

ABSTRACT
Human adenoviruses (Ad) are double-stranded DNA (dsDNA) viruses associated with infectious diseases, but they are better known as tools for gene delivery and oncolytic anticancer therapy. Atomic structures of Ad provide the basis for the development of antivirals and for engineering efforts toward more effective applications. Since 2010, atomic models of human Ad5 have been derived independently from photographic film cryo-electron microscopy (cryo-EM) and X-ray crystallography studies, but discrepancies exist concerning the assignment of cement proteins IIIa, VIII, and IX. To clarify these discrepancies, we employed the technology of direct electron counting to obtain a cryo-EM structure of human Ad5 at 3.2-Å resolution. Our improved structure unambiguously confirms our previous cryo-EM models of proteins IIIa, VIII, and IX and explains the likely cause of conflict in the crystallography models. The improved structure also allows the identification of three new components in the cavity of hexon—the cleaved N terminus of precursor protein VI (pVIn), the cleaved N terminus of precursor protein VII (pVIIn2), and mature protein VI. The binding of pVIIn2—and, by extension, that of genome-condensing pVII—to hexons is consistent with the previously proposed dsDNA genome-capsid coassembly for adenoviruses, which resembles that of single-stranded RNA (ssRNA) viruses but differs from the well-established mechanism of pumping dsDNA into a preformed protein capsid exemplified by tailed bacteriophages and herpesviruses.
IMPORTANCE Adenovirus is a double-edged sword to humans: it is a widespread pathogen but can be used as a bioengineering tool for anticancer and gene therapies. The atomic structure of the virus provides the basis for antiviral and application developments, but conflicting atomic models for the important cement proteins IIIa, VIII, and IX from conventional/film cryo-EM and X-ray crystallography studies have caused confusion. Using cutting-edge cryo-EM technology with electron counting, we improved the structure of human adenovirus type 5 and confirmed our previous models of cement proteins IIIa, VIII, and IX, thus clarifying the inconsistent structures. The improved structure also reveals atomic details of membrane-lytic protein VI and genome-condensing protein VII and supports the previously proposed genome-capsid coassembly mechanism for adenoviruses.
KEYWORDS: human adenovirus, cement protein structure, dsDNA genome packaging, genome-capsid coassembly, endosomal escape
INTRODUCTION
Human adenoviruses (Ad) are widespread and are associated with respiratory, eye, and gastrointestinal infections that might become life-threatening, particularly in young children and immunocompromised individuals (1). General vaccination against Ad is not available because of the existence of more than 60 serotypes of the virus (2). There is also considerable interest in developing derivatives of Ad for therapeutic applications, for example, as gene transfer vectors or oncolytic viruses for cancer therapy (3, 4). Understanding the virion structure of Ad provides crucial insights for the development of antivirals and more effective virus-based therapeutics. The capsid of Ad is composed of three major capsid proteins (hexon, penton base, and fiber) and four minor capsid proteins (IIIa, VI, VIII, and IX; also known as cement proteins). The pseudo-T=25 icosahedral capsid has 12 pentons, each with trimeric fiber proteins, and 240 trimeric hexons. It encloses the 36-kb double-stranded DNA (dsDNA) viral genome along with six additional proteins (IVa2, V, VII, μ, terminal protein, and the viral proteinase). Structures of the isolated Ad hexon, penton base, or fiber protein have been well characterized by X-ray crystallography (5–7). Previous cryo-electron microscopy (cryo-EM) studies of Ad virions at low or intermediate resolution revealed the organization of the major capsid components and the possible distribution of some of the minor capsid proteins (8–11). In 2010, our group reported a 3.6-Å-resolution cryo-EM structure of the human adenovirus type 5 (Ad5) virion, along with new atomic models for proteins IIIa, VIII, and IX (12). At the same time, Reddy et al. published a crystal structure of Ad5 at 3.5-Å resolution, with an atomic model for protein VIII (13), and in 2014, they published a crystal structure at 3.8-Å resolution, with atomic models of IIIa, V, VI, VIII, and IX (14).
While these structures have been highly sought after and their publications were heralded as major advances in the field of structural biology (15), confusions persisted due to the substantial differences between structural interpretations of the cryo-EM map (12) and the X-ray crystallography structure (13, 14). For example, protein IIIa was assigned to densities surrounding the penton base inside the virus in the cryo-EM model (12) but was placed toward the outer surface of the virus in the crystallography studies (14). The C-terminal ∼40 residues of protein IX were modeled to form the characteristic 4-helix bundles in cryo-EM studies (11, 12) but were deemed flexible/invisible in the crystal (14, 16). For protein VIII, although the localizations of the protein in the cryo-EM and crystallography studies roughly agree, major differences exist in the two models (12–14). Such differences have not gone unnoticed; readers have raised concerns regarding the discrepancies between the crystallographic models and prior structural and biochemical results (17, 18).
These discrepancies have created uncertainty, which might impede efforts of structure-based engineering of human Ad. This prospect compelled us to further improve the structure of Ad5 by taking advantage of the state-of-the-art electron-counting cryo-EM technology. The improved structure at 3.2-Å resolution not only confirmed our previous models of proteins IIIa, VIII, and IX from the film data but also allowed us to identify three new protein components in the cavity of hexon—the cleaved N terminus of precursor protein VI (pVIn), the cleaved N terminus of precursor protein VII (pVIIn2), and a segment of mature protein VI. These new discoveries, particularly the unexpected distribution of pVIIn2 and protein VI, provide new insights into adenovirus genome packaging, virion maturation, and endosomal escape during infection.
RESULTS
Overall structure.
We recorded 5,600 electron-counting movies of a freshly prepared Ad5 sample by use of a K2 direct electron detector attached to the end of an energy filter in a Titan Krios electron microscope, and by averaging ∼53,000 particles from these movies, we obtained a three-dimensional (3D) structure at 3.2-Å resolution (Fig. 1 and 2A and Table 1). The larger number of particle images and the much better contrast and higher signal-to-noise ratio of these images than those of our previous film data led to much-better-resolved side chains in the cryo-EM map than those in our previous, 3.6-Å-resolution structure (12). Subtle differences between side chains, such as those among Val, Leu, and Ile, between Phe and Tyr, or between Lys and Arg, are now discernible in our cryo-EM map (Fig. 2B). The identification of amino acids in our map and their matches to protein sequences provide direct evidence in support of our atomic models (Fig. 3). Atomic models derived from this new map for both major capsid proteins (hexon and penton base) and previously modeled minor capsid proteins (IIIa, VIII, and IX) all agree with the previous cryo-EM study (12).
