

Abstract
Key points
Chronic limb ischaemia, characterized by inflammatory mediator release and a low extracellular pH, leads to acid‐sensing ion channel (ASIC) activation and reflexively increases mean arterial pressure; endomorphin release is also increased under inflammatory conditions.
We examined the modulation of ASIC currents by endomorphins in sensory neurons from rats with freely perfused and ligated femoral arteries: peripheral artery disease (PAD) model.
Endomorphins potentiated sustained ASIC currents in both groups of dorsal root ganglion neurons, independent of mu opioid receptor stimulation or G protein activation.
Intra‐arterial administration of lactic acid (to simulate exercising muscle and evoke a pressor reflex), endomorphin‐2 and naloxone resulted in a significantly greater pressor response than lactic acid alone, while administration of APETx2 inhibited endomorphin's enhancing effect in both groups.
These results suggest a novel role for endomorphins in modulating ASIC function to effect lactic acid‐mediated reflex increase in arterial pressure in patients with PAD.
Abstract
Chronic muscle ischaemia leads to accumulation of lactic acid and other inflammatory mediators with a subsequent drop in interstitial pH. Acid‐sensing ion channels (ASICs), expressed in thin muscle afferents, sense the decrease in pH and evoke a pressor reflex known to increase mean arterial pressure. The naturally occurring endomorphins are also released by primary afferents under ischaemic conditions. We examined whether high affinity mu opioid receptor (MOR) agonists, endomorphin‐1 (E‐1) and ‐2 (E‐2), modulate ASIC currents and the lactic acid‐mediated pressor reflex. In rat dorsal root ganglion (DRG) neurons, exposure to E‐2 in acidic solutions significantly potentiated ASIC currents when compared to acidic solutions alone. The potentiation was significantly greater in DRG neurons isolated from rats whose femoral arteries were ligated for 72 h. Sustained ASIC current potentiation was also observed in neurons pretreated with pertussis toxin, an uncoupler of G proteins and MOR. The endomorphin‐mediated potentiation was a result of a leftward shift of the activation curve to higher pH values and a slight shift of the inactivation curve to lower pH values. Intra‐arterial co‐administration of lactic acid and E‐2 led to a significantly greater pressor reflex than lactic acid alone in the presence of naloxone. Finally, E‐2 effects were inhibited by pretreatment with the ASIC3 blocker APETx2 and enhanced by pretreatment with the ASIC1a blocker psalmotoxin‐1. These findings have uncovered a novel role of endomorphins by which the opioids can enhance the lactic acid‐mediated reflex increase in arterial pressure that is MOR stimulation‐independent and APETx2‐sensitive.
Keywords: acid‐sensing ion channels, opioid, muscle ischemia
Key points
Chronic limb ischaemia, characterized by inflammatory mediator release and a low extracellular pH, leads to acid‐sensing ion channel (ASIC) activation and reflexively increases mean arterial pressure; endomorphin release is also increased under inflammatory conditions.
We examined the modulation of ASIC currents by endomorphins in sensory neurons from rats with freely perfused and ligated femoral arteries: peripheral artery disease (PAD) model.
Endomorphins potentiated sustained ASIC currents in both groups of dorsal root ganglion neurons, independent of mu opioid receptor stimulation or G protein activation.
Intra‐arterial administration of lactic acid (to simulate exercising muscle and evoke a pressor reflex), endomorphin‐2 and naloxone resulted in a significantly greater pressor response than lactic acid alone, while administration of APETx2 inhibited endomorphin's enhancing effect in both groups.
These results suggest a novel role for endomorphins in modulating ASIC function to effect lactic acid‐mediated reflex increase in arterial pressure in patients with PAD.
Introduction
Intermittent claudication, a manifestation of limb ischaemia and atherosclerosis in people with peripheral artery disease (PAD), is characterized as leg pain when walking that is typically alleviated by rest (Criqui & Aboyans, 2015). As the median age of the population keeps rising, intermittent claudication will become increasingly prevalent, impacting the quality of life of more and more people. In fact, a recent epidemiological study revealed that between 2000 and 2010 the incidence of PAD increased worldwide by almost 25% (Fowkes et al. 2013). Tissue acidosis of ischaemic muscle is associated with the release of inflammatory mediators such as lactic acid, H+ ions, substance P, arachidonic acid, ATP and serotonin, collectively known as an ‘inflammatory soup’ (Julius & Basbaum, 2001). Lactic acid evokes the exercise pressor reflex (EPR) that arises from contracting skeletal muscle and serves to readjust the cardiovascular system via increases in arterial blood pressure, heart rate and ventilation. Patients with intermittent claudication exhibit an augmented EPR during exercise compared to people with adequately perfused muscle (Hayes et al. 2008). It is known that the build‐up of lactic acid and H+ ions stimulates acid‐sensing ion channels (ASICs) of thin muscle afferents and induces cell depolarization via this chemoreflex.
ASICs are members of the epithelial Na+ channel/degenerin (ENaC/DEG) family of ion channels. There are four genes (ACCN1–4) that code for six subunits (ASIC1a &1b, ASIC2a & 2b, ASIC3 and ASIC4) (Kellenberger & Schild, 2015). ASIC1–3 are expressed mainly in the central and peripheral nervous systems (Baron & Lingueglia, 2015; Kellenberger & Schild, 2015). Dorsal root ganglion (DRG) neurons express primarily ASIC1a and ASIC3 subunits (Kellenberger & Schild, 2015; Sluka & Gregory, 2015). A unique biophysical characteristic of the ASIC3 isoform is that acidification causes a biphasic current. The first component is a rapidly inactivating current, the second a sustained current that persists as long as the pH remains low (Deval et al. 2010; Kellenberger & Schild, 2015). This current profile makes it easy to detect ASIC3 expression against other ASIC isoforms, which only exhibit a fast inactivating current in response to low pH. ASIC currents are modulated by several signalling molecules, including inflammatory mediators. Arachidonic acid and lysophosphatidylcholine, for instance, have been shown to significantly enhance the sustained and peak ASIC3 currents (Smith et al. 2007; Deval et al. 2008; Marra et al. 2016). Furthermore, the basic neuropeptide, dynorphin, can also exert potentiating effects on the ASIC1 isoform (Sherwood & Askwith, 2009).
Ischaemia not only causes the release of inflammatory mediators, but can also increase local opioid neuropeptide release. The naturally occurring opioid peptides, endomorphin 1 (E‐1) and 2 (E‐2), are mu opioid receptor (MOR) agonists that exhibit a high selectivity and affinity for MOR over kappa or delta opioid receptors (Fichna et al. 2007). E‐1 is found predominantly in the brain, while E‐2 is primarily localized in the spinal cord, including primary sensory afferents and dorsal horn (Martin‐Schild et al. 1998; Williams et al. 1999; Fichna et al. 2007). When administered intravenously in rats, both peptides, like other MOR agonists, decrease blood pressure and heart rate (Fichna et al. 2007). Furthermore, E‐1 and E‐2 exert potent analgesic effects in animal inflammatory pain models (e.g. mechanical allodynia, complete Freund adjuvant‐induced), and E‐2 release, in particular, has been reported to increase significantly in these models (Mousa et al. 2002; Fichna et al. 2007; Chen et al. 2015). Given the release of endomorphins at ischaemic sites and lactic acid‐mediated pressor effects, the purpose of the present study was to determine whether these endogenous opioid peptides modulate ASIC currents and, thereby, enhance the lactic acid‐evoked muscle chemoreflex in rats with either freely perfused or ligated femoral arteries (a model of PAD).
Methods
Ethical approval
The Penn State College of Medicine Institutional Animal Care and Use Committee (IACUC) approved the animal experiments described. The authors understand and conform to the principles and regulations described by the Journal of Physiology (Grundy, 2015).
Femoral artery ligation and DRG neuron isolation
Femoral ligation of adult male Sprague‐Dawley rats was performed under anaesthesia (3–5% isoflurane) 3 days prior to DRG neuron isolation. Both femoral arteries were ligated with 5‐0 silk sutures 3 mm distal to the inguinal ligament as previously described (Copp et al. 2015). Under these conditions, blood flow reserve capacity is greatly reduced when compared to normal controls but metabolic demand at rest is sufficiently met (Lash et al. 1995). Additionally, 15–20 μl of DiI (1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindocarbocyanine perchlorate, 3% in DMSO) was injected into the triceps surae muscles. Control (freely perfused, FP) animals did not undergo any sham surgical procedure in order to avoid ASIC gene expression changes previously observed in rats following surgical incision (Kido et al. 2013). Three days after ligation, rats were anaesthetized with CO2 and rapidly decapitated with a laboratory guillotine, and the lumbar (L4–L6) DRG tissue was isolated using ice‐cold Hanks balanced salt solution. The tissue was enzymatically dispersed in a culture flask with Earle's balanced salt solution containing 0.6 mg ml−1 collagenase Type D, 0.35 mg ml−1 trypsin and 0.1 mg ml−1 DNase. The flask was placed in a shaking water bath (35°C) for 40 min, then vigorously shaken for 10 s, and finally centrifuged twice for 6 min at 50 g. Thereafter, the DRG neurons were resuspended in minimum essential medium (MEM) supplemented with 10% fetal bovine serum, 1% glutamine and 1% penicillin‐streptomycin and plated onto poly‐L‐lysine‐coated dishes and stored in a humidified atmosphere (5% CO2/95% air) at 37°C. In some experiments, DRG neurons were pretreated overnight with 0.5 μg μl−1 pertussis toxin (PTX, List Biological Labs., Inc., Campbell, CA, USA) in the supplemented MEM.
