrombocytopenia is associated with poor outcome [8, 10–15]. Second, platelets are well-known players in coagulation and likely to contribute to disseminated intravascular coagulation (DIC). Third, beyond the confines of haemostasis and thrombosis, platelets are now acknowledged as essential actors of the immune response, reacting to infection and disturbed tissue integrity and contributing to inflammation, pathogen killing and tissue repair [16–21]. These advances in platelet biology have opened perspectives on the knowledge of sepsis pathophysiology and on its management. The matter is a complex one as platelets are not only vectors of inflammation contributing to vascular and tissue injury in acute or chronic inflammation [18, 22, 23], but also play an important role in the resolution of inflammation, vascular protection and the repair of damaged tissues. The friend and foe dialogue between platelets and endothelium has been extensively studied and is thought to be relevant to sepsis complications. Here we examine this enlarged spectrum of platelet functions and their relevance to the pathophysiology of multi-organ dysfunction (MOD) and discuss some potential links between these advances and sepsis management.
Sepsis as a dysregulated host response to infection
Recent definition of sepsis [24] emphasizes the non-homoeostatic host response to infection that drives life-threatening organ dysfunctions. Activation of innate immune responses in sepsis realizes a systemic inflammatory condition. The inflammatory phase is characterized by the production of pro-inflammatory mediators and immune cell activation [25–29], and sepsis prognosis is linked to the magnitude and duration of this inflammatory response, high circulating cytokine levels being, for example, associated with poor outcome [30–32]. The triggering of innate immune responses by pathogens and pathogen-associated molecular patterns (PAMPs) has been identified as an early and primary mechanism [2, 31, 33–36]. Interestingly, mechanisms of non-septic systemic-associated inflammatory response syndrome (SIRS) as met in major surgery, severe trauma, extensive burns or pancreatitis may share common features with sepsis-associated SIRS, taking the form of a comparable early inflammatory storm that is triggered by alarmins released by damaged tissues [37]. However, the role played by this hyper-inflammatory phase in the progression of sepsis and its prognostic is to be understood in the context of an accompanying anti-inflammatory response and immunosuppression state, and much effort is made in elaborating a coherent vision of these opposite and complex events [38–43].
Platelets: multifunctional tiny cytoplasmic fragments
Platelets are small (2–4 μm), anucleate, discoid-shaped cytoplasmic fragments released in the bloodstream during the fragmentation of polyploid megakaryocytes in bone marrow sinusoidal blood vessels [44]. In humans, a regulated steady platelet supply and clearance maintains numbers of 150,000–400,000 platelets per microlitre of blood. Platelet production is critically dependent on thrombopoietin (TPO) that acts for an important part on megakaryocyte progenitor proliferation/differentiation and on megakaryocyte maturation [45]. Platelets have a short lifespan, of up to 10 days. They are cleared out from the circulation by mechanisms involving lectin–carbohydrate recognition by splenic and liver macrophages and hepatocytes [46, 47].
Platelets harbour a large variety of mediators stored in a pool of morphologically distinct granules [48]. Granule cargo loading is carried out in megakaryocytes. Platelets also transport mediators, such as serotonin, that they uptake from plasma and can deliver at sites of activation. The cataloguing of platelet-derived mediators reflects the remarkable versatility of platelets in haemostasis, thrombosis and immune responses [49, 50].
The secretion of granule content following platelet activation by agonists is central to platelet functions. Platelet activation induces the expression of membrane proteins and the release of mediators via several mechanisms. Many of these mediators are preformed and stored in granules such as cytokines/chemokines and coagulation factors, others can be synthesized by translational pathways, such as IL-1β, and others are released by yet incompletely defined mechanisms such as CD154. Activated platelets also release vesicles, which include platelet microparticles (PMPs) and exosomes [51]. Platelets represent a major source of circulating MPs [52].
In pathological conditions associated with platelet activation, multiple agonists are generated. In fact, apart from classical strong agonists such as thrombin or collagen, there is an expanding list of agonists that can contribute to platelet activation. These additional platelet agonists have allowed a re-appreciation of mechanisms and role of platelet activation in vascular inflammation and thrombotic events associated with a range of infectious and inflammatory conditions [53].
The archetypal function of platelets is haemostasis. Platelets encounter inhibitory signals that prevent their activation in the healthy vasculature, such as nitric oxide and prostacyclin, which are released by endothelial cells (ECs). Platelets circulate in close proximity to the vessel wall, and the disruption of EC lining overcomes inhibitory signals and drives platelet adherence, activation and aggregation, which temporarily plug the damaged vessel. In this process, platelets also activate and confine coagulation at site of damage, particularly via the exposure of an efficient catalytic phospholipidic surface [54].
Besides binding to damaged vessels and preventing bleeding, platelets support a large spectrum of more recently studied functions, as could be reflected by the diversity of platelet mediators [55–57]. Platelets are activated in conditions that disrupt tissue homoeostasis and exert, directly and indirectly, a complex control over the different stages of inflammation, contributing to pathogen clearance, wound repair and tissue regeneration (Figs. 1, 2). As such, platelets are now acknowledged as essential components of the innate immune response, monitoring and rapidly responding to noxious signals.
Fig. 1.
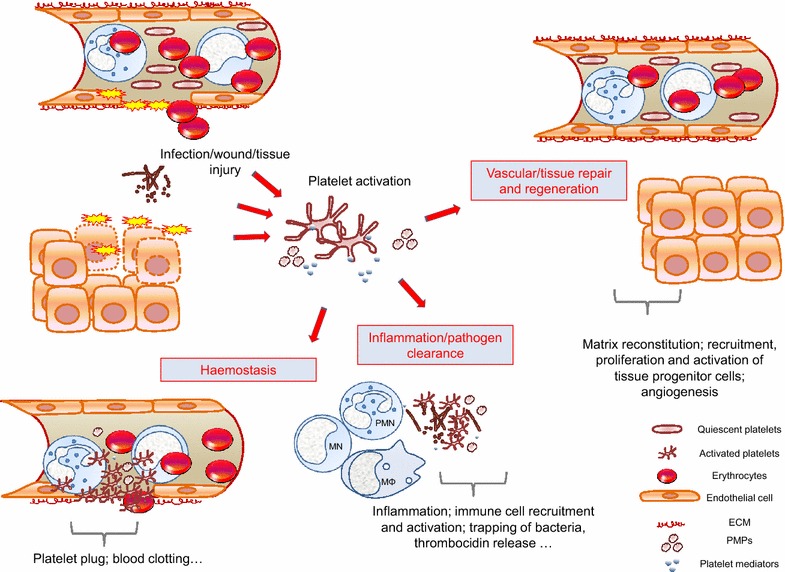
Platelets are integral players in the immune response, linking haemostasis, thrombosis, inflammation, pathogen clearance and tissue repair: a schematic representation. A growing body of evidence highlights a role for platelets beyond the confines of haemostasis and thrombosis. Some of platelet interfaces in innate immune response are schematized. Platelets are activated at sites of infection/tissue injury. Platelets and platelet-derived mediators contribute to arrest bleeding, to clear pathogens directly or indirectly by acting on various steps of the immune response, and to drive vascular/tissue repair by providing matrix building blocks and a multiplicity of signals that remodel matrix, attracting tissue progenitor cells and reconstructing the vascular frame. In doing so, platelets provide a coherent biological response contributing to cure infection and re-establish tissue architecture and homoeostasis. Scales are arbitrary. Platelet-derived microparticles (PMPs) recapitulate several of activated platelet functions. ECM extracellular matrix, MN monocytes, PMN polymorphonuclear neutrophils, MΦ macrophages
Fig. 2.
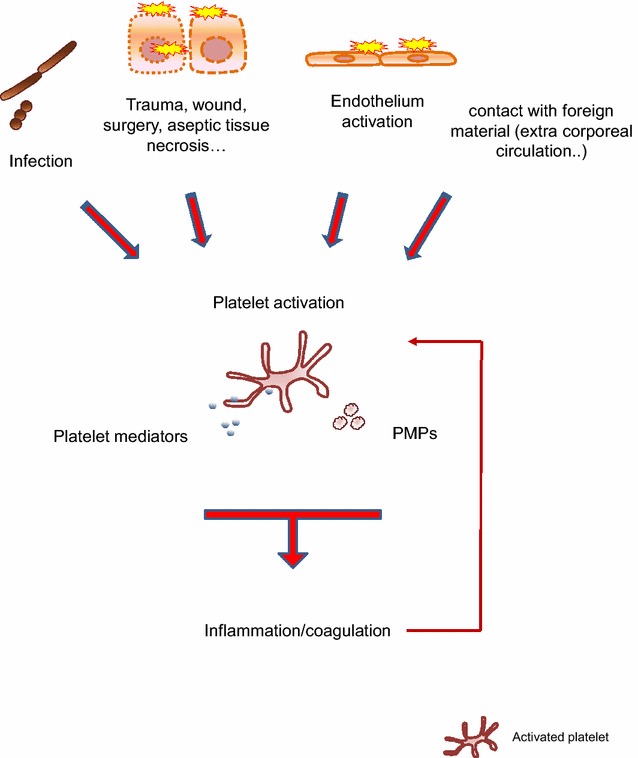
Platelets monitor and are activated in response to noxious signals. Platelets sense and are activated by multiple signals generated in danger situations met by the organism. Interaction with pathogens, endothelial cell/tissue injury and interaction with foreign material activate platelets (see text for details). Platelet activation sparks off a broad range of responses, including the activation of various inflammation and coagulation pathways. Signals generated in inflammation and coagulation can in return activate platelets (thin arrow). PMPs platelet microparticles
Platelets as key players in the inflammatory reaction; critical links with coagulation
Activated platelets secrete a profusion of pro-inflammatory material, cytokines/chemokines, vasoactive amines, eicosanoids, and components of proteolytic cascades that directly or indirectly, through the activation of bystander target cells, fuel inflammation [23, 58, 59]. ECs and leucocytes are prime targets for platelets. Endothelium is a non-adhesive, non-thrombogenic surface in normal conditions; when stimulated by inflammatory mediators, ECs undergo profound changes, collectively designed as “EC activation”, which include the expression of cell adhesion molecules and tissue factor, production of von Willebrand factor, cytokines/chemokines, proteases and vasoactive substances such as nitric oxide. Platelets adhere to activated ECs, following a multi-step process in which glycans play a critical role [60–62]. Inflammation can also alter the protective EC glycocalyx barrier, favouring platelet adhesion [63, 64]. During the adhesion process, platelets can be activated and in turn activate ECs. Platelet activation in inflammation can alter the vascular tone and lead to deleterious effects on vasculature integrity, by increasing vascular barrier permeability and contributing to the generation of cytopathic signals, for example by mediating reactive oxygen species generation by neutrophils [65]; these effects have to be paralleled with the opposed protective role of platelets (below) [66–69]. Leucocytes are a second critical target for platelets, the platelet/leucocyte dialogue being essential in inflammation; here, we focus on neutrophils and monocytes. Platelet/leucocyte interactions are a critical step in leucocyte recruitment, activation and migration in inflammation [70]. Platelet/neutrophil or platelet/monocyte interactions can occur at the EC surface, in clot/thrombi and in circulating blood [18, 70, 71], and platelets direct neutrophil/monocyte migration to sites of tissue injury [72, 73]. Moreover, platelets activate neutrophils and monocytes upon interaction, via several mechanisms, including the triggering of TREM-1 on neutrophils, leading to various pro-inflammatory responses [65, 74–77]. The formation of platelet/leucocyte aggregates in blood depends on platelet activation and is an early phenomenon in sepsis progression. For example, platelet/neutrophils complexes are elevated at early phases, while being reduced in complicated sepsis possibly reflecting peripheral sequestration or sepsis-associated thrombocytopenia [78, 79], and endotoxin administration in humans leads to an increased circulating platelet/neutrophil aggregates that follows a brief decrease [80]. Amplification of inflammation results from the reciprocal activation between platelets and their target cells [66], and circulating monocyte/and neutrophil/platelet aggregates may contribute to disseminate inflammatory signals [81]. Platelets also link several inflammatory cascades; for example, they propagate the activation of the complement system [82]. Commonly, cytokines have an induced expression that is regulated at the transcriptional/translational level. Most of platelet-derived inflammatory mediators are very rapidly released from activated platelets, making platelets instant providers of pro-inflammatory material. Cytokine bioactivity at organs remote from their source is debated as cytokine bioactivity may be hampered in plasma. Platelet transport may protect inflammatory mediators from otherwise degradation. Therefore, platelets play a central role in the inflammatory reaction. Importantly, they also contribute to the control and resolution of inflammation via several mechanisms, including the release of anti-inflammatory cytokines and inflammation pro-resolving mediators [83].
