

Abstract
Key points
In response to exercise, vasodilatation ascends from downstream arterioles into upstream feed arteries (FAs). We hypothesized that the signalling events underlying ascending vasodilatation variy with the intensity and duration of skeletal muscle contraction.
In the gluteus maximus muscle of C57BL/6 mice, brief tetanic contraction evoked rapid onset vasodilatation (ROV) (<1 s) throughout the resistance network. Selective damage to endothelium midway between FAs and primary arterioles eliminated ROV only in FAs. Blocking SKCa and IKCa channels attenuated ROV, implicating hyperpolarization as the underlying signal.
During rhythmic twitch contractions, slow onset vasodilatation (10–15 s) in FAs remained intact following loss of ROV and was eliminated following nitric oxide synthase inhibition.
Tetanic contraction initiates hyperpolarization that conducts along endothelium into FAs. Rhythmic twitch contractions stimulate FA endothelium to release nitric oxide in response to elevated shear stress secondary to metabolic dilatation of arterioles. Complementary endothelial signalling pathways for ascending vasodilatation ensure increased oxygen delivery to active skeletal muscle.
Abstract
In response to exercise, vasodilatation initiated within the microcirculation of skeletal muscle ascends the resistance network into upstream feed arteries (FAs) located external to the tissue. Ascending vasodilatation (AVD) is essential for reducing FA resistance that otherwise restricts blood flow into the microcirculation. In the present study, we tested the hypothesis that signalling events underlying AVD vary with the intensity and duration of muscle contraction. In the gluteus maximus muscle of anaesthetized male C57BL/6 mice (aged 3–4 months), brief tetanic contraction (100 Hz for 500 ms) evoked rapid onset vasodilatation (ROV) in FAs that peaked within 4 s. By contrast, during rhythmic twitch contractions (4 Hz), slow onset vasodilatation (SOV) of FAs began after ∼10 s and plateaued within 30 s. Selectively damaging the endothelium with light‐dye treatment midway between a FA and its primary arteriole eliminated ROV in the FA along with conducted vasodilatation of the FA initiated on the arteriole using ACh microiontophoresis. Superfusion of SKCa and IKCa channel blockers UCL 1684 + TRAM 34 attenuated ROV, implicating endothelial hyperpolarization as the underlying signal. Nevertheless, the SOV of FAs during rhythmic contractions persisted until inhibition of nitric oxide synthase with N ω‐nitro‐l‐arginine methyl ester. Thus, ROV of FAs reflects hyperpolarization of downstream arterioles that conducts along the endothelium into proximal FAs. By contrast, SOV of FAs reflects the local production of nitric oxide by the endothelium in response to luminal shear stress, which increases secondary to arteriolar dilatation downstream. Thus, AVD ensures increased oxygen delivery to active muscle fibres by reducing upstream resistance via complementary signalling pathways that reflect the intensity and duration of muscle contraction.
Keywords: blood flow, endothelium, exercise, microcirculation
Key points
In response to exercise, vasodilatation ascends from downstream arterioles into upstream feed arteries (FAs). We hypothesized that the signalling events underlying ascending vasodilatation variy with the intensity and duration of skeletal muscle contraction.
In the gluteus maximus muscle of C57BL/6 mice, brief tetanic contraction evoked rapid onset vasodilatation (ROV) (<1 s) throughout the resistance network. Selective damage to endothelium midway between FAs and primary arterioles eliminated ROV only in FAs. Blocking SKCa and IKCa channels attenuated ROV, implicating hyperpolarization as the underlying signal.
During rhythmic twitch contractions, slow onset vasodilatation (10–15 s) in FAs remained intact following loss of ROV and was eliminated following nitric oxide synthase inhibition.
Tetanic contraction initiates hyperpolarization that conducts along endothelium into FAs. Rhythmic twitch contractions stimulate FA endothelium to release nitric oxide in response to elevated shear stress secondary to metabolic dilatation of arterioles. Complementary endothelial signalling pathways for ascending vasodilatation ensure increased oxygen delivery to active skeletal muscle.
Abbreviations
- 1A
first‐order arteriole
- 2A
second‐order arteriole
- 3A
third‐order arteriole
- AVD
ascending vasodilatation
- EC
endothelial cell
- EDH
endothelium‐derived hyperpolarization
- FA
feed artery
- FMD
flow‐mediated dilatation
- FITC
fluorescein isothiocyanate
- GM
gluteus maximus muscle
- IKCa
intermediate conductance Ca2+‐activated K+ channel
- I
indomethacin
- ID
internal diameter
- KIR
inward rectifying K+ channels
- L
l‐NAME (N ω‐nitro‐l‐arginine methyl ester)
- LDT
light dye treatment
- MBF
muscle blood flow
- NO
nitric oxide
- PE
phenylephrine
- ROV
rapid onset vasodilatation
- PSS
physiological salt solution
- SKCa
small conductance Ca2+‐activated K+ channel
- SMC
smooth muscle cell
- SNP
sodium nitroprusside
- SOV
slow onset vasodilatation
- T
TRAM 34
- U
UCL 1684
Introduction
In exercising skeletal muscle, arteriolar networks control regional blood flow and capillary perfusion in accordance with the metabolic demands of active muscle fibres. Embedded within the tissue, these microvessels are exquisitely sensitive to changes in their local milieu (Duling & Berne, 1970; Granger et al. 1975; Johnson & Henrich, 1975). Feed arteries (FAs) are located upstream, external to the muscle and positioned to govern the total volume of blood flowing into the muscle. Because FAs can exert half of the total resistance to muscle blood flow (MBF), vasodilatation initiated within a muscle must ascend from arterioles into their FAs to attain peak levels of tissue perfusion and oxygen delivery (Segal & Duling, 1986a; Segal & Jacobs, 2001). Although ascending vasodilatation (AVD) in response to skeletal muscle contractions has been demonstrated in cats (Hilton, 1959; Folkow et al. 1971) and rodents (Segal & Duling, 1986a; VanTeeffelen & Segal, 2006; Sinkler et al. 2016), the onset of AVD has varied from near‐instantaneous to requiring several seconds, suggesting that more than one signalling pathway is involved.
Following a brief contraction of the forearm or knee extensors in humans, MBF increases within the first second post‐contraction (Corcondilas et al. 1964; Tschakovsky et al. 1996; Crecelius et al. 2013; Credeur et al. 2015). Direct observations of the microcirculation in skeletal muscles of rodents reveal almost identical kinetics for such rapid onset vasodilatation (ROV) in arterioles and their FAs in response to a brief tetanic contraction (VanTeeffelen & Segal, 2006; Sinkler et al. 2016). By contrast, slow onset vasodilatation (SOV) in response to rhythmic twitch contractions entails a lag of several seconds before arterioles begin to dilate (Gorczynski et al. 1978; Dodd & Johnson, 1991; VanTeeffelen & Segal, 2000; Fernando et al. 2016). MBF then increases progressively to plateau at a level that corresponds to the intensity of exercise and oxygen demand (Anrep & Von Saalfeld, 1935; Andersen & Saltin, 1985; Armstrong et al. 1987; Musch et al. 1987), reflecting the dilatation of upstream branches of the vascular supply (Folkow et al. 1971; Segal & Duling, 1986a). These spatial and temporal relationships imply that signalling events governing functional vasodilatation during exercise vary with location in the resistance network and with the nature of contractile activity. Whether and how distinct signalling pathways mediate ROV versus SOV during AVD remains to be resolved.
Vasodilatation originating in downstream vessels will increase blood flow velocity through upstream vessels, thereby elevating luminal shear stress, which stimulates the endothelium to release nitric oxide (NO) and elicit flow‐mediated dilatation (FMD). This response requires 8–10 s to begin in arterioles (Koller & Kaley, 1990; Smiesko & Johnson, 1993) and up to ∼40 s in arteries (Lie et al. 1970; Pohl et al. 1986). As shown in arterioles (Koller et al. 1994) and in arteries (Pohl et al. 1986), disrupting the endothelium eliminates FMD, as does the inhibition of autacoid production, particularly NO (Joannides et al. 1995; Kooijman et al. 2008; Wilson et al. 2016), although prostacyclin may also contribute (de Nucci et al. 1988; Koller et al. 1994; Boushel et al. 2002; Schrage et al. 2004). The endothelium is also integral to conducted vasodilatation (Segal & Duling, 1986b). Once initiated through activation of small‐ and intermediate‐conductance Ca2+‐activated K+ channels (SKCa and IKCa, respectively), hyperpolarization spreads through gap junctions from cell to cell along the endothelium and into surrounding smooth muscle cells (SMCs) (Emerson & Segal, 2000a; Bagher & Segal, 2011). Although typically initiated with ACh, endothelium‐dependent hyperpolarization (EDH) has been confirmed for skeletal muscle FAs and implicated in AVD during contractile activity (Segal & Duling, 1986a; Emerson & Segal, 2000b; Segal & Jacobs, 2001). Nevertheless, it is unknown whether the role of cell‐to‐cell conduction or that of FMD in mediating AVD varies with the intensity and duration of skeletal muscle contraction. Therefore, the present study aimed to gain insight into the signals governing MBF regulation in response to a single brief contraction (as would occur with a vertical jump) versus rhythmic activity (as would occur during running and cycling). Using intravital microscopy of the gluteus maximus muscle (GM), which is recruited during both types of activity, we tested the hypothesis that the signalling events underlying AVD of FAs differ between single tetanic versus rhythmic twitch contractions.