FIG 1.

Distribution and structure of minor proteins IIIa, VIII, and IX in the virion of human Ad5. (A and B) Outer surface (A) and inner surface (B) views of human Ad5 capsid. The electron-counting cryo-EM density map of the human Ad5 virion at 3.2-Å resolution is differentially colored according to the identities of the penton base (P), hexons (H1 to H4), and minor proteins IX (purple) (A), IIIa (red) (B), and VIII (magenta) (B). (C to E) Atomic models of minor proteins IX, IIIa, and VIII. Each model is rainbow colored blue to red from the N terminus to the C terminus of the protein chain. The numbers denote protein sequence numbers at boundaries of the model. Three copies of the protein IX N-terminal region and four copies of the protein IX C-terminal region assemble into the triskelion and the 4-helix bundle, respectively. Note the three parallel-one antiparallel configuration of coiled-coil helices in the 4-helix bundle.
FIG 2.
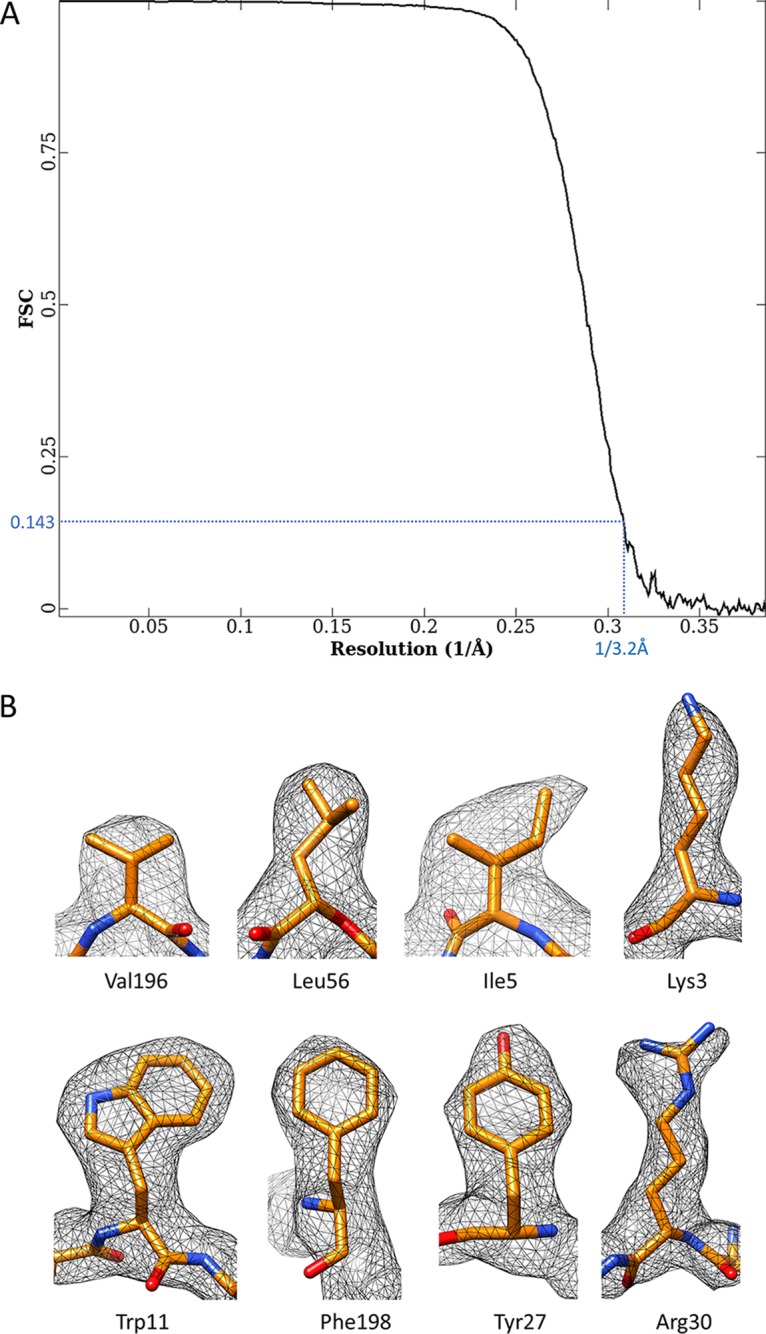
Resolution assessment of the electron-counting cryo-EM structure of the human Ad5 virion. (A) The final resolution of the density map was estimated to be 3.2 Å by the criterion FSC = 0.143 (63). (B) High-resolution features of some protein side chains in the density map of cement protein VIII. Note that subtle differences among Val, Leu, and Ile, between Phe and Tyr, and between Lys and Arg are discernible.
TABLE 1.
Statistics for Phenix real-space refinement of the atomic model
| Parameter | Value |
|---|---|
| Model to map fit (CC around atoms) | 0.8551 |
| Root mean square deviation from ideal value | |
| Bond length (Å) | 0.007 |
| Bond angle (°) | 0.823 |
| All-atom clash score | 5.69 |
| MolProbity score | 1.60 |
| Ramachandran plot statistics | |
| % outliers | 0.02 |
| % allowed | 4.23 |
| % favored | 95.74 |
| Rotamer outliers (%) | 0.03 |
| Cβ deviations | 0 |
FIG 3.

Validating our previous models of proteins IIIa, VIII, and IX with the improved structure of human Ad5 from electron-counting cryo-EM. Example densities (semitransparent surfaces) of proteins IIIa (A), VIII (B), and IX (C) were segmented from the 3.2-Å-resolution cryo-EM reconstruction and fitted with the corresponding atomic models (sticks) to showcase the validity of our model. Some bulky side chains are labeled. Note that only two high-quality helices of the protein IX 4-helix bundle are shown, for clarity. These two helices are antiparallel.
Proteins IIIa and VIII are the minor proteins on the inner surface.