The activation of coagulation and inflammation cascades are consequences of platelet activation, and inflammation and coagulation pathways crosstalk [84]. For example, some platelet mediators have both inflammatory and pro-coagulant properties, such as polyphosphates [85]. Pro-inflammatory cytokines released by platelets can also activate the coagulation cascade at various steps [86]. Conversely, the activation of coagulation by platelets generates a number of inflammatory effectors, such as thrombin. Further, inflammatory mediators can impair anticoagulant and fibrinolysis pathway mechanisms, which may contribute to coagulation dysregulation in sepsis [87–89]. Platelet inflammatory mediators may thus contribute to sepsis coagulopathy [88–91]. DIC is a frequent and major complication of sepsis [41], and various mechanisms concur to involve platelets in DIC; only some can be mentioned. First, platelets support the generation of thrombin. Second, platelet links inflammation and coagulation. Third, platelets are major inducers of the release of pro-thrombotic scaffolds neutrophil extracellular traps (NETs) [92–96].
Notwithstanding, the involvement of PMPs in vascular inflammation and inflammatory disorders, including sepsis, has been emphasized [97–102]. PMPs retain many pro-inflammatory and pro-coagulant features of parent platelets and are thought to disseminate inflammatory and coagulation signals. Although they represent potential pathophysiological players in inflammatory disorders [52, 99, 100, 103, 104], their role in sepsis remains ill-understood.
Platelets in vascular and tissue integrity
In normal wound healing, platelets establish regulatory crosstalks between soluble and cellular actors of tissue repair that concur to the various phases of inflammation and reestablishment of tissue homoeostasis [50, 83, 104, 105]. Platelets accumulate early, are activated at sites of tissue injury and intervene at each stage of tissue repair, the inflammatory, new tissue formation and remodelling stages. In fact, platelet-healing properties are already translated to the clinics [50, 55]. The best studied role of platelets in tissue homoeostasis is the preservation of resting and injured endothelium integrity, a critical point in MOD pathophysiology [71, 106–108]. The importance of platelets is exemplified by the disruption of the endothelium barrier associated with thrombocytopenia [109]. How platelets contribute is incompletely understood. Mechanisms include gap filling, production of EC mitogenic factors and factors enhancing the vascular barrier [71, 107]. On injured endothelium, platelets adhere to the vascular wall at sites of damage and immediate proximity, a first step in a sequence of events that lead to the initiation and the propagation of haemostasis, thrombosis and bleeding arrest [110, 111]. Platelets provide material for endothelium repair, including EC growth-promoting, antiapoptotic mediators, and attractants for progenitor cells endowed with vascular healing properties [104]. They help restoring the disrupted vascular network, providing positive and negative regulators of angiogenesis and stimulating angiogenic mediator production by target cells. Platelets are also important contributors to extracellular matrix (ECM) repair as they are a rich source of ECM components, ECM remodelling proteins, and fibro-competent cell activators. Platelets have however been found to both promote and prevent vascular permeability in inflammation. The differential regulation of vascular permeability by platelets has been studied for a large part in acute lung injury (ALI) models and will be presented in the corresponding section. Importantly, platelets are highly efficient at preventing bleeding in an inflammatory context [76, 107, 112]. The platelet count threshold needed for vasculoprotection in humans in normal or inflammatory conditions is an important question that remains to be answered [107]. More generally, platelets may play a broader role in organ regeneration. Platelets not only prevent blood loss but also provide key signals for matrix architecture reconstruction and for the recruitment, proliferation, survival and differentiation of cells endowed with new tissue formation, such as fibroblasts, smooth muscle cells and tissue-specific progenitors cells [113]. This is remarkably illustrated by the requirement of platelets in liver regeneration [114]. PMPs are also thought to contribute to vascular repair [115]. Hence, platelet activation is both necessary to tissue integrity and undesirable as it generates tissue-damaging signals. The complex network of signals that organize this fine-tuned equilibrium is only recently being biochemically dissected [55, 104]. Many questions remain on this balanced platelet friend or foe contribution, although they are of key importance to the pathophysiology of microvascular dysfunctions, such as in sepsis [68].
Platelets contribute to the innate immune response against infection
The role of platelets in the defence against infection is increasingly stressed [83, 116]. Platelets are now acknowledged as bona fide pathogen sensors interacting directly or indirectly with a number of bacterial, viral, fungal and protozoan pathogens and their products, contributing to their clearance. Platelet interaction with bacteria depends on the nature and concentration of bacteria, interaction time, and involves multiple mechanisms. Toll like receptors-dependent and independent mechanisms, such as those involving Fcγ receptors, complement receptors or glycoproteins GPIIb-IIIa and GPIbα, contribute to platelet–bacteria interactions. Indirect interactions are also involved, such as via the binding of plasma proteins, including fibrinogen, von Willebrand factor, complement proteins or IgG, that bridge pathogens and platelets or via interaction with bacterial toxins. Interaction with pathogens can lead to platelet adhesion or to their activation, aggregation and release of platelet mediators [83, 117–120]. Mechanisms of pathogen clearance by platelets may be direct, through the release of various antimicrobial peptides and indirect via the release of platelet-derived mediators that coordinate chemotaxis and activation of immune cells [83, 116–118, 120, 121]. Infection is commonly associated with tissue injury. Injured and dying cells generate mediators such as alarmins that fuel inflammation [122]. Mediators generated by cell damage such as complement activation products and histones can activate platelets [123, 124]. Notwithstanding, platelets also contribute to the adaptive immune response to infection [17, 18, 22, 125].
The aforementioned role of platelets in the defence against pathogens suggests that they can interfere with the progression of infection. How can these observations be translated to sepsis pathophysiology [120, 124, 126]? Models suggest a protective role of platelets, as, for example, in streptococcal endocarditis, malaria or gram-negative pneumonia [127–129], and thrombocytopenia could be a risk factor for bacterial or fungal infection. Alternatively, platelets could contribute to spreading infection, via the transport of pathogens [130].
Platelets in MOD pathophysiology
Endothelium in MOD: a common pathophysiological denominator
The pathophysiology of sepsis and its complications remains uncertain as much caution has to be applied in extrapolating to clinical sepsis results obtained in rodent models which have their own inherent complexities [131–134]. Within these extrapolation limits, experimental models have, however, yielded significant knowledge. Numerous studies have emphasized the orchestrating role of endothelium in sepsis, and endothelium injury could be one of the primum movens pathophysiological events in sepsis complications [102, 135–148]. Markers of endothelium injury are elevated in sepsis patients, although variably associated with sepsis severity [29, 32, 149]. Inflammation, thrombosis, capillary perfusion alterations are among key features of MOD microvascular alterations [102, 136, 150, 151]. Platelet activation can be detected in sepsis patients and sepsis models, and studies reported association with sepsis severity [79, 124, 152, 153]. Many signals can activate ECs and platelets in sepsis, including pathogens and mediators generated by inflammation and coagulation. Activated platelets may thus contribute to MOD via their role in inflammation and coagulation (Fig. 3) [23, 56, 83, 124, 141, 144, 152, 154].
Fig. 3.
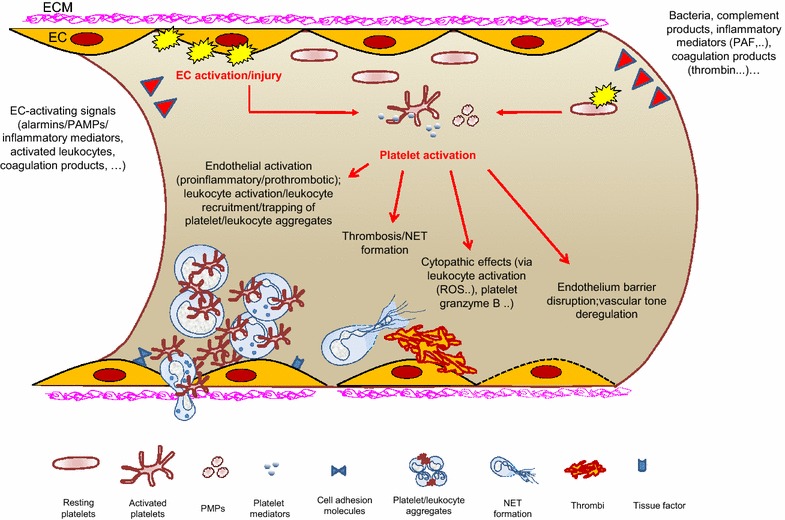
Microvasculature is a critical target of platelet activation in sepsis. Platelets circulate more concentrated close to the vascular wall and sense endothelium disturbances. In sepsis, many cellular and soluble actors concur to activate the endothelium. Activated/injured endothelium is a key driver of platelet activation. Signals generated by infection, inflammation and coagulation can also activate platelets in sepsis. The relative importance of platelet activation by PAMPs in sepsis is not well established. Platelet activation contributes to fuel various pro-inflammatory and pro-coagulant pathways with potential deleterious consequences on endothelium homoeostasis and integrity. Unmitigated platelet activation in sepsis may take a significant part in the complex global scenario that leads to impairment of the endothelium barrier and microcirculatory failure, a leading cause of organ dysfunction in sepsis. Only some pathophysiological events are schematized (see text for details). Scales are arbitrary. ROS reactive oxygen species, EC endothelial cells, NET neutrophils extracellular traps, PMPs platelet microparticles, ECM extracellular matrix
Platelets in acute lung injury (ALI) in sepsis
There are arguments to involve platelets in ALI pathophysiology [155–158]. Dysregulated inflammation and coagulation are central pathophysiological events in ALI and lung vascular endothelium injury is a primary cause of the alteration of the alveolar-capillary barrier leading to pulmonary oedema [159] [160]. Platelets are sequestered early in lung microvascular beds in ALI models and may contribute to the initial insult of lung endothelium [155, 161–164]. EC activation/injury by inflammatory stimuli, PAMPs and alarmins can generate signals mediating platelet accumulation and activation. Entrapment and activation of platelets in pulmonary capillaries will consequently feed the deleterious cascade of pro-inflammatory and pro-coagulant events in the lung [71, 156, 158, 159, 165]. Platelets can also induce apoptosis in the lung in sepsis models [166]. Among these events, platelet/neutrophil interactions have received considerable attention. Neutrophils are of critical importance in MOD, neutrophil influx being a hallmark of ALI and their inappropriate activation leading to tissue damage signals [167]. Platelets play an important role in neutrophil recruitment and activation in the lung, and platelet-mediated neutrophil activation results in the release of cytokines, chemokines, reactive oxygen species and NET generation [71]. Indeed, experimental models highlight the deleterious role of platelet/neutrophil and also platelet/monocyte interactions in the alteration of the alveolar-capillary integrity [71, 108, 168]. Coagulation activation and alveolar fibrin deposition are common findings in ALI, and platelets are thought to be key contributors to the dysregulation of coagulation in ALI, through their role in coagulation and NET generation [169]. Therefore, several studies suggest a platelet involvement in ALI. In fact, platelet depletion, blocking of platelet/neutrophil interaction, NET dismantling or antiplatelet treatments are protective in experimental models [96, 162–164, 170]. Although in vitro experiments show pro-inflammatory and pro-coagulant effects of PMPs, there is little evidence for a specific deleterious role of PMPs in ALI, a study difficult to address due to microparticle identification uncertainties and to the simultaneous presence of microparticles from various origins with heterogeneous functions [158, 171].
Increased vascular permeability is the basis for oedema in inflammation. The concept that platelets protect the basal barrier of alveolar capillaries is supported by experimental evidences, and thrombocytopenia put the lung capillary integrity at risk, particularly in inflammatory conditions. Indeed, severe thrombocytopenia results in increased alveolar-capillary permeability [71, 107, 108]. However, as mentioned above, platelet activation in inflammation can also disrupt endothelium barrier integrity, and platelet depletion is protective in several ALI models [69, 71, 162, 172]. How this dual endothelial barrier-stabilizing versus barrier-destabilizing property of platelets is organized and contribute to ALI progression, as well as the specific role of PMPs, is not understood. Such a differential effect of platelets in controlling endothelial barrier integrity is likely to be based on a complex balance between characteristics of inflammation in vascular beds, early or late phase, magnitude, role of other inflammatory players, i.e. leukocytes, and experimental models used. The changing relative importance during sepsis progression of platelet-activating signals, platelet count and proteome, interactions with leucocytes and ECs, underline the difficulty to dissect these mechanisms [69, 71, 76, 108, 173]. Moreover, platelets may play a positive role in the control and resolution of inflammation in lung injury, a mechanism that is only recently being understood [158]. The genetic background also plays a role in ALI-associated mortality and morbidity [174]. Platelet count is determined by genetic factors, and genetic studies point to an association between low platelet count and acute respiratory distress syndrome (ARDS) risk. Genetic variants within the LRRC16A locus (6p22) are associated with a low platelet count. Interestingly, a low platelet count links a single nucleotide polymorphism within this locus to ARDS risk [175].