Methods
Ethical approval
Experimental protocols were approved by the Animal Care and Use Committee of the University of Missouri and were performed in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals (2011).
Animal care and use
Male C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) at ∼3 months of age and acclimated in animal care facilities of the University of Missouri for at least 1 week prior to study. Mice were maintained under a 12:12 h light/dark cycle at 22–24°C with food and water available ad libitum. One mouse was studied per day. At the time of experimentation, body mass was 28 ± 1 g (n = 20). On the morning of an experiment, a mouse was anaesthetized with pentobarbital sodium (60 mg kg−1) and supplemented as needed (20 mg kg−1) to maintain a stable plane of anaesthesia. Upon completion of experiments (duration, ∼5‐6 h), the mouse was killed with an overdose of pentobarbital (all i.p. injection) followed by cervical dislocation.
Surgical preparation of the GM
The GM was used for the present experiments because it is a powerful extensor of the hip that is recruited during locomotion (Bearden et al. 2004). The mouse GM is comprised of mixed muscle fibre type (Lampa et al. 2004; Manttari & Jarvilehto, 2005), as is typical of human skeletal muscle (Saltin et al. 1977; Lexell & Downham, 1991). To prepare the GM for these experiments, the left hindquarter was shaved and the mouse was placed in a prone position on a warming plate to maintain oesophageal temperature at 37°C. Surgical procedures were performed while viewing through a stereomicroscope. The skin and connective tissue overlying the left GM were carefully removed. Exposed tissue was superfused continuously with bicarbonate‐buffered physiological salt solution (PSS) at 3 ml min−1 comprised of (in mm): 131.9 NaCl, 4.7 KCl (purchased from Fisher Scientific; Pittsburgh, PA, USA), 2 CaCl2, 1.17 MgSO4 and 18 NaHCO3 (purchased from Sigma Chemical Co.; St Louis, MO, USA) equilibrated with 5% CO2/95% N2 (pH 7.4, 35°C). With great care, the muscle was dissected away from its insertion along the lumbar fascia and iliac crest, reflected away from the hindquarter and spread over a transparent rubber pedestal (Sylgard 184; Dow‐Corning; Midland, MI, USA) with the edges pinned to approximate in situ dimensions; muscle thickness is ≤200 μm. The vascular resistance network was thereby exposed, including the FA (i.e. the inferior gluteal artery before it enters the muscle), which becomes the primary arteriole (1A) upon entering the muscle, and downstream second‐ (2A) and third‐ (3A) order branches (Fig. 1). The inferior gluteal motor nerve bundle was then isolated and cut proximally to leave a stump that was aspirated into a borosilicate glass microelectrode filled with PSS, and the interface between the electrode and surrounding tissues was insulated with Kwik‐Cast Sealant (World Precision Instruments Inc., Sarasota, FL, USA) (Sinkler et al. 2016). The completed preparation was transferred to the fixed stage of an intravital microscope (E600FN; Nikon, Melville, NY, USA) mounted on an X‐Y translational stage (Gibraltar; Burleigh Instruments, Fishers, NY, USA) and equilibrated for 30 min. During this period, a map of the resistance network was drawn and observation sites identified for each vessel branch with reference to anatomical landmarks.
Figure 1. Experimental preparation of mouse GM.
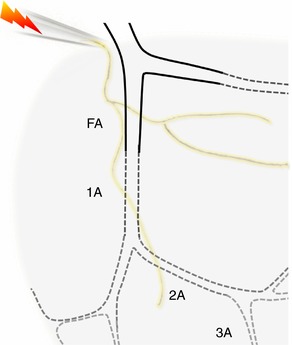
The resistance vessel network supplying the inferior region of the GM includes a FA located external to the muscle (continuous lines) and branches (1A, 2A, 3A) of the arteriolar network embedded within muscle fibres (dotted lines). The motor nerve was secured in a suction electrode and stimulated to evoke muscle contractions while vasomotor responses were recorded for each of the respective branch orders.
Intravital imaging
A FireWire colour charge‐coupled device camera (DFK 21AF04; The Imaging Source, Charlotte, NC, USA) acquired the image via a SLWD 20× objective (Nikon) (numerical aperture = 0.35), stored on a personal computer and displayed on a digital monitor with spatial resolution of ∼1 μm. Digital images were recorded at 30 frames s–1 using custom LabView software (National Instruments, Austin, TX, USA) with an integrated video caliper (developed by Dr Michael J. Davis, University of Missouri). Respective vessel branch orders were studied in each GM preparation as described below in the Experimental protocols. For each vessel studied, the internal diameter (ID; defined as the widest distance between lumen edges) was measured during playback using a video caliper integrated into the software. Only preparations exhibiting robust spontaneous vasomotor tone were studied. At the end of each day, maximal IDs for each observation site were recorded after equilibrating with sodium nitroprusside (SNP) (10−4 m; Sigma) added to the superfusion solution.
Motor nerve stimulation
The motor nerve was stimulated at 30 V with a pulse of 0.1 ms (Sinkler et al. 2016), which corresponded to a 10‐fold greater voltage than the threshold determined for evoking a muscle twitch. The nature of muscle contraction was controlled by electrical stimulation delivered through a SIU5 stimulus isolation unit driven by a square wave stimulator (S48; Grass Instruments, Quincy, MA, USA). For ROV, stimulating at 100 Hz evoked maximal tetanic contraction for 500 ms (Fernando et al. 2016). For SOV, stimulating at 4 Hz for 30 s evoked rhythmic twitch contractions (Jackson et al. 2010); force developed during twitch contractions is one‐third of that developed during tetanic contraction at 100 Hz (Bearden et al. 2004; Fernando et al. 2016). One vessel branch (FA, 1A, 2A or 3A) was observed for each set of contractions (500 ms at 100 Hz + 30 s at 4 Hz) and then the next branch was studied in the same manner. The order in which respective branches were observed was randomized across experiments. For each stimulation, recording was initiated prior to the onset of contraction and maintained throughout recovery until the resting diameter was restored, with an additional 2 min rest before the next contraction. After recording control responses to contractions in each branch, we repeated stimulation protocols during respective experimental interventions. In preliminary time controls (n = 4; data not shown), vasomotor responses in all four branch orders were maintained for four complete sets of stimulation in this manner, confirming that vasomotor tone recovered and baseline resting diameters remained stable for the duration of criterion experiments.
Light‐dye treatment (LDT)
In the presence of oxygen, excitation of a fluorochrome generates reactive oxygen species that can be used to disrupt the integrity of cell membranes and fluorescein is particularly effective in this regard (Rumbaut & Sial, 1999). By conjugating the fluorochrome to a molecule that approximates the size of albumin and introducing it into the circulation, the dye is constrained to the vascular compartment with the generation of reactive oxygen species directed to endothelial cells (ECs) in the region of illumination (Bartlett & Segal, 2000; Budel et al. 2003; Looft‐Wilson et al. 2004). Thus, fluorescein isothiocyanate conjugated to dextran (FITC‐Dextran, 70 kDa; Molecular Probes/ThermoFisher Scientific, Waltham, MA, USA) was dissolved in sterile saline (10 mg ml−1) and injected (10 μl g−1 body mass) into the retro‐orbital sinus (Yardeni et al. 2011) to obtain a circulating concentration of ∼1 mg ml−1 of blood, with the blood volume estimated as ∼10% of body mass (Riches et al. 1973). The site where a FA enters the muscle parenchyma and transitions to become the 1A (Fig. 2) was illuminated (excitation, 460–500 nm) with a 100‐W mercury arc lamp directed through the same objective used for imaging, thereby restricting damage to an intermediate segment ∼300 μm long. Using a photon intensity of ∼0.3 mW (confirmed daily with a PM100D power meter; Thorlabs Inc., Newton, NJ, USA), preliminary experiments established that 7–9 min of illumination effectively damaged ECs while preserving the integrity of surrounding SMCs.
Figure 2. LDT selectively damages endothelium.
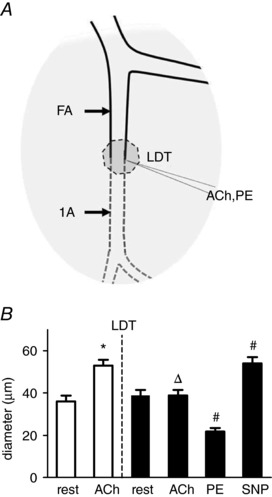
A, LDT was restricted to a segment ∼300 μm long centered where a FA enters the GM to become a 1A. Microiontophoresis delivered ACh or PE (in separate micropipettes) at the midpoint of the LDT segment, where these vasomotor responses were recorded. B, pre‐LDT (open bars), ACh increased the diameter by ∼47%. Post‐LDT (solid bars), selective damage of endothelium was confirmed with a loss of vasodilatation to ACh, whereas constriction to PE and dilatation to SNP confirmed the functional integrity of surrounding SMCs. Summary data are the mean ± SE, n = 5. * P < 0.05 versus rest pre‐LDT (dotted line); Δ P < 0.05 versus ACh response pre‐LDT; # P < 0.05 versus rest post‐LDT.