ASIC cDNA construct isolation
Plasmids expressing ASIC channel proteins were constructed in pCI (Promega, Madison, WI, USA) using standard molecular biology techniques. Briefly, the open reading frame (ORF) for mouse ASIC1a (NM_009597) was amplified from mouse whole brain cDNA (Clontech, Palo Alto, CA, USA). Mouse ASIC2b (MDEG2‐NM_007384) and ASIC3 (DRASIC‐NM_183000) ORFs were amplified from mouse DRG cDNA. Mouse DRG cDNA was prepared from total RNA isolated with the RNeasy kit (Qiagen, Valencia, CA, USA) followed by first strand cDNA synthesis (RT for PCR; Clontech).
Primers used for amplification of each ASIC isoform were:
ASIC1a For, 5′‐GATCGATCACGCGTACCATGGAACTGAAGACCGAGGAG‐3′
ASIC1a Rev, 5′‐ GATCGATCGCGGCCGCTTAGCAGGTAAAGTCCTCAAACG‐3′
ASIC2b For, 5′‐ GATCGATCACGCGTACCATGAGCCGGAGCGGCGGAGC‐3′
ASIC2b Rev, 5′‐ GATCGATCGCGGCCGCTCAGCAGGCAATCTCCTCCAG‐3′
ASIC3 For, 5′‐ GATCGATCACGCGTACCATGAAACCTCCCTCAGGACTG‐3′
ASIC3 Rev, 5′‐ GATCGATCGCGGCCGCCTAGAGCCTTGTCACGAGG‐3′
Sense primers contained MluI sites and antisense primers NotI sites. ORF PCR products and pCI vector were digested with MluI and NotI (New England Biolabs, Ipswich, MA, USA) and products were cloned with T4 ligase using the Fast‐Link DNA Ligation Kit (Epicentre, Madison, WI, USA).
L‐cell transfection
L‐cells (mouse fibroblast cell line) were obtained from ATCC, cultured in Dulbecco's MEM (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin‐streptomycin, 1% glutamine and 10% non‐essential amino acids, and incubated at 37°C in a humidified atmosphere (5% CO2/95% air). The cells were co‐transfected via electroporation with either ASIC1a or ASIC2b or ASIC3 and enhanced green fluorescent protein (EGFP) cDNA constructs. Briefly, a 75 cm2 flask (∼70–80% confluent) was incubated in trypsin for 4–5 min, enzyme activity was stopped with supplementary DMEM and the cell suspension was centrifuged at 900 r.p.m. for 5 min. The cell count was determined with the Cell Countess (Thermo Fisher Scientific, Waltham, MA, USA), and 400 000 cells per 100 μl tip were electroporated with the NEON Electroporator (Thermo Fisher Scientific) with three to four 1125 V pulses for 15–20 ms duration. The electroporation tip contained Opti‐MEM (Thermo Fisher Scientific), one of three ASIC constructs (10–11 μg), EGFP (3 μg) and 2,3‐butanedione monoxime (2 mm). After electroporation, the L‐cells were plated on 35 mm dishes (∼10 000 cells cm−2) containing supplementary DMEM and incubated overnight prior to electrophysiological recordings. In preliminary experiments, exposure of non‐transfected L‐cells (n = 4) to pH 6.0 did not elicit overt native ASIC currents (data not shown).
Western blot experiments were carried out with the Wes system (Protein Simple, San Jose, CA, USA). The microplate was loaded with 0.2 μg μl−1 protein, primary antibodies (listed below) and reagents provided by the manufacturer (including the secondary antibody). Anti‐ASIC1, anti‐ASIC2, anti‐ASIC3 and anti‐actin antibodies were employed at 1:100, 1:200, 1:20 and 1:400, respectively. Actin was employed as a housekeeping gene. The antibodies used were: ASIC1 (Cat. No. N271/44, NeuroMab, Davis, CA, USA), ASIC2 (Cat. No. LS‐C110053, LifeSpan Biosciences, Seattle, WA, USA), ASIC3 (Cat. No. LS‐C313073, LifeSpan Biosciences) and actin (Cat. No. MAB1501, Chemicon/Millipore, Billerica, MA, USA). The secondary antibodies used were: goat anti‐mouse (Cat. No. 042‐205) and goat anti‐rabbit (042‐206), both from Protein Simple.
Electrophysiology
ASIC currents were recorded from DiI‐labelled DRG neurons and EGFP‐expressing L‐cells with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), filtered at 2 kHz with a four‐pole low‐pass Bessel filter, digitized at 10 kHz using an ITC‐18 A/D converter (HEKA Instruments, Inc., Holliston, MA, USA) and acquired with custom‐designed F6 software written by Stephen R. Ikeda (NIH/NIAAA) employing Igor Pro (WaveMetrics, Inc., Lake Oswego, OR, USA). ASIC currents were evoked by switching external recording solutions with a pH of 7.4 to 6.0 employing a custom‐designed gravity‐fed perfusion system that was placed ∼100 μm from the cell of interest. The holding potential (V H) was kept at −80 mV for DRG neurons and −60 mV for L‐cells. The patch pipettes were made from 8250 glass (King Precision Glass Inc., Claremont, CA, USA), pulled on a P‐97 micropipette puller (Sutter Instrument Co., Novato, CA, USA) and coated with Sylgard (Dow Corning). ASIC currents were recorded by switching from a control solution (pH 7.40) to a solution of pH 6.0. After full recovery at pH 7.40, the cells were preincubated for a minimum of 5 min in the control solution containing the test compound (i.e. E‐1 or E‐2). Following the preincubation period, the external solution was switched to one that ranged from pH 6.0 to 7.3 (described below). The endomorphin‐mediated potentiation of the sustained ASIC currents was determined as shown in Fig. 1. We chose the last second of the test pulse to measure current amplitude (Fig. 1 A) as this time period appeared relatively stable. In preliminary experiments, we compared the current amplitude measured at this time point to the last 100 ms of the test pulse. The results indicate that the amplitude obtained at both time points were indistinguishable (data not shown). However, we cannot rule out that the current potentiation may be contaminated by the ASIC3 channel's desensitization component. The pH‐dependent activation curves of the peak transient ASIC3 currents in the absence and presence of either 10 μm E‐2 or E‐1 were obtained by switching from the control solution to a test solution with varying pH values for 10 s as previously described (Yagi et al. 2006). The acid‐induced currents were then normalized to that obtained at pH 5.0. The inactivation curves of the peak transient ASIC3 currents in the absence and presence of either 10 μM E‐2 or E‐1 were acquired with the paradigm shown in Fig. 5 A. The currents were normalized to the maximal peak amplitude acquired at pH 6.0. Both curves were fit to the Hill equation, as described below.
Figure 1. ASIC2b and ASIC1a current activation in L‐cells.
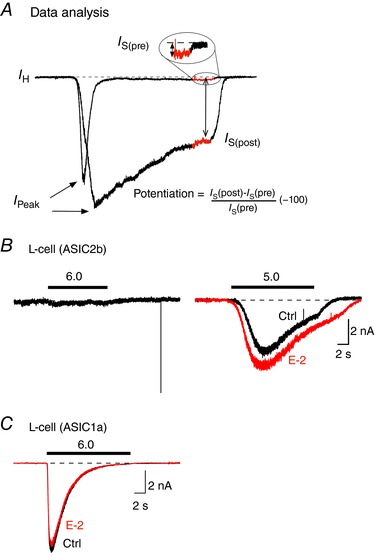
A, the pH‐mediated potentiation of the sustained current (I S) was calculated as I S(post) – I S(pre)/I S(pre) × 100. The measurements for pre (control solution) and post (test solution) currents were acquired as the average current amplitude obtained during the last second of the test pulse (shown in red). B, ASIC current trace of an L‐cell expressing ASIC2b following application of an external solution of pH 6.0 (black trace, left) or 5.0 (black trace, right). After washout, the cell was preincubated in a solution (pH 7.4) containing 10 μm E‐2. Thereafter, the cell was exposed to an external solution (pH 5.0) and E‐2 (10 μm, red trace). C, ASIC1a current trace following exposure to a control solution (pH 6.0, black) and a solution containing 10 μm E‐2 (red) of an L‐cell transfected with ASIC1a cDNA. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 5. Effect of E‐2 on ASIC3 activation and inactivation curves.
