

ABSTRACT
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a group of rare progressive neurodegenerative disorders caused by an abnormally folded prion protein (PrPSc). This is capable of transforming the normal cellular prion protein (PrPC) into new infectious PrPSc. Interspecies prion transmissibility studies performed by experimental challenge and the outbreak of bovine spongiform encephalopathy that occurred in the late 1980s and 1990s showed that while some species (sheep, mice, and cats) are readily susceptible to TSEs, others are apparently resistant (rabbits, dogs, and horses) to the same agent. To study the mechanisms of low susceptibility to TSEs of certain species, the mouse-rabbit transmission barrier was used as a model. To identify which specific amino acid residues determine high or low susceptibility to PrPSc propagation, protein misfolding cyclic amplification (PMCA), which mimics PrPC-to-PrPSc conversion with accelerated kinetics, was used. This allowed amino acid substitutions in rabbit PrP and accurate analysis of misfolding propensities. Wild-type rabbit recombinant PrP could not be misfolded into a protease-resistant self-propagating isoform in vitro despite seeding with at least 12 different infectious prions from diverse origins. Therefore, rabbit recombinant PrP mutants were designed to contain every single amino acid substitution that distinguishes rabbit recombinant PrP from mouse recombinant PrP. Key amino acid residue substitutions were identified that make rabbit recombinant PrP susceptible to misfolding, and using these, protease-resistant misfolded recombinant rabbit PrP was generated. Additional studies characterized the mechanisms by which these critical amino acid residue substitutions increased the misfolding susceptibility of rabbit PrP.
IMPORTANCE Prion disorders are invariably fatal, untreatable diseases typically associated with long incubation periods and characteristic spongiform changes associated with neuronal loss in the brain. Development of any treatment or preventative measure is dependent upon a detailed understanding of the pathogenesis of these diseases, and understanding the mechanism by which certain species appear to be resistant to TSEs is critical. Rabbits are highly resistant to naturally acquired TSEs, and even under experimental conditions, induction of clinical disease is not easy. Using recombinant rabbit PrP as a model, this study describes critical molecular determinants that confer this high resistance to transmissible spongiform encephalopathies.
KEYWORDS: PMCA, PrP, prion, prion propagation, rabbit, susceptibility, TSE
INTRODUCTION
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a family of progressive neurodegenerative disorders distinguished by long incubation periods and characteristic spongiform changes associated with neuronal loss in the central nervous system (CNS). The causative agent of TSEs is an abnormally folded prion protein (PrPSc) capable of transforming the normal cellular prion protein (PrPC) into infectious and transmissible PrPSc (1). TSEs have been described for several mammalian species, appearing either naturally (scrapie in sheep and goats, bovine spongiform encephalopathy [BSE] in cattle, chronic wasting disease [CWD] in cervids, and Creutzfeldt-Jakob disease [CJD] in humans) or by experimental transmission studies (scrapie in mice and hamsters). Although much of the pathogenesis of the prionopathies has been determined in the last 40 years, such as the identification of the etiological agent, there are still many unanswered questions regarding the strain phenomenon and interspecies transmissibility. Prion diseases can be transmitted from one species to some others, albeit with various efficiencies (2). This phenomenon is still inexplicable, as the molecular mechanisms that determine interspecies transmissibility or any transmission barrier remain unknown (3). TSE transmissibility in different animal species, by either experimental challenges or natural infections, together with the BSE outbreak that occurred in the United Kingdom in the late 1980s and 1990s, showed that although some species were readily affected, such as sheep, domestic and captive wild bovids, mice, and cats, other species, such as rabbits, dogs, and horses, did not develop TSEs (4, 5). These findings and the absence of natural prion diseases in the unaffected species led to the belief that they were resistant to TSEs.
However, different in vitro experimental approaches developed to study interspecies transmission barriers proved that the existence of mammalian species resistant to prion infection may have been an incorrect assumption (6). This was shown recently by use of protein misfolding cyclic amplification (PMCA) (7) to generate rabbit prions in vitro and resulted in the first TSE reported for leporids (8) and also for transgenic mice expressing rabbit PrP (9). Although the putative resistance to prion diseases of rabbits was disproven, the apparently low attack rate, long incubation period, and requirement for adaptation through PMCA (10, 11) to achieve infection demonstrated their low susceptibility to TSE infection (9). Thus, any species historically considered resistant to prions may more appropriately be considered to possess low susceptibility (12).
The existence of some PrP-independent, species-specific unknown factors (13) as an explanation for the apparent resistance of some species to TSEs was ruled out recently by successful transmission to transgenic rabbits. The generation of transgenic rabbits expressing ovine PrP and their successful infection with scrapie prions definitively highlighted that the amino acid sequence of rabbit PrPC was responsible for the low susceptibility of rabbits to prion infections (14). Although rabbit PrPC has been studied in detail to identify distinctive structural elements that explain its low susceptibility to prion-like misfolding (15–28), the key elements or amino acid residues causing this behavior remain unknown.
To address this deficit and characterize the mechanisms by which rabbit PrP is resistant to misfolding into a protease-resistant, self-propagating isoform, PMCA of recombinant rabbit PrP (recPMCA) was used. The principal advantage of recPMCA, in contrast to PMCA based on brain homogenates as a source of PrPC (29), is the ability to use different recombinant PrP (rec-PrP) mutants to accurately determine the prion-like misfolding propensity of each mutant (30, 31). The aim of this study was to use rabbit recPMCA to identify and study the effects of specific amino acid residues within the rabbit PrPC sequence that determine its low susceptibility to misfolding. Three promising key amino acid residue substitutions (S107N, M108L, and I202V) have been found that render rabbit recombinant PrP highly susceptible to prion-induced misfolding. However, additional structural studies will be required to further characterize the mechanisms by which these substitutions increase the susceptibility to misfolding of the mutated rabbit PrP's low susceptibility to prion disease.
RESULTS
Resistance of rabbit recombinant PrP to misfolding in vitro.
Different prion strains were chosen to try to misfold rabbit rec-PrP into a protease-resistant, self-propagating isoform. The selection was based on their proven ability to induce misfolding of brain-derived rabbit PrPC by PMCA (Table 1) (8).
TABLE 1.
Seeds used for serial recPMCA rounds performed with rabbit rec-PrP-based substratesc
| Inoculuma | Characteristicsb |
Source | Reference | ||
|---|---|---|---|---|---|
| Species of origin | Actual species | Origin | |||
| Brain-derived inocula | |||||
| RML | Sheep | Mouse | Brain | Rocky Mountain Laboratory | 79 |
| ME7 | Sheep | Mouse | Brain | Francesca Chianini (Moredun, Edinburgh) | 80 |
| CWD | Mule deer | Mule deer | Brain | Jean E. Jewell (University of Wyoming) | 81 |
| Classical BSE | Cattle | Cattle | Brain | Tomás Mayoral (LCV, Madrid, Spain) | 82 |
| Sheep BSE | Cattle | Sheep | Brain | Olivier Andreoletti (INRA, Toulouse, France) | 83 |
| Scrapie (SSBP/1) | Sheep | Sheep | Brain | TSE Resource Centre | 84 |
| PMCA-adapted inocula (propagated in rabbit brain homogenate) | |||||
| De novo RaPrPSc | Rabbit | Rabbit | Brain | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | 8 |
| Rabbit-RML | Sheep | Rabbit | PMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | 8 |
| Rabbit-ME7 | Sheep | Rabbit | PMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | 8 |
| Rabbit-CWD | Mule deer | Rabbit | PMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | 8 |
| Rabbit-BSE | Cattle | Rabbit | PMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | 8 |
| Rabbit-de novo RaPrPSc | Rabbit | Rabbit | PMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | 8 |
| Recombinant inocula | |||||
| recRML-SuperHigh | Mouse | Mouse (recombinant) | recPMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | Unpublished |
| recRML-High | Mouse | Mouse (recombinant) | recPMCA | Joaquín Castilla (CIC bioGUNE, Bilbao, Spain) | Unpublished |
The inoculum column shows the most common name of each prion strain. In the case of those obtained by PMCA, the name indicates the original inoculum and the animal to which it was adapted by PMCA.
Characteristics include the animal species from which the inoculum was isolated, the animal species to which it was adapted, and the origin of the inoculum, i.e., brain if it was obtained directly from the brain of an affected animal, PMCA if it was adapted through serial rounds of PMCA, and recPMCA if it was adapted to a rec-PrP through serial rounds of recPMCA.
In all cases, 0% of tubes were positive for rec-PrPres after 20 recPMCA rounds.
Using recombinant rabbit PrP complemented with Prnp0/0 brain homogenate as the substrate and the first group of inocula listed in Table 1, 20 serial recPMCA rounds were performed at a 1:10 dilution for each seed. Six intraexperimental replicates were used for each seed, and the same number of unseeded tubes was included in all the rounds as a control for spontaneous misfolding or cross-contamination. No protease-resistant misfolded rec-PrP (rec-PrPres) was detected in any of the tubes after 20 serial recPMCA rounds.
Given the absence of rabbit rec-PrPres with the first group of inocula (brain-derived inocula), we used the original inocula obtained by serial PMCA rounds in rabbit brain homogenates (Table 1) (8). These rabbit seeds, besides eliminating the interspecies transmission barrier, had the advantage of already being adapted to the in vitro propagation system.
Two series of 20 rounds of recPMCA were performed with the rabbit PMCA-adapted seeds (interexperimental replicates). Nevertheless, recombinant rabbit PrP could not be misfolded into a protease-resistant prion isoform despite the removal of the species barrier. This suggested that misfolding of rabbit rec-PrP could be impeded by the differences between brain-derived PrP and recombinant PrP, e.g., the latter is devoid of any glycosylation and the glycosylphosphatidylinositol (GPI) anchoring region. For that reason, we chose different recombinant inocula generated previously in the laboratory in an attempt to generate protease-resistant misfolded rabbit rec-PrP. The use of already misfolded rec-PrPs from other species would reduce the potential propagation barrier due to the differences between recombinant and brain-derived PrPs. Therefore, two murine rec-PrPres variants were selected (Table 1), both obtained previously from a murine RML isolate used as a seed in serial recPMCA rounds (data not shown). These two mouse recombinant seeds were selected because, among all the misfolded rec-PrPs available in the laboratory, they were from the species phylogenetically closest to rabbit and because both showed infectivity in vivo (unpublished results).
Identically to that with the previous seeds, 20 serial rounds of recPMCA with six sample replicates failed to misfold rabbit rec-PrPs into a prion-like isoform. Therefore, it was not possible to obtain a proteinase K (PK)-resistant misfolded recombinant rabbit PrP directly by seeding and subjecting samples to serial recPMCA irrespective of the source of the seed or if it was derived from brain homogenate or a recombinant protein.
In vitro misfolding of rabbit recombinant PrPs containing mouse PrP substitutions.
Given the resistance of rabbit rec-PrP to misfolding, other strategies were explored to induce protease-resistant misfolding of recombinant rabbit PrP. Taking into account that mouse rec-PrP could be misfolded previously following the same strategy and that mouse and rabbit PrP primary sequences are very similar (Fig. 1), we generated a set of rabbit rec-PrPs containing each of the mouse PrP individual amino acid substitutions to test their influence on in vitro prion-like misfolding ability. To ensure similar rec-PrP concentrations, as large variations may have an impact on their in vitro misfolding ability, all the substrates were analyzed by Western blotting (Fig. 2). All of them contained similar amounts of rec-PrP and therefore were suitable for comparative in vitro misfolding studies.
FIG 1.

Primary sequence alignment of recombinant rabbit and mouse PrPs showed 11 substitutions in the central region of the protein. Chosen substitutions are highlighted (G99N, S107N, M108L, L137M, Y144W, S173N, V183I, I202V, I204M, I214V, and Q219K) and listed with amino acid numbering corresponding to full-length mouse PrP (identical for rabbit PrP). Discarded substitutions are shown in gray text.