Microiontophoresis
For each experiment using LDT, microiontophoresis delivered the endothelium‐dependent vasodilator ACh or the α1‐adrenoreceptor agonist phenylephrine (PE) at a designated site (Duling et al. 1968). A borosilicate glass micropipette (∼1 μm tip ID) was filled with 1 m ACh or 1 m PE and secured in a holder mounted in a micromanipulator. A silver wire (diameter, 0.010′′) inserted into the micropipette was connected to the positive terminal of a microiontophoresis programmer (Model 260; World Precision Instruments Inc., Sarasota, FL, USA); retain current (400 nA) prevented leakage of the agonist. For the reference electrode, a second silver wire was secured at the edge of the GM and immersed in the superfusion solution. With the tip of the micropipette positioned adjacent to the site of LDT (Fig. 2), either ACh or PE was delivered using a pulse (1 s, 1000 nA) of positive current. Loss of dilatation to ACh confirmed disruption of the endothelium, whereas maintenance of constriction to PE confirmed the integrity of SMCs (Bartlett & Segal, 2000; Budel et al. 2003). To evaluate the effect of LDT on conducted vasodilatation, the ACh micropipette was positioned on the 1A with responses observed in its FA before and after LDT of the intermediate segment (Fig. 3).
Figure 3. LDT of endothelium eliminates conducted vasodilatation.
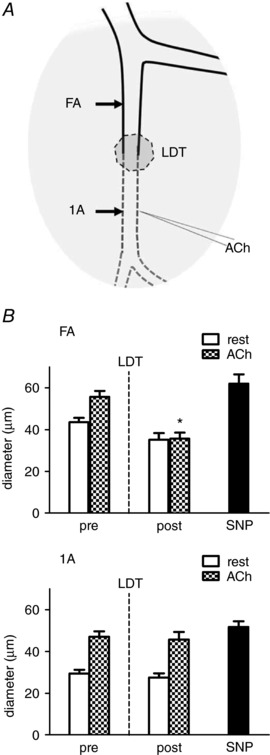
A, micropipette containing ACh was positioned ∼500 μm downstream from LDT segment with its tip adjacent to the 1A. Arrows indicate where 1A (local) and FA (upstream) diameters were recorded. B, top: conducted vasodilatation of FA. Local stimulation of 1A with ACh evoked vasodilatation that conducted upstream into the FA pre‐LDT. Conducted vasodilatation of the FA was eliminated post‐LDT but dilatation to topical SNP was preserved. Bottom: local response of 1A. Both pre‐ and post‐LDT, ACh evoked vasodilatation at the site of 1A stimulation, as did topical SNP. Summary data are the mean ± SE, n = 6. * P < 0.05 versus pre‐LDT.
Experimental protocols
Protocol 1: LDT
In six mice, vasodilatation to GM contractions and ACh microiontophoresis were observed downstream in the 1A and upstream in the FA before and following LDT performed at a site midway between respective segments (Fig. 2). First, ROV (in response to 500 ms contraction at 100 Hz) and SOV (30 s of twitch contractions at 4 Hz) were evaluated in the respective branches under control conditions. Next, ACh was delivered onto the 1A at a site located ∼500 μm downstream from where LDT was to be performed. The response to ACh was evaluated at the site of stimulation (‘local’) and in the FA ∼500 μm upstream from the site of LDT (i.e. 1000 μm upstream from the ACh micropipette) (Fig. 3). The FITC‐dextran was then injected into the circulation and the ACh micropipette was repositioned to the site of LDT, where the local response to ACh was evaluated to confirm EC integrity. LDT was then performed by opening the shutter to the arc lamp for 7–9 min until the endothelium was damaged, as confirmed by loss of the local response to ACh. The ACh micropipette was then replaced with a micropipette containing PE to evaluate the integrity of SMCs at the site of LDT as confirmed by vasoconstriction. Responses of the 1A and FA to tetanic (ROV) and rhythmic (SOV) contractions were then re‐evaluated, as were local and conducted responses to ACh delivered on the 1A. After loss of ROV and conducted vasodilatation in the FA following LDT, the preparation was equilibrated with N ω‐nitro‐l‐arginine methyl ester (l‐NAME) (10−4 m) in the superfusion solution to evaluate the contribution of NO to SOV. Thus, following local disruption of conduction along the endothelium, we tested for NO as the mediator of FMD upstream from the site of LDT (Joannides et al. 1995; Kooijman et al. 2008; Wilson et al. 2016).
Protocol 2: Pharmacology
In 11 mice, ROV and SOV were evoked during observation of the respective branches of the resistance network (Fig. 1) under control conditions with the sequence of branch order randomized across experiments. This procedure was followed by re‐evaluation of ROV and SOV after pharmacological agents were added to the superfusion solution and equilibrated for at least 15 min. The combination of indomethacin (I) (10−5 m) + l‐NAME (L) (10−4 m) was added to inhibit the synthesis of the endothelium‐derived autacoids prostacyclin and NO (de Nucci et al. 1988; Koller et al. 1994; Boushel et al. 2002; Schrage et al. 2004; Crecelius et al. 2013, 2014). The combination of TRAM 34 (T) (10−5 m) + UCL 1684 (U) (10−6 m) was added to block EDH by inhibiting SKCa and IKCa, respectively (Busse et al. 2002; Garland et al. 2011; Behringer & Segal, 2012b). Control experiments confirmed that this combination of U + T inhibited hyperpolarization to ACh (10−5.5 m) in endothelium freshly isolated from a FA (unpublished data in accordance with Behringer & Segal, 2012b). Indomethacin and l‐NAME were purchased from Sigma; UCL 1684 and TRAM 34 were purchased from Tocris Bioscience (Bristol, UK).
Preliminary studies indicated that complete washout of I + L or T + U was difficult to confirm. Thus, the effect of one pair of inhibitors was evaluated first, and then the second pair was added and the order of respective treatments (I + L or T + U) was alternated across experiments. In this manner, each pair of inhibitors was tested alone (i.e. autacoid inhibition or EDH inhibition) and in combination with the other pair of inhibitors. As an internal control for the effect of respective inhibitors on endothelium‐dependent dilatation, responses to increasing [ACh] were tested under each condition in FAs after evaluating ROV and SOV in respective branch orders.
At the end of each day, maximal dilatation was induced by adding SNP (10−4 m) to the superfusion solution and IDmax was recorded for respective branch orders (Sinkler & Segal, 2014; Sinkler et al. 2016).
Muscle force production
In three mice, we determined whether the presence of pharmacological inhibitors affected tetanic force production (Sinkler et al. 2016).
Data acquisition
Following the initial 30 min equilibration, resting internal diameter (IDrest) was recorded for each vessel branch order. Vessel IDs were recorded under each of the experimental conditions and the response of each vessel branch (IDresponse) was measured at designated time points during playback of the video recordings. To facilitate comparisons in the same vessels across pharmacological interventions and among respective branch orders with different resting and maximal (IDmax) diameters, vasodilatation was normalized to the maximal change in diameter during superfusion with 10−4 m SNP (Sinkler & Segal, 2014; Sinkler et al. 2016). Thus, vasodilatation (% maximal response) = (IDresponse – IDrest)/(IDmax – IDrest) × 100%. Wherever normalization was performed, absolute values for IDrest and IDmax are included for reference (Table 1). The amplitude of ROV was quantified as the peak vasodilatation attained following a tetanic contraction and normalized to IDmax of respective branch orders (% max). The amplitude of SOV was quantified (% max) during the plateau of the steady‐state response during rhythmic twitch contractions.
Table 1.
Effect of pharmacological agents on resting diameters
| Branch order | Control | I + L | T + U | I + L + T + U |
|---|---|---|---|---|
| FA | 38 ± 2 | 32 ± 1* | 29 ± 2* | 30 ± 2* |
| 1A | 30 ± 1 | 27 ± 2 | 30 ± 2 | 25 ± 2* |
| 2A | 19 ± 1 | 15 ± 1 | 19 ± 2 | 19 ± 2 |
| 3A | 9 ± 1 | 7 ± 1 | 10 ± 1 | 9 ± 1 |
Resting ID (μm) decreased in FA during I + L, T + U, I + L + T + U and in 1A during I + L + T + U. Resting ID of 2A and 3A were not significantly altered under the respective conditions.
I, indomethacin (10−5 m); L, l‐NAME (10−4 m); T, TRAM 34 (10−5 m); U, UCL 1684 (10−6 m). * P < 0.05 versus control (n = 11 for control, n = 6 for I + L, n = 5 for T + U, n = 10 for I + L + T + U).
Statistical analysis
The effects of LDT at the site of illumination were analysed using repeated one‐way ANOVA with Tukey's tests performed for post hoc comparisons. Differences in vasodilatation among vascular branch orders with respect to the effects of LDT or pharmacological treatments were analysed using two‐way ANOVA with Bonferroni tests for post hoc comparisons. Summary data are presented as the mean ± SE; values for n represent the number of individual mice in each group. P < 0.05 was considered statistically significant.
Results
Protocol 1: LDT
Selective damage of endothelium
To evaluate the selectivity of damage, ACh was delivered at the site of LDT using microiontophoresis. Under control conditions, ACh increased ID by 47% (Fig. 2 A). After performing LDT, an identical ACh stimulus had no effect, confirming EC damage. In turn, microiontophoresis of PE at the same site reduced ID by 44%, whereas superfusion with SNP increased ID to a level not different from the control response to ACh, confirming the functional integrity of SMCs surrounding damaged ECs (Fig. 2 B).