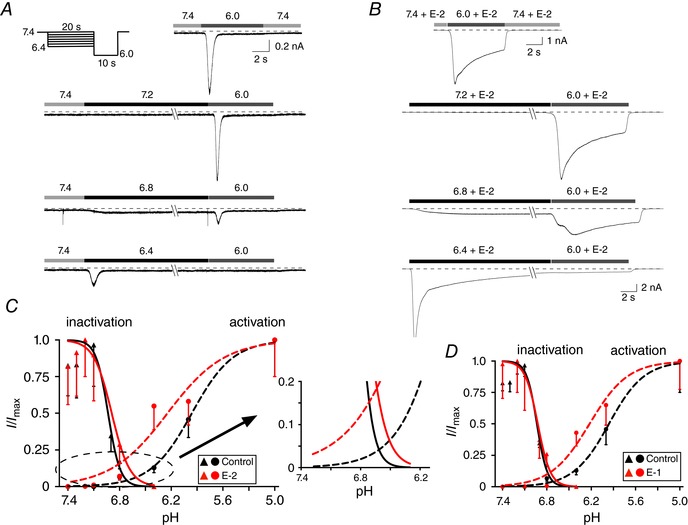
A and B, ASIC3 current traces acquired from a control L‐cell (A) and in the presence of 10 μm E‐2 (B). Activation and inactivation values were obtained by employing the protocol shown in A (top left) with a 20 s condition step between pH 6.4 and 7.4, followed by a 10 s test step to 6.0. The amplitudes of the peak currents from the condition steps were employed for the activation curve and the peak current amplitudes of the test step were used for the inactivation curve (n = 5–16 cells). Activation and inactivation curve points were normalized to pH 5.0 and 7.4, respectively. The smooth curves were obtained by fitting the points to the Hill equation. C, window currents obtained from overlap of activation and inactivation curves before (black) and during (red) E‐2 (10 μm) exposure. The overlapping region has been magnified. D, window currents obtained from overlap of activation and inactivation curves before (black) and during (red) E‐1 (10 μm) exposure. [Color figure can be viewed at wileyonlinelibrary.com]
Recording solutions
The recording solutions with pH ranging from 5.0 to 6.7 were buffered with MES and consisted of (in mm): NaCl 140, KCl 5.4, MES 10, MgCl2 1, CaCl2 5, glucose 10. The solutions with pH ranging from 6.8 to 7.4 were buffered with HEPES and consisted of (in mm): NaCl 140, KCl 5.4, HEPES 10, MgCl2 1, CaCl2 5, and glucose 10. The final pH for each solution was adjusted with NaOH. The pipette solution consisted of (in mm): N‐methyl‐d‐glucamine 80, TEA‐OH 20, CsCl 20, CsOH 40, creatine phosphate 14, Hepes 10, CaCl2 1, Mg‐ATP 4, Na2GTP 0.3 and EGTA 11. The pH was adjusted to 7.2 with CH3SO3H and the osmolality was 293–302 mOsmol kg−1. E‐1 and E‐2 neuropeptides (both from Tocris Cookson, Ellisville, MO, USA) were diluted in the external solution to their final concentrations on the day of the experiment.
Under our recording conditions, application of solutions with pH 6.0 did not activate ASIC2b currents (Fig. 1 B), consistent with other reports (Kellenberger & Schild, 2015). ASIC2b currents were observed with exposure to a solution with pH 5.0 (black). Additionally, when 10 μm E‐2 + pH 5.0 (red) was applied, the ASIC current was enhanced (Fig. 1 B). In this report, solutions with pH ranging from 6.0 to 7.4 were used for experiments with ASIC1a and ASIC3. However, ASIC1a currents did not exhibit any overt modulation when exposed to 10 μm E‐2 + pH 6.0 (Fig. 1 C).
EPR response protocol
In one group of rats, both femoral arteries were ligated 72 h prior to the experiment as described above. On the day of the experiment, rats were anaesthetized with 4% isoflurane in O2. The trachea was cannulated and the lungs were mechanically ventilated. Both carotid arteries and the right jugular vein were cannulated with PE‐50 to measure arterial blood pressure and to administer drugs and fluid, respectively. Heart rate was calculated from the arterial pressure pulse (Spike 2 software; Cambridge Electronics Design, Cambridge, UK). Additionally, the left superficial epigastric artery was cannulated (PE‐8) so that the tip of the catheter was placed near its junction with the femoral artery. In ligated rats, the catheter placed in the superficial epigastric artery was distal to the ligation site. In freely perfused rats, a reversible snare (2‐0 suture) was placed around the left iliac artery and vein (control for the ligation procedure). The snare was tightened before every bolus injection or infusion of drugs. Tightening the snare partially trapped the injectate within the arterial supply of the hindlimb. The snare was released after every infusion then re‐snared prior to bolus injection. The rats were placed and secured in a Kopf stereotaxic frame and a clamp was placed on the pelvis. A precollicular decerebration was performed under isoflurane anaesthesia and all neural tissues rostral to the section were discarded. After the decerebration procedure was completed, the anaesthesia was discontinued and the rat was ventilated mechanically with room air. The rats were given at least 40 min to stabilize after initial procedures.
The rats were subjected to neuromuscular blockade with pancuronium bromide (0.5 mg kg−1) intravenously. Thereafter, a bolus of 100 μl of lactic acid (12 mm) was injected into the superficial epigastric artery catheter and the pressor–cardioaccelerator response was measured. Next, 100 μl of naloxone (100 μg kg−1) was infused for 10 min into the catheter placed in the superficial epigastric artery. Following the naloxone infusion, a bolus of 100 μl lactic acid (12 mm) in combination with E‐2 (10 μm) was injected into the superficial epigastric artery and the pressor–cardioaccelerator response was measured. Finally, 100 μl of APETx2 (200 μg kg−1), an ASIC 3 receptor antagonist, was infused for 10 min into the superficial epigastric artery. After completing the APETx2 infusion, we again injected a bolus of 100 μl lactic acid (12 mm) with E‐2 (10 μm) into the superficial epigastric artery. In another set of experiments, a lactic acid bolus (12 mm) was injected into the artery and the pressor response was measured. Thereafter, both naloxone (100 μg kg−1) and the ASIC1a blocker, PcTx‐1 (1 nm), were infused for 10 min into the superficial epigastric artery. Following the 10 min infusion, a bolus of lactic acid (12 mm) and E‐2 (10 μm) was injected into the epigastric artery. The pressor–cardioaccelerator response was then measured. The interval between each injection of lactic acid was at least 15 min. At the end of each experiment Evan's blue (100 μl) was injected into the superficial epigastric artery to determine if the injectate reached the triceps surae muscles. The data reported here include only those taken from rats in which the dye reached this hindlimb muscle group.
Data analysis
Electrophysiological recordings were analysed with Igor Pro and statistical analysis was performed with Prism (GraphPad Software, La Jolla, CA, USA). The data are expressed as mean ± SD unless otherwise stated. Statistical significance between two groups was performed employing paired and unpaired t tests. Data containing more than two groups (Fig. 7 C) were evaluated with one‐way ANOVA with repeated measures followed by Tukey's post hoc test. This test was chosen to make all possible pairwise comparisons of the unequal group sample sizes. A P value <0.05 was considered statistically significant. The concentration–response relationships were determined by the sequential application of endomorphins in increasing concentrations. Two different concentrations were used with each cell. The results were then pooled and the curves were fit to the Hill equation: B = B MAX/{1 + (IC50/[agonist])nH}, where B is the percentage potentiation, B MAX is the maximum potentiation of the ASIC3 current, IC50 is the half‐maximum concentration, [agonist] is the pH of the test solution and nH is the Hill coefficient. Graphs and current traces were generated with both Igor Pro and Autodesk Graphic software packages.
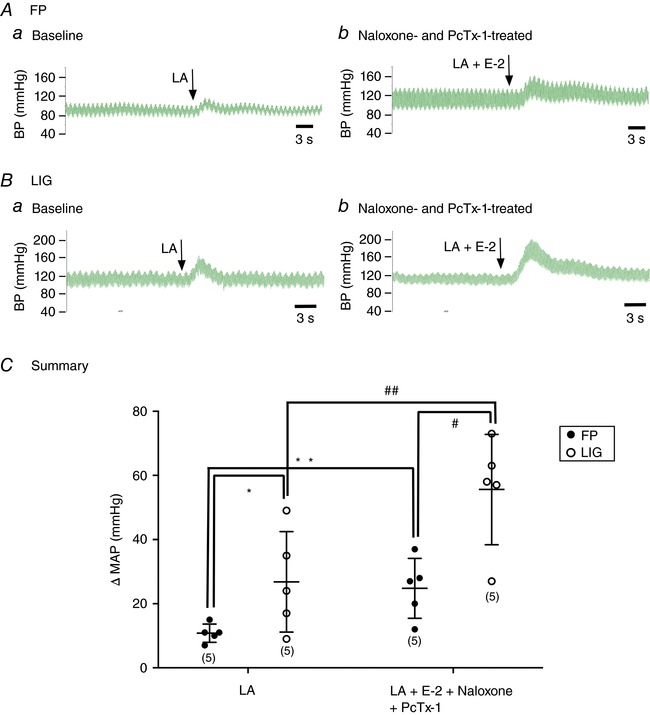
Figure 7. Effect of E‐2 on the lactic acid‐induced reflex increase in mean arterial pressure in FP and LIG rats.