FIG 2.

Biochemical characterization of mutated rabbit rec-PrP-containing substrates showed similar amounts of rec-PrP present. The Western blot shows similar amounts of the different recPMCA substrates based on wild-type rabbit rec-PrPs (wt rPrP) with specific mouse substitutions. Note the presence of a single band of approximately 20 kDa in the absence of protease K (PK) treatment. Wild-type rabbit and mouse rec-PrP-containing substrates are also included. The lower signal for the wild-type mouse rec-PrP (mouse rPrP) can be explained by a lower affinity of the monoclonal antibody SAF84 (1:400) which was used to develop the membranes. Rabbit PrPC (rabbit normal brain homogenate) was run in a separate gel, and the image is shown divided from the other gel by a vertical gray line.
To further facilitate the generation of protease-resistant misfolded recombinant rabbit PrPs carrying the mouse amino acid residue substitutions, a mouse recombinant seed (recRML-High) was chosen to eliminate the transmission barrier due to the differences between mammalian brain-derived and recombinant PrPs. Eight of 11 mutated rabbit rec-PrPs (G99N, S107N, M108L, L137M, Y144W, I202V, I204M, and Q219K) were successfully misfolded into a protease-resistant isoform. One of the mutants (S107N) misfolded spontaneously in one of the control replicates, though in a much later round (R13) than the one induced by the recRML seed (R02) (Fig. 3A). All the misfolded mutated rabbit rec-PrPs showed a consistent PK-resistant band of 16 to 17 kDa, with no obvious differences in migration pattern, when subjected to gel electrophoresis (Fig. 3C).
FIG 3.

Three of 11 recombinant rabbit PrPs with mouse PrP single-residue substitutions were misfolded by recRML and brainRML murine prions in vitro. (A) Graphical representation of the emergence and relative amount of protein misfolding (PrPres) for each round of recPMCA (denoted R01 to R20) as evaluated by Western blotting and SDS-PAGE. The percentages of tubes showing PK-resistant misfolded rec-PrP are indicated in grayscale according to the legend. Different mutated rabbit recombinant proteins were subjected to serial rounds of recPMCA after seeding with recRML (misfolded recombinant mouse PrP [recRML-High]). Note the spontaneous emergence of misfolding for the unseeded S107N variant. WT, wild-type rabbit rec-PrPs. (B) Selected mutated rabbit recombinant proteins were subjected to serial rounds of recPMCA after seeding with brainRML (brain-derived RML). Every experiment contained 4 tubes (intraexperimental replicates). The percentage of positive tubes (tubes showing a protease K-resistant signal after digestion with 80 μg/ml of PK) after each round of recPMCA is noted in grayscale as shown in the legend. (C) Western blot representing the recRML (recombinant origin)- or brainRML (brain origin)-seeded and misfolded mutated rabbit rec-PrPs after PK digestion. Samples were digested with 85 μg/ml of PK. Similar bands of approximately 17 kDa, corresponding to the PK-resistant 90-230 fragment of the prion protein, are shown. Membranes were developed with monoclonal antibody SAF84 (1:400). Rabbit rPrP, undigested recombinant rabbit rec-PrP.
To select substitutions potentially more relevant for lowering the interspecies transmission barrier between rabbit and mouse PrPs, a new set of serial recPMCA rounds was performed. In this case, the substrates that could be misfolded (see above) were seeded with brain-derived RML (brainRML), adding the barrier between recombinant and brain-derived PrPs to the previous interspecies barrier. Prion-like misfolding was induced in three of the eight selected mutated rabbit rec-PrPs (S107N, M108L, and I202V). The S107N substitution once again showed spontaneous misfolding ability (Fig. 3B and C). These results suggest that certain amino acid substitutions probably exert a stronger influence on the interspecies barrier between mouse and rabbit PrPs.
Evaluation of the propagation ability of misfolded mutated rabbit rec-PrPs.
The stability and self-propagation ability of the new rabbit rec-PrPres constructs were evaluated by two different in vitro propagation experiments: (i) 15 serial recPMCA rounds to prove their stable propagation abilities and to remove the original RML seed by dilution and (ii) one recPMCA round using serial dilutions (1:10 to 1:108) of each stabilized rec-PrPres on substrates containing the same mutated rec-PrPs. The amounts of PK-resistant rec-PrPs in each recPMCA product were adjusted by dilution on Prnp0/0 mouse brain homogenate and verified by Western blotting (Fig. 3C). All the rec-PrPres constructs generated based on the distinct mutated rabbit rec-PrPs were able to propagate on substrates containing the same rec-PrPs, with minor differences in propagation capacity (Fig. 4).
FIG 4.

In vitro propagation of mutated rabbit rec-PrPres on substrates containing the same mutated rec-PrPs demonstrates their self-propagation ability. The graph shows the mean (± standard deviation) of the maximum dilution reached by each rec-PrPres seed on a substrate containing the same mutated rec-PrP. The x axis shows rec-PrPres constructs with different origins (recombinant [misfolded by recRML as a seed] or brain [misfolded by brain-derived RML as a seed]). All seeds propagated efficiently in their own substrates (dilutions of 10−4 to 10−8). Mouse RML, wild-type mouse rec-PrP seeded with the original recRML was used as a positive control.
Overcoming the mouse-rabbit transmission barrier by using mutated rabbit rec-PrPres constructs as seeds.
The primary sequences of the new, self-propagating mutated rabbit rec-PrPres constructs differed from that of wild-type rabbit rec-PrP in just one amino acid residue. Therefore, we tried to overcome the transmission barrier by using each of them as seeds. Serial dilutions (1:10 to 1:108) of each rec-PrPres were performed on a wild-type rabbit rec-PrP-containing substrate and submitted to a single 48-h round of recPMCA. Unexpectedly, all of the mutated rec-PrPres constructs were able to induce misfolding on wild-type rabbit rec-PrP in a single PMCA round (Fig. 5A), and all the misfolded wild-type rabbit rec-PrPs derived from the different mutated seeds were propagated by serial recPMCA rounds up to the elimination of the original seed (Fig. 5B).
FIG 5.

All the mutated rabbit rec-PrPres constructs induce prion-like misfolding of wild-type rabbit rec-PrP in vitro. (A) Representation of the mean (± standard deviation) of the maximum dilution reached by each mutated rec-PrPres-based seed on a substrate containing wild-type rabbit rec-PrP. The x axis shows the different mutated rec-PrPres constructs acting as seeds, grouped according to their origins (recombinant [for those misfolded by the recRML seed] or brain [for the ones misfolded using brain-derived RML as a seed]). (B) Western blot representing PK-digested (85 μg/ml) misfolded wild-type rabbit rec-PrPres. A wild-type rabbit rec-PrP-based substrate was seeded with 11 different mutated seeds and subjected to 15 rounds of recPMCA. Similar bands of approximately 17 kDa, corresponding to the PK-resistant 90-230 fragment of the wild-type protein, are shown. Membranes were developed with monoclonal antibody SAF84 (1:400). Rabbit recPrP, undigested recombinant rabbit rec-PrP.
Evaluation of the propagation ability of rabbit rec-PrP with multiple mouse rec-PrP substitutions.
To study the roles of the amino acid residue substitutions that had apparently major effects on rabbit rec-PrP misfolding and on the mouse-rabbit interspecies barrier, we focused on the substitutions that made rabbit rec-PrP more susceptible to misfolding by brain-derived mouse prions (S107N, M108L, and I202V), as they allowed us to overcome both interspecies and recombinant-brain transmission barriers. To assess potential cooperative effects that could completely abrogate the species barrier between mouse and rabbit PrPs, a series of rabbit rec-PrPs were designed with the four possible combinations of these substitutions. Three double-mutated and a single triple-mutated rabbit rec-PrP were produced and recPMCA substrates prepared as described previously, with adjustment of the rec-PrP concentrations by the bicinchoninic acid (BCA) protein assay (Fig. 6A), to assess their in vitro prion-like misfolding abilities.
FIG 6.

Multiple mouse rec-PrP substitutions in rabbit rec-PrP do not reduce the interspecies transmission barrier compared to that with rabbit rec-PrP with single substitutions. (A) Different recPMCA substrates containing single, double, and triple mutant rabbit rec-PrPs were compared by Western blotting. PrP amounts were adjusted by dilution with Prnp0/0 mouse brain homogenate to reduce the variability. The membrane was developed with monoclonal antibody SAF84 (1:400). All samples showed similar amounts of recombinant PrP in the substrates. (B) Diagram of rec-PrPres generation over the course of serial in vitro propagation of the rabbit recombinant single, double, and triple mutant rec-PrPs, using recRML as the seed. Generation of rec-PrPres was evaluated by PK digestion and Western blotting (with monoclonal antibody SAF84) for each recPMCA round (rounds 1 to 10). Wild-type rabbit (rRaWt) and wild-type mouse (rMoWt) rec-PrP-containing substrates were included as negative and positive controls, respectively. The percentages of tubes showing PK-resistant misfolded rec-PrP are indicated in grayscale according to the legend. The triple mutant rabbit rec-PrP construct did not show enhanced misfolding ability compared to that of single mutant rec-PrPs. The rabbit rec-PrPs with double substitutions gave rise to rec-PrPres in later rounds than those for rabbit rec-PrPs with single substitutions, especially the I202V variant, suggesting that single substitutions combined do not act in a cooperative manner to reduce the transmission barrier.
Neither double-mutated rabbit rec-PrP nor even triple-mutated rabbit rec-PrP, the most similar in primary sequence to recRML, showed enhanced misfolding ability with respect to rec-PrPs with a single amino acid mutation (S107N or I202V) (Fig. 6B). This indicates that the three substitutions did not act cooperatively to promote rabbit rec-PrP misfolding or to lower the interspecies transmission barrier between mouse and rabbit. Moreover, the rabbit rec-PrPs with double substitutions gave rise to rec-PrPres in later rounds than those for rabbit rec-PrPs with single substitutions, especially the I202V substitution, suggesting that rather than acting in a cooperative manner, the single substitutions may interfere with each other, reducing the effect that each one produces alone on the proneness of rabbit rec-PrP to misfold into a protease-resistant isoform.
Defining the roles of substitutions that had greater effects on the misfolding ability of rabbit rec-PrP.
To determine if the absence of the substituted amino acid or the presence of the new residue favors rabbit rec-PrP misfolding, we further studied positions 107, 108, and 202. To minimize alterations of the tertiary PrP structure, we chose alternative substitutions in these positions based on the amino acids present in other species' PrPs. From the alignment of the primary PrP sequences of 12 mammalian species (mouse [Mus musculus], bank vole [Myodes glareolus], Syrian hamster [Mesocricetus auratus], sheep [Ovis aries; with ARQ polymorphisms], cattle [Bos taurus], mule deer [Odocoileus hemionus], horse [Equus ferus caballus], pig [Sus scrofa], human [Homo sapiens; with polymorphism M129M], dog [Canis lupus familiaris], cat [Felis silvestris catus], and mink [Neovison vison]), we chose not to make any alternative substitution at position 107, because it is either a serine or an asparagine residue in all the species analyzed. Thus, alternative substitutions were detected and performed for positions 108 and 202 only, where the following changes were made: M108I and I202M (the only side chains found in other species analyzed).
In the case of the M108I substitution, both seeds (recRML and brainRML) were able to induce prion-like misfolding as in the case of the M108L substitution, which could be expected given the structural similarity of both amino acids (Fig. 7). Although these results may suggest the absence of methionine at position 108 as the key event for modulation of the interspecies barrier, the similarity of isoleucine (Ile) and leucine (Leu) does not completely exclude the possibility that the key event is the presence of a branched-chain amino acid residue, such as Ile, Leu, or even valine (Val).