Platelets and acute kidney injury (AKI) in sepsis
Acute kidney injury (AKI), a major sepsis complication, is accompanied by hemodynamic disturbances such as decreased glomerular filtration rate and microcirculation alterations [146, 176–180]. The extent of apoptosis and necrosis in tubular lesions is debated. Subtle, heterogeneous, potentially reversible, cytopathic and adaptive cellular events (metabolic changes, mitochondrial dysfunction, autophagy, cell cycle arrest, etc.) may characterize tubular lesions in sepsis AKI [181–184]. Beyond the classic paradigm of renal hypoperfusion, the role of immune response pathways and particularly inflammation in AKI progression is increasingly stressed [1, 178, 185–195]. Alarmins, PAMPs, inflammatory mediators and leukocytes can activate ECs in the renal microcirculation bed, leading to inflammation/thrombosis, metabolic alterations, oxidative stress, concurring to microvascular dysfunction. Due to the close dependence between TECs and tubular microvascularization, compromise blood flow and inflammation in the microvascular beds can lead to tubular epithelial cells (TECs) injury, driving inflammation, mitochondrial/metabolic alterations and various adaptive responses, including cell cycle arrest. Alarmins, PAMPs and inflammatory mediators may also impact TECs after being filtered, and TECs are active participants in kidney inflammation [192, 196–198].
In the highly vascularized kidney, platelet/endothelium interactions can be postulated to be of specific importance. In an AKI model in which selective kidney endothelial injury is realized, there are evidences for platelet contribution [199]. Platelets will be arrested and activated on the kidney endothelium activated by circulating deleterious signals. Inflammation-mediated alteration of EC glycocalyx can also favour platelet adhesion [141, 146, 200–203]. Platelets can also be activated by ischaemic blood flow disturbances in the septic kidney. Therefore, and although much remains to be understood, platelets may be pathophysiological players in sepsis AKI. On the other hand, as mentioned above, platelets contribute to the resolution of inflammation and vasculature integrity. Important questions remain with reference to the identification of soluble and cellular effectors that contribute to the resolution of inflammation and tubular regeneration in the kidney [204]. Microparticles, and PMPs more specifically, are elevated in sepsis and sepsis complicated by AKI [101, 102, 193]. However, their specific role remains to be addressed.
Platelets and organ-to-organ crosstalk in sepsis
Despite the importance of the deleterious organ-to-organ communication in sepsis, underlying mechanisms are only beginning to be unravelled. Inflammatory signals are implicated in these communications [205]. Can platelets vectorize the exchange of pro-inflammatory and/or pro-coagulant signals that link injuries in distant organs? Interestingly, the activation of platelets at remote sites may mediate lung injury, as shown in mesenteric ischaemia/reperfusion models [206]. Platelets can mediate remote kidney damage induced by pneumonia [207]. Among platelet-derived mediators that could convey such a deleterious action, platelet factor 4 (CXCL4) and CD154 have been identified [208, 209]. When activated, platelets express CD154 and release a soluble form of CD154 [22, 210]; CD154 may bear a particular responsibility as, for example, the CD154/CD40 dyad plays a deleterious role in ALI, including pancreatitis-associated lung injury [211, 212], and as it could be brought to lung microcirculation via PMPs. Further, platelet CD154 mediates neutrophil recruitment in septic lung injury [213]. Although these results suggest a role for platelets, the extent and relative contribution of platelets, platelet-derived mediators, PMPs or circulating platelet/leucocyte aggregates in conveying deleterious signals at distance in patients with sepsis is unknown.
Platelet count in sepsis and the dilemma of platelet transfusion
Platelet count and dynamics of platelet count as determinants of clinical outcome in sepsis patients
Thrombocytopenia is common in sepsis and more generally in critically ill patients and has long been recognized as an independent risk factor for mortality in ICU patients and a sensitive marker for disease severity; the severity of sepsis is a risk factor for thrombocytopenia [6, 8–15, 214–221]. For these reasons, the platelet count is included in the ICU severity of illness scoring system. Platelet count kinetics is often biphasic in ICU patients, characterized by a moderate initial decrease in the first days followed by a rise [11, 216, 222]. Early thrombocytopenia and new-onset thrombocytopenia during ICU hospitalization are associated with a poor prognostic; the magnitude and duration of thrombocytopenia and the absence of relative increase in the platelet count have been linked to the poor outcome [6, 9, 11, 216, 221–227]. In a large recent study, which included 931 sepsis patients, a low platelet count at admission in the ICU was associated with an increased mortality risk [29]. Notably, patients with low platelet counts were more severely ill at ICU admission. Understanding pathophysiological links between platelet count alterations and clinical outcomes is therefore an important issue for the intensive care physician.
The multiple causes of thrombocytopenia in sepsis patients
The association between thrombocytopenia and clinical outcome does not establish causality, and identifying the causes of thrombocytopenia is essential to patient management. Management of the underlying condition is a primary focus, and an important issue is platelet transfusion. Platelet transfusion may be ineffectual and deleterious in patients with, for example, intravascular platelet activation and have their own risks [228–230]. In a recent report, sepsis was identified as associated with ineffectual platelet transfusion, as evaluated by inadequate platelet count increase [231].
Several mechanisms, acting alone or in combination, can be responsible for a low platelet count in sepsis, and all steps of platelet life may be concerned. Decreased platelet production in the bone marrow can result from pre-existing conditions or from the inhibitory effect of pathogen toxins, drugs or inflammatory mediators on haematopoiesis. Peripheral mechanisms are essential causes of thrombocytopenia [15, 214, 218, 227, 232, 233]. The reduction in platelet half-life and their consumption/destruction may be linked to the many events of platelet activation occurring in sepsis, intravascular coagulopathy and immune mechanisms. Drug-induced thrombocytopenia, hemophagocytosis, bleeding, hemodilution are also major explanatory factors. Laboratory artefact of pseudothrombocytopenia can be encountered, and assessing the reality of thrombocytopenia is an important point [230].
Systematic investigations with routinely available tests can help to delineate mechanisms of thrombocytopenia [218, 232, 234]. An early rise of reticulated platelets follows endotoxin administration in humans, and the percentage of immature platelet fraction that evaluates thrombopoietic rate could be a useful tool to witness early bone marrow reaction predicting sepsis development [80, 235]. Apart from altering platelet count, sepsis and sepsis medications can also result in platelet function defect, adding another pathophysiological interface [236, 237]. A detailed description of these mechanisms and diagnostic/management guidance has been excellently reviewed and is beyond the scope of the present work [154, 222, 230, 233, 238–242]. A difficulty in approaching thrombocytopenia and its management is related to the paradox of platelets being potentially both deleterious and beneficial during sepsis course. In a first point of view, platelet count reduction is related to sepsis via consumption mechanisms including pathogen and pathogen product-mediated activation, induction of apoptosis, lysis and increased phagocytic clearance. Acute infections often lead to thrombocytopenia [58], and bloodstream infection is associated with lower platelet counts [221]. Coagulopathy, particularly DIC, platelet sequestration by leucocytes and by inflammatory vascular beds are also commonly stressed mechanisms of thrombocytopenia. Through these mechanisms, platelets can be perceived as bystanders whose destruction is related to the severity of infection and to the characteristics of the host response to the infectious challenge. In that case, the use of platelet transfusion may be perceived as being detrimental, via the fuelling of inflammation and coagulation. On the other hand, platelets are active players in pathogen clearance, leading to the possibility that a low platelet count and platelet function alteration may first favour infection. Further, platelets also protect vascular integrity; hence, maintaining an adequate threshold of platelet count seems a necessary target to prevent bleeding. In fact, platelet transfusions are mostly used to prevent or treat bleeding [228]. Conciliating such a paradox of platelets being both deleterious and beneficial is a challenging point for platelet-targeted therapeutic interventions in sepsis.
Can platelets represent therapeutic targets and diagnostic tools in sepsis?
The clinical management of sepsis remains a difficult challenge, and pathophysiological advances have not yet been translated into effective therapeutic protocols [2]. Notably, strategies to counteract the runaway pro-inflammatory state in sepsis, such as inhibition of specific inflammatory mediators, have given disappointing results [243]. However, current knowledge on sepsis pathophysiology, highlighting multiple humoral and cellular factors in the inappropriate inflammatory response to infection, suggests that therapies targeting a single mediator will not demonstrate effectiveness [41]. Additional complexity is linked to individual disease susceptibilities and medical comorbidities that would necessitate individual approaches. Accumulating evidence therefore speaks for an integrated approach of sepsis treatment based on a better knowledge of its natural history.
The recently described involvement of platelets at the crossroads of several immune response pathways has led to the assumption that platelets or platelet-derived effectors represent therapeutic targets in sepsis. Platelet activation can drive multiple inflammatory and coagulation pathways, and targeting platelets offer the theoretical perspective of targeting simultaneously several deleterious pathways. Although the clinical relevance of animal models has many drawbacks, it is of interest that platelet depletion, inhibition of platelet functions and antiplatelet drugs show protection in experimental ALI or AKI [124]. P2Y12 inhibitors reduce inflammatory and pro-thrombotic mechanisms after endotoxin administration in humans [244]. Several observational and retrospective clinical studies have shown that antiplatelet agents such as acetylsalicylic acid, platelet P2Y12 inhibitor clopidogrel or GPIIb/IIIa antagonists reduce mortality or complications in critically ill patients [245–255]. However, some studies are conflicting [249, 252, 256] (Table 1). There is therefore a strong need for large randomized controlled clinical trials to investigate the effects of antiplatelet therapy in sepsis. The complexity of such studies relates in part to the heterogeneity of sepsis patients in terms of nature of the causal germ, site and severity of infection, multiple comorbidities, gender, age and genetic background. There is also individual variability in the concentration of antiplatelet agents that efficiently inhibits platelet function. A defective response to clopidogrel or aspirin treatment may concern up to 30 or 40% individuals, respectively [257–259]. Also, antiplatelet treatments have differential effects on platelet functions. Platelets treated with aspirin can still be activated by strong agonists, such as thrombin or ADP. Hence, in a full-blown pro-inflammatory/pro-coagulant condition as met in sepsis, it remains to be determined whether platelet activation is efficiently inhibited by antiplatelet treatments. Platelets are an important blood reservoir of pro-inflammatory molecules and may contribute to the “cytokine storm” that characterizes sepsis. However, many cellular players, including leucocytes and EC, also produce such mediators, and the relative contribution of platelets is not understood. In a recent study, antiplatelet therapy did not significantly reduce plasma pro-inflammatory cytokines levels in sepsis patients [260]. Antiplatelet agents have also been shown to have indirect off-platelet effects, a mechanism which importance is not yet established [261]. Finally, the impairment of platelet function may have undesirable consequences, such as bleeding or the blunting of platelet protective functions.
Table 1.