Loss of conducted vasodilatation following selective damage to endothelium with LDT
Under control conditions, local stimulation of a 1A with the ACh micropipette positioned ∼500 μm downstream from the midpoint of the LDT segment (Fig. 3 A) evoked local vasodilatation and this response conducted into the FA as recorded 1000 μm upstream (i.e. ∼500 upstream from the midpoint of LDT) (Fig. 3 B). Following LDT, local dilatation to ACh was intact in the 1A, whereas conducted vasodilatation into the FA was abolished, thereby confirming the endothelium as the cellular pathway for conduction (Fig. 3 B). Addition of SNP to the superfusion solution evoked maximal dilatation at both sites, confirming the ability of SMCs to relax irrespective of EC damage.
Loss of AVD for ROV following endothelial damage
A 500‐ms tetanic contraction resulted in ROV that peaked within ∼4 s for 1As and FAs (Fig. 4 A). Following LDT, ROV persisted in the 1A but was eliminated in the FA. Nevertheless, the addition of SNP dilated both sites maximally. Thus, LDT eliminated AVD for ROV and conducted vasodilatation to ACh in the same manner.
Figure 4. LDT of endothelium eliminates AVD for ROV but not for SOV until NO synthase is inhibited.
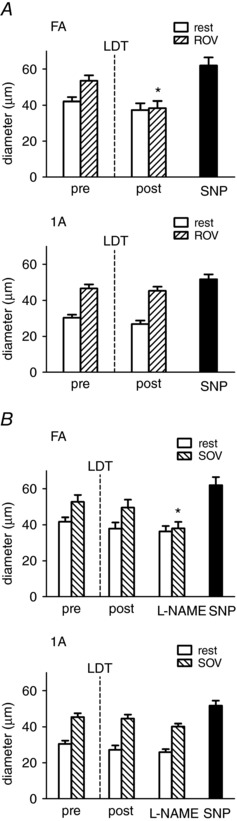
The GM was stimulated (as in Fig. 1) to evoke contractions pre‐ and post‐LDT (as in Fig. 3). A, top: in FA, tetanic contraction (500 ms, 100 Hz) evoked ROV pre‐LDT; this response was eliminated post‐LDT, whereas dilatation to topical SNP was preserved. Bottom: in 1A, ROV remained intact following LDT along with dilatation to topical SNP. B, for rhythmic twitch contractions (4 Hz, 30 s), SOV remained intact in both FA (top) and 1A (bottom) following LDT. Following addition of l‐NAME, SOV was eliminated in FA but not in 1A. Summary data are the mean ± SE, n = 6. * P < 0.05 versus pre‐LDT.
Preservation of AVD for SOV following LDT until inhibition of NO synthase
Slow onset vasodilatation began within 10–15 s of initiating twitch contractions and plateaued by 30 s in all branch orders. Following LDT, SOV was not different from control in either the 1A or the FA. However, following equilibration with l‐NAME in the superfusion solution, SOV was abolished in the FA yet persisted in the 1A within the muscle (Fig. 4 B). These findings indicate differential regulation of respective segments along a common path for blood flow according to anatomical location (Fig. 2).
Protocol 2: Networks and pharmacology
Vessel diameters, vasomotor tone and pharmacological interventions
Internal diameters decreased (Fig. 5 A) and spontaneous vasomotor tone increased (Fig. 5 B) from proximal FA to distal 3A branches. Equilibration with either I + L (to inhibit autacoids) or T + U (to inhibit EDH) constricted FAs significantly, as did the combination of I + L + T + U (Table 1). For 1A, only the combination of all four agents produced constriction. By contrast, combined inhibition did not affect the resting diameters of 2A or 3A branches. Thus, vasomotor tone was enhanced only in proximal branches during superfusion with these pharmacological agents, consistent with differential regulation of microvessel diameter according to branch order.
Figure 5. Vessel diameters and vasomotor tone along the resistance network.
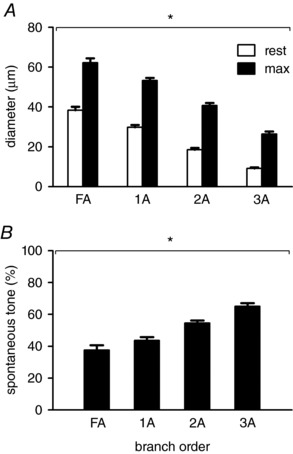
A, Internal diameters decreased and B, spontaneous vasomotor tone (calculated as [(IDmax – IDrest)/IDmax] × 100%) increased with vessel branch order (FA, 38 ± 3%; 1A, 44 ± 2%; 2A, 55 ± 2%; 3A, 66 ± 2%). Summary data are the mean ± SE; n = 11. * P < 0.05, main effect of branch order.
We used ACh as a well‐defined stimulus for endothelium‐dependent vasodilatation in the microcirculation (Emerson & Segal, 2000b; Budel et al. 2003; Looft‐Wilson et al. 2004; Sinkler & Segal, 2014) that acts through Gq protein‐coupled muscarinic receptors to raise [Ca2+]i, thereby activating both EDH (via SKCa and IKCa) and vasodilator autacoid production. In FAs, inhibition of respective signalling pathways shifted the [ACh]‐response curve to the right (Fig. 6). The effects of these inhibitors on arterioles was difficult to resolve (data not shown), which we attribute to differences in their regulation as explained earlier.
Figure 6. Inhibiting endothelium‐derived autacoids and EDH shifts FA response to [ACh].
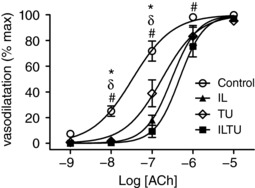
Superfusion of I + L to inhibit autacoids (NO + prostaglandins) and superfusion of T + U to inhibit SKCa and IKCa shifted the [ACh]‐response curve to the right, with the effect of I + L + T + U > I + L > T + U. Summary data are the mean ± SE (abbreviations and n as in Table 1). * P < 0.05, IL versus control, δ P < 0.05, TU versus control, # P < 0.05, ILTU versus control (abbreviations and n as in Table 1). Values are normalized to maximal vasodilatation with SNP to evaluate reactivity.
Tetanic contraction evoked ROV in all branch orders (Fig. 7). Under control conditions, ROV began within 1 s post‐contraction and attained peak values within 4 s in all vessel branch orders. To compare relative responses across vessels that differ in size (Fig. 5), vasodilatation was normalized to respective maximal dilatations with SNP (see Methods). In proximal FAs and 1As, inhibition of SKCa and IKCa with T + U attenuated ROV relatively more than inhibition of autacoids with I + L (Fig. 7). The combination of all four inhibitors attenuated peak dilatation most effectively in FAs and also doubled the time required to reach peak dilatation (∼8 s) across branch orders (Fig. 7). In turn, near‐lack of effect of these inhibitors on dilatation of 2As and 3As indicates that signals (e.g. metabolites) arising from surrounding muscle fibres prevail in these downstream branches.
Figure 7. Inhibiting autacoids and EDH attenuates ROV amplitude and time course most effectively in proximal branches.
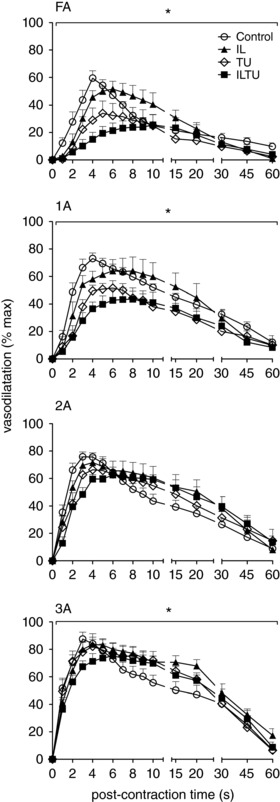
In response to 500 ms tetanic contraction, ROV peaked within 4 s under control conditions for all branch orders. For FA and 1A branches, addition of T + U had a relatively greater effect than I + L; combining I + L + T + U further attenuated dilatation at the same time as delaying the peak response to ∼8 s throughout the network. Summary data are the mean ± SE (abbreviations and n as in Table 1). * P < 0.05, main effect of inhibitors. Values are normalized to respective maximal dilatations with SNP to compare relative responses of branch orders that differ in diameter (Fig. 5).
For rhythmic twitch contractions, normalized vasodilatations increased with vessel branch order, however none of the pharmacological treatments affected SOV (Fig. 8; vessels as in Table 1). A key difference between Protocol 1 and Protocol 2 is that the endothelium remained intact in Protocol 2. Thus, even during the inhibition of KCa channels (and thereby EDH), cell‐to‐cell signalling through gap junctions along the endothelium could sustain AVD via alternative (albeit attenuated) pathways for initiating hyperpolarization; for example, activation of inward rectifying K+ channels (KIR) and/or Na+/K+ ATPase (Edwards et al. 1998; Crecelius et al. 2014).
Figure 8. Slow onset vasodilatation was unaffected by inhibiting autacoids or EDH.