FIG 7.

Alternative substitutions at positions 108 and 202 of rabbit rec-PrP show the relevance of the presence and absence of branched-chain amino acids, respectively, to alterations of misfolding proneness. The histogram shows rec-PrPres generation over the course of serial in vitro propagation of rabbit recombinant mutated PrPs with alternative substitutions, using recombinant RML and brain-derived RML as seeds. Generation of rec-PrPres was evaluated by PK digestion and Western blotting (with monoclonal antibody SAF84) for each recPMCA round (rounds 1 to 20). The percentages of tubes showing PK-resistant misfolded rec-PrP are indicated in grayscale according to the legend. For the M108I substitution, both recRML and brainRML were able to induce misfolding. Rabbit rec-PrP with the alternative substitution I202M was not misfolded by brainRML, and the rec-PrPres induced by recRML was generated in later rounds than those for rabbit I202V rec-PrP.
In contrast, rabbit rec-PrP with the alternative substitution I202M was not misfolded into a protease-resistant isoform by brainRML, and the rec-PrPres induced by recRML was generated in later rounds than those with rabbit I202V rec-PrP (Fig. 7). This result suggests a lower misfolding proneness for the I202M variant than for the I202V variant, pointing toward the absence of Ile as the critical factor. Its effect seems to be independent of the biochemical properties of the side chain, since replacement by Val, also a branched-chain amino acid, was able to alter its effect.
In vitro misfolding ability of mouse rec-PrPs containing substitutions different from those in rabbit rec-PrP.
After evaluating the molecular determinants in the context of rabbit PrP for the mouse-rabbit interspecies transmission barrier, the effects of the same substitutions were examined in the context of mouse PrP. Eleven mouse rec-PrPs were generated with single amino acid substitutions based on alignment with the rabbit rec-PrP sequence (Fig. 1). The amount of rec-PrP in each of the substrates complemented with Prnp0/0 mouse brain homogenate was normalized using the BCA protein assay (Fig. 8A).
FIG 8.

In vitro propagation ability of mouse rec-PrP containing single amino acid substitutions from rabbit rec-PrP shows poor correlation with that for the same substitutions in a rabbit rec-PrP context. (A) Eleven different recPMCA substrates containing single mutant mouse rec-PrPs were compared by Western blotting. PrP amounts were adjusted by dilution with Prnp0/0 mouse brain homogenate to reduce the variability, and the membrane was developed with monoclonal antibody SAF83 (1:400). WT, wild type. All the samples were run in the same gel, but the blot was cropped, as indicated by the vertical dotted line, to avoid displaying unrelated samples. (B) Representation of the means (± standard deviations) of the maximum dilutions reached by the recRML seed on substrates containing wild-type and different mutated mouse rec-PrPs. Although all the mutated mouse rec-PrPs showed more resistance to recRML propagation than the wild-type mouse rec-PrP did, only N99G rec-PrP completely failed to propagate.
Despite all the mutated mouse rec-PrPs showing a significant barrier to recRML propagation, none of them reached the propagation levels of the wild-type mouse rec-PrP-containing substrate, and the rec-PrP with the N99G substitution blocked propagation completely (Fig. 8B). However, by serial recPMCA rounds, it was possible to overcome this barrier, and mouse N99G rec-PrPres could be generated after four serial recPMCA rounds (results not shown).
Overall, the results do not show a perfect correlation between the positions that promoted rabbit rec-PrP misfolding by mouse prions and the positions that hindered recRML propagation in the context of mouse rec-PrP. The only exception might be the substitution at position 99, which allowed rabbit rec-PrP misfolding in early rounds of recPMCA and also showed a strong hindrance of recRML propagation in the context of mouse rec-PrP. However, this correlation was not observed for the rest of the substitution positions (Fig. 3A and 8B).
In vitro misfolding ability of different mutated rabbit rec-PrPs after seeding with prions from other species.
Based on the results obtained, the S107N, M108L, and I202V substitutions emerged as those most influential on the transmission barrier between mouse and rabbit, as they were the only ones able to be converted to rec-PrPres directly by brain-derived RML. To assess whether these substitutions were acting specifically on this particular species barrier or if they increased the rabbit rec-PrP misfolding proneness in general, a series of in vitro propagation studies were conducted.
The three selected mutated rabbit rec-PrPs were subjected to serial recPMCA rounds using prion strains from different species as seeds. The seeds were chosen due to their ability to misfold brain-derived rabbit PrP (8) but not wild-type recombinant rabbit PrP (Table 1). Thus, mouse strain ME7 was selected as an alternative murine strain. To test seeds from other species, ovine scrapie (SSBP/1), sheep-adapted BSE (shBSE), and classical BSE were chosen. The rec-PrP amounts in all the substrates were adjusted using the BCA protein assay and evaluated by Western blotting (Fig. 9).
FIG 9.
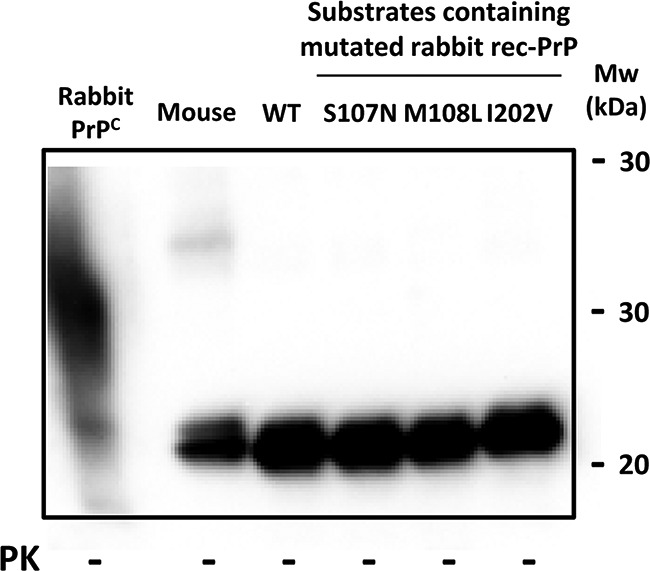
Western blot comparing different rec-PrP-based substrates for recPMCA, showing similar amounts of rec-PrP. Different recPMCA substrates containing single mutant rabbit rec-PrPs (S107N, M108L, and I202V) were compared by Western blotting. Similar amounts of rec-PrPs were used after adjustment with Prnp0/0 mouse brain homogenate to reduce the variability. All the samples showed very similar rec-PrP amounts in the substrate. The membrane was developed with monoclonal antibody SAF84 (1:400).
As expected, mouse rec-PrP showed the highest susceptibility to misfolding by all the seeds tested. It was able to propagate the brain-derived mouse ME7 strain in only a single round of recPMCA (Fig. 10). The mutated rabbit rec-PrPs were also misfolded into a protease-resistant isoform by the murine, ovine, and bovine seeds used. Unlike the results of previous experiments, a misfolded rec-PrPres was also observed at round 20 of recPMCA with an unseeded mouse rec-PrP-containing substrate, which was probably a spontaneously misfolded rec-PrPres (Fig. 10). Similar to previous results obtained by use of the mouse RML seed, substrates containing rabbit rec-PrPs with S107N and I202V substitutions showed earlier generation of rec-PrPres as well as a higher rec-PrPres-positive tube percentage. Rabbit I202V rec-PrP was especially efficient at propagating sheep scrapie and mouse ME7, a mouse-adapted scrapie isolate, suggesting a preference for scrapie and scrapie-derived strains. However, the other mutated rabbit rec-PrPs did not show a predilection for any particular strain or species. Again, rec-PrPres was detected at late recPMCA rounds for rabbit S107N rec-PrP, most likely because of a spontaneous misfolding event, in agreement with its consistent formation in different experiments.
FIG 10.

Mutated rabbit rec-PrPs that are more susceptible to misfolding by RML are also susceptible to prions from other species. The diagram shows rec-PrPres generation over the course of serial in vitro propagation of ME7, scrapie (SSBP/1), sheep BSE (ShBSE), and classical BSE inocula on substrates containing mutated rabbit rec-PrPs (S107N, M108L, and I202V) selected because of their higher propensity to be misfolded by mouse RML. Generation of rec-PrPres was evaluated by protease K (PK) digestion and Western blotting (with monoclonal antibody SAF84) for each recPMCA round (rounds 1 to 20). Wild-type rabbit (RaWt) and wild-type mouse (MoWt) rec-PrP-containing substrates were included as negative and positive controls, respectively. Note that wild-type mouse recombinant PrP was the most susceptible to misfolding by all the seeds used, as expected, while recombinant rabbit PrP was again unable to be misfolded. Regarding the mutated rabbit rec-PrPs, despite all being misfolded by all of the seeds used, the I202V mutant showed, in most cases, the greatest percentage of misfolded tubes and earlier affected rounds, suggesting that it is the most susceptible to misfolding by all of the seeds used. The percentages of tubes showing PK-resistant misfolded rec-PrP are indicated in greyscale according to the legend.
Evaluation of the tendency of different rec-PrPs to populate a β-state in the presence of chaotropic agents.
The formation of PrPSc requires a structural rearrangement of the mostly α-helical PrPC to β-sheet-rich PrPSc (32). Thus, the stability of the native conformation of PrPC may influence its proneness to misfolding. Furthermore, under mild destabilizing conditions, such as acidic pH and treatment with chaotropic agents, PrP can acquire a conformational state that is mostly β-sheet structured, i.e., the β-state. Hornemann and collaborators proposed that this may be an intermediate form between the native and unfolded states of the PrP folding pathway (33). Additionally, a study by Khan and collaborators showed that the addition of chaotropic agents to a recombinant PrP solution has a great influence on the fraction of rec-PrP that acquires the β-state and that the proneness to convert to the β-state is closely related to the susceptibility to prion infection (34). We applied the technique developed in the latter study to determine if the prion-like misfolding proneness of the mutated rabbit rec-PrP most susceptible to all the seeds tested (I202V mutant) correlated with an increased conversion to the β-state upon treatment with chaotropic agents. Syrian hamster rec-PrP was used as a control for the technique, as well as wild-type mouse and rabbit rec-PrPs as susceptible and resistant controls, respectively. Although the original technique was performed using urea as the chaotropic agent, due to technical issues we decided to use another well-established chaotropic agent, guanidinium chloride (GdnHCl). The switch of chaotropic agent was because the use of GdnHCl allowed the addition of more diluted protein, facilitating adjustment of rec-PrP concentrations. We first evaluated if the effects of conversion to the β-state were equal upon treatment with urea or GdnHCl by using hamster rec-PrP. The β-state acquisition patterns were similar in both cases, with most of the hamster rec-PrP converted to the β-state at pH 4 and 4.5, between 2 M and 4 M in the case of urea and between 1 M and 2 M in the case of GdnHCl (Fig. 11A). Equally, hamster rec-PrP at pH 5 had a very low rate of conversion to the β-state with either agent, all in agreement with the work of Khan and collaborators (34).
FIG 11.