As reported previously, two cement proteins—IIIa and VIII—were identified on the inner surface of the Ad5 capsid (12). On the inner surface around each 5-fold axis, five copies of protein IIIa cement the gaps between the penton base and peripenton hexons (Fig. 1B and D), amounting to a total of 60 copies per virion. Two copies of protein VIII are located in each asymmetric unit of the capsid—one close to the 3-fold axis and the other close to the 5-fold axis, contacting protein IIIa (Fig. 1B)—totaling 120 copies per virion. Despite their quasi-equivalent locations, the two protein VIII molecules have identical structures. In contrast, in the crystallography study (14), densities around the 5-fold axes were modeled as proteins V and VI instead of IIIa; two protein VIII molecules were assigned to densities at positions similar to protein VIII locations in our cryo-EM structure, but their models are much simpler. Overall, the densities for the internal cement proteins have the best quality in our cryo-EM map, with prominent side chain features for de novo atomic modeling, as evidenced in Fig. 3A and B (see Movies S1 and S2 in the supplemental material). The excellent agreement of amino acid residues between the atomic models of proteins IIIa and VIII and their corresponding densities eliminates any doubts about their assignments.
Protein IX is the minor protein on the outer surface.
Sequence analysis indicated that the C-terminal region of protein IX is a long α-helix with a strong propensity for coiled-coil formation (11). In our prior model, protein IX has an extended configuration with an N-terminal triskelion domain and a C-terminal coiled-coil helix joined by a long flexible loop (Fig. 1C). An external interaction network (Fig. 3 in reference 12) is formed among 240 molecules of protein IX, through interactions involving its two ends: the N termini of three protein IX molecules join to form a triskelion and the C termini of four protein IX molecules bundle to form a 4-helix-bundle coiled coil (Fig. 1C and 4). This model was verified in the present study by the improved density maps for both the triskelion and coiled-coil domains, as highlighted by the consistency between the atomic model and cryo-EM densities of the bulky side chains of Tyr14, Trp22, Arg26, Arg92, and Arg96 (Fig. 3C; Movie S3). In contrast, in the crystallographic structure, although each triskelion was correctly identified as a homotrimer of the protein IX N-terminal segment, the polarity of the protein chain (N to C) was opposite that in our cryo-EM model, leading to incorrect sequence registration and the suggestion of a flexible (and thus invisible) C-terminal coiled-coil helix bundle (14, 16).
FIG 4.

Cement protein IX forms the triskelions and 4-helix bundles on the outer surface of Ad5 capsid. (A) Distribution of four protein IX molecules in each asymmetric unit of Ad5 capsid. The C-terminal regions of four protein IX molecules assemble into a 4-helix bundle, while their N-terminal regions are distributed in four triskelion structures. Quasi-equivalent hexons H1 to H4 are labeled. (B) Densities connecting the N-terminal triskelion domain of protein IX to its C-terminal coiled-coil helix in the 4-helix bundle are discernible by lowering the display threshold. The density map is viewed from the same orientation as that for panel A, with the densities of hexons H2 and H3 hidden for clarity. Only two of the four triskelions and their linker densities to the 4-helix bundle are shown, for clarity. Note that the brown density links the triskelion to a helix from the top of the 4-helix bundle, while the magenta density (and the other two not shown) links the triskelion to a helix from the bottom of the 4-helix bundle. (C) Polarities of the four helices in the 4-helix bundle. Because protein side chains in an α-helix should point slightly toward the N-terminal end of the helix, the polarities of the four helices can be determined unambiguously based on the orientations of side chain densities, as denoted by dashed lines. The four helices were determined to be three parallel and one antiparallel.
In our model, each 4-helix bundle is formed by the C-terminal helices of four protein IX molecules, three parallel and one antiparallel (Fig. 1C and 4A), consistent with a previous low-resolution cryo-EM study of Ad5 with peptide tag/Fab-labeled protein IX (19). In stark contrast to the extended configuration of the triskelion and helix bundle domains of protein IX in human Ad5, the cryo-EM structure of bovine adenovirus type 3 reveals a parallel 3-helix bundle immediately above the triskelion (17), which can be explained by the shorter connecting loop between its coiled-coil helix and triskelion domains than that in human Ad5 (20). The incorrect polarity assignment of the protein IX triskelion in the crystallography model led to the proposal of an invisible 3-helix bundle positioned above the triskelion, similar to the arrangement observed in bovine adenovirus type 3, while the visible 4-helix bundle was interpreted as a subdomain of protein IIIa containing two helix-turn-helix segments (14, 16). Aside from the well-resolved side chain densities in two helices of the 4-helix bundle (Fig. 3C), the following two additional pieces of evidence also support the validity of our protein IX model. First, we observed four threads of densities, three from the bottom and one from the top, connecting the four helices in each helix bundle to four neighboring protein IX triskelion branches, directly supporting our proposed arrangement of a 4-helix bundle containing three parallel and one antiparallel helix of the protein IX C terminus (Fig. 4B). Second, as a general rule of protein modeling, the side chains in an α-helix should point slightly toward the N-terminal end of the helix. Based on this rule, the polarity of one of the four helices, as established by the directions of pointing of side chain densities (dashed lines in Fig. 4C), differs from that of the other three in the 4-helix bundle. Therefore, the interpretation of the observed 4-helix bundle as two helix-turn-helix segments (i.e., two helices have inverted orientations relative to those of the other two) of protein IIIa in the crystallography study (14) is incompatible with this observation.
Identification and modeling of proteins VI, pVIn, and pVIIn2 in the inner cavity of hexon.
Unlike the above-described minor proteins that play structural roles in virion assembly, minor proteins VI and VII (and their precursor proteins, pVI and pVII) play critical functional roles in endosomal escape during viral entry (21, 22) and in genome condensation (23), respectively. Unfortunately, the mechanisms behind the functions of VI and VII have been elusive due to the lack of structural information. The improved quality of our new structure has now allowed us to identify parts of proteins VI and VII and to build atomic models de novo, offering clues about how they work.
Both proteins VI and VII are located in the inner cavities of hexons, based on high-resolution features of their side chains in our cryo-EM map (Fig. 5A to D). The segment from positions 109 to 143 of protein VI (sequence numbering as in pVI) crosses the opening of the inner cavity of each hexon (Fig. 5D and E). Lining the wall of the hexon cavity are two copies of the cleaved N terminus of pVI (pVIn)—a side product of pVI maturation catalyzed by the adenovirus proteinase (AVP). Region 5-33 of the 33-amino-acid (aa) pVIn segment was modeled with the N-terminal 4 residues being flexible (Fig. 5D and E). The identification of VI and pVIn in the inner cavity of hexon is consistent with previous biochemical data showing that pVI has a high affinity for hexon (24, 25) and that pVIn binds at the base of the hexon cavity in mature virions (26).