Summary of cohort studies on antiplatelet agents and sepsis
| Authors | Study year | Study type and setting | Patient number | Antiplatelet agent | Patients | Study conclusions | Potential limitations |
|---|---|---|---|---|---|---|---|
| Wang et al. [268] | 2016 | Meta analysis of cohort studies | 14,612 | ASA, clopidogrel, ticlopidine | ICU patients with ARDS predisposing conditions | Reduced mortality and lower incidence of ARDS | Non-sepsis patients included Treatment bias of antiplatelet agents |
| Kor et al. [269] | 2012–2014 | Multicenter, double-blind, placebo-controlled, randomized clinical trial | 390 | ASA | Patients with elevated risk for developing ARDS in the emergency department | ASA did not reduce the risk of ARDS and 28-day or 1-year survival | Non-sepsis patients included Low rate of ARDS development |
| Wiewel et al. [260] | 2011–2014 | Prospective observational study with propensity matching | 972 | Mostly ASA | Sepsis within 24 h after admission in 2 mixed medical/surgical ICU | Antiplatelet therapy was not associated with alterations in the presentation or outcome of sepsis or the host response | Treatment bias of ASA Inadequate patient number and power |
| Osthoff et al. [270] | 2001–2013 | Retrospective cohort study with propensity matching | 689 | ASA | Patients with S. aureus and E. coli bloodstream infection admitted in a single medical/surgical ICU | Low-dose ASA at the time of bloodstream infection was strongly associated with a reduced short-term mortality in patients with S. aureus bloodstream infection | Treatment bias of ASA at the time of enrolment Severity at presentation was not included in the analysis model Inadequate patient number and power |
| Tsai et al. [255] | 2000–2010 | A nation-wide population-based cohort and nested case–control study | 683,421 | ASA, clopidogrel, ticlopidine | Sepsis | Antiplatelet agents were associated with a survival benefit in sepsis patients | Claims database |
| Chen et al. [253] | 2006–2012 | Secondary analysis of prospective cohort with propensity matching | 1149 | ASA | Patients admitted in a mixed ICU for at least 2 days | Decreased risk of ARDS | Non-sepsis patients included Treatment bias of ASA |
| Boyle et al. [271] | 2010–2012 | Prospective observational study | 202 | ASA | ICU patients requiring invasive mechanical ventilation | Reduced risk of ICU mortality | Treatment bias of ASA Non-sepsis patients included |
| Valerio-Rojas et al. [249] | 2007–2009 | Retrospective cohort with propensity matching | 651 | ASA, clopidogrel | ICU patients with sepsis | No decrease in hospital mortality but decreased incidence of ARDS | Inadequate patient number and power Unmeasured bias and confounding |
| Otto et al. [251] | 2013 | Retrospective cohort | 886 | ASA, clopidogrel | Surgical ICU patients with sepsis and a minimum length of stay of 48 h and a history of atherosclerotic vascular diseases | ASA treatment reduced the ICU and hospital mortality. Combination of ASA with clopidogrel did not show any significant effect on mortality. Clopidogrel alone might have a similar benefit | Unmeasured bias and confounding |
| Sossdorf et al. [250] | 2013 | Retrospective cohort | 979 | ASA | Septic patients admitted to a surgical ICU | Decreased mortality with ASA or NSAIDs was associated with decreased hospital mortality. No benefit when ASA and NSAIDs are given together | Unmeasured bias and confounding |
| Eisen et al. [248] | 2000–2009 | Retrospective cohort study with propensity matching | 7945 | ASA | ICU patients with SIRS/sepsis on ASA at the time of SIRS/sepsis | ASA was associated with survival | Treatment bias of ASA at the time of enrolment and confounders |
| O’Neal et al. [272] | 2006–2008 | Cross-sectional analysis of a prospective cohort | 575 | ASA and Statin | Patients admitted in a mixed ICU for at least 2 days | ASA was not associated with the diagnosis of ALI/ARDS, sepsis or hospital mortality | Treatment bias of ASA Unmeasured bias and confounding Non-sepsis patients included |
| Erlich et al. [246] | 2006 | Retrospective cohort | 161 | ASA, clopidogrel, ticlopidine | Adult patients admitted in a medical ICU with a major risk factor for ALI | Reduced incidence of ALI/ARDS | Treatment bias of ASA Non-sepsis patients included |
| Kor et al. [256] | 2009 | Second analysis of prospective multicenter observational study | 3855 | ASA | Consecutive, adult, non-surgical patients with at least one major risk factor for ALI | ASA was not associated with ICU or hospital mortality and ICU or hospital lengths | Treatment bias of ASA Non-sepsis patients included Unmeasured bias and confounding |
| Storey et al. [273] | 2006–2008 | Post hoc analysis PLATO study | 18,421 | Ticagrelor vs clopidogrel | Patients with acute coronary syndrome | Reduced mortality following pulmonary infection and sepsis in acute coronary syndrome with ticagrelor | Unmeasured bias and confounding |
| Winning et al. [245] | 2007–2009 | Retrospective cohort | 615 | ASA, clopidogrel | Consecutive patients admitted in a mixed ICU | Reduction in organ failure and mortality in critically ill patients with pre-existing medication | Non-sepsis patients included Treatment bias of ASA |
| Winning et al. [274] | 2002–2007 | Retrospective cohort | 224 | ASA, clopidogrel ticlopidine | Patients admitted for CAP not receiving statins and using antiplatelet drugs for more than 6 months | Reduction in need of intensive care treatment and length of hospital stay | Unmeasured bias and confounding |
| Gross et al. [275] | 2001–2005 | Retrospective cohort | 417,648 | Clopidogrel | All adult (≥ 18 years) Medicaid beneficiaries in Kentucky | Increased CAP incidence and no significant reduction in severity | Claims database |
ASA Acetylsalicylic acid, ARDS acute respiratory distress syndrome, ALI acute lung injury, CAP community-acquired pneumonia, NSAID non-steroidal anti-inflammatory drug
As mentioned above, elucidating mechanisms of thrombocytopenia in sepsis are essential with reference to transfusion. Platelet transfusion is mostly used to prevent/treat bleeding [228, 229]. The risk of bleeding increases with the severity of thrombocytopenia [222]. The threshold of platelet count ensuring protection may be higher in sepsis patients, reflecting the severity of the disturbance of the vascular beds. Commonly advocated threshold of platelet count is in the range of 10–50 × 109/L, depending on clinical situations, additional bleeding risks, evidence for central thrombocytopenia. The risk of bleeding is, however, not straightforwardly linked to the depth of thrombocytopenia, in the context of a sustained production of platelets, and additional parameters in the critically ill patient may interfere; indeed, the risk of bleeding is also increased for platelet counts between 50 and 100 × 109/L [8, 9, 222, 229]. In fact, there is a poor evidence-based clinical benefit of platelet transfusion in the non-bleeding ICU patient [154, 228–230, 242]. The lack of a clear understanding of thrombocytopenia causes makes the risk/benefit assessment difficult, as there is a theoretical risk to aggravate the underlying pathophysiology [229]. The main regulator of platelet production, TPO, is elevated in sepsis and related to the platelet count [262, 263], which may be linked to the reduction in platelet mass or stimulation of TPO production by inflammatory mediators. Experimental models show that TPO neutralization reduces the severity of organ damage [264]. However, in the clinics, the potential benefit of TPO administration in thrombocytopenic patients in sepsis has been recently suggested [265]. At this stage, results from randomized controlled trials remain necessary to evaluate TPO interest in sepsis.
If the interpretation of thrombocytopenia in sepsis patients is made difficult by the multiplicity of underlying mechanisms, the platelet count by itself may hold valuable information [242]. The platelet count may represent a surrogate marker of the severity of organ dysfunction. A low platelet count occurring even early in sepsis patients is indeed recognized as a sign of poor prognostic; however, a single platelet count at admission may have little pertinence [266], and the kinetics of platelet counts appears to have a deeper meaning. Two alterations of this kinetics have been shown to be of clinical interest in sepsis patients, suggesting that they must be given specific attention. Both the magnitude of the drop in platelet count rather that thrombocytopenia per se, and the non-resolution of thrombocytopenia are strong predictors of mortality in sepsis [9, 15, 267]. The onset and dynamics of thrombocytopenia have been stressed as potential diagnostic approaches in ICU patients [222].
Conclusion
Platelets play key roles in various aspects of the immune response, suggesting that they take a significant part in sepsis pathophysiology. Therapeutic control of platelet functions would offer the perspective of targeting simultaneously several deleterious pathways in sepsis. The difficult extrapolation of experimental models to clinical sepsis and the conflicting results of clinical studies do not allow us today to introduce an antiplatelet agent in clinical practice. However, septic critically ill patients treated with long-term antiplatelet agent may benefit from the continuation of their treatment in the absence of bleeding risk, avoiding a rebound of platelet reactivity. The multiple facets of platelet involvement in sepsis therefore represent substantial challenges to the clinician and call for a deeper understanding of the relative importance of platelet contribution to determine their ultimate clinical significance.
Authors’ contributions
AD and JR conceived, designed and coordinated this review. SL, JV, CR, CC and AO helped to critically revise the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Funding
JV acknowledges support from a Marie Curie international outgoing fellowship within the 7th European Community Framework Programme.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- PAMPs
pathogen-associated molecular patterns
- DIC
disseminated intravascular coagulation
- EC
endothelial cell
- ICU
intensive care unit
- MOD
multi-organ dysfunction
- PMPs
platelet microparticles
- SIRS
systemic inflammatory response syndrome
- TEC
tubular epithelial cell
- AKI
acute kidney injury
- ECM
extracellular matrix
- NET
neutrophil extracellular traps
- TPO
thrombopoietin
- TREM-1
triggering receptor expressed on myeloid cells 1
Footnotes
A correction to this article is available online at https://doi.org/10.1186/s13613-018-0378-6.
Contributor Information
Antoine Dewitte, Email: antoine.dewitte@chu-bordeaux.fr.
Sébastien Lepreux, Email: sebastien.lepreux@chu-bordeaux.fr.
Julien Villeneuve, Email: julienvilleneuve81@gmail.com.
Claire Rigothier, Email: claire.rigothier@chu-bordeaux.fr.
Christian Combe, Email: christian.combe@chu-bordeaux.fr.
Alexandre Ouattara, Email: alexandre.ouattara@chu-bordeaux.fr.
Jean Ripoche, Email: jean.ripoche@u-bordeaux.fr.
References
- 1.Singer M, De Santis V, Vitale D, Jeffcoate W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364(9433):545–548. doi: 10.1016/S0140-6736(04)16815-3. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(21):2063. doi: 10.1056/NEJMc1312359. [DOI] [PubMed] [Google Scholar]
- 3.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40(4):463–475. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41(5):1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am J Respir Crit Care Med. 2016;193(3):259–272. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 6.Baughman RP, Lower EE, Flessa HC, Tollerud DJ. Thrombocytopenia in the intensive care unit. Chest. 1993;104(4):1243–1247. doi: 10.1378/chest.104.4.1243. [DOI] [PubMed] [Google Scholar]
- 7.Drews RE, Weinberger SE. Thrombocytopenic disorders in critically ill patients. Am J Respir Crit Care Med. 2000;162(2 Pt 1):347–351. doi: 10.1164/ajrccm.162.2.ncc3-00. [DOI] [PubMed] [Google Scholar]
- 8.Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, et al. Thrombocytopenia and prognosis in intensive care. Crit Care Med. 2000;28(6):1871–1876. doi: 10.1097/00003246-200006000-00031. [DOI] [PubMed] [Google Scholar]
- 9.Strauss R, Wehler M, Mehler K, Kreutzer D, Koebnick C, Hahn EG. Thrombocytopenia in patients in the medical intensive care unit: bleeding prevalence, transfusion requirements, and outcome. Crit Care Med. 2002;30(8):1765–1771. doi: 10.1097/00003246-200208000-00015. [DOI] [PubMed] [Google Scholar]
- 10.Smith-Erichsen N. Serial determinations of platelets, leucocytes and coagulation parameters in surgical septicemia. Scand J Clin Lab Invest Suppl. 1985;178:7–14. [PubMed] [Google Scholar]