The β-state acquisition proneness of rabbit I202V rec-PrP upon treatment with chaotropic agents was not altered with respect to that of wild-type rabbit rec-PrP. (A) Fractions of hamster rec-PrP that acquired the β-state upon treatment with increasing concentrations of urea or GdnHCl at pH 4, pH 4.5, and pH 5. The y axis shows the fractions of rec-PrP that acquired the β-state under specific pH conditions. The x axis shows increasing concentrations of the chaotropic agent used in each case. Measurement of the β-state fraction was performed by circular dichroism assay at pH 4, 4.5, and 5, as the β-state proneness is higher under more acidic conditions. The β-state acquisition patterns were very similar with both chaotropic agents, with most of the hamster rec-PrP being in the β-state at pH 4 and 4.5 between 2 M and 4 M in the case of urea and between 1 M and 2 M in the case of GdnHCl. (B) Fractions of hamster, mouse, rabbit, and rabbit I202V rec-PrPs that acquired the β-state upon treatment with increasing concentrations of GdnHCl at pH 4, pH 4.5, and pH 5. The y axes show the fractions of rec-PrP that acquired the β-state under various pH conditions. The x axes show the increasing concentrations of GdnHCl used. Measurement of the β-state fraction was performed by circular dichroism assay at pH 4, 4.5, and 5. The measurements are grouped by pH, since the rec-PrPs which showed higher levels of β-state acquisition were the ones that showed greater fractions of β-state under pH conditions closer to neutral. In all the plots, trend lines are included as a guide. No differences in β-state acquisition proneness were detected between mouse, rabbit, and rabbit I202V rec-PrPs, indicating that the higher misfolding ability shown previously for mouse and rabbit I202V rec-PrPs than that for wild-type rabbit rec-PrP is not associated with their β-state acquisition proneness.
When we created denaturing curves for wild-type mouse, wild-type rabbit, and rabbit I202V rec-PrPs by using GdnHCl at the same three pH values, we observed that at pH 4 all the rec-PrPs showed a great tendency to populate the β-state, but at pH 4.5 and 5 only hamster rec-PrP showed conversion to the β-state (Fig. 11B). This suggests that the β-state is thermodynamically more accessible for hamster rec-PrP, which is therefore the most susceptible to misfolding induced by prions. For mouse, rabbit, and rabbit I202V rec-PrPs, however, no differences in population of the β-state were detected, indicating that the higher misfolding ability previously shown for mouse and rabbit I202V rec-PrPs than for wild-type rabbit rec-PrP is not associated with their proneness to interconvert to the β-state (Fig. 11B). The increased tendency of rabbit I202V rec-PrP to misfolding induced by different prion strains seems to be independent of the stability of its native conformation. This suggests that the effect of the I202V substitution on rabbit rec-PrP is related to subtle, local conformational rearrangements rather than to a global tertiary structural disturbance.
DISCUSSION
The molecular determinants of prion disease species transmission barriers remain mostly elusive, with the exception of similarity between the PrP primary sequences of the prion donor and acceptor individuals somehow favoring transmissibility, most likely through structural compatibility (35). To determine the locations within the prion protein responsible for rendering rabbit PrP particularly resistant to misfolding, recPMCA was chosen because it allows examination of any mutant. Unlike brain-derived rabbit PrPC, for which spontaneous conversion was observed, unseeded rabbit rec-PrP never gave rise to spontaneous rec-PrPres, despite several attempts (Table 1) (8). Although the secondary and tertiary structures of rec-PrP are very similar to those of brain-derived PrPC (36), there are important differences in terms of posttranslational modifications, such as the lack of GPI anchoring and the absence of glycosylation, that might influence the transmission barrier (37–40). Accordingly, when both human rec-PrP and brain-derived human PrPC were mixed within the same PMCA substrate, an inhibitory effect was observed, probably due to the interaction of rec-PrP with the seed, which blocked interactions with PrPC (41).
Elimination of the putative interspecies transmission barrier by use of seeds previously obtained by PMCA with rabbit brain homogenate (8) failed to overcome the high resistance to misfolding shown by rabbit rec-PrP. The differences between the structures of recombinant and brain-derived PrPs may be more significant than the differences between PrPs from different species. Therefore, we reduced the differences between recombinant and brain-derived PrPs as much as possible by using mouse rec-PrPres as the seed, with a primary amino acid sequence as close to that of the rabbit version as possible. Once again, this failed to induce the formation of rec-PrPres from wild-type rabbit rec-PrP. The low conversion efficiency of rec-PrPs, given the absence of posttranslational modifications that could restrict the acquisition of self-propagating conformations (42), together with the increased intrinsic resistance to misfolding shown by rabbit rec-PrP (43), may explain our inability to induce wild-type rabbit rec-PrPres.
Assuming that the primary structure of rabbit PrP was responsible for resistance of conversion to PrPres, the misfolding capacity of 11 mutated recombinant rabbit PrPs was examined. Mutant generation and assessment of the resultant misfolding ability have been used previously in several in vitro (40, 44–46) and ex vivo (15, 47, 48) models to study transmission barriers and to elucidate the most relevant amino acids that determine each barrier. Surprisingly, for 8 of the 11 mutated rabbit rec-PrPs, a single mouse-derived residue was sufficient to render them susceptible to misfolding induced by recombinant RML. However, as more than a single recPMCA round was required for their generation, it indicated that none of the mutations was solely responsible for the interspecies barrier, although all of them lowered it. Upon addition of the recombinant-brain transmission barrier by using brain-derived RML, 3 of the 8 permissive mutated rec-PrPs, the S107N, M108L, and I202V mutants, gave rise to misfolded rec-PrPres, suggesting that they might exert the strongest influence on the interspecies barrier between mouse and rabbit or make rabbit rec-PrP more prone to prion-induced misfolding.
We do not have a definitive explanation for why some mutated PrPs could be misfolded only by a recombinant seed (recRML), not by those of brain origin (brainRML). However, the presence or absence of the GPI anchor and glycosylation might be determinants. The absence of the GPI anchor has a strong influence over strain propagation in vivo (49, 50) and in vitro (38, 51). In addition, the role of glycosylation in modifying strains and the transmission barrier has been reported (50). For these reasons, it is reasonable to think that these differences between seed (derived from brain) and substrate (recombinant) may be more influential for some PrP mutants (those unable to be misfolded by brainRML) than for others (those able to be misfolded by brainRML). Another potential explanation for this differential behavior among mutated PrPs is that the seeds used (brain versus recombinant origin) had significantly different titles and just the most efficient PrP mutants were able to be misfolded with the low-title seed, putatively the brain-derived one.
To determine if the substitutions that facilitated rabbit rec-PrP misfolding induced by RML also make mouse rec-PrP more resistant, the inverse 11 substitutions were performed in mouse rec-PrP, and the misfolding ability of the resultant constructs was evaluated. There was very poor correlation between the more critical substitutions in the rabbit rec-PrP context and those in the mouse rec-PrP context. This is in agreement with the theory that transmission barriers are not controlled solely by key differences in primary amino acid sequences but by the conformational compatibility of the seed and the substrate PrPs and that these are affected differently by the same substitutions depending on the surrounding residues (52). However, some correlation was present, as the substitutions that showed the weakest (amino acid positions 183 and 214) and strongest (position 202) effects on the transmission barriers were conserved in both rec-PrPs, suggesting that the latter is possibly the most influential.
Previous reports on the key positions that might determine PrP misfolding susceptibility in other models or species enabled comparisons with our results. Vorberg and collaborators (15) studied the mouse-rabbit transmission barrier determinants in cell culture. Unable to observe misfolding of wild-type rabbit PrP expressed in a persistently RML-infected mouse neuroblastoma cell line, they generated a set of mouse-rabbit chimeric PrPs and evaluated their misfolding ability. Rabbit PrP with a mouse central region (residues 111 to 177) was completely resistant to misfolding by RML, as was mouse PrP with a rabbit amino-terminal region (residues 1 to 111). However, mouse PrP with a rabbit PrP carboxyl-terminal region (residues 177 to 254) dramatically reduced but did not prevent PrPres formation (15). These results suggest that amino acid differences in any region of the protein can influence transmissibility, as observed by recPMCA, in which substitutions affecting the barrier were detected all along the protein. To further explore the individual amino acid residues that most significantly affected the misfolding ability of mouse PrP, Vorberg et al. performed single substitutions. Among the N99G, N107S, and L108M substitutions, only those at positions 99 and 108 impeded the formation of PrPres, although a mouse PrP mutant carrying both N107S and L108M substitutions abrogated the blocking effect exerted by the L108M substitution alone. This demonstrates once again that tertiary structural matching may be more important than PrP sequence similarity for determining transmission barriers. The flexible amino-terminal region of PrP has long been implicated in intra- and intermolecular interactions with the structured domain of the protein, the cell surface, or natural ligands (53). Furthermore, the relevance of this region to PrPres formation has been shown previously (54), as well as its participation in interspecies transmission (55). During PrPSc formation, this domain undergoes a dramatic conformational change, from being flexible and totally exposed in PrPC to becoming partially protected against protease digestion in PrPSc (56), and any substitutions impeding this dramatic conformational change may explain the strong effect caused by amino-terminal substitutions. In this regard, a serine residue at position 107, present almost exclusively in Leporidae and macaques, may be responsible for rabbits' high resistance to prion diseases or for macaques' low susceptibility to CWD infection, to which closely related primates—such as squirrel monkeys—are highly susceptible (57).
Vorberg and collaborators performed substitutions on residues 137 and 144 (in the central region of PrP), but these did not show any relevant influence on the mouse PrP misfolding capacity, in agreement with the results shown here. They also mutated residue 173, showing a blockade of RML propagation, in contrast to the results obtained by recPMCA. The amino acid residue at position 173 and adjacent residues, located in the loop between β-sheet 2 and α-helix 2 (also known as the rigid loop), have attracted the attention of many researchers given their possible involvement in the transmission barrier between several species (58–63, 85–89), as well as in PrPres formation in vitro (45, 64). The resolution of the rabbit PrP structure by X-ray crystallography revealed a quite rigid and well-structured loop more similar to that of cervids than the flexible mouse PrP β2-α2 loop (34). The structural resolution of other PrPs by nuclear magnetic resonance (NMR) studies suggested that amino acid changes within the rigid loop, despite keeping the global structure of the protein, caused alterations in the hydrogen bonds that may influence the protein-protein interactions required for conversion of PrPC to PrPSc (53). However, other NMR-based studies also suggested that the low susceptibility to prion diseases showed by rabbits or horses may rely not just on the presence of the rigid loop but also on its interaction with α-helix 3 (65).
Substitutions performed in the carboxyl-terminal region, at positions 202, 204, and 219, influenced the transmission barrier. In particular, residue 202 had a strong modulating effect on rabbit rec-PrP misfolding ability. These results are in general agreement with the proposed critical effect of α-helix 3 and β2-α2 loop interactions in the PrPC-to-PrPSc conversion initiation (65–70). Moreover, the positions found to be relevant for rabbit rec-PrP misfolding have been shown to be important in other species as well. For example, the V203I mutation in human PrP, located at a position equivalent to position 202 in rabbit PrP, is associated with a familial form of CJD (71), and positions equivalent or adjacent to positions 204 and 219 were found to be relevant for small ruminants' resistance or susceptibility to scrapie (66, 72).