FIG 5.

Identification and modeling of proteins VI, pVIn, and pVIIn2 in the inner cavity of hexon. (A) Facet of Ad5 capsid viewed from inside the capsid. The density map is differentially colored in the same way as in Fig. 1A and B, except that previously unmodeled densities are highlighted in yellow. Note that the unmodeled densities are mainly distributed in the cavities of hexons. (B and C) A “remnant map” was calculated by removing densities of hexon, penton base, and proteins IIIa, VIII, and IX, leaving previously unmodeled densities alone. The density map in panel B is flipped over in panel C to show the cup-shaped densities hiding deeply in the cavity of each hexon, which are attributable to the disordered N- and/or C-terminal region of protein VI. (D) Zoomed-in view of hexon H2. The location of this hexon is marked with a dashed hexagon in panel A. Three subunits of the hexon trimer and the minor proteins in the cavity of the hexon are differentially colored. The hexon trimer is displayed at a threshold of 3δ (δ is the standard deviation), pVIn (cyan or magenta) and pVIIn2 (gold) at a threshold of 2.75δ, and mature VI (green) at a threshold of 1.75δ. (E) Atomic models (sticks) of pVIn, pVIIn2, and VI fitted to their corresponding cryo-EM densities (semitransparent surfaces), with some landmark residues labeled. Only one of the two equivalent copies of pVIn is shown. The schematics at the bottom show maturational cleavages in pVI and pVII. Each precursor protein is represented as a bar, with the polypeptide length (in amino acids) indicated in the center. Consensus cleavage sites are denoted by arrows, and the nonconsensus site is denoted by an arrowhead. (F) Disordered densities hidden deeply in the cavity of hexon H2. The surface presentation of the hexon was calculated by use of atomic models of the hexon trimer, and one half was removed to expose the inner cavity of the hexon. The nomenclature “hexon base,” “hexon tower,” and “β-constriction site” is used following that in the literature (5). Disordered densities (yellow) in the cavity were segmented from the cryo-EM density map and displayed at a threshold of 0.9δ. A model of protein VI (green ribbon) crossing the opening of the cavity is also shown. The two dashed lines denote that the disordered densities are speculated to be attributable to the unmodeled N- and C-terminal regions of protein VI.
Unexpectedly, a segment spanning aa 14 to 24 of the cleaved N terminus of pVII, which we named pVIIn2, was identified for a density also lining the wall of the hexon cavity and occupying a third position equivalent to those of the two pVIn molecules (Fig. 5D and E). Our structural observation of pVIIn2 spanning only aa 14 to 24 (instead of aa 1 to 24) of pVII is consistent with a mass spectrometric measurement showing that pVII is cleaved for a second time, at a nonconsensus cleavage site at aa 13/14, in addition to the consensus site at aa 24/25 recognized by AVP (27).
Although the models of VI, pVIn, and pVIIn2 are relatively short, they are supported by consistency between the model and well-resolved side chain densities in each molecule, as illustrated in Fig. 5E. Particularly, the similarly shaped and equivalently positioned pVIn and pVIIn2 densities are differentiated based on bulky side chains of Arg16, Phe17, Lys20, and Phe22 in pVIIn2 contrasting with the nonbulky Gly26, Thr27, Ser28, and Ser31 residues in the corresponding region of pVIn (Fig. 5E). These results contradict both the protein VI and pVIn models in the crystallography study (14), in two ways: (i) part of the protein IIIa density was mistaken for protein VI in the crystallography structure, as described above in the section concerning protein IIIa; and (ii) for pVIn, the polarity of the protein chain in the crystallography model is opposite that in the cryo-EM model reported here.
The protein VI density we modeled is only a small fraction of the full-length mature protein VI (aa 109 to 143 versus aa 34 to 239 as numbered in pVI), suggesting that both its N- and C-terminal regions are flexible. Since the ordered part of protein VI blocks the opening of the hexon cavity, with both ends (Pro109 and Thr143 in Fig. 5E) on the inner side of the cavity (Fig. 5D), it is conceivable that the flexible N- and C-terminal regions are both inside the cavity. This notion is supported by the existence of disordered densities in the deep cavity of hexons (Fig. 5F). Particularly, the density at the dead end of the cavity is as strong as that of hexon proteins (Fig. 5C). The N-terminal region of mature protein VI has an amphipathic α-helix that was demonstrated to function as a membrane penetrator for endosomal escape during viral infection (21, 22). The deduced location of the protein VI N terminus, situated deep inside the hexon cavity, may have implications for protein VI's function in endosomal escape for adenoviruses.
Interactions of pVIn and pVIIn2 with hexon.
The newly observed pVIn and pVIIn2 segments have similar but distinguishable interactions with hexon subunits (Fig. 6). There are MSGG and MFGG sequences at the C termini of pVIn and pVIIn2, respectively (Fig. 5E), both of which are part of the (M/I/L)XGG-X consensus site recognized by AVP in the corresponding precursor protein (28). In both pVIn and pVIIn2, the two glycine residues fold back toward the N terminus (Fig. 5E), and either a serine (Ser31 of pVIn) or a phenylalanine (Phe22 of pVIIn2) residue points to an equivalent pocket in the hexon wall, giving rise to the similar appearances of the two molecules in this region (Fig. 6B to E). However, the density of the bulky Phe22 residue in pVIIn2 is readily distinguishable from that of the Ser31 residue in pVIn, and their interactions with hexon residues in the pocket are distinct from one another. For pVIn, the polar side chain of Ser31 forms a hydrogen bond with the side chain of hexon residue Arg872, which in turn is further stabilized by hydrogen bonding with Glu921 of the same hexon subunit (Fig. 6D). For pVIIn2, the side chain of Phe22 inserts into a hydrophobic pocket formed by Leu368, Leu372, Leu647, Leu919, Leu870, Phe712, and Ala679 of the hexon subunit, while the Arg872 side chain gating the pocket adopts a conformation different from that in the pVIn-binding state due to steric effects (Fig. 6E). A second main binding site shared by pVIn and pVIIn2 is near the top edge of the hexon wall. Ile25 of pVIn and Leu15 of pVIIn2 bind to an equivalent hydrophobic pocket formed by Leu24, Leu28, Val29, Ala32, Leu41, and Phe45 of one hexon subunit and Leu630 and Leu634 of a second subunit (Fig. 6B, C, and F).
FIG 6.