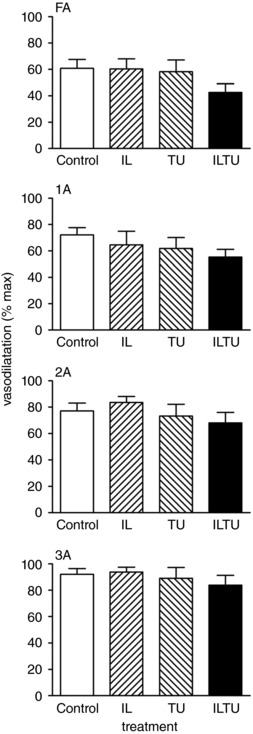
During 4 Hz rhythmic contractions, SOV increased from FA to 3A (P < 0.05). Superfusion of I + L or T + U had no effect on SOV in any branch order. The combination of I + L + T + U tended to decrease SOV in FA and 1A; however, the effect was not statistically significant. Summary data are the mean ± SE (abbreviations as in Table 1; n = 9 for Control, n = 4 for I + L, n = 5 for T + U, n = 9 for I + L + T + U). Values are normalized to respective maximal dilatations with SNP to compare the relative responses of vessel branch orders that differ in size (Fig. 5).
Muscle force production
Active force per cross‐sectional area of the GM during 500 ms tetanic contractions was 90 ± 5 mN mm−2 (n = 3). During superfusion with T + U alone or in combination with I + L, active force was 92 ± 1 and 92 ± 3 mN mm−2, respectively. Thus, pharmacological treatments did not interfere with force production, thereby excluding changes in skeletal muscle function as an explanation for any changes in vasodilatation.
Discussion
The present study has resolved fast and slow components of AVD for exercising skeletal muscle. Our findings indicate that a single tetanic contraction initiates ROV through hyperpolarizing arterioles embedded within the muscle. The electrical signal spreads through gap junctions along the endothelium and into SMCs, thereby ascending into FAs external to the muscle. Thus, via conducted vasodilatation (Segal, 1991; Bagher & Segal, 2011), rapid AVD increases MBF promptly in response to a robust contraction, facilitating the initial delivery of oxygen during the transition from rest to exercise. Although slower in onset, metabolic dilatation of arterioles during rhythmic contractions increases blood flow through proximal FAs, thereby stimulating NO release in response to elevated luminal shear stress predicted to occur in response to dilatation of arterioles downstream. Thus, through FMD (Lie et al. 1970; Pohl et al. 1986; Davies, 1995), slow AVD increases oxygen delivery in accordance with the prevailing demands of active muscle fibres. In such a manner, signalling events underlying endothelium‐dependent vasodilatation vary with the nature of contractile activity (Fig. 9). Even when endothelial conduction and flow‐mediated signalling of FAs are inhibited, metabolic dilatation of arterioles persists. Such redundancy among signalling pathways ensures that blood flow and oxygen delivery increase in response to the energetic demands of active muscle fibres.
Figure 9. Summary of rapid and slow ascending vasodilatation.
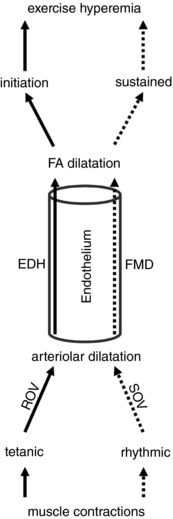
A tetanic contraction initiates ROV in arterioles via EDH, with hyperpolarization (and vasodilatation) conducted along the endothelium into the proximal FA. Rhythmic contractions give rise to SOV in arterioles through metabolic vasodilatation, resulting in FMD of the FA mediated by NO release in response to elevated shear stress.
With exercise onset and an increase in metabolic demand of active muscle fibres, the locus of blood flow control ascends the vascular supply, from distal arterioles regulating capillary perfusion and oxygen extraction into proximal arterioles and FAs that govern total blood flow and oxygen delivery (Folkow et al. 1971; Granger et al. 1976; Segal, 2005). A longstanding question centres on how vasodilatation ascends the resistance network from arterioles embedded within the tissue into the proximal arterial supply, remote from the direct effects of vasodilator metabolites. Two mechanisms have been proposed to mediate AVD: (i) initiation of an electrical signal in the microcirculation that spreads upstream from cell to cell along the vessel wall (Hilton, 1959) and (ii) initiation of FMD following elevated luminal shear stress (Lie et al. 1970; Pohl et al. 1986; Davies, 1995) in response to the fall in microvascular resistance downstream (Segal & Duling, 1986a; Seals et al. 2011). Although FMD of the brachial artery via reactive hyperaemia of the forearm microcirculation provides an index of endothelial function (Yeboah et al. 2007; Seals et al. 2011; Thijssen et al. 2011), in vivo evidence for endothelial conduction as a mechanism for rapid AVD has been elusive. By manipulating the nature of skeletal muscle contraction in conjunction with local disruption of the endothelium and selective pharmacological interventions, the present experiments resolve complementary roles for each of these signalling pathways for the first time.
Rapid AVD reflects endothelial conduction of hyperpolarization
Subsequent to the original experiments identifying a rapid increase in MBF following a handgrip contraction (Corcondilas et al. 1964), remarkable consistency has been demonstrated for the kinetics of ROV in the human forearm (Carlson et al. 2008; Casey & Joyner, 2012; Crecelius et al. 2013, 2014) and knee extensors (Credeur et al. 2015). Thus, the onset of ROV begins within 1 s post‐contraction, blood flow peaks within 4–5 s and the response amplitude increases with the intensity of contraction. Although evaluated as flow changes through a proximal artery, these measures in humans reflect integrated vasodilator responses in the microcirculation downstream. Remarkably, the kinetics of ROV observed for individual microvessels of rodent skeletal muscles in response to tetanic contraction approximate the time course shown for MBF in humans. Intravital imaging of the hamster retractor muscle (VanTeeffelen & Segal, 2006) and the mouse GM (Jackson et al. 2010; Sinkler et al. 2016) have shown that arterioles and FAs begin dilatation within 1 s post‐contraction; dilatation peaks within ∼4 s and its amplitude increases with contraction duration (100 to 1000 ms). These findings illustrate that invasive experiments performed using rodents provide mechanistic insight into functional responses recorded in humans.
ACh initiates EDH through SKCa and IKCa activation (Busse et al. 2002; Garland et al. 2011; Behringer & Segal, 2012b). Once initiated, hyperpolarization spreads from cell to cell through gap junctions along the endothelium and into SMCs as the signal travels along the vessel wall (Bagher & Segal, 2011). In turn, hyperpolarizing the membrane of SMCs elicits vasodilatation by inhibiting voltage gated Ca2+ channels, thereby reducing [Ca2+]i (Nelson et al. 1990). A key observation in the present experiments is the loss of ROV in a FA, whereas ROV in the 1A downstream remained intact (Fig. 4). Thus, local disruption of the endothelium abolished ROV of the FA in a manner identical to its effect on conducted vasodilatation (Fig. 3). These data confirm the endothelium as the cellular pathway for conducted vasodilatation from arterioles into proximal FAs and thereby identify its integral role to rapid AVD.
To resolve whether vasodilatation was ascending from distal to proximal branches within resistances network, we studied the dynamics of ROV for 3A, 2A, 1A and FA branches supplying the GM (Sinkler et al. 2016). These data revealed that ROV begins sooner and peaks earlier in the smallest arterioles located furthest downstream, then progresses upstream from 3A → 2A → 1A → FA. This spatiotemporal gradient for ROV is consistent with electrical signal transmission along the endothelium (Behringer & Segal, 2012a). Thus, ascending ROV is manifest within the branching hierarchy of microvascular networks. Moreover, conducted vasodilatation may be initiated in capillaries (Segal, 1991; Song & Tyml, 1993; Berg et al. 1997; Beach et al. 1998), where the endothelium is intimately associated with muscle fibres (Murrant et al. 2017). Recent findings in the cerebral circulation support this interpretation at the same time as highlighting an integral role for K+ channels (Longden et al. 2017).
The activation of SKCa and IKCa results in K+ efflux to produce EDH (Busse et al. 2002; Garland et al. 2011; Behringer & Segal, 2012b). In support of rapid AVD reflecting K+ channel activation, pharmacological inhibitors of EDH attenuated peak ROV in FAs by half and blunted peak dilatation throughout the resistance network (Fig. 7). Although our focus was on SKCa and IKCa, complementary signalling events can hyperpolarize ECs. For example, a 5–10 mm rise in [K+]o activates KIR channels to promote hyperpolarization and conducted vasodilatation (Jantzi et al. 2006). Elevation of [K+]o can also initiate hyperpolarization via stimulating Na+/K+ ATPase activity (Edwards et al. 1998). Even if these responses are initiated in SMCs, the signal is transmitted directly to ECs via myoendothelial gap junctions (Emerson & Segal, 2000a). In the mouse cremaster preparation, genetic deletion of SKCa attenuated rapid dilatation of arterioles in response to 1 s contraction of muscle fibres (at 100 Hz), whereas deletion of the gap junction protein connexin40 had a similar effect (Milkau et al. 2010). In human subjects, intra‐arterial infusion of ouabain (to inhibit Na+/K+ ATPase) in combination with Ba2+ (to inhibit KIR) attenuated rapid vasodilatation in the forearm (Crecelius et al. 2013). It is reassuring that these inhibitors had similar effects on rapid dilatations of arterioles to muscle fibre contraction in the hamster cremaster muscle (Armstrong et al. 2007). In the dog gastrocnemius muscle, a 1 s tetanic contraction elevated K+ in the venous effluent with a time course that explained the initial decrease vascular resistance (Mohrman & Sparks, 1974). Although K+ efflux from repolarizing skeletal muscle fibres can activate Na+/K+ ATPase and KIR channels in nearby microvessels, the link between muscle fibre contraction and KCa activation in the endothelium remains to be clarified. Nevertheless, the present findings indicate that a single tetanic contraction evokes EDH in arterioles that conducts along the endothelium into proximal FAs (Fig. 9). This signalling pathway for ascending ROV (i.e. rapid AVD) can increase blood flow into skeletal muscle commensurate with the onset of activity.