The key to rabbits' resistance to prion diseases has been sought mainly through structural studies of the rabbit wild-type PrP and a small number of mutations suggested to be relevant in the above-mentioned studies. The three-dimensional structures of the central and carboxyl-terminal regions of wild-type rabbit PrP (residues 124 to 228) (PDB accession numbers 2F3J and 3O79), the S173N mutant (residues 124 to 228) (PDB accession number 2JOH), and the I214V mutant (residues 124 to 228) (PDB accession number 2JOM), resolved by NMR or X-ray crystallography, have attracted the attention of many structural biologists. Wild-type rabbit PrP has a well-structured β2-α2 loop and a particularly large continuous positively charged area. A network of hydrogen bonds was also detected that could confer wild-type rabbit PrP with enhanced structural stability. All these features were altered in the mutated forms studied, which showed (i) an increase in the β2-α2 loop flexibility that changed its interaction with α-helix 3, (ii) an altered distribution of surface charges, and (iii) a reduction in the hydrogen bond network, all of which rendered them less stable (19). Molecular dynamics simulation studies comparing the rabbit PrP structure with those of PrPs from mouse, human, dog, and horse revealed other unique characteristics. The most relevant structures that could contribute to rabbit PrP stability are salt bridges: D177-R163, which bonds the β2-α2 loop with α-helix 2, and D201-R155, which bonds α-helices 1 and 3 (16, 17). Similar studies comparing rabbit wild-type PrP and S173N and I214V mutated forms showed that either mutation significantly altered the salt bridge network and the hydrogen bond network, which increased structural instability. Thus, all these studies point toward distinctive intramolecular interactions being responsible for the stability of rabbit PrP, with a major contribution of the interactions established between the β2-α2 loop and α-helices 2 and 3 (16, 17, 24). Importantly, these studies also reveal that point mutations can cause unpredictable changes in regions away from the mutation site itself. The results of the present study and those of others clearly show that many amino acid residues within rabbit PrP may participate in its low prion-like misfolding proneness. The unpredictable structural effects of point mutations in distant protein regions (22) highlight the need to investigate as many mutated rabbit PrP structures as possible and to correlate them with misfolding proneness or transmissibility studies in vivo, in cell cultures, or in vitro in systems such as the one used here. To investigate the possible mechanism(s) by which each particular residue alters the susceptibility of recombinant rabbit PrP to misfolding induced by prions, we focused on the three substitutions among the 11 generated that exerted the strongest effect on rec-PrPres formation propensity. Only the S107N, M108L, and I202V substitutions allowed direct conversion of rabbit rec-PrP seeded with brain-derived RML, abrogating the transmission barrier, probably due to differences between brain-derived and recombinant PrPs in addition to the interspecies transmission barrier. Moreover, their importance is supported by previous studies, while other substitutions, such as the N99G or L137M mutation, have not been studied previously or have been shown not to be critical in other systems (15).
Amino acid residue substitutions that increase the susceptibility of rabbit rec-PrP to RML may cause a completely different effect with respect to susceptibility to other strains. Therefore, it was necessary to determine if the effects of the S107N, M108L, and I202V substitutions were comparable with other mouse prion strains or prions from other species. As expected, mouse rec-PrP was the most susceptible to misfolding by all the prions tested, in agreement with its susceptibility in vivo. In fact, it propagated ME7 mouse prion strain in a single recPMCA round despite the barrier between brain-derived and recombinant PrPs. From the three substitutions, just the asparagine at position 107 is shared by mice, sheep, and cattle, which may explain the ability of this mutated rabbit rec-PrP to propagate scrapie and BSE. In contrast, the M108L and I202V substitutions, which introduce an additional amino acid residue difference between rabbit PrP and the one from sheep or cattle, suggest a mechanism less dependent on sequence identity. The results obtained with the I202V mutant are of particular interest because, apart from being the one most readily misfolded by all the seeds, it showed a greater propagation ability with scrapie or mouse-adapted scrapie, in contrast to the other mutated PrPs. These results suggest that structural rearrangement caused by the I202V substitution may make the mutant PrP more structurally similar to PrPs susceptible to scrapie.
Overall, the previous results indicate that the selected amino acid substitutions cause a general increase in the propensity for prion-induced misfolding of rabbit rec-PrP. Because the high stability of the native conformation of rabbit PrP has been suggested to be the cause of its low misfolding susceptibility, we wondered if rabbit I202V rec-PrP would show a lower stability than that of wild-type PrP, thereby explaining its propensity for prion-induced misfolding. Although it is not known if the stability of the native conformation is related to in vivo susceptibility, Khan and collaborators showed that the proneness of β-state acquisition of certain PrPs was related to the in vivo susceptibility for at least five mammalian species (34). To determine if the I202V substitution was acting through the same mechanism, its accessibility to the β-state was measured. Although it was applicable, the measurement method using GdnHCl seemed to be less sensitive in our hands than the one developed originally. When the method was applied to hamster, mouse, rabbit, and rabbit I202V rec-PrPs, in contrast to what was described by Khan and collaborators, mouse rec-PrP was no more prone to β-state acquisition than rabbit rec-PrP, indicating that either the lower sensitivity of our method was not able to detect any differences or the β-state acquisition propensity does not necessarily correlate with in vivo susceptibility. Thus, the I202V substitution seems not to affect the global stability of rabbit rec-PrP, and even if it did, the reason for the increase in rabbit rec-PrP misfolding proneness might not be related to stability in the presence of chaotropic agents.
In essence, we found eight amino acid residues in rabbit rec-PrP that are probably involved in its low susceptibility to misfolding into a protease-resistant isoform. Three of those residues (S107, M108, and I202) appear to have a stronger influence, as their replacement by mouse rec-PrP residues allowed the propagation of brain-derived mouse prions. Moreover, these substitutions made rabbit rec-PrP susceptible to prion strains from different species, proving that their effect is not limited to the mouse-rabbit interspecies transmission barrier. However, the mechanism of action could not be unraveled definitively, although any effect on the global stability of the native rabbit rec-PrP structure was ruled out. Despite clearly showing that the recPMCA technique allowed the detection of three residues in the PrP of a mammalian species with very low susceptibility to TSE as the most influential in modulating its prion-like misfolding ability, the results should be validated in animal models to determine the correlation with in vivo data.
MATERIALS AND METHODS
Preparation of purified recombinant PrPs.
Bacterial expression of the hamster, mouse, and rabbit recombinant PrPs (rec-PrPs) was performed using expression vectors prepared by standard molecular biology techniques. Specifically, pOPIN vectors developed by the Oxford Protein Production Facility UK (OPPF) were used to introduce the wild-type hamster, mouse, and rabbit PRNP genes by homologous recombination, using the In-Fusion cloning method (73). Briefly, the pOPIN E vector was digested with the NcoI and PmeI restriction enzymes (New England Laboratories) according to the protocol specified by OPPF for using the OPPF pOPIN vector suite for HTP In-Fusion cloning and then purified for homologous recombination with a fragment containing the open reading frame (ORF) (positions 23 to 231) of the hamster, mouse, or rabbit PRNP gene, previously obtained by PCR with the following oligonucleotides: 5′-AGGAGATATACCATGAAGAAGCGGCCAAAGCCTGG-3′ and 5′-GATGGTGATGGTGATGTTAGGACCTTCTTCCATC-3′ for hamster PrP, 5′-AGGAGATATACCATGAAAAAGCGGCCAAAGCCTGAA-3′ and 5′-GTGATGGTGATGTTAGGATCTTCTCCCGTCGTAATA-3′ for mouse PrP, and 5′-AGGAGATATACCATGAAGAAGCGGCCGAAGCCTGG-3′ and 5′-GTGATGGTGATGTTAGCCGGCCGCCCTCTGGTAGGC-3′ for rabbit PrP. All the genetic constructs containing rabbit PrP with mouse substitutions and mouse PrP with rabbit substitutions were created by site-directed PCR mutagenesis, which first uses internal primers for the specific substitution and then uses external primers which hybridize with the pOPIN vector (5′-CCGCGGGGGGACGGCTGCC-3′ and 5′-GAACAGAGGTGCGTCTGGTG-3′ for amplification of the mutant template. On considering the individual substitutions (Fig. 1), a few of them were easily discarded primarily due to their position. For example, the four amino acid differences found in the octapeptide repeat (OR) region, which despite being involved in the pathology of TSEs has been shown not to be essential for prion infection or propagation (74), were no longer taken into consideration. The differences shown in the alignment at the carboxyl terminus of the protein were similarly discarded, as this is the GPI anchoring region in vivo and is absent in recombinant PrP. The primers bearing the desired point mutations are listed in Table 2. Double mutants of rabbit rec-PrP for residues 107 and 108 were generated using the same strategy and mutagenic primers, but with the rabbit I202V mutant as a template. The triple (S107N, M108L, and I202V) mutant rabbit PrP was generated using mutagenic primers for the S107N, M108L mutant (Table 2) and a plasmid bearing the I202V substitution as the template.
TABLE 2.
Primers designed for site-directed mutagenesis of rabbit recombinant PrP with substitutions from mouse PrP and of mouse recombinant PrP with substitutions from rabbit PrP
| Encoded mutation | Primer sequencea |
|
|---|---|---|
| Forward | Reverse | |
| Rabbit G99N rec-PrP | CACAACCAGTGGAACAAGCCCAGTAAG | CTTACTGGGCTTGTTCCACTGGTTGTG |
| Rabbit S107N rec-PrP | GCCGAAAACCAACATGAAGCACGTGG | CCACGTGCTTCATGTTGGTTTTCGGC |
| Rabbit M108L rec-PrP | CCGAAAACCAGCCTGAAGCACGTGGC | GCCACGTGCTTCAGGCTGGTTTTCGG |
| Rabbit L137M rec-PrP | CATGAGCAGGCCCATGATCCACTTCGGC | GCCGAAGTGGATCATGGGCCTGCTCATG |
| Rabbit Y144W rec-PrP | CTTCGGCAACGACTGGGAGGACCGCTAC | GTAGCGGTCCTCCCAGTCGTTGCCGAAG |
| Rabbit S173N rec-PrP | CAACCAGAACAACTTCGTGCACGACTG | CAGTCGTGCACGAAGTTGTTCTGGTTG |
| Rabbit V183I rec-PrP | CAACATCACGATTAAGCAGCACACGGTG | CACCGTGTGCTGCTTAATCGTGATGTTG |
| Rabbit I202V rec-PrP | CGAGACCGACGTCAAGATCATGGAG | CTCCATGATCTTGACGTCGGTCTCG |
| Rabbit I204M rec-PrP | CGACATCAAGATGATGGAGCGCGTG | CACGCGCTCCATCATCTTGATGTCG |
| Rabbit I214V rec-PrP | GCAGATGTGCGTCACGCAGTACC | GGTACTGCGTGACGCACATCTGC |
| Rabbit Q219K rec-PrP | CACGCAGTACCAGAAGGAGTCCCAGG | CCTGGGACTCCTTCTGGTACTGCGTG |
| Rabbit M108I rec-PrP | GCCGAAAACCAGCATCAAGCACGTGGCCGGGG | CCCCGGCCACGTGCTTGATGCTGGTTTTCGGC |
| Rabbit I202M rec-PrP | CACCGAGACCGACATGAAGATCATGG | CCATGATCTTCATGTCGGTCTCGGTG |
| Mouse N99G rec-PrP | CATAATCAGTGGGGCAAGCCCAGC | GCTGGGCTTGCCCCACTGATTATG |
| Mouse N107S rec-PrP | CCAAAAACCTCCCTCAAGCATGTGG | CCACATGCTTGAGGGAGGTTTTTGG |
| Mouse L108M rec-PrP | CCAAAAACCAACATGAAGCATGTGGCAG | CTGCCACATGCTTCATGTTGGTTTTTGG |
| Mouse M137L rec-PrP | GAGCAGGCCCCTCATCCATTTTGG | CCAAAATGGATGAGGGGCCTGCTC |
| Mouse W144Y rec-PrP | GCAACGACTACGAGGACCGCTAC | GTAGCGGTCCTCGTAGTCGTTGC |
| Mouse N173S rec-PrP | CAGTACAGCAACCAGAACAGCTTCGTGCACGACTGCGTC | GACGCAGTCGTGCACGAAGCTGTTCTGGTTGCTGTACTG |
| Mouse I183V rec-PrP | CGACTGCGTCAATATCACCGTCAAGCAGCACACGGTCACC | GGTGACCGTGTGCTGCTTGACGGTGATATTGACGCAGTCG |
| Mouse V202I rec-PrP | GAACTTCACCGAGACCGATATAAAGATGATGGAGCGCG | CGCGCTCCATCATCTTTATATCGGTCTCGGTGAAGTTC |
| Mouse M204I rec-PrP | GATGTGAAGATCATGGAGCGCGTG | CACGCGCTCCATGATCTTCACATC |
| Mouse V214I rec-PrP | GCAGATGTGCATCACCCAGTACC | GGTACTGGGTGATGCACATCTGC |
| Mouse K219Q rec-PrP | CAGTACCAGCAGGAGTCCCAGG | CCTGGGACTCCTGCTGGTACTG |
| Rabbit S107N M108L rec-PrP | GCCGAAAACCAACCTGAAGCACGTGG | CCACGTGCTTCAGGTTGGTTTTCGGC |
Underlining indicates the codon that is modified, and bold type indicates the specific nucleotides that have changed.