- 11.Akca S, Haji-Michael P, de Mendonca A, Suter P, Levi M, Vincent JL. Time course of platelet counts in critically ill patients. Crit Care Med. 2002;30(4):753–756. doi: 10.1097/00003246-200204000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Crowther MA, Cook DJ, Meade MO, Griffith LE, Guyatt GH, Arnold DM, et al. Thrombocytopenia in medical-surgical critically ill patients: prevalence, incidence, and risk factors. J Crit Care. 2005;20(4):348–353. doi: 10.1016/j.jcrc.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Moreau D, Timsit JF, Vesin A, Garrouste-Orgeas M, de Lassence A, Zahar JR, et al. Platelet count decline: an early prognostic marker in critically ill patients with prolonged ICU stays. Chest. 2007;131(6):1735–1741. doi: 10.1378/chest.06-2233. [DOI] [PubMed] [Google Scholar]
- 14.Hui P, Cook DJ, Lim W, Fraser GA, Arnold DM. The frequency and clinical significance of thrombocytopenia complicating critical illness: a systematic review. Chest. 2011;139(2):271–278. doi: 10.1378/chest.10-2243. [DOI] [PubMed] [Google Scholar]
- 15.Venkata C, Kashyap R, Farmer JC, Afessa B. Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. J Intensive Care. 2013;1(1):9. doi: 10.1186/2052-0492-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semple JW, Freedman J. Platelets and innate immunity. Cell Mol Life Sci. 2010;67(4):499–511. doi: 10.1007/s00018-009-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 18.Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol. 2012;34(1):5–30. doi: 10.1007/s00281-011-0286-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost. 2014;12(11):1764–1775. doi: 10.1111/jth.12730. [DOI] [PubMed] [Google Scholar]
- 20.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123(18):2759–2767. doi: 10.1182/blood-2013-11-462432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu XR, Zhang D, Oswald BE, Carrim N, Wang X, Hou Y, et al. Platelets are versatile cells: new discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci. 2016;53(6):409–430. doi: 10.1080/10408363.2016.1200008. [DOI] [PubMed] [Google Scholar]
- 22.Dewitte A, Tanga A, Villeneuve J, Lepreux S, Ouattara A, Desmouliere A, et al. New frontiers for platelet CD154. Exp Hematol Oncol. 2015;4:6. doi: 10.1186/s40164-015-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114(3):449–458. doi: 10.1160/TH14-12-1067. [DOI] [PubMed] [Google Scholar]
- 24.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3) JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Investig. 2003;112(4):460–467. doi: 10.1172/JCI200319523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care. 2010;14(1):R15. doi: 10.1186/cc8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93(3):329–342. doi: 10.1189/jlb.0912437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parlato M, Cavaillon JM. Host response biomarkers in the diagnosis of sepsis: a general overview. Methods Mol Biol. 2015;1237:149–211. doi: 10.1007/978-1-4939-1776-1_15. [DOI] [PubMed] [Google Scholar]
- 29.Claushuis TA, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PM, Hoogendijk AJ, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. 2016;127(24):3062–3072. doi: 10.1182/blood-2015-11-680744. [DOI] [PubMed] [Google Scholar]
- 30.Hatherill M, Tibby SM, Turner C, Ratnavel N, Murdoch IA. Procalcitonin and cytokine levels: relationship to organ failure and mortality in pediatric septic shock. Crit Care Med. 2000;28(7):2591–2594. doi: 10.1097/00003246-200007000-00068. [DOI] [PubMed] [Google Scholar]
- 31.Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29(7 Suppl):S99–S106. doi: 10.1097/00003246-200107001-00032. [DOI] [PubMed] [Google Scholar]
- 32.Mikacenic C, Hahn WO, Price BL, Harju-Baker S, Katz R, Kain KC, et al. Biomarkers of endothelial activation are associated with poor outcome in critical illness. PLoS ONE. 2015;10(10):e0141251. doi: 10.1371/journal.pone.0141251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5(1):36–44. doi: 10.4161/viru.25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta. 2017;1863(10 Pt B):2564–2573. doi: 10.1016/j.bbadis.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208(13):2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munford RS, Pugin J. Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med. 2001;163(2):316–321. doi: 10.1164/ajrccm.163.2.2007102. [DOI] [PubMed] [Google Scholar]
- 39.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 40.Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. 2008;29(4):617–625. doi: 10.1016/j.ccm.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, et al. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev. 2013;93(3):1247–1288. doi: 10.1152/physrev.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274(1):330–353. doi: 10.1111/imr.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Machlus KR, Italiano JE., Jr The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. 2013;201(6):785–796. doi: 10.1083/jcb.201304054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hitchcock IS, Kaushansky K. Thrombopoietin from beginning to end. Br J Haematol. 2014;165(2):259–268. doi: 10.1111/bjh.12772. [DOI] [PubMed] [Google Scholar]
- 46.Josefsson EC, Dowling MR, Lebois M, Kile BT. The regulation of platelet life span. In: Michelson AD, editor. Platelets. Cambridge: Academic Press; 2013. pp. 51–66. [Google Scholar]
- 47.Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Regulating billions of blood platelets: glycans and beyond. Blood. 2015;126(16):1877–1884. doi: 10.1182/blood-2015-01-569129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rendu F, Brohard-Bohn B. The platelet release reaction: granules’ constituents, secretion and functions. Platelets. 2001;12(5):261–273. doi: 10.1080/09537100120068170. [DOI] [PubMed] [Google Scholar]
- 49.Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. 2004;103(6):2096–2104. doi: 10.1182/blood-2003-08-2804. [DOI] [PubMed] [Google Scholar]
- 50.Nurden AT, Nurden P, Sanchez M, Andia I, Anitua E. Platelets and wound healing. Front Biosci. 2008;13:3532–3548. doi: 10.2741/2947. [DOI] [PubMed] [Google Scholar]
- 51.Nieuwland R, Sturk A. Platelet-derived microparticles. In: Michelson AD, editor. Platelets. Cambridge: Academic Press; 2013. pp. 403–413. [Google Scholar]
- 52.Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets. 2017;28(3):214–221. doi: 10.1080/09537104.2016.1265924. [DOI] [PubMed] [Google Scholar]
- 53.Morrell CN, Maggirwar SB. Recently recognized platelet agonists. Curr Opin Hematol. 2011;18(5):309–314. doi: 10.1097/MOH.0b013e3283497dfb. [DOI] [PubMed] [Google Scholar]
- 54.Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22(9):1381–1389. doi: 10.1161/01.ATV.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 55.Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost. 2011;105(Suppl 1):S13–S33. doi: 10.1160/THS10-11-0720. [DOI] [PubMed] [Google Scholar]
- 56.Garraud O, Hamzeh-Cognasse H, Pozzetto B, Cavaillon JM, Cognasse F. Bench-to-bedside review: platelets and active immune functions-new clues for immunopathology? Crit Care. 2013;17(4):236. doi: 10.1186/cc12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nurden AT. The biology of the platelet with special reference to inflammation, wound healing and immunity. Front Biosci (Landmark Ed). 2018;01(23):726–751. doi: 10.2741/4613. [DOI] [PubMed] [Google Scholar]
- 58.Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol. 2015;194(12):5579–5587. doi: 10.4049/jimmunol.1500259. [DOI] [PubMed] [Google Scholar]
- 59.Manne BK, Xiang SC, Rondina MT. Platelet secretion in inflammatory and infectious diseases. Platelets. 2017;28(2):155–164. doi: 10.1080/09537104.2016.1240766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen J, Lopez JA. Interactions of platelets with subendothelium and endothelium. Microcirculation. 2005;12(3):235–246. doi: 10.1080/10739680590925484. [DOI] [PubMed] [Google Scholar]
- 61.Siegel-Axel DI, Gawaz M. Platelets and endothelial cells. Semin Thromb Hemost. 2007;33(2):128–135. doi: 10.1055/s-2007-969025. [DOI] [PubMed] [Google Scholar]
- 62.Etulain J, Schattner M. Glycobiology of platelet-endothelial cell interactions. Glycobiology. 2014;24(12):1252–1259. doi: 10.1093/glycob/cwu056. [DOI] [PubMed] [Google Scholar]
- 63.Kolarova H, Ambruzova B, Svihalkova Sindlerova L, Klinke A, Kubala L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediat Inflamm. 2014;2014:694312. doi: 10.1155/2014/694312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt EP, Kuebler WM, Lee WL, Downey GP. Adhesion molecules: master controllers of the circulatory system. Compr Physiol. 2016;6(2):945–973. doi: 10.1002/cphy.c150020. [DOI] [PubMed] [Google Scholar]
- 65.Page C, Pitchford S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int Immunopharmacol. 2013;17(4):1176–1184. doi: 10.1016/j.intimp.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 66.May AE, Seizer P, Gawaz M. Platelets: inflammatory firebugs of vascular walls. Arterioscler Thromb Vasc Biol. 2008;28(3):s5–s10. doi: 10.1161/ATVBAHA.107.158915. [DOI] [PubMed] [Google Scholar]
- 67.Lowenberg EC, Meijers JC, Levi M. Platelet-vessel wall interaction in health and disease. Neth J Med. 2010;68(6):242–251. [PubMed] [Google Scholar]
- 68.Stokes KY, Granger DN. Platelets: a critical link between inflammation and microvascular dysfunction. J Physiol. 2012;590(Pt 5):1023–1034. doi: 10.1113/jphysiol.2011.225417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. 2013;112(11):1506–1519. doi: 10.1161/CIRCRESAHA.113.300512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets. 2015;26(6):507–520. doi: 10.3109/09537104.2015.1064881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Middleton EA, Weyrich AS, Zimmerman GA. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol Rev. 2016;96(4):1211–1259. doi: 10.1152/physrev.00038.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234–1238. doi: 10.1126/science.1256478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pornbacher M, Lauber K, Krombach F, et al. Platelets guide leukocytes to their sites of extravasation. PLoS Biol. 2016;14(5):e1002459. doi: 10.1371/journal.pbio.1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peters MJ, Dixon G, Kotowicz KT, Hatch DJ, Heyderman RS, Klein NJ. Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br J Haematol. 1999;106(2):391–399. doi: 10.1046/j.1365-2141.1999.01553.x. [DOI] [PubMed] [Google Scholar]
- 75.Haselmayer P, Grosse-Hovest L, von Landenberg P, Schild H, Radsak MP. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood. 2007;110(3):1029–1035. doi: 10.1182/blood-2007-01-069195. [DOI] [PubMed] [Google Scholar]
- 76.Gros A, Ollivier V, Ho-Tin-Noe B. Platelets in inflammation: regulation of leukocyte activities and vascular repair. Front Immunol. 2014;5:678. doi: 10.3389/fimmu.2014.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet interaction with innate immune cells. Transfus Med Hemother. 2016;43(2):78–88. doi: 10.1159/000444807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gawaz M, Fateh-Moghadam S, Pilz G, Gurland HJ, Werdan K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Investig. 1995;25(11):843–851. doi: 10.1111/j.1365-2362.1995.tb01694.x. [DOI] [PubMed] [Google Scholar]
- 79.Russwurm S, Vickers J, Meier-Hellmann A, Spangenberg P, Bredle D, Reinhart K, et al. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock. 2002;17(4):263–268. doi: 10.1097/00024382-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 80.Stohlawetz P, Folman CC, von dem Borne AE, Pernerstorfer T, Eichler HG, Panzer S, et al. Effects of endotoxemia on thrombopoiesis in men. Thromb Haemost. 1999;81(4):613–617. [PubMed] [Google Scholar]
- 81.Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001;104(13):1533–1537. doi: 10.1161/hc3801.095588. [DOI] [PubMed] [Google Scholar]
- 82.Ioannou A, Kannan L, Tsokos GC. Platelets, complement and tissue inflammation. Autoimmunity. 2013;46(1):1–5. doi: 10.3109/08916934.2012.722144. [DOI] [PubMed] [Google Scholar]
- 83.Stocker TJ, Ishikawa-Ankerhold H, Massberg S, Schulz C. Small but mighty: platelets as central effectors of host defense. Thromb Haemost. 2017;117(4):651–661. doi: 10.1160/TH16-12-0921. [DOI] [PubMed] [Google Scholar]
- 84.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45. doi: 10.1038/nri3345. [DOI] [PubMed] [Google Scholar]
- 85.Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139(6):1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131(4):417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 87.Esmon CT. Coagulation inhibitors in inflammation. Biochem Soc Trans. 2005;33(Pt 2):401–405. doi: 10.1042/BST0330401. [DOI] [PubMed] [Google Scholar]
- 88.Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. 2010;2(3):e2010024. doi: 10.4084/mjhid.2010.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. 2017;149:38–44. doi: 10.1016/j.thromres.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 90.Simmons J, Pittet JF. The coagulopathy of acute sepsis. Curr Opin Anaesthesiol. 2015;28(2):227–236. doi: 10.1097/ACO.0000000000000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davis RP, Miller-Dorey S, Jenne CN. Platelets and coagulation in infection. Clin Transl Immunol. 2016;5(7):e89. doi: 10.1038/cti.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma AC, Kubes P. Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J Thromb Haemost. 2008;6(3):415–420. doi: 10.1111/j.1538-7836.2007.02865.x. [DOI] [PubMed] [Google Scholar]