Slow AVD reflects NO release during FMD and the actions of vasodilator metabolites
In arterioles of the rat mesentery and cremaster muscle, there is a delay of ∼8 s between the increase in flow velocity and the onset of dilatation (Smiesko et al. 1989; Koller & Kaley, 1990). In the femoral artery of dogs, the latency between increasing flow and the onset of dilatation was 10–40 s (Lie et al. 1970; Pohl et al. 1986) and a lag of 10–15 s has been reported for FMD of the brachial artery in humans (Thijssen et al. 2011). In the present experiments, rhythmic contractions of the GM required 10–15 s to elicit SOV and continued to elicit slow AVD of the FA above the site of LDT. In light of studies showing FMD to be mediated by NO (Joannides et al. 1995; Kooijman et al. 2008; Wilson et al. 2016), we tested whether SOV of the FA following LDT was NO‐dependent by inhibiting NO synthase. Because this was found to be effective (Fig. 4), slow AVD in the absence of endothelial conduction can be explained by dilatation of downstream arterioles increasing blood flow velocity (and luminal shear stress) through the FA upstream (Segal & Duling, 1986a), thereby stimulating NO release. Nevertheless, dilatation persisted in arterioles (1A) of the GM even when SOV of the FA was eliminated (Protocol 1).
Prostacyclin also has been reported to contribute to FMD in arterioles (Koller et al. 1994) and the combination of inhibiting both NO synthase and cyclooxygenase has been used to test for endothelium‐dependent dilatation in humans (Boushel et al. 2002; Schrage et al. 2004; Crecelius et al. 2013). We therefore used indomethacin in combination with l‐NAME (I + L) for Protocol 2 to inhibit respective autacoids. During rhythmic contractions, there was no significant effect of I + L or T + U alone or in combination on SOV in any branch order (Fig. 8). This outcome raises the question of how vasodilatation occurs when SKCa and IKCa are inhibited together with NO synthase and cyclooxygenase. Because these inhibitors had no effect on the force produced during GM contraction (see Results), active muscle fibres continued to produce vasodilator metabolites. Accordingly, K+ release from muscle fibres can still activate Na+/K+ ATPase and/or KIR in microvessels embedded within the tissue and thereby give rise to hyperpolarization that conducted along the intact endothelium, albeit with a slower time course and attenuated vasomotor response versus control conditions. Furthermore, production of metabolic vasodilators (e.g. adenosine and inorganic phosphate) and progressive changes in tissue pO2, pCO2 and pH (Granger et al. 1975; Stowe et al. 1975; Hilton et al. 1978; Berne et al. 1983) should remain effective. By contrast, ROV cannot be explained by such changes in the local milieu, which require longer to develop (Corcondilas et al. 1964; Clifford, 2007). The present findings thereby substantiate the conclusion that signalling events initiating exercise hyperaemia (ROV) are distinct from those that maintain the steady‐state response (SOV) (Saltin et al. 1998; Clifford & Hellsten, 2004; Joyner & Casey, 2015). Furthermore, our data collectively illustrate spatiotemporal gradients, as well as redundancy in the contributions of ROV, FMD and metabolic vasodilatation to blood flow control in resistance networks supplying active skeletal muscle.
Critique of methods and experimental design
A limitation of the GM preparation is movement of the tissue in association with muscle fibre contraction, which makes it impossible to record corresponding electrical events in ECs and SMCs, particularly during a tetanic contraction and ROV. Having demonstrated that LDT disrupts the conduction of hyperpolarization (and vasodilatation) along the endothelium of isolated pressurized FAs (Emerson & Segal, 2000b), we used LDT to inhibit conducted vasodilatation as a reference for its effects on rapid AVD (Protocol 1). Under these conditions, NO synthase inhibition alone inhibited slow AVD, demonstrating FMD as an alternative signalling pathway for reducing upstream resistance when conduction along the endothelium is interrupted. It should be recognized that elevated shear stress in FAs subsequent to dilatation of downstream arterioles was not measured directly in these experiments and is inferred from previous studies (Lie et al. 1970; Pohl et al. 1986). Moreover, with conduction (and rapid AVD) abolished following LDT, the effect of arteriolar dilatation on elevating FA shear stress should enhance this stimulus for NO release.
As a further test regarding the roles of electrical and autacoid signalling, we delivered selective and validated pharmacological agents via the superfusion solution (Protocol 2), thereby affecting all branches of the network (FA, 1A, 2A and 3A) in addition to other cells within the tissue. Because FAs are external to the tissue and exposed directly to the superfusion solution, inhibition of EDH and autacoid signalling shifted their [ACh]‐response curve (Fig. 6). However, we were unable to identify such an effect for intramuscular arterioles. Nevertheless, the inability to resolve the effects of inhibiting KCa channels and autacoids on arteriolar responses to ACh does not preclude interpreting the actions of these inhibitors on ROV, particularly in proximal vessels (Fig. 7). Their lesser effect on distal branches in conjunction with a lack of effect on SOV further illustrates the multifaceted nature of functional vasodilatation and its differential regulation along the resistance network.
The peptide neurotoxins apamin (MW, 2027) + charybdotoxin (MW, 4296) are used as inhibitors of SKCa and IKCa, respectively (Busse et al. 2002; Garland et al. 2011; Behringer & Segal, 2012b). By contrast, we used respective synthetic channel blockers UCL 1684 (MW, 654) + TRAM 34 (MW, 345) to more readily access arterioles within the tissue after confirming their ability to block EDH (see Methods). Although there was no effect of pharmacological agents on SOV, T + U alone and in combination with I + L attenuated ROV in proximal branches. The attenuation of peak ROV amplitude observed in the present study is consistent with findings in humans, where Ba2+ + ouabain were administered to block the hyperpolarizing effect of elevated [K+]o on outward current of KIR and Na+/K+ activity (Crecelius et al. 2013, 2014). Our experiments focused on SKCa and IKCa as the basis of EDH and there may well be redundancy among respective signalling pathways involving K+ channels. A key finding is that our combination of inhibitors eliminated the spatiotemporal gradient for ROV that prevails under control conditions (Sinkler et al. 2016), thereby delaying responses throughout the network (Fig. 7). This outcome implies that the initial endothelial signalling events integral to co‐ordinating AVD among downstream and upstream branches become dysfunctional when K+ signalling is attenuated.
Although the evaluation of ROV and AVD is limited to non‐invasive, indirect measurements in proximal arteries of humans, direct observations of the microvessels actually controlling these responses are enabled using animal models. Similar signalling events are manifest in humans (Casey & Joyner, 2012; Crecelius et al. 2013, 2014; Credeur et al. 2015) and rodents (VanTeeffelen & Segal, 2006; Jackson et al. 2010; Sinkler et al. 2016). This conclusion is supported by the almost identical time course of ROV, as well as its sensitivity to the intensity of muscle contraction and to the complementary actions of experimental interventions. Nevertheless, a key difference between respective approaches entails the delivery of pharmacological agents. Addition to the superfusion solution as performed here presents agents to all cells in the tissue. Thus, an equilibration period (typically 15–20 min) ensured their access to arterioles embedded within the muscle. By contrast, intra‐arterial infusion in humans delivers agents via the bloodstream, which may promote a more direct effect on the endothelium. Finding that respective techniques have produced consistent results emphasizes the value of complementary approaches to resolving mechanisms underlying MBF regulation during exercise.
Summary
Using intravital imaging of the mouse GM, the present study has resolved distinct components of AVD by manipulating the nature of skeletal muscle contraction. Brief tetanic contraction evokes ROV in arterioles through initiating EDH. The electrical signal (hyperpolarization) conducts along the endothelium from within the muscle into proximal FAs located external to the tissue. In such a manner, rapid AVD serves to lower upstream resistance and increase MBF commensurate with the onset of activity and thereby facilitate the transition from rest. By contrast, rhythmic twitch contractions lead to an increase in flow through FAs secondary to functional dilatation of arterioles embedded within the muscle fibres. Slow AVD is mediated by NO release from FA endothelium in response to elevated shear stress. It serves to promote MBF in accordance with the prevailing level of contractile activity and persists following disruption of endothelial conduction. In such a manner, the mechanism of endothelium‐dependent vasodilatation can vary with the intensity and duration of muscle contraction (Fig. 9). Functional dilatation of arterioles persists even when electrical and flow‐dependent signalling of the endothelium are inhibited, confirming the redundancy of complementary signalling pathways that effect blood flow increases in response to the energetic demands of active muscle fibres.
Additional information
Competing interests
The authors declare that they have no competing interests. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the American Heart Association.