The expression vectors were transformed into Escherichia coli Rosetta (DE3) competent cells (EMD Millipore) by using standard molecular biology procedures allowing the expression of the corresponding recombinant proteins. Briefly, the bacterial pellet obtained after the induction of 1 liter of culture with IPTG (isopropyl-β-d-thiogalactopyranoside; final concentration, 1 mM) (Gold Biotechnology) was resuspended in 50 ml of lysis buffer (50 mM Tris-HCl [Fisher Bioreagents], 5 mM EDTA [Sigma-Aldrich], 1% Triton X-100 [Amresco], 1 mM phenylmethylsulfonyl fluoride [PMSF] [Sigma-Aldrich], 100 μg/ml lysozyme [Sigma-Aldrich], pH adjusted to 8.0) and then incubated for 30 min with stirring at 200 rpm at room temperature in the presence of 100 U/ml DNase (Sigma-Aldrich) and 20 mM MgCl2 (Sigma-Aldrich). The lysate was centrifuged at 4°C for 1 h at 8,500 × g (Sorvall ST 16R; Thermo Scientific), and the resulting pellet was resuspended in 50 ml of washing buffer (20 mM Tris-HCl [Fisher Bioreagents], 150 mM NaCl [Sigma-Aldrich], 1 mM EDTA [Sigma-Aldrich], 1% Sarkosyl [Sigma-Aldrich], pH adjusted to 8.0). The lysate was centrifuged under the conditions described above, and the pellet was dissolved in 6 ml of inclusion buffer (20 mM Tris-HCl [Fisher Bioreagents], 0.5 M NaCl [Sigma-Aldrich], 6 M guanidine hydrochloride [GdnHCl] [Fisher Scientific], pH adjusted to 8.0) and incubated at 37°C with stirring overnight in order to dissolve the inclusion bodies and solubilize the recombinant protein in the medium. The samples were centrifuged at 4°C for 1 h at 8,500 × g, and the supernatants were filtered through a 0.20-μm-pore-size filter (Minisart; Sartorius Stedim) before purification.
Purification of the recombinant proteins was performed with a histidine affinity column (5-ml HisTrap crude FF; GE Healthcare Amersham). The column was equilibrated with 35 ml of binding buffer (20 mM Tris-HCl [Fisher Bioreagents], 500 mM NaCl [Sigma-Aldrich], 5 mM imidazole [Sigma-Aldrich], 2 M GdnHCl [Fisher Scientific], pH adjusted to 8.0), and the filtered sample containing the soluble recombinant PrP was loaded onto the column by use of needles (22-gauge, 1 1/4 in. long; Terumo). The column was washed with 75 ml of binding buffer and the recombinant protein eluted with 30 ml of elution buffer (20 mM Tris-HCl [Fisher Bioreagents], 500 mM NaCl [Sigma-Aldrich], 500 mM imidazole [Sigma-Aldrich], 2 M GdnHCl [Fisher Scientific], pH adjusted to 8.0). The eluted proteins were denatured by addition of GdnHCl (Sigma-Aldrich) to a final concentration of 6 M and then concentrated to 4 to 5 mg/ml by use of 10-kDa centrifugal filter units (Amicon Ultra-15; Millipore). The recombinant proteins were stored at −20°C until required. The quality and purity were assessed by Coomassie blue staining after electrophoresis in a 4 to 15% or 4 to 12% SDS-PAGE gel.
The proteins used for β-state acquisition proneness measurements by circular dichroism (CD) were subjected to a second purification step based on size exclusion chromatography in order to obtain more homogeneous and pure samples. Proteins were purified by affinity chromatography as described previously, concentrated in Amicon Ultra-15 10-kDa centrifugal filter units (Millipore), and dialyzed (SnakeSkin dialysis tubing [10K molecular size cutoff, 22 mm]; Thermo-Pierce) for 6 to 8 h at 4°C against phosphate-buffered saline (PBS) with 2 M GdnHCl. The dialysis buffer was changed to PBS for 6 to 8 h, to PBS at pH 6 for another 6 to 8 h, and finally to sodium acetate at pH 4 (5 mM sodium acetate [Sigma-Aldrich], 67 mM NaCl [Sigma-Aldrich], 5 mM EDTA [Sigma-Aldrich]) for the last 6 to 8 h. The dialyzed protein was centrifuged at 19,000 × g for 15 min at 4°C (Sorvall ST 16R; Thermo Scientific), and the supernatant was filtered through a 0.20-μm-pore-size filter (Minisart; Sartorius Stedim). A HiLoad 16/600 Superdex 75pg (GE Healthcare) size exclusion chromatography column assembled on an AKTÄ fast-performance liquid chromatography (FPLC) system (GE Healthcare) was equilibrated with 1.2 volumes of degassed sodium acetate buffer and the dialyzed protein injected. Chromatography was performed with a constant buffer flow of 1 to 2 ml/min, controlled by Unicorn Manager software (Amersham Biosciences), until the elution of all the protein (approximately 1.5 column volumes). The protein in the eluted fractions was detected by measuring the absorbance at 280 nm, and all the protein-containing fractions were mixed and concentrated with Amicon Ultra-15 10-kDa centrifugal filter units (Millipore) to a 1- to 2-ml final volume and stored at 4°C for up to 3 days.
Preparation of PMCA substrates.
Perfused brains from PrP knockout (Prnp0/0) mice (75) were homogenized at 10% in conversion buffer (CB) as described previously (10). Homogenates were cleared by centrifugation at 19,000 × g for 1 h at 4°C. At the same time, rec-PrPs purified as described above were diluted 1:5 in PBS and refolded by dialysis against PBS for 1 h at 4°C, using Slide-A-Lyzer dialysis cassettes (Thermo Scientific).
After centrifugation at 19,000 × g for 15 min at 4°C, the soluble rec-PrP was mixed 1:10 with the brain homogenates described above. The concentration of rec-PrP was measured by its absorbance at 280 nm and confirmed by bicinchoninic acid (BCA) assay (Thermo-Pierce). PMCA substrates were aliquoted in 200-μl PCR tubes and stored at −80°C until required.
Prion strains and isolates.
A panel of TSE agents, including mouse, sheep, cattle, mule deer, and rabbit prions (Table 1), were prepared from infected brain tissues as 10% (wt/vol) homogenates in PBS with protease inhibitors (Roche). The homogenate stocks were aliquoted and stored at −80°C until required.
The strains adapted previously to PMCA or recPMCA were obtained in our laboratory by the following procedure. Each of the original prion isolates was mixed 1:10 (vol/vol) with a 10% (wt/vol) brain homogenate expressing the PrP of interest in CB or with a 10% (wt/vol) Prnp0/0 mouse brain homogenate containing the rec-PrP of interest, and 10 to 15 serial rounds of PMCA or recPMCA were performed at a 1:10 dilution (76).
In vitro propagation of prions by PMCA.
The in vitro prion propagation studies were performed based on modified versions of the PMCA described previously (10, 11, 77). Briefly, recombinant protein-based PMCAs were conducted in 0.2-ml tubes with a final volume of 50 μl, using a Misonix Q-700 sonicator with a microplate system (Qsonica) and incubation cycles of 30 min, followed by sonication pulses of 15 s at 50 to 60% power. The whole process was performed at an average temperature of 39°C, regulated by a circulating water bath. For misfolded rec-PrP generation, successive rounds of recPMCA in which prion strains were diluted 1:10 in the corresponding substrates were performed (31). After a 24-h PMCA round, the resultant sample from the first round was diluted 1:10 in fresh substrate, and the process was repeated for 10 to 20 rounds of recPMCA. To evaluate propagation ability by serial dilution of seeds, dilutions were subjected to a single 48-h recPMCA round, with favoring of the reproducibility of results by use of 1-mm zirconia-silica beads (BioSpec Products) (29).
Biochemical characterization of in vitro- and in vivo-generated prions. (i) Protease K digestion.
recPMCA-treated samples were incubated with 25 μg/ml of protease K (PK) (Roche) for 1 h at 42°C and with constant agitation at 450 rpm (Thermomixer Comfort; Eppendorf) as described previously (30). Samples were mixed previously 1:1 (vol/vol) with 10% Sarkosyl (Sigma-Aldrich) digestion buffer, and the digestion was stopped by adding Laemmli electrophoresis buffer (NuPAGE; Invitrogen Life Technologies).
(ii) PK-resistant PrP detection.
Protein immunodetection by Western blotting was performed after separation of proteins by SDS-PAGE. TGX Criterion 4 to 15% gels (Bio-Rad) were used. The recombinant proteins were electroblotted onto polyvinylidene difluoride (PVDF) membranes (Turbo PVDF Trans-Blot transfer pack; Bio-Rad) and developed with SAF83 or SAF84 primary antibody (1:400) (Cayman Chemical) and horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G secondary antibody (IgG-HRP; Santa Cruz Biotechnology). The immunoreactive bands were visualized by chemiluminescence assay, using a Super Signal West Pico kit (Thermo Scientific Pierce), and the digital images were displayed by use of a FluorChem Q imager (Alpha Innotech).
Comparative analysis of β-state acquisition propensities of rec-PrPs upon chaotropic agent treatment followed by CD.
The concentrations of proteins purified by affinity chromatography and size exclusion chromatography were measured at 280 nm by use of a NanoDrop 2000 spectrophotometer (Thermo Scientific) and then diluted in sodium acetate buffer without EDTA to reach a concentration of 20.4 μM in the case of those incubated with GdnCl (0 to 4 M) (Sigma-Aldrich) and a concentration of 39.4 μM for those incubated with urea (0 to 8 M) (Sigma-Aldrich). Stock solutions of 1 M sodium acetate at pH 4, 6 M NaCl, and 9.25 M urea and GdnHCl were prepared. The urea was treated with Amberlite resin (Sigma-Aldrich) to eliminate reactive cyanate ions that could trigger irreversible carbamylation of protein amine groups and interfere with the CD measurements. Using the purified rec-PrPs and the stock solutions, 30 samples containing a 5 or 10 μM concentration of each rec-PrP in 5 mM sodium acetate and 67 mM NaCl buffer were prepared with increasing urea or GdnHCl concentrations, respectively, at three different pH values (4, 4.5, and 5). The samples were incubated for 5 days at room temperature prior to measurement on a Jasco J-810 spectropolarimeter in a 2-mm quartz cuvette (Suprasil 110-QS, 2 mm; Hellma Analytics) at 25°C.
Circular dichroism measurements and calculations to determine the fraction of rec-PrP at β-state.
The method developed by Khan and collaborators for quantitative measurement by CD of the β-state population upon increasing chaotropic concentrations and acidic pH was used (34). The existence of a mix of monomers and octamers as described by Baskakov and collaborators (78) makes a simple three-state model (native, unfolded, and β-state) inaccurate for the analysis of biphasic PrP unfolding curves induced by increasing concentrations of urea, whereas a four-state equilibrium model (native, unfolded, monomeric β-state, and oligomeric β-state) would also fail to yield a satisfactory fitting. Therefore, Khan and collaborators chose an approximation that does not make assumptions on the oligomerization state of the PrP and takes advantage of the distinguishable β-sheet-like CD spectrum presented by β-state PrP, in contrast to the α-helical spectrum of the native state and the random coil of the unfolded state. Given that the CD spectrum consists of the sum of the contributions of the conformational states of all the proteins in solution, the molar ellipticity (θ) observed for a certain sample can be expressed by the following equation: θ220,obs = θ220,native [native] + θ220,β-state [β-state] + θ220,unfolded [unfolded], where [native], [β-state], and [unfolded] are the fractions of PrP in each state and θ220,native, θ220,β-state and θ220,unfolded are the molar ellipticities corresponding to the conformations. This can be simplified by normalizing the CD data to apparent fractions, where the values of the apparent fractions of the native and unfolded states are 1 and 0, respectively, as follows:
Thus, the equation can be expressed as follows: Fap220 = [native] + Z220[β-state], where Fap220 is the normalized experimental CD signal, Z220 is the normalized signal of the β-state at 220 nm, and [native] and [β-state] are the fractional concentrations of the native and β-state conformations, respectively.