Binding sites of pVIn, pVIIn2, and protein VI in the cavity of hexon. (A) Atomic model of hexon H2, corresponding to the density map shown in Fig. 5D. The two insets show zoomed-in views of the boxed regions, showing β-augmentation interactions among hexon subunit, pVIn, and VI. (B and C) Overall views of the binding sites of pVIn (B) and pVIIn2 (C) in the hexon wall. (D and E) Comparison of pVIn (D) and pVIIn2 (E) C-terminal binding to two equivalent pockets in the hexon wall. The images are zoomed-in views of the boxed regions in panels B and C, respectively. Note the similar shapes of pVIn and pVIIn2 in this region, but the prominently distinguishable pVIn Ser31 and pVIIn2 Phe22 densities, as well as distinct interactions with labeled hexon residues. (F) Second binding site shared by pVIn and pVIIn2 in the capsid wall. This site of a hydrophobic pocket is bound by pVIIn2 Leu15, as shown in the boxed region in panel C and the zoomed-in view here, or by pVIn Ile25, as shown in panel B.
Besides the two binding sites along the hexon wall shared with pVIIn2, the N-terminal region of pVIn is also stabilized by binding in a groove at the rim of the hexon cavity (Fig. 6A). The groove is contributed by all three subunits in the hexon trimer. This observation is consistent with the previously proposed binding site of pVIn spanning hexon residues 32 to 65, which was mapped by hydrogen-deuterium-exchange mass spectrometry (26). Furthermore, the two short β-strands of pVIn, segment 16-18 and segment 11-13, are augmented with β-strand 49-51 in a hexon subunit. For one of the two pVIn molecules, the β-augmentation is further extended to β-strands 120-123 and 113-115 in protein VI (Fig. 6A).
The above-described VI, pVIn, and pVIIn2 models were derived from densities in the cavity of hexon H2 because those densities have the best quality. On fitting these models derived from hexon H2 into densities in the cavities of the other three hexons (H1, H3, and H4), we found that pVIn and pVIIn compete for the three binding sites in each hexon, which is expected considering their similar binding sites along the hexon wall. In other words, the well-defined arrangement of two pVIn and one pVIIn segment in hexon H2 seems to be an exception rather than the rule. The competitive binding of pVIn and pVIIn2 also explains the puzzling stoichiometry of ∼350 copies of protein VI (29, 30) binding to 240 copies of hexon trimers in each virion: this copy number of protein VI is too large if only one protein VI molecule binds each hexon but too small if three protein VI molecules bind each hexon.
DISCUSSION
Cryo-EM has come of age as a tool for deciphering molecular interactions in large complexes. Electron counting enabled by direct electron detection cameras has significantly improved the contrast of cryo-EM images and the quality of 3D reconstructions (31, 32). The improved cryo-EM structure of human Ad5 from electron-counting cryo-EM presented in this study confirms our previous cryo-EM models of cement proteins IIIa, VIII, and IX (12), thus clarifying the contradicting interpretations in the literature regarding their structural roles (13, 14). More significantly, the improved cryo-EM reconstruction has allowed us to identify and model three protein fragments related to functionally important minor proteins VI and VII inside human Ad5.
Protein VI (or its precursor protein, pVI) is a multifunctional protein in the life cycle of adenovirus. Prior to capsid assembly, pVI escorts hexons into the cell nucleus (33), and after a capsid is assembled, pVI is involved in the activation of AVP (34, 35) for maturational cleavage of many precursor proteins, including itself (36). Most importantly, protein VI is responsible for endosomal escape of the virus during cell entry (21, 22). Release of protein VI from the virion for endosomal escape was proposed to occur through partial disassembly of the capsid at the vertex region, triggered by the low-pH conditions inside the endosome (21). The trail of literature seems to suggest that this proposal was likely influenced by the incorrect assignment of protein VI to the protein IIIa densities beneath the peripenton hexons (9) (see Results). Our localization of mature protein VI inside all hexons and hidden deeply in the cavity makes the mechanism of its release intriguing. It remains to be seen how the low pH in the endosome triggers the release of the membrane-lytic protein VI, leading to rupture of the endosomal membrane and escape of the capsid.
Protein VII is a major core protein, totaling 600 to 800 copies per virion as measured by mass spectrometry (29, 30). Sequence analysis of protein VII revealed the presence of four highly basic domains separated by several predicted α-helices, leading to the speculation of a histone/protamine-like structure and function of protein VII (23). In vitro nuclease digestion of the Ad genome suggested that protein VII likely works in a unit of six to condense the viral dsDNA genome to form a chromatin-like structure as in eukaryotic cells (23, 37). It is hard to conceive how such a condensed genome with protein VII can pass through a portal complex, such as those in tailed dsDNA bacteriophages and herpesviruses (i.e., the headful dsDNA genome packaging mechanism) (38–40). Such a portal complex was proposed to exist in adenovirus (41) but has yet to be identified. To the contrary, our data are consistent with the coassembly mechanism proposed previously (42), although we cannot rule out the possibility of headful DNA packaging in adenoviruses. Such a coassembly mechanism utilizes the genome as a scaffold for capsid assembly, resembling that of many single-stranded RNA (ssRNA) viruses (43–45). The binding of pVIIn2, and by extension the precursor protein pVII, in the cavities of all hexons bears similarity to the extensive interactions between ssRNA viral genomes and the capsid inner surface (43): while the dsDNA viral genome is being condensed by pVII and other core proteins into a chromatin-like structure, the N termini of pVII molecules at the periphery of this genomic core provide numerous anchor points for capsomers to assemble around it. After assembly, the AVP activated by the dsDNA and the cleaved C-terminal segment of pVI (46, 47) cleaves off the N-terminal segment of pVII (and probably other packaging-related minor core proteins, such as L1 52/55k [48, 49]) to free up the genomic core from tethering to the capsid shell so that it can be released in the next round of infection. Indeed, immature adenovirus particles, represented by a protease-impaired temperature-sensitive Ad2 ts1 mutant (50, 51) grown at a nonpermissive temperature, have difficulties in uncoating and genome release (52, 53). Two cryo-EM structures of such immature Ad2 particles both revealed that the capsid was tightly connected to the genomic core (52, 54). Should such a coassembly mechanism withstand the test of future experiments, it would represent a novel mechanism for dsDNA virus assembly that is drastically distinct from the well-established mechanism of pumping dsDNA into a preformed protein capsid exemplified by many tailed dsDNA bacteriophages and herpesviruses (38–40).