- 93.Ghasemzadeh M, Hosseini E. Platelet-leukocyte crosstalk: linking proinflammatory responses to procoagulant state. Thromb Res. 2013;131(3):191–197. doi: 10.1016/j.thromres.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 94.Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. 2016;7:271. doi: 10.3389/fimmu.2016.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Swystun LL, Liaw PC. The role of leukocytes in thrombosis. Blood. 2016;128(6):753–762. doi: 10.1182/blood-2016-05-718114. [DOI] [PubMed] [Google Scholar]
- 96.McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129(10):1357–1367. doi: 10.1182/blood-2016-09-741298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nieuwland R, Berckmans RJ, McGregor S, Boing AN, Romijn FP, Westendorp RG, et al. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95(3):930–935. [PubMed] [Google Scholar]
- 98.George FD. Microparticles in vascular diseases. Thromb Res. 2008;122(Suppl 1):S55–S59. doi: 10.1016/S0049-3848(08)70020-3. [DOI] [PubMed] [Google Scholar]
- 99.Italiano JE, Jr, Mairuhu AT, Flaumenhaft R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol. 2010;17(6):578–584. doi: 10.1097/MOH.0b013e32833e77ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reid VL, Webster NR. Role of microparticles in sepsis. Br J Anaesth. 2012;109(4):503–513. doi: 10.1093/bja/aes321. [DOI] [PubMed] [Google Scholar]
- 101.Tokes-Fuzesi M, Woth G, Ernyey B, Vermes I, Muhl D, Bogar L, et al. Microparticles and acute renal dysfunction in septic patients. J Crit Care. 2013;28(2):141–147. doi: 10.1016/j.jcrc.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 102.Souza AC, Yuen PS, Star RA. Microparticles: markers and mediators of sepsis-induced microvascular dysfunction, immunosuppression, and AKI. Kidney Int. 2015;87(6):1100–1108. doi: 10.1038/ki.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ripoche J. Blood platelets and inflammation: their relationship with liver and digestive diseases. Clin Res Hepatol Gastroenterol. 2011;35(5):353–357. doi: 10.1016/j.clinre.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 104.Gawaz M, Vogel S. Platelets in tissue repair: control of apoptosis and interactions with regenerative cells. Blood. 2013;122(15):2550–2554. doi: 10.1182/blood-2013-05-468694. [DOI] [PubMed] [Google Scholar]
- 105.Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. 2015;29(3):153–162. doi: 10.1016/j.blre.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359(12):1261–1270. doi: 10.1056/NEJMra0800887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ho-Tin-Noe B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. 2011;9(Suppl 1):56–65. doi: 10.1111/j.1538-7836.2011.04317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weyrich AS, Zimmerman GA. Platelets in lung biology. Annu Rev Physiol. 2013;75:569–591. doi: 10.1146/annurev-physiol-030212-183752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kitchens CS, Weiss L. Ultrastructural changes of endothelium associated with thrombocytopenia. Blood. 1975;46(4):567–578. [PubMed] [Google Scholar]
- 110.Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K, Deckmyn H. Platelets at work in primary hemostasis. Blood Rev. 2011;25(4):155–167. doi: 10.1016/j.blre.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 111.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327–358. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 112.Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, Zhao BQ, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111(10):4958–4964. doi: 10.1182/blood-2007-11-123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mazzucco L, Borzini P, Gope R. Platelet-derived factors involved in tissue repair-from signal to function. Transfus Med Rev. 2010;24(3):218–234. doi: 10.1016/j.tmrv.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 114.Rafii S, Cao Z, Lis R, Siempos II, Chavez D, Shido K, et al. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol. 2015;17(2):123–136. doi: 10.1038/ncb3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morel O, Toti F, Morel N, Freyssinet JM. Microparticles in endothelial cell and vascular homeostasis: are they really noxious? Haematologica. 2009;94(3):313–317. doi: 10.3324/haematol.2008.003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. 2014;12(6):426–437. doi: 10.1038/nrmicro3269. [DOI] [PubMed] [Google Scholar]
- 117.Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost. 2011;9(6):1097–1107. doi: 10.1111/j.1538-7836.2011.04264.x. [DOI] [PubMed] [Google Scholar]
- 118.Kerrigan SW. The expanding field of platelet-bacterial interconnections. Platelets. 2015;26(4):293–301. doi: 10.3109/09537104.2014.997690. [DOI] [PubMed] [Google Scholar]
- 119.de Stoppelaar SF, Claushuis TA, Schaap MC, Hou B, van der Poll T, Nieuwland R, et al. Toll-like receptor signalling is not involved in platelet response to streptococcus pneumoniae in vitro or in vivo. PLoS ONE. 2016;11(6):e0156977. doi: 10.1371/journal.pone.0156977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections—complex interactions with bacteria. Front Immunol. 2015;6:82. doi: 10.3389/fimmu.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol. 2006;4(6):445–457. doi: 10.1038/nrmicro1425. [DOI] [PubMed] [Google Scholar]
- 122.Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Investig. 2012;122(8):2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952–1961. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.de Stoppelaar SF, van ‘t Veer C, van der Poll T. The role of platelets in sepsis. Thromb Haemost. 2014;112(4):666–677. doi: 10.1160/TH14-02-0126. [DOI] [PubMed] [Google Scholar]
- 125.Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive immunity. Cell Immunol. 2005;238(1):1–9. doi: 10.1016/j.cellimm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 126.Middleton E, Rondina MT. Platelets in infectious disease. Hematol Am Soc Hematol Educ Program. 2016;2016(1):256–261. doi: 10.1182/asheducation-2016.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dankert J, van der Werff J, Zaat SA, Joldersma W, Klein D, Hess J. Involvement of bactericidal factors from thrombin-stimulated platelets in clearance of adherent viridans streptococci in experimental infective endocarditis. Infect Immun. 1995;63(2):663–671. doi: 10.1128/iai.63.2.663-671.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McMorran BJ, Marshall VM, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, et al. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science. 2009;323(5915):797–800. doi: 10.1126/science.1166296. [DOI] [PubMed] [Google Scholar]
- 129.de Stoppelaar SF, van ‘t Veer C, Claushuis TA, Albersen BJ, Roelofs JJ, van der Poll T. Thrombocytopenia impairs host defense in gram-negative pneumonia-derived sepsis in mice. Blood. 2014;124(25):3781–3790. doi: 10.1182/blood-2014-05-573915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kahn F, Hurley S, Shannon O. Platelets promote bacterial dissemination in a mouse model of streptococcal sepsis. Microbes Infect. 2013;15(10–11):669–676. doi: 10.1016/j.micinf.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 131.Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007;81(1):137–143. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]
- 132.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Investig. 2009;119(10):2868–2878. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ward PA. New approaches to the study of sepsis. EMBO Mol Med. 2012;4(12):1234–1243. doi: 10.1002/emmm.201201375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fink MP. Animal models of sepsis. Virulence. 2014;5(1):143–153. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vincent JL. Microvascular endothelial dysfunction: a renewed appreciation of sepsis pathophysiology. Crit Care. 2001;5(2):S1–S5. doi: 10.1186/cc1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 137.Bateman RM, Sharpe MD, Ellis CG. Bench-to-bedside review: microvascular dysfunction in sepsis–hemodynamics, oxygen transport, and nitric oxide. Crit Care. 2003;7(5):359–373. doi: 10.1186/cc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60(1):49–57. doi: 10.1016/S0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 139.Warkentin TE, Aird WC, Rand JH. Platelet-endothelial interactions: sepsis, HIT, and antiphospholipid syndrome. Hematol Am Soc Hematol Educ Program. 2003;2003:497–519. doi: 10.1182/asheducation-2003.1.497. [DOI] [PubMed] [Google Scholar]
- 140.Matsuda N, Hattori Y. Vascular biology in sepsis: pathophysiological and therapeutic significance of vascular dysfunction. J Smooth Muscle Res. 2007;43(4):117–137. doi: 10.1540/jsmr.43.117. [DOI] [PubMed] [Google Scholar]
- 141.Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83(3):536–545. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- 142.Gando S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med. 2010;38(2 Suppl):S35–S42. doi: 10.1097/CCM.0b013e3181c9e31d. [DOI] [PubMed] [Google Scholar]
- 143.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med. 2011;3(88):88ps25. doi: 10.1126/scitranslmed.3002011. [DOI] [PubMed] [Google Scholar]
- 144.Tyml K. Critical role for oxidative stress, platelets, and coagulation in capillary blood flow impairment in sepsis. Microcirculation. 2011;18(2):152–162. doi: 10.1111/j.1549-8719.2010.00080.x. [DOI] [PubMed] [Google Scholar]
- 145.Boisrame-Helms J, Kremer H, Schini-Kerth V, Meziani F. Endothelial dysfunction in sepsis. Curr Vasc Pharmacol. 2013;11(2):150–160. [PubMed] [Google Scholar]
- 146.De Backer D, Orbegozo Cortes D, Donadello K, Vincent JL. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence. 2014;5(1):73–79. doi: 10.4161/viru.26482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Opal SM, van der Poll T. Endothelial barrier dysfunction in septic shock. J Int Med. 2015;277(3):277–293. doi: 10.1111/joim.12331. [DOI] [PubMed] [Google Scholar]
- 148.Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, et al. The endothelium in sepsis. Shock. 2016;45(3):259–270. doi: 10.1097/SHK.0000000000000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Xing K, Murthy S, Liles WC, Singh JM. Clinical utility of biomarkers of endothelial activation in sepsis–a systematic review. Crit Care. 2012;16(1):R7. doi: 10.1186/cc11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ait-Oufella H, Maury E, Lehoux S, Guidet B, Offenstadt G. The endothelium: physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010;36(8):1286–1298. doi: 10.1007/s00134-010-1893-6. [DOI] [PubMed] [Google Scholar]
- 151.Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369(18):1726–1734. doi: 10.1056/NEJMra1208943. [DOI] [PubMed] [Google Scholar]
- 152.Gawaz M, Dickfeld T, Bogner C, Fateh-Moghadam S, Neumann FJ. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. 1997;23(4):379–385. doi: 10.1007/s001340050344. [DOI] [PubMed] [Google Scholar]
- 153.Hurley SM, Lutay N, Holmqvist B, Shannon O. The dynamics of platelet activation during the progression of streptococcal sepsis. PLoS ONE. 2016;11(9):e0163531. doi: 10.1371/journal.pone.0163531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Levi M. Platelets in critical illness. Semin Thromb Hemost. 2016;42(3):252–257. doi: 10.1055/s-0035-1570080. [DOI] [PubMed] [Google Scholar]
- 155.Heffner JE, Sahn SA, Repine JE. The role of platelets in the adult respiratory distress syndrome. Culprits or bystanders? Am Rev Respir Dis. 1987;135(2):482–492. doi: 10.1164/arrd.1987.135.2.482. [DOI] [PubMed] [Google Scholar]
- 156.Bozza FA, Shah AM, Weyrich AS, Zimmerman GA. Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am J Respir Cell Mol Biol. 2009;40(2):123–134. doi: 10.1165/rcmb.2008-0241TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reilly JP, Christie JD. Linking genetics to ARDS pathogenesis: the role of the platelet. Chest. 2015;147(3):585–586. doi: 10.1378/chest.14-2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yadav H, Kor DJ. Platelets in the pathogenesis of acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2015;28:ajplung 00266. doi: 10.1152/ajplung.00266.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 160.Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol. 2013;75:593–615. doi: 10.1146/annurev-physiol-030212-183756. [DOI] [PubMed] [Google Scholar]
- 161.Kiefmann R, Heckel K, Schenkat S, Dorger M, Wesierska-Gadek J, Goetz AE. Platelet-endothelial cell interaction in pulmonary micro-circulation: the role of PARS. Thromb Haemost. 2004;91(4):761–770. doi: 10.1160/TH03-11-0685. [DOI] [PubMed] [Google Scholar]