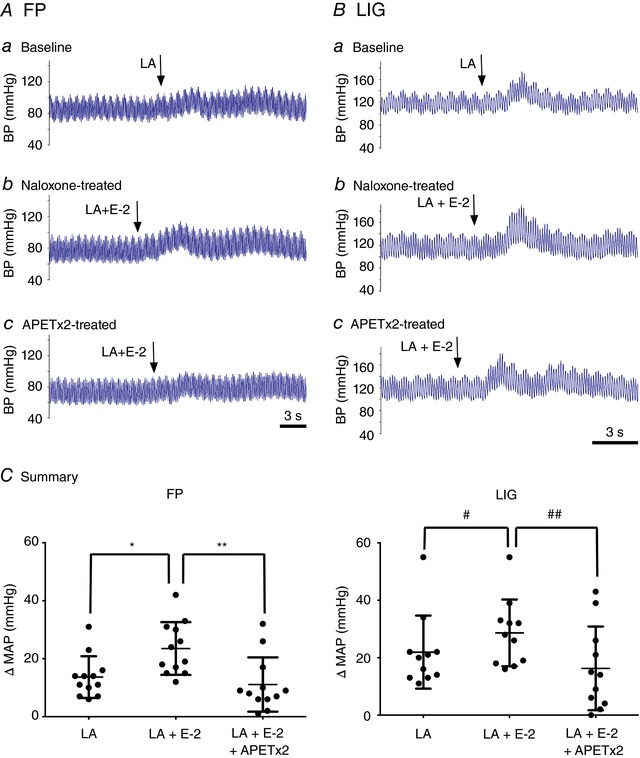
A and B, blood pressure (BP) responses to a lactic acid (LA, 12 mm) infusion in a rat with FP (Aa, n = 12) or LIG (Ba, n = 11) arteries. After a return to baseline, naloxone (100 μg kg−1) was infused for 10 min and then LA + E‐2 (10 μm) were co‐infused (Ab and Bb). Once BP returned to control levels, APETx2 (100 μg kg−1) was administered and then LA + E‐2 (10 μm) were reintroduced. C, summary plots showing the mean change (±SD) in arterial pressure (MAP) following infusion of LA alone, LA + E‐2 (+Naloxone) and LA + E‐2 (+APETx2). * P = 0.0004 and # P = 0.045 compared to LA alone, respectively; ** P = 0.005 and ## P = 0.008 compared to LA + E‐2, respectively, using repeated one‐way ANOVA followed by a Tukey's post hoc test. [Color figure can be viewed at wileyonlinelibrary.com]
Results
Potentiation of sustained ASIC currents by endomorphins in DRG neurons of FP and LIG rats and in L‐cells heterologously expressing ASIC3
The present study examined the modulation of ASIC currents by endomorphins, especially E‐2, under normal and ischaemic conditions in DRG neurons innervating triceps surae muscle. Figure 2 A shows an ASIC current activated after exposure to an external solution of pH 6.0 (black trace) in a DRG neuron isolated from a control rat with FP hindlimbs. The ASIC current displayed the typical sustained current under acidic conditions. After a return to baseline, the neuron was pre‐exposed to an external solution (pH 7.4) containing 10 μm E‐2 for 5 min. Thereafter, the solution was switched to one with a pH of 6.0 + 10 μm E‐2 (red trace). Under these conditions, the transient peak and sustained currents were enhanced by about 48 and 71%, respectively. The ASIC currents illustrated in Fig. 2 B are those isolated from a rat 72 h after femoral artery ligation (LIG). Following exposure to 10 μm E‐2 + pH 6.0, a 150% increase of the sustained current (red trace) was observed when compared to that of pH 6.0 alone (black trace), while there was a minimal effect on the transient peak current. Note that the endomorphin‐mediated enhancement of transient peak currents was not always observed. In some neurons, endomorphin exposure resulted in either current enhancement or block or no effect (discussed below). The dot plot in Fig. 2 G shows that E‐2 exhibited a significantly (P = 0.025) greater effect on the sustained ASIC currents in DiI‐labelled DRG neurons from rats with ligated arteries than FP animals. Figure 2 C and D shows ASIC currents recorded from DiI‐labelled DRG neurons of FP and LIG animals before and during E‐1 (10 μm) application, respectively. In this set of experiments, E‐1 potentiated ASIC currents 0.76‐fold [0.11–1.41, 95% confidence interval (CI)] and 1.94‐fold (0.40–3.47, 95% CI) from FP and LIG rats, respectively. The statistical comparison of the mean E‐1‐mediated ASIC current potentiation revealed comparable effects for both groups of DRG neurons (P = 0.058).
Figure 2. Modulation of ASIC currents by endomorphin peptides in DiI‐labelled DRG neurons isolated from freely perfused (FP) and femoral‐ligated (LIG) rats and in L‐cells transfected with ASIC3.
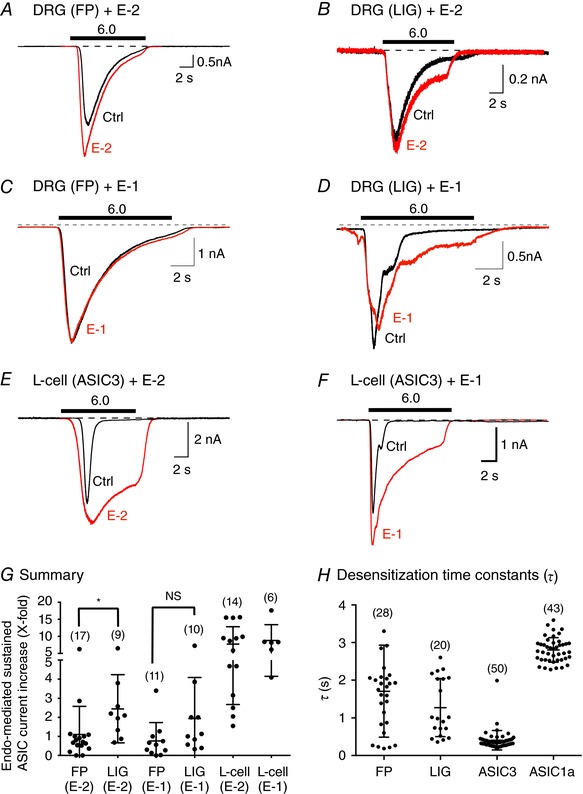
A and C, ASIC current traces from FP DRG neurons before (Ctrl, black) and after E‐2 or E‐1 (10 μm, red) exposure. B and D, ASIC current traces from LIG DRG neurons before (Ctrl, black) and after E‐2 or E‐1 (10 μm, red) exposure. E and F, ASIC currents in L‐cells transfected with ASIC3 before (Ctrl, black) and after E‐2 (10 μm, red) or E‐1 (10 μm, red) exposure. The solid bars above the traces represent a 10 s exposure to the pH 6.0 test solution. The cells were pre‐exposed to E‐2 or E‐1 for 5 min (pH 7.4) just prior to exposure to the test solutions (pH 6.0). The holding potential (V H) for DRG neurons and L‐cells was −80 and −60 mV, respectively. G, summary dot plot with mean (±SD) of the E‐2‐ and E‐1‐mediated sustained ASIC current potentiation. * P = 0.025 using unpaired t test; NS, not significant (P = 0.058) using unpaired t test. H, mean (±SD) desensitization time constant (τ) values of the rapidly inactivating ASIC currents obtained from DRG neurons (FP and LIG) and ASIC3‐transfected L‐cells. Numbers in parentheses indicate the number of recordings. [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether the enhancement of sustained ASIC currents by endomorphins involved MOR signalling pathways, a separate group of DRG neurons was pretreated with PTX to uncouple MOR from heterotrimeric G proteins. Exposure of PTX‐treated DRG neurons to E‐2 (10 μm, pH 6.0) resulted in potentiation of sustained ASIC currents. The mean percentage increase of ASIC currents was 114 ± 87% (n = 5), a value that was not significantly different (P = 0.757) from DRG neurons of FP rats not treated with PTX (101 ± 75%, n = 12). These results suggest that the endomorphin‐mediated enhancement of sustained ASIC currents was independent of MOR stimulation.