MATERIALS AND METHODS
Virus culture and purification.
To obtain homogeneous samples for high-resolution cryo-EM structure determination, we chose to purify mature virions from the viral culture medium instead of from the cytoplasm of infected cells, as generally practiced, to avoid contamination of any immature or partially mature particles. The E1B gene-attenuated oncolytic virus Ad-ΔE1B19/55 (55), containing the wild-type Ad5 capsid, was inoculated into confluent 293T cells (ATCC CRL-3216) at a multiplicity of infection (MOI) of 0.01. At 3 days postinfection, the supernatant was collected and clarified by centrifugation at 10,000 × g for 15 min. Viral particles were pelleted by centrifugation at 100,000 × g for 1 h, resuspended in a Tris-buffered saline (TBS) solution containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 1 mM MgCl2, and purified in a CsCl density gradient by centrifugation at 100,000 × g overnight. The virus band was collected, dialyzed overnight in TBS, and concentrated with a Millipore Amicon ultrafiltration unit with a 100-kDa nominal molecular size limit. A final volume of 30 μl of concentrated sample was typically obtained by starting from 1 liter of virus culture.
Cryo-EM sample preparation and data collection.
To prepare cryo-EM samples, an aliquot of 2.5 μl of sample was applied to a Quantifoil R2/1 Cu grid, blotted with filter paper, and plunge frozen in liquid ethane by use of an FEI Vitrobot. Cryo-EM images were collected with Leginon (56) on an FEI Titan Krios electron microscope equipped with a Gatan imaging filter (GIF) and a K2 Summit direct detection camera. The K2 camera was operated in superresolution mode. A nominal magnification of ×81,000 was used, giving a pixel size of 0.85 Å/pixel at the sample level. A slit width of 20 eV was set for the energy filter. Movies were recorded with an electron dose rate below 8 e− per physical pixel per second. An accumulated dose of 25 e−/Å2 on the sample was fractionated into a movie stack of 32 image frames.
Data processing and structure determination.
For each of the total of 5,608 movies recorded, the frames were aligned for drift correction by a previously described method (32). All the frames except the first one were averaged to produce micrographs. The defocus value was set to −2 μm during the imaging session and was determined with CTFFIND3 (57) to be in the range of −0.5 to −3 μm in the images. Particles were picked with Ethan (58) and preprocessed with EMAN (59). Center and orientation parameters of each particle were determined and refined iteratively with the common-line-based method, using the IMIRS software package (60, 61). 3D reconstructions were carried out with the GPU program eLite3D (62). Resolution was determined based on the criterion FSC = 0.143, where FSC is the Fourier shell correlation (63). Density map rendering and figure preparation were carried out with UCSF Chimera (64).
Atomic model building.
Atomic model building for hexon, penton base, and proteins IIIa, VIII, and IX started with fitting our previous atomic model (Protein Data Bank [PDB] ID 3IYN) (12) into the density map in Chimera, followed by iterative manual adjustment in Coot (65) and real-space refinement in Phenix (66). Atomic models of pVIn, pVIIn2, and VI were built ab initio with Coot and refined with Phenix. Assignment of pVIIn2 was not inferred from any previous knowledge but was a result of systematic screening/elimination of other possible protein sequences in adenovirus mature virions. Each protein sequence was tested multiple times at both the N and C termini. We found that only the pVIIn2 sequence fit satisfactorily in the resolved side chain densities.
Accession number(s).
The cryo-EM density map and the atomic models were deposited in the Electron Microscopy Data Bank (EMDB) and PDB under accession numbers EMD-7034 and 6B1T, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Robin Parks and Kathy Poulin for providing reagents that were used during the pursuit of this study.
This project was supported in part by grants from the National Institutes of Health (grants GM071940, DE025567, and AI094386) and indirectly through the National Center for Advancing Translational Science (NCATS) (UCLA CTSI grant UL1TR001881). We acknowledge the use of instruments at the Electron Imaging Center for Nanomachines, supported by UCLA and by instrumentation grants from the NIH (grants 1S10OD018111 and 1U24GM116792) and the NSF (grant DBI-1338135).
ADDENDUM
A paper from the Reddy group reporting a cryo-EM structure of adenovirus D26 (67) appeared recently. Their new assignments for minor proteins IIIa, VIII, and IX correct the previous crystal models and are now consistent with our assignments; however, their model of minor protein pVIn has a sequence polarity opposite that presented here, and pVIIn2 and mature VI were not identified in the cavity of hexon.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00850-17.