To solve for [β-state], PrP unfolding was monitored at a second wavelength, allowing the establishment of a two-equation system. For that purpose, the normalized β-state signal Zλ needs to be different from Z220 and also from the CD signals of the native and unfolded forms. Based on the CD spectra of the same PrP in the native, unfolded, and β-states, they chose a 229-nm wavelength to generate the second equation, as follows: Fap229 = [native] + Z229[β-state].
By combining the equations for 220 and 229 nm, it is possible to solve the fraction of PrP that acquires the β-state at a certain pH, as follows: [β-state] = (Fap220 − Fap229)/(Z220 − Z229).
CD spectra were collected between 280 and 198 nm, with 2 accumulations per sample, a 4-s integration time, 0.2 nm of data pitch, a bandwidth of 4 nm, and a scan speed of 20 nm/s. The measurements at 220 and 229 nm for each different chaotropic agent concentration were used to plot the denaturation curves at different pH values. From these data, the fraction in β-state was calculated for each protein. The same sample was used for each pH, with modification via dialysis of the rec-PrP in a 10-kDa dialysis cassette (Slide-A-Lyzer dialysis cassette [10K molecular size cutoff]; Thermo Scientific) against sodium acetate buffer at pH 4, 4.5, or 5 at 4°C for 8 h.
For this work, the study was performed with GdnHCl instead of urea as the chaotropic agent, with checking first that the denaturation curves were equivalent for both chaotropic agents in the case of hamster rec-PrP at pH 4, 4.5, and 5.
ACKNOWLEDGMENTS
This work was supported financially by Spanish government grants AGL2015-65046-C2-1-R and PCIN-2013-065 and Basque government grant 2014111157.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank the following for their support: the IKERBasque Foundation, CIC bioGUNE for the vivarium and maintenance, Patricia Piñeiro and Maite Pérez for technical support, OPPF for plasmid pOPIN E, and Tomás Mayoral, Olivier Andréoletti, and Jean Jewell for the BSE, sheep BSE, and CWD brain tissue samples, respectively.
We declare that we have no conflicts of interest.
J.C., N.F.-B., and H.E. conceived the study. H.E., S.R.E., C.H., and J.M.C. performed most of the experiments, and H.E., G.O., and O.M. performed circular dichroism experiments. J.C., N.F.-B., H.E., M.P.D., and F.C. analyzed and evaluated the results. J.C., H.E., N.F.-B., M.P.D., and F.C. wrote and reviewed the manuscript.
REFERENCES
- 1.Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Dickinson AG. 1976. Scrapie in sheep and goats. Front Biol 44:209–241. [PubMed] [Google Scholar]
- 3.Sweeting B, Khan MQ, Chakrabartty A, Pai EF. 2010. Structural factors underlying the species barrier and susceptibility to infection in prion disease. Biochem Cell Biol 88:195–202. doi: 10.1139/O09-172. [DOI] [PubMed] [Google Scholar]
- 4.Sigurdson CJ, Miller MW. 2003. Other animal prion diseases. Br Med Bull 66:199–212. doi: 10.1093/bmb/66.1.199. [DOI] [PubMed] [Google Scholar]
- 5.Bian J, Khaychuk V, Angers RC, Fernandez-Borges N, Vidal E, Meyerett-Reid C, Kim S, Calvi CL, Bartz JC, Hoover EA, Agrimi U, Richt JA, Castilla J, Telling GC. 2017. Prion replication without host adaptation during interspecies transmissions. Proc Natl Acad Sci U S A 114:1141–1146. doi: 10.1073/pnas.1611891114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Borges N, Chianini F, Erana H, Vidal E, Eaton SL, Pintado B, Finlayson J, Dagleish MP, Castilla J. 2012. Naturally prion resistant mammals: a utopia? Prion 6:425–429. doi: 10.4161/pri.22057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saa P, Castilla J, Soto C. 2005. Cyclic amplification of protein misfolding and aggregation. Methods Mol Biol 299:53–65. [DOI] [PubMed] [Google Scholar]
- 8.Chianini F, Fernandez-Borges N, Vidal E, Gibbard L, Pintado B, de Castro J, Priola SA, Hamilton S, Eaton SL, Finlayson J, Pang Y, Steele P, Reid HW, Dagleish MP, Castilla J. 2012. Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A 109:5080–5085. doi: 10.1073/pnas.1120076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vidal E, Fernandez-Borges N, Pintado B, Erana H, Ordonez M, Marquez M, Chianini F, Fondevila D, Sanchez-Martin MA, Andreoletti O, Dagleish MP, Pumarola M, Castilla J. 2015. Transgenic mouse bioassay: evidence that rabbits are susceptible to a variety of prion isolates. PLoS Pathog 11:e1004977. doi: 10.1371/journal.ppat.1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castilla J, Saa P, Hetz C, Soto C. 2005. In vitro generation of infectious scrapie prions. Cell 121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 11.Saa P, Castilla J, Soto C. 2006. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 281:35245–35252. doi: 10.1074/jbc.M603964200. [DOI] [PubMed] [Google Scholar]
- 12.Chianini F, Fernández-Borges N, Eraña H, Pang Y, Vidal E, Eaton S, Finlayson J, Dagleish M, Castilla J. 2013. Prion-resistant or prion-susceptible species, this is the question. Virulence 4:333–334. doi: 10.4161/viru.24456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. 2010. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934. doi: 10.1021/bi100370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarradin P, Viglietta C, Limouzin C, Andreoletti O, Daniel-Carlier N, Barc C, Leroux-Coyau M, Berthon P, Chapuis J, Rossignol C, Gatti JL, Belghazi M, Labas V, Vilotte JL, Beringue V, Lantier F, Laude H, Houdebine LM. 2015. Transgenic rabbits expressing ovine PrP are susceptible to scrapie. PLoS Pathog 11:e1005077. doi: 10.1371/journal.ppat.1005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vorberg I, Groschup MH, Pfaff E, Priola SA. 2003. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J Virol 77:2003–2009. doi: 10.1128/JVI.77.3.2003-2009.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J. 2009. Studies on the structural stability of rabbit prion probed by molecular dynamics simulations. J Biomol Struct Dyn 27:159–162. doi: 10.1080/07391102.2009.10507305. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J. 2010. Studies on the structural stability of rabbit prion probed by molecular dynamics simulations of its wild-type and mutants. J Theor Biol 264:119–122. doi: 10.1016/j.jtbi.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 18.Nisbet RM, Harrison CF, Lawson VA, Masters CL, Cappai R, Hill AF. 2010. Residues surrounding the glycosylphosphatidylinositol anchor attachment site of PrP modulate prion infection: insight from the resistance of rabbits to prion disease. J Virol 84:6678–6686. doi: 10.1128/JVI.02709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen Y, Li J, Xiong M, Peng Y, Yao W, Hong J, Lin D. 2010. Solution structure and dynamics of the I214V mutant of the rabbit prion protein. PLoS One 5:e13273. doi: 10.1371/journal.pone.0013273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J. 2011. Comparison studies of the structural stability of rabbit prion protein with human and mouse prion proteins. J Theor Biol 269:88–95. doi: 10.1016/j.jtbi.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 21.Julien O, Chatterjee S, Bjorndahl TC, Sweeting B, Acharya S, Semenchenko V, Chakrabartty A, Pai EF, Wishart DS, Sykes BD, Cashman NR. 2011. Relative and regional stabilities of the hamster, mouse, rabbit, and bovine prion proteins toward urea unfolding assessed by nuclear magnetic resonance and circular dichroism spectroscopies. Biochemistry 50:7536–7545. doi: 10.1021/bi200731e. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez-Funez P, Zhang Y, Sanchez-Garcia J, Jensen K, Zou WQ, Rincon-Limas DE. 2011. Pulling rabbits to reveal the secrets of the prion protein. Commun Integr Biol 4:262–266. doi: 10.4161/cib.4.3.15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweeting B, Brown E, Khan MQ, Chakrabartty A, Pai EF. 2013. N-terminal helix-cap in alpha-helix 2 modulates beta-state misfolding in rabbit and hamster prion proteins. PLoS One 8:e63047. doi: 10.1371/journal.pone.0063047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Zhang Y. 2014. Molecular dynamics studies on the NMR and X-ray structures of rabbit prion proteins. J Theor Biol 342:70–82. doi: 10.1016/j.jtbi.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Wang F, Zhang Y. 2015. Molecular dynamics studies on the NMR structures of rabbit prion protein wild type and mutants: surface electrostatic charge distributions. J Biomol Struct Dyn 33:1326–1335. doi: 10.1080/07391102.2014.947325. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Wang F. 2016. A review on the salt bridge ASP177-ARG163 (O-N) of wild-type rabbit prion protein. J Biomol Struct Dyn 34:1020–1028. doi: 10.1080/07391102.2015.1064832. [DOI] [PubMed] [Google Scholar]
- 27.Yu Z, Huang P, Yu Y, Zheng Z, Huang Z, Guo C, Lin D. 2016. Unique properties of the rabbit prion protein oligomer. PLoS One 11:e0160874. doi: 10.1371/journal.pone.0160874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malevanets A, Chong PA, Hansen DF, Rizk P, Sun Y, Lin H, Muhandiram R, Chakrabartty A, Kay LE, Forman-Kay JD, Wodak SJ. 2017. Interplay of buried histidine protonation and protein stability in prion misfolding. Sci Rep 7:882. doi: 10.1038/s41598-017-00954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Borges N, de Castro J, Castilla J. 2009. In vitro studies of the transmission barrier. Prion 3:220–223. doi: 10.4161/pri.3.4.10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elezgarai SR, Fernández-Borges N, Erana H, Sevillano A, Moreno J, Harrathi C, Saá P, Gil D, Kong Q, Requena JR, Andreoletti O, Castilla J. 2017. Generation of a new infectious recombinant prion: a model to understand Gerstmann-Sträussler-Scheinker syndrome. Sci Rep 7:9584. doi: 10.1038/s41598-017-09489-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernández-Borges N, Eraña H, Elezgarai SR, Harrathi C, Venegas V, Castilla J. 2017. A quick method to evaluate the effect of the amino acid sequence in the misfolding proneness of the prion protein. In Lawson V. (ed), Prions. Methods in molecular biology, vol 1658. Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 32.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. 1991. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Stockel J, Mehlhorn I, Groth D, Baldwin MA, Prusiner SB, James TL, Cohen FE. 1997. Physical studies of conformational plasticity in a recombinant prion protein. Biochemistry 36:3543–3553. doi: 10.1021/bi961965r. [DOI] [PubMed] [Google Scholar]
- 34.Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, Chakrabartty A. 2010. Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci U S A 107:19808–19813. doi: 10.1073/pnas.1005267107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore RA, Vorberg I, Priola SA. 2005. Species barriers in prion diseases—brief review. Arch Virol 2005(Suppl):187–202. [DOI] [PubMed] [Google Scholar]
- 36.Hornemann S, Schorn C, Wuthrich K. 2004. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 5:1159–1164. doi: 10.1038/sj.embor.7400297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishina KA, Deleault NR, Mahal SP, Baskakov I, Luhrs T, Riek R, Supattapone S. 2006. The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 45:14129–14139. doi: 10.1021/bi061526k. [DOI] [PubMed] [Google Scholar]