To better study the direct modulation of ASIC sustained currents by endomorphins, the L‐cell line was transfected with ASIC3 cDNA. Unlike DRG neurons, L‐cells do not natively express MORs (Andria & Simon, 1999) or ASICs (see above). Thus, exposure of ASIC3‐expressing L‐cells to endomorphins would exclude activation of MOR‐activated G protein signalling cascades and provide us with an unambiguous null background. The currents shown in Fig. 2 E and F are those obtained from transfected L‐cells before (pH 6.0, black) and during E‐2 (10 μm + pH 6.0, red) or E‐1 (10 μm + pH 6.0, red) exposure, respectively. The summary plot (Fig. 2 G) shows a dramatically greater potentiation of ASIC3 currents in heterologously expressing L‐cells than in DRG neurons (note gap on y‐axis). This larger potentiation is a result of higher homomeric ASIC3 expression levels or a more homogenous channel population inherent in heterologous expression systems.
Desensitization time constants (τ) of the rapidly inactivating ASIC3 and ASIC1a currents in the continued presence of protons are approximately 0.3 s for the former and vary for the latter ranging from 1.2 to 3.4 s (Grunder & Pusch, 2015; Kellenberger & Schild, 2015). We measured τ in DRG neurons, which express both ASIC3 and ASIC1a, and compared these values to τ in ASIC3‐ and ASIC1a‐expressing L‐cells. Figure 2 H shows that the mean τ values from FP and LIG DRG neurons were not significantly different (P = 0.166) for the two groups. Since a functional ASIC is composed of three channel isoforms (Kellenberger & Schild, 2015), the scatter observed in DRG neurons is probably the result of varying ASIC3 and ASIC1a ratios that constitute the whole‐cell ASIC current (see below). By comparison, τ values for L‐cells expressing ASIC3 or ASIC1a were relatively uniform.
ASIC expression profile in DRG neurons following femoral ligation
In a separate group of rats, expression levels of ASIC subunits in DRG neurons were measured to determine whether the increase of the endomorphin‐mediated potentiation of ASIC currents in neurons from LIG rats resulted from changes in ASIC expression levels. Figure 3 A–C shows Western blots for ASIC1‐3 in five control (FP) and four experimental rats 72 h after femoral artery ligation (LIG). Expression values for each lane were normalized to actin. Figure 3 A shows two bands for ASIC1 subunits, which are indicative of channel glycosylation (Johnson et al. 2001; Kadurin et al. 2008; Jing et al. 2011, 2012). Thus, both bands were used to determine the expression level. Arterial ligation led to a significant (P = 0.007) decrease of ASIC1 expression in DRG tissue when compared to control animals. On the other hand, ASIC3 expression levels significantly (P = 0.009) increased (Fig. 3 C). The mean ASIC2 expression levels (Fig. 3 B) did not change significantly (P = 0.253), although the ligated group exhibited greater variability of protein expression.
Figure 3. Effect of femoral artery ligation on ASIC1‐3 isoform expression levels.
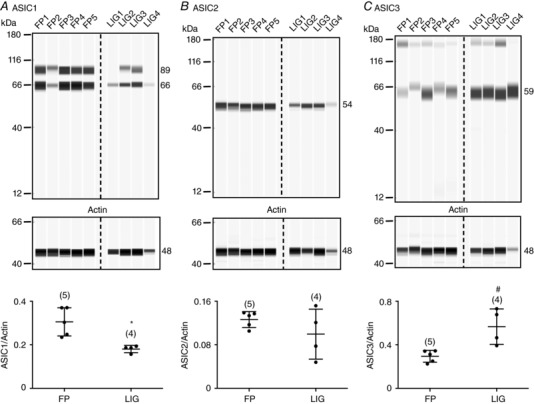
Western blots for ASIC1 (A), ASIC2 (B) and ASIC3 (C) in DRG tissue isolated from rats that were freely perfused (FP1–FP5) or with ligated femoral arteries (LIG1–LIG4). The 66 and 89 kDa bands in A represent the non‐glycosylated and glycosylated forms of ASIC1, respectively. Each lane (loaded with 0.2 μg μl−1 protein) represents one animal. Actin was used as the loading control. The anti‐ASIC1, anti‐ASIC2, anti‐ASIC3 and anti‐actin antibodies were used at a dilution of 1:100, 1:200, 1:20 and 1:400, respectively. The plots on the bottom show the mean (±SD) values for ASIC/actin ratios for FP and LIG rats. * P = 0.007 and # P = 0.009 for ASIC1 and ASIC3, respectively, employing t tests.
Pharmacological and biophysical properties of ASIC3 currents exposed to endomorphins
The pharmacological profiles of the E‐2‐ and E‐1‐mediated sustained ASIC3 current potentiation in L‐cells were determined next. Figure 4 shows representative ASIC3 current traces after a 10 s sequential exposure to pH 6.0 alone, with 0.3 and 10 μm E‐2 (Fig. 4 A) or E‐1 (Fig. 4 B). Similar to experiments above, the cells were pre‐exposed for 5 min to the corresponding endomorphin concentration (pH 7.4) prior to application of test solutions. The E‐2 and E‐1 concentration–response curves are plotted in Fig. 4 C. The data points were fit to the Hill equation, and the EC50 values (±95% CIs) and Hill coefficients (±SEM) obtained were 4.1 (2.2–7.5) μm and 1.61 ± 0.84 for E‐2 and 1.9 (0.8–4.3) μm and 1.40 ± 0.52 for E‐1, respectively.
Figure 4. E‐2 and E‐1 concentration‐response relationships in ASIC3‐expressing L‐cells.
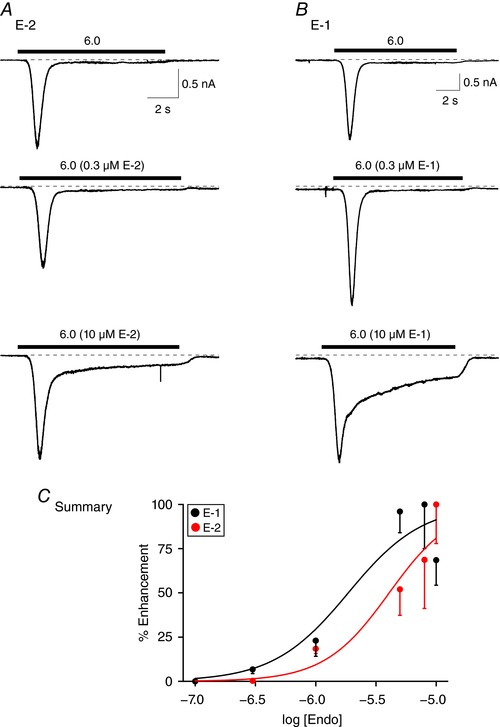
ASIC3 current traces obtained before (pH 6.0) and during application of 0.3 and 10 μm E‐2 in the same cell (pH 6.0, A) or E‐1 in the same cell (pH 6.0, B). The cells were pretreated with either E‐2 or E‐1 (pH 7.4) 5 min prior to agonist exposure. C, endomorphin concentration–response relationships for E‐2 (red) and E‐1 (black) peptides. Each point represents the mean (±SEM, n = 3–10 cells) enhancement (%) of the sustained ASIC3 current. The smooth curves were obtained by fitting the points to the Hill equation. [Color figure can be viewed at wileyonlinelibrary.com]
The next series of experiments examined the effect of E‐2 and E‐1 on the biophysical characteristics of ASIC3 currents in L‐cells. Figure 5 A and B shows representative current traces evoked by the pH step protocol (Yagi et al. 2006) to obtain the activation values and fit to the Hill equation before (black circles) and during E‐2 application (red circles, Fig. 5 C). The pH50 activation values and Hill coefficients (±SEM) were 5.96 and 1.81 ± 0.94 and 6.29 and 1.67 ± 0.68 for control and E‐2‐exposed L‐cells, respectively. The pH‐dependent inactivation curves for control (black triangles) and E‐2‐exposed cells (red triangles) are also plotted in Fig. 5 C. The pH50 inactivation values and Hill coefficients (±SEM) were 6.93 and 6.20 ± 1.46 and 6.89 and 4.15 ± 3.57 for control and E‐2‐exposed cells, respectively. The window currents (overlap of the activation and inactivation curves) have been magnified (Fig. 5 C, right) to illustrate the E‐2‐mediated shift of both curves. These results indicate that E‐2 exposure potentiated ASIC3 currents by shifting the inactivation curve slightly rightward to lower pH values and shifting the activation curve leftward to higher pH values. E‐1 caused similar effects on the ASIC3 window currents (Fig. 5 D).
External Ca2+ ions reduce endomorphin‐mediated sustained ASIC3 current enhancement
Previous studies have shown an interplay between extracellular Ca2+ and H+ ions (Immke & McCleskey, 2001, 2003). It is thought that both ions compete for the same binding site on ASIC3. Thus, we next examined whether external Ca2+ ion levels modulate the endomorphin potentiating effect of ASIC3 sustained currents. Figure 7 shows ASIC3 currents activated by pH 6.0 + 1.25 mm external Ca2+ (baseline), followed by application of 10 μm E‐2 (Fig. 6 A) or E‐1 (Fig. 6 B) at pH 6.0 + 1.25 mm Ca2+ and 0 Ca2+. It can be seen that as external Ca2+ levels decrease, the endomorphin‐mediated potentiating effect increases. The summary plot shown in Fig. 6 C indicates that decreasing Ca2+ ions from 1.25 mm to 0 (i.e. ‘nominal’) leads to an approximate doubling of the potentiating effects of endomorphins on ASIC3 sustained currents.