- 162.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Investig. 2006;116(12):3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Investig. 2009;119(11):3450–3461. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ortiz-Munoz G, Mallavia B, Bins A, Headley M, Krummel MF, Looney MR. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood. 2014;124(17):2625–2634. doi: 10.1182/blood-2014-03-562876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Katz JN, Kolappa KP, Becker RC. Beyond thrombosis: the versatile platelet in critical illness. Chest. 2011;139(3):658–668. doi: 10.1378/chest.10-1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, et al. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLoS ONE. 2012;7(7):e41549. doi: 10.1371/journal.pone.0041549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Brown KA, Brain SD, Pearson JD, Edgeworth JD, Lewis SM, Treacher DF. Neutrophils in development of multiple organ failure in sepsis. Lancet. 2006;368(9530):157–169. doi: 10.1016/S0140-6736(06)69005-3. [DOI] [PubMed] [Google Scholar]
- 168.Zarbock A, Ley K. The role of platelets in acute lung injury (ALI) Front Biosci (Landmark Ed) 2009;01(14):150–158. doi: 10.2741/3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. 2003;31(4 Suppl):S213–S220. doi: 10.1097/01.CCM.0000057846.21303.AB. [DOI] [PubMed] [Google Scholar]
- 170.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Investig. 2012;122(7):2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.McVey M, Tabuchi A, Kuebler WM. Microparticles and acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;303(5):L364–L381. doi: 10.1152/ajplung.00354.2011. [DOI] [PubMed] [Google Scholar]
- 172.Asaduzzaman M, Lavasani S, Rahman M, Zhang S, Braun OO, Jeppsson B, et al. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care Med. 2009;37(4):1389–1396. doi: 10.1097/CCM.0b013e31819ceb71. [DOI] [PubMed] [Google Scholar]
- 173.Cloutier N, Pare A, Farndale RW, Schumacher HR, Nigrovic PA, Lacroix S, et al. Platelets can enhance vascular permeability. Blood. 2012;120(6):1334–1343. doi: 10.1182/blood-2012-02-413047. [DOI] [PubMed] [Google Scholar]
- 174.Reddy AJ, Kleeberger SR. Genetic polymorphisms associated with acute lung injury. Pharmacogenomics. 2009;10(9):1527–1539. doi: 10.2217/pgs.09.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wei Y, Wang Z, Su L, Chen F, Tejera P, Bajwa EK, et al. Platelet count mediates the contribution of a genetic variant in LRRC16A to ARDS risk. Chest. 2015;147(3):607–617. doi: 10.1378/chest.14-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.De Backer D, Creteur J, Preiser JC, Dubois MJ, Vincent JL. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002;166(1):98–104. doi: 10.1164/rccm.200109-016OC. [DOI] [PubMed] [Google Scholar]
- 177.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351(2):159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 178.Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow in experimental septic acute renal failure. Kidney Int. 2006;69(11):1996–2002. doi: 10.1038/sj.ki.5000440. [DOI] [PubMed] [Google Scholar]
- 179.Shum HP, Yan WW, Chan TM. Recent knowledge on the pathophysiology of septic acute kidney injury: a narrative review. J Crit Care. 2016;31(1):82–89. doi: 10.1016/j.jcrc.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 180.Post EH, Kellum JA, Bellomo R, Vincent JL. Renal perfusion in sepsis: from macro- to microcirculation. Kidney Int. 2017;91(1):45–60. doi: 10.1016/j.kint.2016.07.032. [DOI] [PubMed] [Google Scholar]
- 181.Wan L, Bagshaw SM, Langenberg C, Saotome T, May C, Bellomo R. Pathophysiology of septic acute kidney injury: what do we really know? Crit Care Med. 2008;36(4 Suppl):S198–S203. doi: 10.1097/CCM.0b013e318168ccd5. [DOI] [PubMed] [Google Scholar]
- 182.Lerolle N, Nochy D, Guerot E, Bruneval P, Fagon JY, Diehl JL, et al. Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med. 2010;36(3):471–478. doi: 10.1007/s00134-009-1723-x. [DOI] [PubMed] [Google Scholar]
- 183.Jacobs R, Honore PM, Joannes-Boyau O, Boer W, De Regt J, De Waele E, et al. Septic acute kidney injury: the culprit is inflammatory apoptosis rather than ischemic necrosis. Blood Purif. 2011;32(4):262–265. doi: 10.1159/000330244. [DOI] [PubMed] [Google Scholar]
- 184.Takasu O, Gaut JP, Watanabe E, To K, Fagley RE, Sato B, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187(5):509–517. doi: 10.1164/rccm.201211-1983OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S. Renal blood flow in sepsis. Crit Care. 2005;9(4):R363–R374. doi: 10.1186/cc3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Prowle JR, Ishikawa K, May CN, Bellomo R. Renal blood flow during acute renal failure in man. Blood Purif. 2009;28(3):216–225. doi: 10.1159/000230813. [DOI] [PubMed] [Google Scholar]
- 187.Chvojka J, Sykora R, Karvunidis T, Radej J, Krouzecky A, Novak I, et al. New developments in septic acute kidney injury. Physiol Res. 2010;59(6):859–869. doi: 10.33549/physiolres.931936. [DOI] [PubMed] [Google Scholar]
- 188.Murugan R, Karajala-Subramanyam V, Lee M, Yende S, Kong L, Carter M, et al. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010;77(6):527–535. doi: 10.1038/ki.2009.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kellum JA. Impaired renal blood flow and the ‘spicy food’ hypothesis of acute kidney injury. Crit Care Med. 2011;39(4):901–903. doi: 10.1097/CCM.0b013e31820f70bb. [DOI] [PubMed] [Google Scholar]
- 190.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22(6):999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 191.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2(2):1303–1353. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gomez H, Ince C, De Backer D, Pickkers P, Payen D, Hotchkiss J, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3–11. doi: 10.1097/SHK.0000000000000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Umbro I, Gentile G, Tinti F, Muiesan P, Mitterhofer AP. Recent advances in pathophysiology and biomarkers of sepsis-induced acute kidney injury. J Infect. 2015;72:131–142. doi: 10.1016/j.jinf.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 194.Gomez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016;22(6):546–553. doi: 10.1097/MCC.0000000000000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bellomo R, Kellum JA, Ronco C, Wald R, Martensson J, Maiden M, et al. Acute kidney injury in sepsis. Intensive Care Med. 2017;43(6):816–828. doi: 10.1007/s00134-017-4755-7. [DOI] [PubMed] [Google Scholar]
- 196.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Investig. 2011;121(11):4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zarbock A, Gomez H, Kellum JA. Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr Opin Crit Care. 2014;20(6):588–595. doi: 10.1097/MCC.0000000000000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Glodowski SD, Wagener G. New insights into the mechanisms of acute kidney injury in the intensive care unit. J Clin Anesth. 2015;27(2):175–180. doi: 10.1016/j.jclinane.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 199.Schwarzenberger C, Sradnick J, Lerea KM, Goligorsky MS, Nieswandt B, Hugo CP, et al. Platelets are relevant mediators of renal injury induced by primary endothelial lesions. Am J Physiol Renal Physiol. 2015;308(11):F1238–F1246. doi: 10.1152/ajprenal.00535.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wiesinger A, Peters W, Chappell D, Kentrup D, Reuter S, Pavenstadt H, et al. Nanomechanics of the endothelial glycocalyx in experimental sepsis. PLoS ONE. 2013;8(11):e80905. doi: 10.1371/journal.pone.0080905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol. 2015;80(3):389–402. doi: 10.1111/bcp.12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care. 2015;28(19):26. doi: 10.1186/s13054-015-0741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Martin L, Koczera P, Zechendorf E, Schuerholz T. The endothelial glycocalyx: new diagnostic and therapeutic approaches in sepsis. Biomed Res Int. 2016;2016:3758278. doi: 10.1155/2016/3758278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;14(67):293–307. doi: 10.1146/annurev-med-050214-013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Doi K, Rabb H. Impact of acute kidney injury on distant organ function: recent findings and potential therapeutic targets. Kidney Int. 2016;89(3):555–564. doi: 10.1016/j.kint.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 206.Lapchak PH, Kannan L, Ioannou A, Rani P, Karian P, Dalle Lucca JJ, et al. Platelets orchestrate remote tissue damage after mesenteric ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2012;302(8):G888–G897. doi: 10.1152/ajpgi.00499.2011. [DOI] [PubMed] [Google Scholar]
- 207.Singbartl K, Bishop JV, Wen X, Murugan R, Chandra S, Filippi MD, et al. Differential effects of kidney-lung cross-talk during acute kidney injury and bacterial pneumonia. Kidney Int. 2011;80(6):633–644. doi: 10.1038/ki.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lapchak PH, Ioannou A, Kannan L, Rani P, Dalle Lucca JJ, Tsokos GC. Platelet-associated CD40/CD154 mediates remote tissue damage after mesenteric ischemia/reperfusion injury. PLoS ONE. 2012;7(2):e32260. doi: 10.1371/journal.pone.0032260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lapchak PH, Ioannou A, Rani P, Lieberman LA, Yoshiya K, Kannan L, et al. The role of platelet factor 4 in local and remote tissue damage in a mouse model of mesenteric ischemia/reperfusion injury. PLoS ONE. 2012;7(7):e39934. doi: 10.1371/journal.pone.0039934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391(6667):591–594. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 211.Frossard JL, Kwak B, Chanson M, Morel P, Hadengue A, Mach F. Cd40 ligand-deficient mice are protected against cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology. 2001;121(1):184–194. doi: 10.1053/gast.2001.25483. [DOI] [PubMed] [Google Scholar]
- 212.Hashimoto N, Kawabe T, Imaizumi K, Hara T, Okamoto M, Kojima K, et al. CD40 plays a crucial role in lipopolysaccharide-induced acute lung injury. Am J Respir Cell Mol Biol. 2004;30(6):808–815. doi: 10.1165/rcmb.2003-0197OC. [DOI] [PubMed] [Google Scholar]
- 213.Rahman M, Zhang S, Chew M, Ersson A, Jeppsson B, Thorlacius H. Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann Surg. 2009;250(5):783–790. doi: 10.1097/SLA.0b013e3181bd95b7. [DOI] [PubMed] [Google Scholar]
- 214.Stephan F, Hollande J, Richard O, Cheffi A, Maier-Redelsperger M, Flahault A. Thrombocytopenia in a surgical ICU. Chest. 1999;115(5):1363–1370. doi: 10.1378/chest.115.5.1363. [DOI] [PubMed] [Google Scholar]
- 215.Mavrommatis AC, Theodoridis T, Orfanidou A, Roussos C, Christopoulou-Kokkinou V, Zakynthinos S. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit Care Med. 2000;28(2):451–457. doi: 10.1097/00003246-200002000-00027. [DOI] [PubMed] [Google Scholar]
- 216.Nijsten MW, ten Duis HJ, Zijlstra JG, Porte RJ, Zwaveling JH, Paling JC, et al. Blunted rise in platelet count in critically ill patients is associated with worse outcome. Crit Care Med. 2000;28(12):3843–3846. doi: 10.1097/00003246-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 217.Sharma B, Sharma M, Majumder M, Steier W, Sangal A, Kalawar M. Thrombocytopenia in septic shock patients–a prospective observational study of incidence, risk factors and correlation with clinical outcome. Anaesth Intensive Care. 2007;35(6):874–880. doi: 10.1177/0310057X0703500604. [DOI] [PubMed] [Google Scholar]
- 218.Thiolliere F, Serre-Sapin AF, Reignier J, Benedit M, Constantin JM, Lebert C, et al. Epidemiology and outcome of thrombocytopenic patients in the intensive care unit: results of a prospective multicenter study. Intensive Care Med. 2013;39(8):1460–1468. doi: 10.1007/s00134-013-2963-3. [DOI] [PubMed] [Google Scholar]
- 219.Williamson DR, Lesur O, Tetrault JP, Nault V, Pilon D. Thrombocytopenia in the critically ill: prevalence, incidence, risk factors, and clinical outcomes. Can J Anaesth. 2013;60(7):641–651. doi: 10.1007/s12630-013-9933-7. [DOI] [PubMed] [Google Scholar]
- 220.Aydemir H, Piskin N, Akduman D, Kokturk F, Aktas E. Platelet and mean platelet volume kinetics in adult patients with sepsis. Platelets. 2015;26(4):331–335. doi: 10.3109/09537104.2012.701027. [DOI] [PubMed] [Google Scholar]
- 221.Thiery-Antier N, Binquet C, Vinault S, Meziani F, Boisrame-Helms J, Quenot JP. Is thrombocytopenia an early prognostic marker in septic shock? Crit Care Med. 2016;44(4):764–772. doi: 10.1097/CCM.0000000000001520. [DOI] [PubMed] [Google Scholar]
- 222.Greinacher A, Selleng K. Thrombocytopenia in the intensive care unit patient. Hematol Am Soc Hematol Educ Program. 2010;2010:135–143. doi: 10.1182/asheducation-2010.1.135. [DOI] [PubMed] [Google Scholar]
- 223.Brun-Buisson C, Doyon F, Carlet J, Dellamonica P, Gouin F, Lepoutre A, et al. Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA. 1995;274(12):968–974. doi: 10.1001/jama.1995.03530120060042. [DOI] [PubMed] [Google Scholar]
- 224.Martin CM, Priestap F, Fisher H, Fowler RA, Heyland DK, Keenan SP, et al. A prospective, observational registry of patients with severe sepsis: the Canadian Sepsis Treatment and Response Registry. Crit Care Med. 2009;37(1):81–88. doi: 10.1097/CCM.0b013e31819285f0. [DOI] [PubMed] [Google Scholar]
- 225.Selleng S, Malowsky B, Strobel U, Wessel A, Ittermann T, Wollert HG, et al. Early-onset and persisting thrombocytopenia in post-cardiac surgery patients is rarely due to heparin-induced thrombocytopenia, even when antibody tests are positive. J Thromb Haemost. 2010;8(1):30–36. doi: 10.1111/j.1538-7836.2009.03626.x. [DOI] [PubMed] [Google Scholar]