Author contributions
SYS and SSS conceived and designed the experiments. SYS performed the experiments in the laboratory of SSS. Both authors analysed and interpreted the data. SYS prepared the figures and drafted the manuscript. SSS edited the manuscript and figures. All authors have approved the final version of the manuscript submitted for publication and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by the National Institutes of Health grant R37‐HL041026 from the United States Public Health Service. SYS was supported by predoctoral fellowship 15PRE22840000 from the Midwest Affiliate of the American Heart Association.
Translational perspective
The present study illustrates an integral role for the endothelium in ascending vasodilatation (AVD) mediated through both electrical (conducted) and autacoid (via shear stress) signalling. Conditions accompanied by endothelial dysfunction include ageing (Seals et al. 2011), obesity (Bender & Laughlin, 2015; Kraemer‐Aguiar et al. 2015) and diabetes (Laughlin, 2016); each is associated with limited exercise capacity. In the mouse gluteus maximus muscle as studied here, both impaired ROV and restricted MBF during rhythmic contractions occur with advanced age (Jackson et al. 2010). These findings implicate a blunted ability to evoke dilatation in proximal segments of the resistance network. Ageing also impairs conducted vasodilatation (Bearden et al. 2004), probably as a result of the attenuated spread of hyperpolarization along the endothelium (Behringer et al. 2013). That ageing impairs flow‐mediated dilatation (FMD) in association with reduced nitric oxide bioavailability is documented in rats (Muller‐Delp et al. 2002), as well as humans (Seals et al. 2011). In accordance with our present findings, the difficulty in transitioning from rest to activity during advanced age may reflect impairment in the rapid component of AVD (conduction of hyperpolarization) in conjunction with increased sympathetic activation of vascular smooth muscle (Carlson et al. 2008; Jackson et al. 2010; Casey & Joyner, 2012; Sinkler et al. 2016). In turn, difficulty in sustaining activity may reflect impairment in the slow component of AVD as a consequence of endothelial dysfunction and impaired FMD (Kelm, 2002; Landmesser & Drexler, 2007; Seals et al. 2011). Under such conditions, proximal (feed artery) resistance can restrict muscle blood flow. The selective application of LDT to impair endothelial conduction in vivo has proven integral to resolving respective components of AVD. We thereby identify complementary signalling pathways for selective and strategic approaches to promote oxygen delivery and physical activity.
Acknowledgements
Charmain Fernando evaluated force production of the GM. Charles Norton and Charmain Fernando provided critical reviews of the manuscript.
Edited by: Scott Powers & Harold Schultz
References
- Andersen P & Saltin B (1985). Maximal perfusion of skeletal muscle in man. J Physiol 366, 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrep GV & Von Saalfeld E (1935). The blood flow through the skeletal muscle in relation to its contraction. J Physiol 85, 375–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RB, Delp MD, Goljan EF & Laughlin MH (1987). Distribution of blood flow in muscles of miniature swine during exercise. J Appl Physiol 62, 1285–1298. [DOI] [PubMed] [Google Scholar]
- Bagher P & Segal SS (2011). Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf) 202, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett IS & Segal SS (2000). Resolution of smooth muscle and endothelial pathways for conduction along hamster cheek pouch arterioles. Am J Physiol Heart Circ Physiol 278, H604–H612. [DOI] [PubMed] [Google Scholar]
- Beach JM, McGahren ED & Duling BR (1998). Capillaries and arterioles are electrically coupled in hamster cheek pouch. Am J Physiol Heart Circ Physiol 275, H1489–H1496. [DOI] [PubMed] [Google Scholar]
- Bearden SE, Payne GW, Chisty A & Segal SS (2004). Arteriolar network architecture and vasomotor function with ageing in mouse gluteus maximus muscle. J Physiol 561, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2012a). Spreading the signal for vasodilatation: implications for skeletal muscle blood flow control and the effects of ageing. J Physiol 590, 6277–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2012b). Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+‐activated K+ channels. Circ Res 110, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Shaw RL, Westcott EB, Socha MJ & Segal SS (2013). Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+‐activated K+ channel activation. Arterioscler Thromb Vasc Biol 33, 1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender SB & Laughlin MH (2015). Modulation of endothelial cell phenotype by physical activity: impact on obesity‐related endothelial dysfunction. Am J Physiol Heart Circ Physiol 309, H1–H8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg BR, Cohen KD & Sarelius IH (1997). Direct coupling between blood flow and metabolism at the capillary level in striated muscle. Am J Physiol Heart Circ Physiol 272, H2693–H2700. [DOI] [PubMed] [Google Scholar]
- Berne RM, Knabb RM, Ely SW & Rubio R (1983). Adenosine in the local regulation of blood flow: a brief overview. Fed Proc 42, 3136–3142. [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M & Kjaer M (2002). Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budel S, Bartlett IS & Segal SS (2003). Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: unmasking an NO wave. Circ Res 93, 61–68. [DOI] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte P & Weston A (2002). EDHF: bringing the concepts together. Trends Pharmacol Sci 23, 374–380. [DOI] [PubMed] [Google Scholar]
- Carlson RE, Kirby BS, Voyles WF & Dinenno FA (2008). Evidence for impaired skeletal muscle contraction‐induced rapid vasodilation in aging humans. Am J Physiol Heart Circ Physiol 294, H1963–H1970. [DOI] [PubMed] [Google Scholar]
- Casey DP & Joyner MJ (2012). Influence of α‐adrenergic vasoconstriction on the blunted skeletal muscle contraction‐induced rapid vasodilation with aging. J Appl Physiol 113, 1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS (2007). Skeletal muscle vasodilatation at the onset of exercise. J Physiol 583, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS & Hellsten Y (2004). Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97, 393–403. [DOI] [PubMed] [Google Scholar]
- Corcondilas A, Koroxenidis GT & Shepherd JT (1964). Effect of a brief contraction of forearm muscles on forearm blood flow. J Appl Physiol 19, 142–146. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2013). Mechanisms of rapid vasodilation after a brief contraction in human skeletal muscle. Am J Physiol Heart Circ Physiol 305, H29–H40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Luckasen GJ, Larson DG & Dinenno FA (2014). KIR channel activation contributes to onset and steady‐state exercise hyperemia in humans. Am J Physiol Heart Circ Physiol 307, H782–H791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credeur DP, Holwerda SW, Restaino RM, King PM, Crutcher KL, Laughlin MH, Padilla J & Fadel PJ (2015). Characterizing rapid‐onset vasodilation to single muscle contractions in the human leg. J Appl Physiol 118, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF (1995). Flow‐mediated endothelial mechanotransduction. Physiol Rev 75, 519–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nucci G, Gryglewski RJ, Warner TD & Vane JR (1988). Receptor‐mediated release of endothelium‐derived relaxing factor and prostacyclin from bovine aortic endothelial cells is coupled. Proc Natl Acad Sci U S A 85, 2334–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd LR & Johnson PC (1991). Diameter changes in arteriolar networks of contracting skeletal muscle. Am J Physiol Heart Circ Physiol 260, H662–H670. [DOI] [PubMed] [Google Scholar]
- Duling BR & Berne RM (1970). Longitudinal gradients in periarteriolar oxygen tension. A possible mechanism for the participation of oxygen in local regulation of blood flow. Circ Res 27, 669–678. [DOI] [PubMed] [Google Scholar]
- Duling BR, Berne RM & Born GVR (1968). Microiontophoretic application of vasoactive agents to the microcirculation of the hamster cheek pouch. Microvasc Res 1, 158–173. [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000a). Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87, 474–479. [DOI] [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000b). Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 86, 94–100. [DOI] [PubMed] [Google Scholar]
- Fernando CA, Liu Y, Sowa G & Segal SS (2016). Attenuated rapid onset vasodilation with greater force production in skeletal muscle of caveolin‐2−/‐ mice. Am J Physiol Heart Circ Physiol 311, H415–H425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkow B, Sonnenschein RR & Wright DL (1971). Loci of neurogenic and metabolic effects on precapillary vessels of skeletal muscle. Acta Physiol Scand 81, 459–471. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczynski RJ, Klitzman B & Duling BR (1978). Interrelations between contracting striated muscle and precapillary microvessels. Am J Physiol Heart Circ Physiol 235, H494–H504. [DOI] [PubMed] [Google Scholar]
- Granger HJ, Goodman AH & Cook BH (1975). Metabolic models of microcirculatory regulation. Fed Proc 34, 2025–2030. [PubMed] [Google Scholar]
- Granger HJ, Goodman AH & Granger DN (1976). Role of resistance and exchange vessels in local microvascular control of skeletal muscle oxygenation in the dog. Circ Res 38, 379–385. [DOI] [PubMed] [Google Scholar]