Figure 6. Effect of varying external Ca2+ concentration on the endomorphin‐mediated enhancement of sustained ASIC currents.

A, ASIC3 current traces in an L‐cell exposed to pH 6.0 + 1.25 mm Ca2+, pH 6.0 + 1.25 mm Ca2+ + 10 μm E‐2 and pH 6.0 + 0 Ca2+ + 10 μm E‐2. B, same as A, but with E‐1. C, summary plot showing the endomorphin‐mediated ASIC3 current enhancement with varying external Ca2+ concentrations. Each point represents the mean (±SD, n = 5–19 cells) sustained ASIC current enhancement for E‐1 (black) and E‐2 (red). [Color figure can be viewed at wileyonlinelibrary.com]
E‐2 enhances the LA‐induced increase in arterial blood pressure in rats
In this set of experiments, we sought to provide evidence that endomorphins modulate the lactic acid‐mediated evoked muscle chemoreflex in FP and LIG rats. Figure 7 Aa shows a reflex increase of 11% in blood pressure (BP) from 88 to 98 mmHg following intra‐arterial lactic acid (12 mm) injection – a concentration comparable to rat plasma levels reached after strenuous exercise (Madureira & Hasson‐Voloch, 1988). After BP returned to control levels, the MOR blocker naloxone (100 μg kg−1) was infused. Following a 10 min incubation period, lactic acid (12 mm) and E‐2 (10 μm) were co‐administered and BP increased from 77 to 94 mmHg (a 17% increase, Fig. 7 Ab). Once baseline was restored, the ASIC3 blocker, APETx2 (200 μg kg−1), was administered for approximately 10 min and followed by administration of both lactic acid and E‐2. Figure 7 Ac shows that BP increased from 75 to 84 mmHg (a 9% increase). A similar approach was used in a group of LIG rats. Figure 7 Ba shows that lactic acid (12 mm) infusion alone increased BP by 21%, from 123 to 144 mmHg. Additionally, when both lactic acid and E‐2 (10 μm) were infused after naloxone treatment, BP increased from 118 to 151 mmHg (a 33% increase, Fig. 7 Bb). Finally, the co‐administration of lactic acid and E‐2 following APETx2 infusion led to a BP increase from 123 to 144 mmHg (a 21% increase, Fig. 7 Bc). The summary dot plots for FP and LIG rats in Fig. 7 C show that E‐2 significantly (P < 0.05) enhanced the lactic acid‐mediated increase in BP, while APETx2 pretreatment significantly (P < 0.05) blocked the E‐2‐mediated enhancement of BP.
In a separate group of rats with FP and LIG femoral arteries, we examined the effect of the ASIC1a blocker, PcTx‐1 (Mazzuca et al. 2007), on the lactic acid‐mediated BP increase before and during E‐2 administration. Figure 8 Aa shows that in a rat with FP arteries, BP increased by 16% from 92 to 107 mmHg following lactic acid (12 mm) intra‐arterial administration. After BP stabilization, naloxone and PcTx‐1 (1 nm) were infused for 10 min and then both lactic acid and E‐2 (10 μm) were administered. BP increased from 115 to 139 mmHg (a 21% increase, Fig. 8 Ab). Figure 8 Ba shows the BP response to the lactic acid bolus infusion in a rat with ligated arteries. Prior to lactic acid infusion, BP was 118 mmHg and increased 23% to 145 mmHg with lactic acid. Next, the rat was pretreated with PcTx‐1 and naloxone for 10 min. Afterwards, lactic acid infusion led to a 53% increase of BP from 117 to 180 mmHg. The summary plot shown in Fig. 8 C indicates that E‐2 significantly (P < 0.05) enhanced the lactic acid‐mediated BP response in FP and LIG rats following blockade of both ASIC1a and MOR. The plot also shows that the lactic acid‐mediated BP increase was significantly (P < 0.05) higher in rats with LIG femoral arteries than in FP rats.
Figure 8. Effect of E‐2 on the lactic acid‐induced reflex increase in mean arterial pressure in FP and LIG rats after ASIC1a channel block.
A and B, blood pressure (BP) responses to a lactic acid (LA, 12 mm) infusion in a rat with FP (Aa, n = 5) or LIG (Ba, n = 5) arteries. After return to baseline, naloxone (100 μg kg−1) and PcTx‐1 (1 nm) were infused for 10 min and then LA + E‐2 (10 μm) were co‐infused (Ab and Bb). C, summary plots showing the mean change (±SD) in arterial pressure (MAP) following infusion of LA alone and LA + E‐2, pretreated with naloxone and PcTx‐1. * P = 0.006 and # P = 0.012 compared to FP rats using unpaired t test; ** P = 0.036 and ## P = 0.004 compared to LA alone, using paired t test. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Twenty years ago, the first ASIC isoform (ASIC1a) was cloned (Waldmann et al. 1997) and E‐1 and E‐2 were discovered in the brain (Zadina et al. 1997). Since then, the known roles these proteins play have expanded from nociception to cardiovascular homeostasis (Abboud & Benson, 2015; Lingueglia & Lazdunski, 2015). Ischaemia of the lower limbs is a major manifestation of patients with PAD, and lactic acid build‐up is a likely contributor to intermittent claudication. The release of endomorphins at inflammatory sites suggests their role in peripheral pain control and pressor responses. The present study focused on endomorphin modulation of ASIC3 at three levels: heterologously expressed in L cells; ASIC current recordings in DRG neurons; and an in vivo correlate of the lactic acid‐mediated reflex pressor response in FP and LIG rats (a model of PAD). Our findings represent a novel interaction between endogenous opioid peptides and ASIC3 that probably augments the lactic acid‐mediated muscle chemoreflex in PAD patients.
The results of the present study show that E‐1 and E‐2 potentiated acid‐activated ASIC3 sustained currents, with minimal effects on ASIC1a currents and no effect on ASIC2 channels – at least in solutions with pH greater than 6.0. Other reports have shown ASIC current potentiation by peptides (Sherwood & Askwith, 2009; Kellenberger & Schild, 2015; Lingueglia & Lazdunski, 2015) or fatty acids, such as arachidonic acid or lysophosphatidylcholine, both released under ischaemic conditions (Smith et al. 2007; Deval et al. 2008; Marra et al. 2016). The modulation of ASIC currents in all cases is presumed to be a direct interaction with the channels rather than through signalling events. For example, ASIC1 transient peak currents were reported to be potentiated by the kappa opioid receptor agonists, big dynorphin and its fragment dynorphin A (Sherwood & Askwith, 2009). Both opioid peptides enhanced the channel activity in ASIC1‐transfected oocytes and cortical neurons without activating kappa opioid receptors or G proteins. Similarly, sustained ASIC3 currents have been shown to be potentiated by the neuropeptides FF(NPFF) and FMRFamide in rat DRG neurons (Askwith et al. 2000; Kellenberger & Schild, 2015).
Our results are difficult to reconcile with those previously reported in rat DRG neurons in which morphine or [d‐Ala2,N‐Me‐Phe4,Gly5‐ol]‐enkephalin (DAMGO) blocked the peak ASIC current via MOR stimulation (Cai et al. 2014). The opioid‐mediated block was shown to be PTX‐ and naloxone‐sensitive. In that study, the effect of opioids on the sustained ASIC currents was not examined (Cai et al. 2014). As mentioned above, under our recording conditions, the effect of endomorphins on peak ASIC currents was inconsistent. That is, either E‐1 or E‐2 exposure led to an increase or decrease or no effect on the peak currents. We found that endomorphins modulated ASIC currents in DRG neurons pretreated overnight with PTX, potentiated sustained currents in ASIC3‐expressing L‐cells (which do not express MOR) and enhanced the LA‐mediated increase in arterial blood pressure in the presence of naloxone. It is possible that the chemical characteristics of morphine and DAMGO interfere with their ability to directly modulate ASICs while the small size of endomorphins enables them to interact with the channels and affect the sustained currents. Further studies are needed to determine the interacting domains between endomorphins and ASIC3.
The pharmacological profile of the endomorphin‐mediated sustained ASIC3 current potentiation in transfected L‐cells ranged from 2 to 4 μm, values that are greater than those reported for MOR activation (Fichna et al. 2007). Both endomorphins, whether released peripherally by primary afferents or exogenously administered, are rapidly degraded (Fichna et al. 2007). In the present study, the interstitial endomorphin levels were not determined. Nevertheless, in some animal models of ischaemia and inflammation, endomorphin release has been reported to increase (Mousa et al. 2002, Fichna et al. 2007). It is possible that in ischaemic tissue endomorphin levels can reach greater than normal concentrations due to greater release by primary afferents and lower activity of enzymes that degrade E‐1 and E‐2. The end result would be that endomorphins exert analgesic actions. However, our results suggest that endomorphins may elicit pain in ischaemic muscle through potentiation of ASIC3 channels of primary afferents that are stimulated by the drop in tissue pH.