- 38.Kim JI, Surewicz K, Gambetti P, Surewicz WK. 2009. The role of glycophosphatidylinositol anchor in the amplification of the scrapie isoform of prion protein in vitro. FEBS Lett 583:3671–3675. doi: 10.1016/j.febslet.2009.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imamura M, Kato N, Yoshioka M, Okada H, Iwamaru Y, Shimizu Y, Mohri S, Yokoyama T, Murayama Y. 2011. Glycosylphosphatidylinositol anchor-dependent stimulation pathway required for generation of baculovirus-derived recombinant scrapie prion protein. J Virol 85:2582–2588. doi: 10.1128/JVI.02098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faburay B, Tark D, Kanthasamy AG, Richt JA. 2014. In vitro amplification of scrapie and chronic wasting disease PrP(res) using baculovirus-expressed recombinant PrP as substrate. Prion 8:393–403. doi: 10.4161/19336896.2014.983753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan J, Zhan YA, Abskharon R, Xiao X, Martinez MC, Zhou X, Kneale G, Mikol J, Lehmann S, Surewicz WK, Castilla J, Steyaert J, Zhang S, Kong Q, Petersen RB, Wohlkonig A, Zou WQ. 2013. Recombinant human prion protein inhibits prion propagation in vitro. Sci Rep 3:2911. doi: 10.1038/srep02911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noble GP, Wang DW, Walsh DJ, Barone JR, Miller MB, Nishina KA, Li S, Supattapone S. 2015. A structural and functional comparison between infectious and non-infectious autocatalytic recombinant PrP conformers. PLoS Pathog 11:e1005017. doi: 10.1371/journal.ppat.1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Funez P, Zhang Y, Casas-Tinto S, Xiao X, Zou WQ, Rincon-Limas DE. 2010. Sequence-dependent prion protein misfolding and neurotoxicity. J Biol Chem 285:36897–36908. doi: 10.1074/jbc.M110.174391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr, Caughey B. 1995. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 92:3923–3927. doi: 10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bossers A, Belt P, Raymond GJ, Caughey B, de Vries R, Smits MA. 1997. Scrapie susceptibility-linked polymorphisms modulate the in vitro conversion of sheep prion protein to protease-resistant forms. Proc Natl Acad Sci U S A 94:4931–4936. doi: 10.1073/pnas.94.10.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan X, Huang JJ, Zhou Z, Chen J, Liang Y. 2014. How does domain replacement affect fibril formation of the rabbit/human prion proteins. PLoS One 9:e113238. doi: 10.1371/journal.pone.0113238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Priola SA, Chesebro B. 1995. A single hamster PrP amino acid blocks conversion to protease-resistant PrP in scrapie-infected mouse neuroblastoma cells. J Virol 69:7754–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Priola SA, Chabry J, Chan K. 2001. Efficient conversion of normal prion protein (PrP) by abnormal hamster PrP is determined by homology at amino acid residue 155. J Virol 75:4673–4680. doi: 10.1128/JVI.75.10.4673-4680.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahal SP, Jablonski J, Suponitsky-Kroyter I, Oelschlegel AM, Herva ME, Oldstone M, Weissmann C. 2012. Propagation of RML prions in mice expressing PrP devoid of GPI anchor leads to formation of a novel, stable prion strain. PLoS Pathog 8:e1002746. doi: 10.1371/journal.ppat.1002746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aguilar-Calvo P, Xiao X, Bett C, Erana H, Soldau K, Castilla J, Nilsson KP, Surewicz WK, Sigurdson CJ. 2017. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep 7:43295. doi: 10.1038/srep43295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McNally KL, Ward AE, Priola SA. 2009. Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 83:4469–4475. doi: 10.1128/JVI.02412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagiwara K, Hara H, Hanada K. 2013. Species-barrier phenomenon in prion transmissibility from a viewpoint of protein science. J Biochem 153:139–145. doi: 10.1093/jb/mvs148. [DOI] [PubMed] [Google Scholar]
- 53.Billeter M, Riek R, Wider G, Hornemann S, Glockshuber R, Wuthrich K. 1997. Prion protein NMR structure and species barrier for prion diseases. Proc Natl Acad Sci U S A 94:7281–7285. doi: 10.1073/pnas.94.14.7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lawson VA, Priola SA, Wehrly K, Chesebro B. 2001. N-terminal truncation of prion protein affects both formation and conformation of abnormal protease-resistant prion protein generated in vitro. J Biol Chem 276:35265–35271. doi: 10.1074/jbc.M103799200. [DOI] [PubMed] [Google Scholar]
- 55.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 56.Stahl N, Baldwin MA, Teplow DB, Hood L, Gibson BW, Burlingame AL, Prusiner SB. 1993. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 57.Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Cervenakova L, Favara C, Gardner D, Long D, Parnell M, Striebel J, Priola SA, Ward A, Williams ES, Race R, Chesebro B. 2009. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis 15:1366–1376. doi: 10.3201/eid1509.090253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Westaway D, Goodman PA, Mirenda CA, McKinley MP, Carlson GA, Prusiner SB. 1987. Distinct prion proteins in short and long scrapie incubation period mice. Cell 51:651–662. doi: 10.1016/0092-8674(87)90134-6. [DOI] [PubMed] [Google Scholar]
- 59.Westaway D, Zuliani V, Cooper CM, Da Costa M, Neuman S, Jenny AL, Detwiler L, Prusiner SB. 1994. Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev 8:959–969. doi: 10.1101/gad.8.8.959. [DOI] [PubMed] [Google Scholar]
- 60.Bartz JC, McKenzie DI, Bessen RA, Marsh RF, Aiken JM. 1994. Transmissible mink encephalopathy species barrier effect between ferret and mink: PrP gene and protein analysis. J Gen Virol 75:2947–2953. doi: 10.1099/0022-1317-75-11-2947. [DOI] [PubMed] [Google Scholar]
- 61.Kurt TD, Telling GC, Zabel MD, Hoover EA. 2009. Trans-species amplification of PrP(CWD) and correlation with rigid loop 170N. Virology 387:235–243. doi: 10.1016/j.virol.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 62.Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. 2005. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci U S A 102:646–650. doi: 10.1073/pnas.0409008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. 1996. NMR structure of the mouse prion protein domain PrP(121-231). Nature 382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 64.Bossers A, de Vries R, Smits MA. 2000. Susceptibility of sheep for scrapie as assessed by in vitro conversion of nine naturally occurring variants of PrP. J Virol 74:1407–1414. doi: 10.1128/JVI.74.3.1407-1414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perez DR, Damberger FF, Wuthrich K. 2010. Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol 400:121–128. doi: 10.1016/j.jmb.2010.04.066. [DOI] [PubMed] [Google Scholar]
- 66.Chakroun N, Prigent S, Dreiss CA, Noinville S, Chapuis C, Fraternali F, Rezaei H. 2010. The oligomerization properties of prion protein are restricted to the H2H3 domain. FASEB J 24:3222–3231. doi: 10.1096/fj.09-153924. [DOI] [PubMed] [Google Scholar]
- 67.Chen J, Thirumalai D. 2013. Helices 2 and 3 are the initiation sites in the PrP(C) → PrP(SC) transition. Biochemistry 52:310–319. doi: 10.1021/bi3005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hafner-Bratkovic I, Jerala R. 2011. Disulfide mapping reveals the domain swapping as the crucial process of the structural conversion of prion protein. Prion 5:56–59. doi: 10.4161/pri.5.2.16232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eghiaian F, Daubenfeld T, Quenet Y, van Audenhaege M, Bouin AP, van der Rest G, Grosclaude J, Rezaei H. 2007. Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proc Natl Acad Sci U S A 104:7414–7419. doi: 10.1073/pnas.0607745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prigent S, Rezaei H. 2011. PrP assemblies: spotting the responsible regions in prion propagation. Prion 5:69–75. doi: 10.4161/pri.5.2.16383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peoc'h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Lauprêtre N, Laplanche J. 2000. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 15:482. [DOI] [PubMed] [Google Scholar]
- 72.Vaccari G, Di Bari MA, Morelli L, Nonno R, Chiappini B, Antonucci G, Marcon S, Esposito E, Fazzi P, Palazzini N, Troiano P, Petrella A, Di Guardo G, Agrimi U. 2006. Identification of an allelic variant of the goat PrP gene associated with resistance to scrapie. J Gen Virol 87:1395–1402. doi: 10.1099/vir.0.81485-0. [DOI] [PubMed] [Google Scholar]
- 73.Berrow NS, Alderton D, Sainsbury S, Nettleship J, Assenberg R, Rahman N, Stuart DI, Owens RJ. 2007. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res 35:e45. doi: 10.1093/nar/gkm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yam AY, Gao CM, Wang X, Wu P, Peretz D. 2010. The octarepeat region of the prion protein is conformationally altered in PrP(Sc). PLoS One 5:e9316. doi: 10.1371/journal.pone.0009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 1994. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 76.Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C. 2008. Cell-free propagation of prion strains. EMBO J 27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saborio GP, Permanne B, Soto C. 2001. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 78.Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. 2002. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- 79.Chandler RL. 1961. Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet i:1378–1379. [DOI] [PubMed] [Google Scholar]
- 80.Dickinson AG, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. J Comp Pathol 79:363–366. doi: 10.1016/0021-9975(69)90051-6. [DOI] [PubMed] [Google Scholar]
- 81.Williams ES, Young S. 1992. Spongiform encephalopathies in Cervidae. Rev Sci Tech 11:551–567. doi: 10.20506/rst.11.2.611. [DOI] [PubMed] [Google Scholar]
- 82.Wilesmith JW, Wells GA, Cranwell MP, Ryan JB. 1988. Bovine spongiform encephalopathy: epidemiological studies. Vet Rec 123:638–644. [PubMed] [Google Scholar]
- 83.Foster JD, Hope J, Fraser H. 1993. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet Rec 133:339–341. doi: 10.1136/vr.133.14.339. [DOI] [PubMed] [Google Scholar]
- 84.Stack MJ, Chaplin MJ, Clark J. 2002. Differentiation of prion protein glycoforms from naturally occurring sheep scrapie, sheep-passaged scrapie strains (CH1641 and SSBP1), bovine spongiform encephalopathy (BSE) cases and Romney and Cheviot breed sheep experimentally inoculated with BSE using two monoclonal antibodies. Acta Neuropathol 104:279–286. [DOI] [PubMed] [Google Scholar]
- 85.Gorfe AA, Caflisch A. 2007. Ser170 controls the conformational multiplicity of the loop 166-175 in prion proteins: implication for conversion and species barrier. FASEB J 21:3279–3287. doi: 10.1096/fj.07-8292com. [DOI] [PubMed] [Google Scholar]
- 86.Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Fernandez-Borges N, Schwarz P, Castilla J, Wuthrich K, Aguzzi A. 2010. A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest 120:2590–2599. doi: 10.1172/JCI42051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kurt TD, Jiang L, Bett C, Eisenberg D, Sigurdson CJ. 2014. A proposed mechanism for the promotion of prion conversion involving a strictly conserved tyrosine residue in the beta2-alpha2 loop of PrPC. J Biol Chem 289:10660–10667. doi: 10.1074/jbc.M114.549030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kurt TD, Jiang L, Fernandez-Borges N, Bett C, Liu J, Yang T, Spraker TR, Castilla J, Eisenberg D, Kong Q, Sigurdson CJ. 2015. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest 125:1485–1496. doi: 10.1172/JCI79408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wen Y, Li J, Yao W, Xiong M, Hong J, Peng Y, Xiao G, Lin D. 2010. Unique structural characteristics of the rabbit prion protein. J Biol Chem 285:31682–31693. doi: 10.1074/jbc.M110.118844. [DOI] [PMC free article] [PubMed] [Google Scholar]