- Hilton SM (1959). A peripheral arterial conducting mechanism underlying dilatation of the femoral artery and concerned in functional vasodilatation in skeletal muscle. J Physiol 149, 93–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton SM, Hudlicka O & Marshall JM (1978). Possible mediators of functional hyperaemia in skeletal muscle. J Physiol 282, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DN, Moore AW & Segal SS (2010). Blunting of rapid onset vasodilatation and blood flow restriction in arterioles of exercising skeletal muscle with ageing in male mice. J Physiol 588, 2269–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ & Welsh DG (2006). Inward rectifying potassium channels facilitate cell‐to‐cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol 291, H1319–H1328. [DOI] [PubMed] [Google Scholar]
- Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C & Luscher TF (1995). Nitric oxide is responsible for flow‐dependent dilatation of human peripheral conduit arteries in vivo. Circulation 91, 1314–1319. [DOI] [PubMed] [Google Scholar]
- Johnson PC & Henrich HA (1975). Metabolic and myogenic factors in local regulation of the microcirculation. Fed Proc 34, 2020–2024. [PubMed] [Google Scholar]
- Joyner MJ & Casey DP (2015). Regulation of increased blood flow (hyperemia) to muscles during exercise: a hierarchy of competing physiological needs. Physiol Rev 95, 549–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm M (2002). Flow‐mediated dilatation in human circulation: diagnostic and therapeutic aspects. Am J Physiol Heart Circ Physiol 282, H1–H5. [DOI] [PubMed] [Google Scholar]
- Koller A & Kaley G (1990). Endothelium regulates skeletal muscle microcirculation by a blood flow velocity‐sensing mechanism. Am J Physiol Heart Circ Physiol 258, H916–H920. [DOI] [PubMed] [Google Scholar]
- Koller A, Sun D, Huang A & Kaley G (1994). Corelease of nitric oxide and prostaglandins mediates flow‐dependent dilation of rat gracilis muscle arterioles. Am J Physiol Heart Circ Physiol 267, H326–H332. [DOI] [PubMed] [Google Scholar]
- Kooijman M, Thijssen DH, de Groot PC, Bleeker MW, van Kuppevelt HJ, Green DJ, Rongen GA, Smits P & Hopman MT (2008). Flow‐mediated dilatation in the superficial femoral artery is nitric oxide mediated in humans. J Physiol 586, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer‐Aguiar LG, de Miranda ML, Bottino DA, Lima Rde A, de Souza M, Balarini Mde M, Villela NR & Bouskela E (2015). Increment of body mass index is positively correlated with worsening of endothelium‐dependent and independent changes in forearm blood flow. Front Physiol 6, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampa SJ, Potluri S, Norton AS & Laskowski MB (2004). A morphological technique for exploring neuromuscular topography expressed in the mouse gluteus maximus muscle. J Neurosci Methods 138, 51–56. [DOI] [PubMed] [Google Scholar]
- Landmesser U & Drexler H (2007). Endothelial function and hypertension. Curr Opin Cardiol 22, 316–320. [DOI] [PubMed] [Google Scholar]
- Laughlin MH ( 2016). Physical activity‐induced remodeling of vasculature in skeletal muscle: role in treatment of type 2 diabetes. J Appl Physiol 120, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexell J & Downham DY (1991). The occurrence of fibre‐type grouping in healthy human muscle: a quantitative study of cross‐sections of whole vastus lateralis from men between 15 and 83 years. Acta Neuropathol 81, 377–381. [DOI] [PubMed] [Google Scholar]
- Lie M, Sejersted OM & Kiil F (1970). Local regulation of vascular cross section during changes in femoral arterial blood flow in dogs. Circ Res 27, 727–737. [DOI] [PubMed] [Google Scholar]
- Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill‐Eubanks D & Nelson MT (2017). Capillary K+‐sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looft‐Wilson RC, Payne GW & Segal SS (2004). Connexin expression and conducted vasodilation along arteriolar endothelium in mouse skeletal muscle. J Appl Physiol 97, 1152–1158. [DOI] [PubMed] [Google Scholar]
- Manttari S & Jarvilehto M (2005). Comparative analysis of mouse skeletal muscle fibre type composition and contractile responses to calcium channel blocker. BMC Physiol 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkau M, Kohler R & de Wit C (2010). Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J 24, 3572–3579. [DOI] [PubMed] [Google Scholar]
- Mohrman DE & Sparks HV (1974). Role of potassium ions in the vascular response to a brief tetanus. Circ Res 35, 384–390. [DOI] [PubMed] [Google Scholar]
- Muller‐Delp JM, Spier SA, Ramsey MW & Delp MD (2002). Aging impairs endothelium‐dependent vasodilation in rat skeletal muscle arterioles. Am J Physiol Heart Circ Physiol 283, H1662–H1672. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Lamb IR & Novielli NM (2017). Capillary endothelial cells as coordinators of skeletal muscle blood flow during active hyperemia. Microcirculation 24, e12348. [DOI] [PubMed] [Google Scholar]
- Musch TI, Friedman DB, Pitetti KH, Haidet GC, Stray‐Gundersen J, Mitchell JH & Ordway GA (1987). Regional distribution of blood flow of dogs during graded dynamic exercise. J Appl Physiol 63, 2269–2277. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF & Standen NB (1990). Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol 259, C3–C18. [DOI] [PubMed] [Google Scholar]
- Pohl U, Holtz J, Busse R & Bassenge E (1986). Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension 8, 37–44. [DOI] [PubMed] [Google Scholar]
- Riches AC, Sharp JG, Thomas DB & Smith SV (1973). Blood volume determination in the mouse. J Physiol 228, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaut RE & Sial AJ (1999). Differential phototoxicity of fluorescent dye‐labeled albumin conjugates. Microcirculation 6, 205–213. [PubMed] [Google Scholar]
- Saltin B, Henriksson J, Nygaard E, Andersen P & Jansson E (1977). Fiber types and metabolic potentials of skeletal muscles in sedentary man and endurance runners. Ann NY Acad Sci 301, 3–29. [DOI] [PubMed] [Google Scholar]
- Saltin B, Radegran G, Koskolou MD & Roach RC (1998). Skeletal muscle blood flow in humans and its regulation during exercise. Acta Physiol Scand 162, 421–436. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ & Dinenno FA (2004). Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL & Donato AJ (2011). Aging and vascular endothelial function in humans. Clin Sci (Lond) 120, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS (1991). Microvascular recruitment in hamster striated muscle: role for conducted vasodilation. Am J Physiol Heart Circ Physiol 261, H181–H189. [DOI] [PubMed] [Google Scholar]
- Segal SS (2005). Regulation of blood flow in the microcirculation. Microcirculation 12, 33–45. [DOI] [PubMed] [Google Scholar]
- Segal SS & Duling BR (1986a). Communication between feed arteries and microvessels in hamster striated muscle: segmental vascular responses are functionally coordinated. Circ Res 59, 283–290. [DOI] [PubMed] [Google Scholar]
- Segal SS & Duling BR (1986b). Flow control among microvessels coordinated by intercellular conduction. Science 234, 868–870. [DOI] [PubMed] [Google Scholar]
- Segal SS & Jacobs TL (2001). Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J Physiol 536, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkler SY, Fernando CA & Segal SS (2016). Differential α‐adrenergic modulation of rapid onset vasodilatation along resistance networks of skeletal muscle in old versus young mice. J Physiol 594, 6987–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkler SY & Segal SS (2014). Aging alters reactivity of microvascular resistance networks in mouse gluteus maximus muscle. Am J Physiol Heart Circ Physiol 307, H830–H839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiesko V & Johnson PC (1993). The arterial lumen is controlled by flow‐related shear stress. Physiology 8, 34–38. [Google Scholar]
- Smiesko V, Lang DJ & Johnson PC (1989). Dilator response of rat mesenteric arcading arterioles to increased blood flow velocity. Am J Physiol Heart Circ Physiol 257, H1958–H1965. [DOI] [PubMed] [Google Scholar]
- Song H & Tyml K (1993). Evidence for sensing and integration of biological signals by the capillary network. Am J Physiol Heart Circ Physiol 265, H1235–H1242. [DOI] [PubMed] [Google Scholar]
- Stowe DF, Owen TL, Anderson DK, Haddy FJ & Scott JB (1975). Interaction of O2 and CO2 in sustained exercise hyperemia of canine skeletal muscle. Am J Physiol Heart Circ Physiol 229, H28–H33. [DOI] [PubMed] [Google Scholar]
- Thijssen DH, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, Parker B, Widlansky ME, Tschakovsky ME & Green DJ (2011). Assessment of flow‐mediated dilation in humans: a methodological and physiological guideline. Am J Physiol Heart Circ Physiol 300, H2–H12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschakovsky ME, Shoemaker JK & Hughson RL (1996). Vasodilation and muscle pump contribution to immediate exercise hyperemia. Am J Physiol Heart Circ Physiol 271, H1697–H1701. [DOI] [PubMed] [Google Scholar]
- VanTeeffelen JW & Segal SS (2000). Effect of motor unit recruitment on functional vasodilatation in hamster retractor muscle. J Physiol 524, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanTeeffelen JW & Segal SS (2006). Rapid dilation of arterioles with single contraction of hamster skeletal muscle. Am J Physiol Heart Circ Physiol 290, H119–H127. [DOI] [PubMed] [Google Scholar]
- Wilson C, Lee MD & McCarron JG (2016). Acetylcholine released by endothelial cells facilitates flow‐mediated dilatation. J Physiol 594, 7267–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardeni T, Eckhaus M, Morris HD, Huizing M & Hoogstraten‐Miller S (2011). Retro‐orbital injections in mice. Lab Anim (NY) 40, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeboah J, Crouse JR, Hsu FC, Burke GL & Herrington DM (2007). Brachial flow‐mediated dilation predicts incident cardiovascular events in older adults: the Cardiovascular Health Study. Circulation 115, 2390–2397. [DOI] [PubMed] [Google Scholar]