We also examined the mechanism by which E‐1 and E‐2 potentiate sustained ASIC3 currents. Our data indicate that the activation curve was shifted to more basic values than seen with lactic acid alone. Similar findings were previously shown to occur with arachidonic acid and lysophosphatidylcholine (Deval et al. 2008; Marra et al. 2016). Both of these compounds are released under ischaemic and inflammatory conditions. It should be mentioned that arachidonic acid can also potentiate ASIC1 and ASIC2 as well (Smith et al. 2007). Unlike these studies, however, our results show that the inactivation curve was shifted to lower pH values. The consequence was a greater increase of window currents than that obtained from a shift in the activation curve alone. Therefore, the endomorphin‐mediated potentiation results from earlier channel opening and longer inactivation periods. This suggests that in ischaemic tissue, the already augmented lactic acid‐mediated chemoreflex is activated at pH levels closer to 7.4 in the presence of endomorphins, leading to further sensitization of primary afferents.
In a series of studies, McCleskey and colleagues described the relationship between external Ca2+ concentration and changes in H+ ion concentrations and its effect on ASIC3 current kinetics. They showed that decreasing external Ca2+ concentration shifted the pH‐dependent activation to lower values and single channel conductance increased at lower Ca2+ concentrations (Immke & McCleskey, 2003). It was postulated that at physiological pH (7.40), Ca2+ ions bind to a single site near the channel's pore and block Na+ conductance. As pH drops, the binding affinity for Ca2+ decreases such that the divalent ion is released and Na+ ion conductance resumes. They also provided evidence that lactic acid acts as a Ca2+ ion ‘chelator’ such that under exercising conditions, the buildup of lactic acid relieves Ca2+ block and increases ASIC3 activity (Immke & McCleskey, 2001). The authors suggested that lactic acid can sensitize ASIC currents by mildly decreasing extracellular Ca2+ ions (Immke & McCleskey, 2001). In the present study, we found that the endomorphin potentiation of ASIC3 currents increased more than 2‐fold when external Ca2+ was lowered from 1.25 mm to 0 (‘nominal’) Ca2+. As mentioned previously, endomorphin release is increased under inflammatory conditions. Given that interstitial lactic acid levels can increase up to 6‐fold in animals with ischaemic skeletal muscle (Hagberg, 1985), it is possible that ASIC currents can be enhanced due to both chelation of Ca2+ ions by lactic acid and E‐2‐mediated potentiation. This would suggest that the chemoreflex of ischaemic muscle can be exaggerated further by lactic acid through Ca2+ chelation and potentiation of ASIC3 channels by the enhanced endomorphin release.
We used an in vivo model of femoral ligation to determine the ASIC current modulation by endomorphins in DiI‐labelled DRG neurons in an ischaemic state. Our results indicate that under ischaemic conditions, endomorphins exert a greater potentiation effect on ASIC currents in DRG neurons isolated from rats with ligated arteries than those obtained from freely perfused animals. These results parallel those of the Western blotting assays of DRG tissue isolated from rats with ligated femoral arteries in which ASIC3 levels increased significantly, while the ASIC1 levels were significantly downregulated. Note that the Western blots reflect ASIC expression at the surface and cytoplasm. A similar study previously found that ASIC3 levels were higher in rats with ligated femoral arteries, while the levels of ASIC1 were not explored (Liu et al. 2010). While a functional ASIC is composed of three channel isoforms, it appears that in this animal model, ASIC3 channel isoforms make a greater contribution to the total ASIC currents in DRG neurons innervating muscle. As noted above, while statistically not significant, we also observed a shift of tau for the LIG group toward a value of ASIC3‐expressing L cells.
Lactic acid produced by muscle or administered to the epigastric artery evokes a reflex pressor response by stimulating group III and IV afferents (Liu et al. 2010). Previous studies have shown that arterial injection of lactic acid in rats and cats leads to an increase in BP, an effect that is exaggerated in animals that have undergone femoral ligation for 72 h (Hayes et al. 2008; Liu et al. 2010; Xing et al. 2012). The non‐selective ASIC blocker, amiloride, and the ASIC3 channel toxin, APETx2, can attenuate the lactic acid pressor effects (Hayes et al. 2008; Liu et al. 2010; Xing et al. 2012). The intravenous administration of endomorphins generally results in a decrease of both BP and heart rate through MOR stimulation (Fichna et al. 2007). Our results showed the expected pressor effects from intra‐arterial administration of lactic acid in rats with FP and LIG femoral arteries. Administration of both lactic acid and E‐2 significantly enhanced the effect observed with lactic acid alone, an effect that was greater in LIG rats. This enhancement in rats with ligated femoral arteries was presumably a reflection of the increased ASIC3 expression levels and decreased ASIC1a protein. Since the rats were pretreated with naloxone, the endomorphin's ability to stimulate MOR was removed. Administration of APETx2 attenuated the E‐2‐mediated enhancement of the lactic acid pressor response, while pretreatment with PcTx‐1 significantly enhanced the response. Together with our in vitro findings, these results are consistent with our hypothesis that the increase in BP following E‐2 and lactic acid administration was a result of potentiation of ASIC3 activation.
The results of this study seem to indicate that endomorphins may exhibit algesic properties under ischaemic conditions through a direct interaction with ASIC3 channels by potentiating the sustained currents in an acidic milieu. This is difficult to reconcile with the fact that endomorphin release is associated with pain relief via MOR stimulation in DRG neurons. One possible explanation by which endomorphins modulate ASIC currents is as follows: under normal conditions, an exercising muscle with adequate blood flow will release lactic acid and the interstitial space will become acidic. This triggers E‐2 release at the periphery and presynaptically within the dorsal horn. Lactic acid chelates Ca2+ ions (Immke & McCleskey, 2003) facilitating ASIC activity, Na+ entry and depolarization. Presynaptically, endomorphins are released by the primary afferent (Martin‐Schild et al. 1998; Fichna et al. 2007). E‐2, in turn, will bind to MOR pre‐ and post‐synaptically so that the ascending pain pathway is inhibited – providing pain relief. On the other hand, during conditions of chronic arterial insufficiency, there is an accumulation of inflammatory mediators including lactic acid (Julius & Basbaum, 2001). Our results suggest that under ischaemic conditions, ASIC3 expression increases while ASIC1 expression is downregulated (Fig. 3). Over time, the interstitial pH remains low and the higher levels of lactic acid chelate Ca2+ ions, thereby facilitating ASIC activation, increasing Na+ influx and leading to hyperexcitability and an enhanced lactic acid‐induced increase in BP (Figs 7 and 8). At the same time, E‐2 release is increased at the periphery and presynpatically, which leads to analgesia. Given that endomorphins are high affinity and selective MOR agonists, under these defined conditions, their continued presence may lead to receptor internalization. Additionally, E‐2 can directly potentiate ASIC3 channel currents (Fig. 2) which, given our data, can lead to an augmented lactic acid‐mediated increase in blood pressure. Similarly, at the dorsal horn, internalized MORs are not stimulated by E‐2 and the ascending pain pathway is uninterrupted. It is worth mentioning that some patients with PAD are prescribed potent opioid analgesics such as oxycodone to treat intermittent claudication (Samolsky‐Dekel et al. 2010). Thus, it is tempting to further speculate that oral intake of such opiates coupled with increased E‐2 levels lead to longer‐lasting MOR internalization and development of tolerance. With continued opiate use, the patient may experience opioid‐induced hyperalgesia, presumably as a result of E‐2‐induced potentiation and sensitization of ASIC3. We should emphasize that such a scenario is highly speculative, but lays the groundwork for future studies to delineate the opiate‐mediated modulation of ASIC currents under ischaemic conditions.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
MF performed electrophysiological recordings, qRT‐PCR and Western blotting assays in VRV's laboratory; JKD and PBH performed Western blotting assays, L‐cell transfection and qRT‐PCR assays in VRV's laboratory; JKD performed the imaging experiments in VRV's laboratory. JSK performed the in vivo BP measurements and surgical procedures in MPK's laboratory; and HLP performed the cloning and plasmid generation in Dr Stephen R. Ikeda's laboratory (Laboratory of Molecular Physiology, NIAAA/NIH). All authors contributed to the conception and design of experiments, as well as data analysis. VRV drafted the manuscript and all authors provided editorial comments. All authors approved the final version of the manuscript, all persons listed as authors qualify for authorship, and all those who qualify for authorship are included.
Funding
Funding was received via NIH RO1 AR‐059397 (VR‐V and MPK) and NIH PO1‐HL134609 (MPK).