- 226.Vandijck DM, Blot SI, De Waele JJ, Hoste EA, Vandewoude KH, Decruyenaere JM. Thrombocytopenia and outcome in critically ill patients with bloodstream infection. Heart Lung. 2010;39(1):21–26. doi: 10.1016/j.hrtlng.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 227.Lim SY, Jeon EJ, Kim HJ, Jeon K, Um SW, Koh WJ, et al. The incidence, causes, and prognostic significance of new-onset thrombocytopenia in intensive care units: a prospective cohort study in a Korean hospital. J Korean Med Sci. 2012;27(11):1418–1423. doi: 10.3346/jkms.2012.27.11.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Lieberman L, Bercovitz RS, Sholapur NS, Heddle NM, Stanworth SJ, Arnold DM. Platelet transfusions for critically ill patients with thrombocytopenia. Blood. 2014;123(8):1146–1151. doi: 10.1182/blood-2013-02-435693. [DOI] [PubMed] [Google Scholar]
- 229.Pene F, Benoit DD. Thrombocytopenia in the critically ill: considering pathophysiology rather than looking for a magic threshold. Intensive Care Med. 2013;39(9):1656–1659. doi: 10.1007/s00134-013-3022-9. [DOI] [PubMed] [Google Scholar]
- 230.Greinacher A, Selleng S. How I evaluate and treat thrombocytopenia in the intensive care unit patient. Blood. 2016;128(26):3032–3042. doi: 10.1182/blood-2016-09-693655. [DOI] [PubMed] [Google Scholar]
- 231.Ning S, Barty R, Liu Y, Heddle NM, Rochwerg B, Arnold DM. Platelet transfusion practices in the icu: data from a large transfusion registry. Chest. 2016;150(3):516–523. doi: 10.1016/j.chest.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 232.Antier N, Quenot JP, Doise JM, Noel R, Demaistre E, Devilliers H. Mechanisms and etiologies of thrombocytopenia in the intensive care unit: impact of extensive investigations. Ann Intensive Care. 2014;4:24. doi: 10.1186/s13613-014-0024-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Larkin CM, Santos-Martinez MJ, Ryan T, Radomski MW. Sepsis-associated thrombocytopenia. Thromb Res. 2016;141:11–16. doi: 10.1016/j.thromres.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 234.Pigozzi L, Aron JP, Ball J, Cecconi M. Understanding platelet dysfunction in sepsis. Intensive Care Med. 2016;42(4):583–586. doi: 10.1007/s00134-015-4012-x. [DOI] [PubMed] [Google Scholar]
- 235.De Blasi RA, Cardelli P, Costante A, Sandri M, Mercieri M, Arcioni R. Immature platelet fraction in predicting sepsis in critically ill patients. Intensive Care Med. 2013;39(4):636–643. doi: 10.1007/s00134-012-2725-7. [DOI] [PubMed] [Google Scholar]
- 236.Levi M. Platelets at a crossroad of pathogenic pathways in sepsis. J Thromb Haemost. 2004;2(12):2094–2095. doi: 10.1111/j.1538-7836.2004.01004.x. [DOI] [PubMed] [Google Scholar]
- 237.Yaguchi A, Lobo FL, Vincent JL, Pradier O. Platelet function in sepsis. J Thromb Haemost. 2004;2(12):2096–2102. doi: 10.1111/j.1538-7836.2004.01009.x. [DOI] [PubMed] [Google Scholar]
- 238.Goyette RE, Key NS, Ely EW. Hematologic changes in sepsis and their therapeutic implications. Semin Respir Crit Care Med. 2004;25(6):645–659. doi: 10.1055/s-2004-860979. [DOI] [PubMed] [Google Scholar]
- 239.Arnold DM, Lim W. A rational approach to the diagnosis and management of thrombocytopenia in the hospitalized patient. Semin Hematol. 2011;48(4):251–258. doi: 10.1053/j.seminhematol.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 240.Thiele T, Selleng K, Selleng S, Greinacher A, Bakchoul T. Thrombocytopenia in the intensive care unit-diagnostic approach and management. Semin Hematol. 2013;50(3):239–250. doi: 10.1053/j.seminhematol.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 241.Smock KJ, Perkins SL. Thrombocytopenia: an update. Int J Lab Hematol. 2014;36(3):269–278. doi: 10.1111/ijlh.12214. [DOI] [PubMed] [Google Scholar]
- 242.Thachil J, Warkentin TE. How do we approach thrombocytopenia in critically ill patients? Br J Haematol. 2017;177(1):27–38. doi: 10.1111/bjh.14482. [DOI] [PubMed] [Google Scholar]
- 243.Remick DG. Cytokine therapeutics for the treatment of sepsis: why has nothing worked? Curr Pharm Des. 2003;9(1):75–82. doi: 10.2174/1381612033392567. [DOI] [PubMed] [Google Scholar]
- 244.Thomas MR, Outteridge SN, Ajjan RA, Phoenix F, Sangha GK, Faulkner RE, et al. Platelet P2Y12 inhibitors reduce systemic inflammation and its prothrombotic effects in an experimental human model. Arterioscler Thromb Vasc Biol. 2015;35(12):2562–2570. doi: 10.1161/ATVBAHA.115.306528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Winning J, Neumann J, Kohl M, Claus RA, Reinhart K, Bauer M, et al. Antiplatelet drugs and outcome in mixed admissions to an intensive care unit. Crit Care Med. 2010;38(1):32–37. doi: 10.1097/CCM.0b013e3181b4275c. [DOI] [PubMed] [Google Scholar]
- 246.Erlich JM, Talmor DS, Cartin-Ceba R, Gajic O, Kor DJ. Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: a population-based cohort study. Chest. 2011;139(2):289–295. doi: 10.1378/chest.10-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Losche W, Boettel J, Kabisch B, Winning J, Claus RA, Bauer M. Do aspirin and other antiplatelet drugs reduce the mortality in critically ill patients? Thrombosis. 2012;2012:720254. doi: 10.1155/2012/720254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Eisen DP, Reid D, McBryde ES. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med. 2012;40(6):1761–1767. doi: 10.1097/CCM.0b013e318246b9df. [DOI] [PubMed] [Google Scholar]
- 249.Valerio-Rojas JC, Jaffer IJ, Kor DJ, Gajic O, Cartin-Ceba R. Outcomes of severe sepsis and septic shock patients on chronic antiplatelet treatment: a historical cohort study. Crit Care Res Pract. 2013;2013:782573. doi: 10.1155/2013/782573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sossdorf M, Otto GP, Boettel J, Winning J, Losche W. Benefit of low-dose aspirin and non-steroidal anti-inflammatory drugs in septic patients. Crit Care. 2013;17(1):402. doi: 10.1186/cc11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Otto GP, Sossdorf M, Boettel J, Kabisch B, Breuel H, Winning J, et al. Effects of low-dose acetylsalicylic acid and atherosclerotic vascular diseases on the outcome in patients with severe sepsis or septic shock. Platelets. 2013;24(6):480–485. doi: 10.3109/09537104.2012.724482. [DOI] [PubMed] [Google Scholar]
- 252.Akinosoglou K, Alexopoulos D. Use of antiplatelet agents in sepsis: a glimpse into the future. Thromb Res. 2014;133(2):131–138. doi: 10.1016/j.thromres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 253.Chen W, Janz DR, Bastarache JA, May AK, O’Neal HR, Jr, Bernard GR, et al. Prehospital aspirin use is associated with reduced risk of acute respiratory distress syndrome in critically ill patients: a propensity-adjusted analysis. Crit Care Med. 2015;43(4):801–807. doi: 10.1097/CCM.0000000000000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Toner P, McAuley DF, Shyamsundar M. Aspirin as a potential treatment in sepsis or acute respiratory distress syndrome. Crit Care. 2015;19:374. doi: 10.1186/s13054-015-1091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Tsai MJ, Ou SM, Shih CJ, Chao PW, Wang LF, Shih YN, et al. Association of prior antiplatelet agents with mortality in sepsis patients: a nationwide population-based cohort study. Intensive Care Med. 2015;41(5):806–813. doi: 10.1007/s00134-015-3760-y. [DOI] [PubMed] [Google Scholar]
- 256.Kor DJ, Erlich J, Gong MN, Malinchoc M, Carter RE, Gajic O, et al. Association of prehospitalization aspirin therapy and acute lung injury: results of a multicenter international observational study of at-risk patients. Crit Care Med. 2011;39(11):2393–2400. doi: 10.1097/CCM.0b013e318225757f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Gum PA, Kottke-Marchant K, Poggio ED, Gurm H, Welsh PA, Brooks L, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88(3):230–235. doi: 10.1016/S0002-9149(01)01631-9. [DOI] [PubMed] [Google Scholar]
- 258.Nguyen TA, Diodati JG, Pharand C. Resistance to clopidogrel: a review of the evidence. J Am Coll Cardiol. 2005;45(8):1157–1164. doi: 10.1016/j.jacc.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 259.Macchi L, Sorel N, Christiaens L. Aspirin resistance: definitions, mechanisms, prevalence, and clinical significance. Curr Pharm Des. 2006;12(2):251–258. doi: 10.2174/138161206775193064. [DOI] [PubMed] [Google Scholar]
- 260.Wiewel MA, de Stoppelaar SF, van Vught LA, Frencken JF, Hoogendijk AJ, Klein Klouwenberg PM, et al. Chronic antiplatelet therapy is not associated with alterations in the presentation, outcome, or host response biomarkers during sepsis: a propensity-matched analysis. Intensive Care Med. 2016;42(3):352–360. doi: 10.1007/s00134-015-4171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Muhlestein JB. Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Thromb Haemost. 2010;103(1):71–82. doi: 10.1160/TH09-03-0177. [DOI] [PubMed] [Google Scholar]
- 262.Zakynthinos SG, Papanikolaou S, Theodoridis T, Zakynthinos EG, Christopoulou-Kokkinou V, Katsaris G, et al. Sepsis severity is the major determinant of circulating thrombopoietin levels in septic patients. Crit Care Med. 2004;32(4):1004–1010. doi: 10.1097/01.CCM.0000121433.61546.A0. [DOI] [PubMed] [Google Scholar]
- 263.Lupia E, Goffi A, Bosco O, Montrucchio G. Thrombopoietin as biomarker and mediator of cardiovascular damage in critical diseases. Mediat Inflamm. 2012;2012:390892. doi: 10.1155/2012/390892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Cuccurullo A, Greco E, Lupia E, De Giuli P, Bosco O, Martin-Conte E, et al. Blockade of thrombopoietin reduces organ damage in experimental endotoxemia and polymicrobial sepsis. PLoS ONE. 2016;11(3):e0151088. doi: 10.1371/journal.pone.0151088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wu Q, Ren J, Wu X, Wang G, Gu G, Liu S, et al. Recombinant human thrombopoietin improves platelet counts and reduces platelet transfusion possibility among patients with severe sepsis and thrombocytopenia: a prospective study. J Crit Care. 2014;29(3):362–366. doi: 10.1016/j.jcrc.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 266.Van Deuren M, Neeleman C, Van ‘t Hek LG, Van der Meer JW. A normal platelet count at admission in acute meningococcal disease does not exclude a fulminant course. Intensive Care Med. 1998;24(2):157–161. doi: 10.1007/s001340050538. [DOI] [PubMed] [Google Scholar]
- 267.Agrawal S, Sachdev A, Gupta D, Chugh K. Platelet counts and outcome in the pediatric intensive care unit. Indian J Crit Care Med. 2008;12(3):102–108. doi: 10.4103/0972-5229.43678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wang L, Li H, Gu X, Wang Z, Liu S, Chen L. Effect of antiplatelet therapy on acute respiratory distress syndrome and mortality in critically ill patients: a meta-analysis. PLoS ONE. 2016;11(5):e0154754. doi: 10.1371/journal.pone.0154754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kor DJ, Carter RE, Park PK, Festic E, Banner-Goodspeed VM, Hinds R, et al. Effect of aspirin on development of ARDS in at-risk patients presenting to the emergency department: the LIPS-A randomized clinical trial. JAMA. 2016;315(22):2406–2414. doi: 10.1001/jama.2016.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Osthoff M, Sidler JA, Lakatos B, Frei R, Dangel M, Weisser M, et al. Low-dose acetylsalicylic acid treatment and impact on short-term mortality in Staphylococcus aureus bloodstream infection: a propensity score-matched cohort study. Crit Care Med. 2016;44(4):773–781. doi: 10.1097/CCM.0000000000001554. [DOI] [PubMed] [Google Scholar]
- 271.Boyle AJ, Di Gangi S, Hamid UI, Mottram LJ, McNamee L, White G, et al. Aspirin therapy in patients with acute respiratory distress syndrome (ARDS) is associated with reduced intensive care unit mortality: a prospective analysis. Crit Care. 2015;19:109. doi: 10.1186/s13054-015-0846-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.O’Neal HR, Jr, Koyama T, Koehler EA, Siew E, Curtis BR, Fremont RD, et al. Prehospital statin and aspirin use and the prevalence of severe sepsis and acute lung injury/acute respiratory distress syndrome. Crit Care Med. 2011;39(6):1343–1350. doi: 10.1097/CCM.0b013e3182120992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Storey RF, James SK, Siegbahn A, Varenhorst C, Held C, Ycas J, et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets. 2014;25(7):517–525. doi: 10.3109/09537104.2013.842965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Winning J, Reichel J, Eisenhut Y, Hamacher J, Kohl M, Deigner HP, et al. Anti-platelet drugs and outcome in severe infection: clinical impact and underlying mechanisms. Platelets. 2009;20(1):50–57. doi: 10.1080/09537100802503368. [DOI] [PubMed] [Google Scholar]
- 275.Gross AK, Dunn SP, Feola DJ, Martin CA, Charnigo R, Li Z, et al. Clopidogrel treatment and the incidence and severity of community acquired pneumonia in a cohort study and meta-analysis of antiplatelet therapy in pneumonia and critical illness. J Thromb Thrombolysis. 2013;35(2):147–154. doi: 10.1007/s11239-012-0833-4. [DOI] [PMC free article] [PubMed] [Google Scholar]