REFERENCES
- 1.Ghebremedhin B. 2014. Human adenovirus: viral pathogen with increasing importance. Eur J Microbiol Immunol 4:26–33. doi: 10.1556/EuJMI.4.2014.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang GH, Xu WB. 2013. Recent advance in new types of human adenovirus. Bing Du Xue Bao 29:342–348. [PubMed] [Google Scholar]
- 3.Appaiahgari MB, Vrati S. 2015. Adenoviruses as gene/vaccine delivery vectors: promises and pitfalls. Expert Opin Biol Ther 15:337–351. doi: 10.1517/14712598.2015.993374. [DOI] [PubMed] [Google Scholar]
- 4.Rosewell Shaw A, Suzuki M. 2016. Recent advances in oncolytic adenovirus therapies for cancer. Current Opin Virol 21:9–15. doi: 10.1016/j.coviro.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts MM, White JL, Grutter MG, Burnett RM. 1986. Three-dimensional structure of the adenovirus major coat protein hexon. Science 232:1148–1151. doi: 10.1126/science.3704642. [DOI] [PubMed] [Google Scholar]
- 6.van Raaij MJ, Mitraki A, Lavigne G, Cusack S. 1999. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401:935–938. doi: 10.1038/44880. [DOI] [PubMed] [Google Scholar]
- 7.Zubieta C, Schoehn G, Chroboczek J, Cusack S. 2005. The structure of the human adenovirus 2 penton. Mol Cell 17:121–135. doi: 10.1016/j.molcel.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 8.Stewart PL, Burnett RM, Cyrklaff M, Fuller SD. 1991. Image reconstruction reveals the complex molecular organization of adenovirus. Cell 67:145–154. doi: 10.1016/0092-8674(91)90578-M. [DOI] [PubMed] [Google Scholar]
- 9.Stewart PL, Fuller SD, Burnett RM. 1993. Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J 12:2589–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabry CM, Rosa-Calatrava M, Conway JF, Zubieta C, Cusack S, Ruigrok RW, Schoehn G. 2005. A quasi-atomic model of human adenovirus type 5 capsid. EMBO J 24:1645–1654. doi: 10.1038/sj.emboj.7600653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saban SD, Silvestry M, Nemerow GR, Stewart PL. 2006. Visualization of alpha-helices in a 6-angstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J Virol 80:12049–12059. doi: 10.1128/JVI.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H, Jin L, Koh SB, Atanasov I, Schein S, Wu L, Zhou ZH. 2010. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329:1038–1043. doi: 10.1126/science.1187433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy VS, Natchiar SK, Stewart PL, Nemerow GR. 2010. Crystal structure of human adenovirus at 3.5 A resolution. Science 329:1071–1075. doi: 10.1126/science.1187292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy VS, Nemerow GR. 2014. Structures and organization of adenovirus cement proteins provide insights into the role of capsid maturation in virus entry and infection. Proc Natl Acad Sci U S A 111:11715–11720. doi: 10.1073/pnas.1408462111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison SC. 2010. Looking inside adenovirus. Science 329:1026–1027. doi: 10.1126/science.1194922. [DOI] [PubMed] [Google Scholar]
- 16.Reddy VS, Nemerow GR. 2014. Reply to Campos: revised structures of adenovirus cement proteins represent a consensus model for understanding virus assembly and disassembly. Proc Natl Acad Sci U S A 111:E4544–E4545. doi: 10.1073/pnas.1417014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng L, Huang X, Li X, Xiong W, Sun W, Yang C, Zhang K, Wang Y, Liu H, Huang X, Ji G, Sun F, Zheng C, Zhu P. 2014. Cryo-EM structures of two bovine adenovirus type 3 intermediates. Virology 450–451:174–181. doi: 10.1016/j.virol.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Vellinga J, van den Wollenberg DJ, van der Heijdt S, Rabelink MJ, Hoeben RC. 2005. The coiled-coil domain of the adenovirus type 5 protein IX is dispensable for capsid incorporation and thermostability. J Virol 79:3206–3210. doi: 10.1128/JVI.79.5.3206-3210.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabry CM, Rosa-Calatrava M, Moriscot C, Ruigrok RW, Boulanger P, Schoehn G. 2009. The C-terminal domains of adenovirus serotype 5 protein IX assemble into an antiparallel structure on the facets of the capsid. J Virol 83:1135–1139. doi: 10.1128/JVI.01808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campos SK. 2014. New structural model of adenoviral cement proteins is not yet concrete. Proc Natl Acad Sci U S A 111:E4542–E4543. doi: 10.1073/pnas.1415364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol 79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maier O, Galan DL, Wodrich H, Wiethoff CM. 2010. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 402:11–19. doi: 10.1016/j.virol.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sung MT, Cao TM, Coleman RT, Budelier KA. 1983. Gene and protein sequences of adenovirus protein VII, a hybrid basic chromosomal protein. Proc Natl Acad Sci U S A 80:2902–2906. doi: 10.1073/pnas.80.10.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews DA, Russell WC. 1995. Adenovirus protein-protein interactions: molecular parameters governing the binding of protein VI to hexon and the activation of the adenovirus 23K protease. J Gen Virol 76:1959–1969. doi: 10.1099/0022-1317-76-8-1959. [DOI] [PubMed] [Google Scholar]
- 25.Graziano V, McGrath WJ, Suomalainen M, Greber UF, Freimuth P, Blainey PC, Luo G, Xie XS, Mangel WF. 2013. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. I. Binding to DNA and to hexon of the precursor to protein VI, pVI, of human adenovirus. J Biol Chem 288:2059–2067. doi: 10.1074/jbc.M112.377150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snijder J, Benevento M, Moyer CL, Reddy V, Nemerow GR, Heck AJ. 2014. The cleaved N-terminus of pVI binds peripentonal hexons in mature adenovirus. J Mol Biol 426:1971–1979. doi: 10.1016/j.jmb.2014.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanche F, Monegier B, Faucher D, Duchesne M, Audhuy F, Barbot A, Bouvier S, Daude G, Dubois H, Guillemin T, Maton L. 2001. Polypeptide composition of an adenovirus type 5 used in cancer gene therapy. J Chromatogr A 921:39–48. doi: 10.1016/S0021-9673(01)00896-2. [DOI] [PubMed] [Google Scholar]
- 28.Webster A, Russell S, Talbot P, Russell WC, Kemp GD. 1989. Characterization of the adenovirus proteinase: substrate specificity. J Gen Virol 70:3225–3234. doi: 10.1099/0022-1317-70-12-3225. [DOI] [PubMed] [Google Scholar]
- 29.van Oostrum J, Burnett RM. 1985. Molecular composition of the adenovirus type 2 virion. J Virol 56:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehmberg E, Traina JA, Chakel JA, Chang RJ, Parkman M, McCaman MT, Murakami PK, Lahidji V, Nelson JW, Hancock WS, Nestaas E, Pungor E Jr. 1999. Reversed-phase high-performance liquid chromatographic assay for the adenovirus type 5 proteome. J Chromatogr B Biomed Sci Appl 732:411–423. doi: 10.1016/S0378-4347(99)00316-3. [DOI] [PubMed] [Google Scholar]