Edited by: Don Bers & Kathleen Morgan
References
- Andria ML & Simon EJ (1999). Localization of promoter elements in the human mu‐opioid receptor gene and regulation by DNA methylation. Mol Brain Res 70, 54–65. [DOI] [PubMed] [Google Scholar]
- Abboud FM & Benson CJ (2015). ASICs and cardiovascular homeostasis. Neuropharmacology 94, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askwith CC, Cheng C, Ikuma M, Benson C, Price MP & Welsh MJ (2000). Neuropeptide FF and FMRFamide potentiate acid‐evoked currents from sensory neurons and proton‐gated DEG/ENaC channels. Neuron 26, 133–141. [DOI] [PubMed] [Google Scholar]
- Baron A & Lingueglia E (2015). Pharmacology of acid‐sensing ion channels—Physiological and therapeutical perspectives. Neuropharmacology 94, 19–35. [DOI] [PubMed] [Google Scholar]
- Cai Q, Qiu C‐Y, Qiu F, Liu T‐T, Qu Z‐W, Liu Y‐M & Hu W‐P (2014). Morphine inhibits acid‐sensing ion channel currents in rat dorsal root ganglion neurons. Brain Res 1554, 12–20. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang K, Yang T, Wang W, Mei XP, Zhu C, Wang W, Zhang FX & Li YQ (2015). Downregulation of spinal endomorphin‐2 correlates with mechanical allodynia in a rat model of tibia cancer. Neuroscience 286, 151–161. [DOI] [PubMed] [Google Scholar]
- Copp SW, Stone AJ, Li J & Kaufman MP (2015). Role played by interleukin‐6 in evoking the exercise pressor reflex in decerebrate rats: effect of femoral artery ligation. Am J Physiol Heart Circ Physiol 309, H166–H173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criqui MH & Aboyans V (2015). Epidemiology of peripheral artery disease. Circ Res 116, 1509–1526. [DOI] [PubMed] [Google Scholar]
- Deval E, Gasull X, Noel J, Salinas M, Baron A, Diochot S & Linguella E (2010). Acid‐sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther 128, 549–558. [DOI] [PubMed] [Google Scholar]
- Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V, Jodar M, Lazdunski M & Lingueglia E (2008). ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J 27, 3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichna J, Janecka A, Costentin J & Do Rego JC (2007). The endomorphin system and its evolving neurophysiological role. Pharmacol Rev 59, 88–123. [DOI] [PubMed] [Google Scholar]
- Fowkes FGR, Rudan D, Rudan I, Aboyans V, Denenberg JO & McDermott MM (2013). Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet 382, 1329–1340. [DOI] [PubMed] [Google Scholar]
- Grunder S & Pusch M (2015). Biophysical properties of acid‐sensing ion channels (ASICs). Neuropharmacology 94, 9–18. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H (1985). Intracellular pH during ischemia in skeletal muscle: relationship to membrane potential, extracellular pH, tissue lactic acid and ATP. Pflugers Arch 404, 342–347. [DOI] [PubMed] [Google Scholar]
- Hayes SG, McCord JL, Rainier J, Liu Z & Kaufman MP (2008). Role played by acid‐sensitive ion channels in evoking the exercise pressor reflex. Am J Physiol Heart Circ Physiol 295, H1720–H1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke DC & McCleskey EW (2001). Lactate enhances the acid‐sensing Na+ channel on ischemia‐sensing neurons. Nat Neurosci 4, 869–870. [DOI] [PubMed] [Google Scholar]
- Immke DC & McCleskey EW (2003). Protons open acid‐sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron 37, 75–84. [DOI] [PubMed] [Google Scholar]
- Jing L, Chu XP, Jiang YQ, Collier DM, Wang B, Jiang Q, Snyder PM & Zha XM (2012). N‐glycosylation of acid‐sensing ion channel 1a regulates its trafficking and acidosis‐induced spine remodeling. J Neurosci 32, 4080–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Jiang YQ, Jiang Q, Wang B, Chu XP & Zha XM (2011). The interaction between the first transmembrane domain and the thumb of ASIC1a is critical for its N‐glycosylation and trafficking. PLoS ONE 6, e26909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Jin K, Minami M, Chen D & Simon RP (2001). Global ischemia induces expression of acid‐sensing ion channel 2a in rat brain. J Cereb Blood Flow Metab 21, 734–740. [DOI] [PubMed] [Google Scholar]
- Julius D & Basbaum AI (2001). Molecular mechanisms of nociception. Nature 413, 203–210. [DOI] [PubMed] [Google Scholar]
- Kadurin I, Golubovic A, Leisle L, Schindelin H & Grunder S (2008). Differential effects of N‐glycans on surface expression suggest structural differences between the acid‐sensing ion channel (ASIC) 1a and ASIC1b. Biochem J 412, 469–475. [DOI] [PubMed] [Google Scholar]
- Kellenberger S & Schild L (2015). International Union of Basic and Clinical Pharmacology. XCI. Structure, function, and pharmacology of acid‐sensing ion channels and the epithelial Na+ channel. Pharmacol Rev 67, 1–35. [DOI] [PubMed] [Google Scholar]
- Kido K, Gautam M, Benson CJ, Gu H & Brennan TJ (2013). Effect of deep tissue incision on pH responses of afferent fibers and dorsal root ganglia innervating muscle. Anesthesiology 119, 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash JM, Nixon JC & Unthank JL (1995). Exercise training effects on collateral and microvascular resistances in rat model of arterial insufficiency. Am J Physiol 268, H125–H137. [DOI] [PubMed] [Google Scholar]
- Lingueglia E & Lazdunski M (2015). Acid‐sensing ion channels in the nervous system. Foreward. Neuropharmacology 94, 1. [DOI] [PubMed] [Google Scholar]
- Liu J, Gao Z & Li J (2010). Femoral artery occlusion increases expression of ASIC3 in dorsal root ganglion neurons. Am J Physiol Heart Circ Physiol 299, H1357–H1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madureira G & Hasson‐Voloch A (1988). Lactate utilization and influx in resting and working rat red muscle. Comp Biochem Physiol A Comp Physiol 89, 693–698. [DOI] [PubMed] [Google Scholar]
- Marra S, Ferru‐Clement R, Breuil V, Delaunay A, Christin M, Friend V, Sebille S, Cognard C, Ferreira T, Roux C, Euller‐Ziegler L, Noel J, Linguella E & Deval E (2016). Non‐acidic activation of pain‐related acid‐sensing ion channel 3 by lipids. EMBO J 35, 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Schild S, Gerall AA, Kastin AJ & Zadina JE (1998). Endomorphin‐2 is an endogenous opioid in primary sensory afferent fibers. Peptides 19, 1783–1789. [DOI] [PubMed] [Google Scholar]
- Mazzuca M, Heurteaux C, Alloui A, Diochot S, Baron A, Voilley N, Blondeau N, Escoubas P, Gelot A, Cupo A, Zimmer A, Zimmer AM, Eschalier A & Lazdunski M (2007). A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nature Neurosci 10, 943–945. [DOI] [PubMed] [Google Scholar]
- Mousa SA, Machelska H, Schafer M & Stein C (2002). Immunohistochemical localization of endomorphin‐1 and endomorphin‐2 in immune cells and spinal cord in a model of inflammatory pain. J Neuroimmunol 126, 5–15. [DOI] [PubMed] [Google Scholar]
- Samolsky‐Dekel BG, Melotti RM, Gargiulo M, Freyrie A, Stella A & Di Nino G (2010). Pain management in peripheral arterial obstructive disease: oral slow‐release oxycodone versus epidural L‐bupivacaine. Eur J Vasc Endovasc Surg 39, 774–778. [DOI] [PubMed] [Google Scholar]
- Sherwood TW & Askwith CC (2009). Dynorphin opioid peptides enhance acid‐sensing ion channel 1a activity and acidosis‐induced neuronal death. J Neurosci 29, 14371–14380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA & Gregory NS (2015). The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 94, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ES, Cadiou H & McNaughton PA (2007). Arachidonic acid potentiates acid‐sensing ion channels in rat sensory neurons by a direct action. Neuroscience 145, 686–698. [DOI] [PubMed] [Google Scholar]
- Williams CA, Wu SY, Dun SL, Kwok EH & Dun NJ (1999). Release of endomorphin‐2 like substances from the rat spinal cord. Neurosci Lett 273, 25–28. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C & Lazdunski M (1997). A proton‐gated cation channel involved in acid‐sensing. Nature 386, 173–177. [DOI] [PubMed] [Google Scholar]
- Xing J, Lu J & Li J (2012). Acid‐sensing ion channel subtype 3 function and immunolabelling increases in skeletal muscle sensory neurons following femoral artery occlusion. J Physiol 590, 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi J, Wenk HN, Naves LA & McCleskey EW (2006). Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ Res 99, 501–509. [DOI] [PubMed] [Google Scholar]
- Zadina JE, Hackler L, Ge LJ & Kastin AJ (1997). A potent and selective endogenous agonist for the mu‐opiate receptor. Nature 386, 499–502. [DOI] [PubMed] [Google Scholar]