- 31.Milazzo AC, Cheng A, Moeller A, Lyumkis D, Jacovetty E, Polukas J, Ellisman MH, Xuong NH, Carragher B, Potter CS. 2011. Initial evaluation of a direct detection device detector for single particle cryo-electron microscopy. J Struct Biol 176:404–408. doi: 10.1016/j.jsb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. 2013. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wodrich H, Guan T, Cingolani G, Von Seggern D, Nemerow G, Gerace L. 2003. Switch from capsid protein import to adenovirus assembly by cleavage of nuclear transport signals. EMBO J 22:6245–6255. doi: 10.1093/emboj/cdg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster A, Hay RT, Kemp G. 1993. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell 72:97–104. doi: 10.1016/0092-8674(93)90053-S. [DOI] [PubMed] [Google Scholar]
- 35.Mangel WF, McGrath WJ, Toledo DL, Anderson CW. 1993. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 361:274–275. doi: 10.1038/361274a0. [DOI] [PubMed] [Google Scholar]
- 36.Mangel WF, San Martin C. 2014. Structure, function and dynamics in adenovirus maturation. Viruses 6:4536–4570. doi: 10.3390/v6114536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corden J, Engelking HM, Pearson GD. 1976. Chromatin-like organization of the adenovirus chromosome. Proc Natl Acad Sci U S A 73:401–404. doi: 10.1073/pnas.73.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang W, Chang J, Jakana J, Weigele P, King J, Chiu W. 2006. Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature 439:612–616. doi: 10.1038/nature04487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. 2006. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 40.Catalano CE. (ed). 2005. Viral genome packaging machines: genetics, structure, and mechanism. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 41.Christensen JB, Byrd SA, Walker AK, Strahler JR, Andrews PC, Imperiale MJ. 2008. Presence of the adenovirus IVa2 protein at a single vertex of the mature virion. J Virol 82:9086–9093. doi: 10.1128/JVI.01024-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Arcos R. 2005. Interaction of the adenovirus major core protein precursor, pVII, with the viral DNA packaging machinery. Virology 334:194–202. doi: 10.1016/j.virol.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 43.Dai X, Li Z, Lai M, Shu S, Du Y, Zhou ZH, Sun R. 2017. In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature 541:112–116. doi: 10.1038/nature20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rolfsson O, Middleton S, Manfield IW, White SJ, Fan B, Vaughan R, Ranson NA, Dykeman E, Twarock R, Ford J, Cheng Kao C, Stockley PG. 2016. Direct evidence for packaging signal-mediated assembly of bacteriophage MS2. J Mol Biol 428:431–448. doi: 10.1016/j.jmb.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verlinden Y, Cuconati A, Wimmer E, Rombaut B. 2000. Cell-free synthesis of poliovirus: 14S subunits are the key intermediates in the encapsidation of poliovirus RNA. J Gen Virol 81:2751–2754. doi: 10.1099/0022-1317-81-11-2751. [DOI] [PubMed] [Google Scholar]
- 46.Graziano V, Luo G, Blainey PC, Perez-Berna AJ, McGrath WJ, Flint SJ, San Martin C, Xie XS, Mangel WF. 2013. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. II. Adenovirus proteinase is activated in an unusual one-dimensional biochemical reaction. J Biol Chem 288:2068–2080. doi: 10.1074/jbc.M112.407312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blainey PC, Graziano V, Perez-Berna AJ, McGrath WJ, Flint SJ, San Martin C, Xie XS, Mangel WF. 2013. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. IV. Viral proteinase slides along DNA to locate and process its substrates. J Biol Chem 288:2092–2102. doi: 10.1074/jbc.M112.407460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Condezo GN, Marabini R, Ayora S, Carazo JM, Alba R, Chillon M, San Martin C. 2015. Structures of adenovirus incomplete particles clarify capsid architecture and show maturation changes of packaging protein L1 52/55k. J Virol 89:9653–9664. doi: 10.1128/JVI.01453-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez-Berna AJ, Mangel WF, McGrath WJ, Graziano V, Flint J, San Martin C. 2014. Processing of the L1 52/55k protein by the adenovirus protease: a new substrate and new insights into virion maturation. J Virol 88:1513–1524. doi: 10.1128/JVI.02884-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weber J. 1976. Genetic analysis of adenovirus type 2. III. Temperature sensitivity of processing viral proteins. J Virol 17:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeh-Kai L, Akusjarvi G, Alestrom P, Pettersson U, Tremblay M, Weber J. 1983. Genetic identification of an endoproteinase encoded by the adenovirus genome. J Mol Biol 167:217–222. doi: 10.1016/S0022-2836(83)80044-8. [DOI] [PubMed] [Google Scholar]
- 52.Perez-Berna AJ, Marabini R, Scheres SH, Menendez-Conejero R, Dmitriev IP, Curiel DT, Mangel WF, Flint SJ, San Martin C. 2009. Structure and uncoating of immature adenovirus. J Mol Biol 392:547–557. doi: 10.1016/j.jmb.2009.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez-Berna AJ, Ortega-Esteban A, Menendez-Conejero R, Winkler DC, Menendez M, Steven AC, Flint SJ, de Pablo PJ, San Martin C. 2012. The role of capsid maturation on adenovirus priming for sequential uncoating. J Biol Chem 287:31582–31595. doi: 10.1074/jbc.M112.389957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silvestry M, Lindert S, Smith JG, Maier O, Wiethoff CM, Nemerow GR, Stewart PL. 2009. Cryo-electron microscopy structure of adenovirus type 2 temperature-sensitive mutant 1 reveals insight into the cell entry defect. J Virol 83:7375–7383. doi: 10.1128/JVI.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J, Cho JY, Kim JH, Jung KC, Yun CO. 2002. Evaluation of E1B gene-attenuated replicating adenoviruses for cancer gene therapy. Cancer Gene Ther 9:725–736. doi: 10.1038/sj.cgt.7700494. [DOI] [PubMed] [Google Scholar]
- 56.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. 2005. Automated molecular microscopy: the new Leginon system. J Struct Biol 151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 57.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 58.Kivioja T, Ravantti J, Verkhovsky A, Ukkonen E, Bamford D. 2000. Local average intensity-based method for identifying spherical particles in electron micrographs. J Struct Biol 131:126–134. doi: 10.1006/jsbi.2000.4279. [DOI] [PubMed] [Google Scholar]
- 59.Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 60.Liang Y, Ke EY, Zhou ZH. 2002. IMIRS: a high-resolution 3D reconstruction package integrated with a relational image database. J Struct Biol 137:292–304. doi: 10.1016/S1047-8477(02)00014-X. [DOI] [PubMed] [Google Scholar]
- 61.Liu H, Cheng L, Zeng S, Cai C, Zhou ZH, Yang Q. 2008. Symmetry-adapted spherical harmonics method for high-resolution 3D single-particle reconstructions. J Struct Biol 161:64–73. doi: 10.1016/j.jsb.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 62.Zhang X, Zhang X, Zhou ZH. 2010. Low cost, high performance GPU computing solution for atomic resolution cryoEM single-particle reconstruction. J Struct Biol 172:400–406. doi: 10.1016/j.jsb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 64.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 65.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu X, Veesler D, Campbell MG, Barry ME, Asturias FJ, Barry MA, Reddy VS. 2017. Cryo-EM structure of human adenovirus D26 reveals the conservation of structural organization among human adenoviruses. Sci Adv 3:e1602670. doi: 10.1126/sciadv.1602670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.