

ABSTRACT
High-risk human papillomaviruses (HPVs) link their life cycle to epithelial differentiation and require activation of DNA damage pathways for efficient replication. HPVs modulate the expression of cellular transcription factors, as well as cellular microRNAs (miRNAs) to control these activities. One miRNA that has been reported to be repressed in HPV-positive cancers of the cervix and oropharynx is miR-424. Our studies show that miR-424 levels are suppressed in cell lines that stably maintain HPV 31 or 16 episomes, as well as cervical cancer lines that contain integrated genomes such as SiHa. Introduction of expression vectors for miR-424 reduced both the levels of HPV genomes in undifferentiated cells and amplification upon differentiation. Our studies show that the levels of two putative targets of miR-424 that function in DNA damage repair, CHK1 and Wee1, are suppressed in HPV-positive cells, providing an explanation for why this microRNA is targeted in HPV-positive cells.
IMPORTANCE We describe here for the first time a critical role for miR-424 in the regulation of HPV replication. HPV E6 and E7 proteins suppress the levels of miR-424, and this is important for controlling the levels of CHK1, which plays a central role in viral replication.
KEYWORDS: amplification, HPV, microRNA, DNA damage repair, CHK1, check point
INTRODUCTION
Human papillomaviruses (HPVs) are the causative agents of cervical and other anogenital cancers (1). HPVs are classified as either high-risk types (such as HPV16, -18, -31, and -35), which are associated with the development of cancers of the anogenital tract and oropharynx or low-risk groups (such as HPV6 and HPV11) that are infrequently detected in malignancies. HPV virions infect cells in the basal layer of stratified epithelia to establish persistent infections. In infected basal cell cells, genomes are maintained as low-copy episomes that replicate in synchrony with cellular chromosomes. Productive viral replication or amplification, however, occurs only upon epithelial differentiation in suprabasal layers (2, 3). High-risk HPVs encode two major oncoproteins, E6 and E7, that play critical roles in cell transformation and provide important regulatory functions during the differentiation-dependent life cycle. One major target of E6 is the E3 ligase E6-associated protein E6AP, which forms trimeric complexes with p53, resulting in its rapid turnover (4–6). Similarly, E7 binds and induces the degradation of pRb, which results in the constitutive activation of E2F family members, leading to altered cell cycle control and differentiation (7, 8). Additional important targets of these viral proteins include the ataxia telangiectasia-mutated (ATM) and the ATM and Rad3-related (ATR) DNA repair pathways that play critical roles in HPV replication (9, 10). E6 and E7 regulate these pathways by altering the expression and/or activities of transcription factors, posttranslational modifying enzymes, and microRNAs (miRNAs) (11–13).
MicroRNAs are small, noncoding RNAs that can bind to sequences in the 3′ untranslated regions (3′ UTRs) of mRNAs to inhibit translation or to induce mRNA degradation (14). More than 1,000 human miRNAs have been identified, and these play roles in regulating development and differentiation of a variety of tissues, including the epithelia. Many viruses, such as herpesviruses (15) and retroviruses, encode their own miRNAs (16). In contrast, HPVs do not encode their own miRNAs (17) but instead manipulate the expression of host miRNAs to regulate their viral life cycle (18, 19).
The expression of a number of cellular miRNAs is altered through the action of the HPV E6 and E7 proteins. For instance, miR-203 levels are decreased by both E6 and E7 in differentiating HPV-positive epithelial cells (18, 20), resulting in the retention of high levels of p63 that are critical for maintaining cells active in the cell cycle and for efficient viral genome amplification (19). HPV proteins also reduce the expression of miR-145, which regulates the levels of the cellular transcription factor KLF-4, that in turn regulates cell cycle progression in differentiating cells (21). In addition, miR-145 directly targets elements in the E1 and E2 ORFS to control genome amplification. A number of studies have examined miRNA expression in HPV-positive cell lines (18, 20, 22–24); however, there is significant variability in the spectrum of miRNAs reported to be altered, and this is likely the result of multiple miRNAs targeting the same cellular factors or pathways. In both cervical cancers and HPV-positive oropharyngeal cancers the levels of miR-424 are decreased, and this may be important for progression to malignancy (23, 25). One potential target of miR-424 is the kinase CHK1 (26), which is an important downstream effector of the ataxia telangiectasia and Rad3-related protein (ATR) DNA damage response. Since previous studies identified an important role for the ATR pathway in HPV pathogenesis, we investigated what role, if any, miR-424 has in the viral life cycle.
RESULTS
To investigate what role miR-424 might play in the HPV life cycle, we first examined the levels in human foreskin keratinocytes (HFKs) in comparison to matched HFKs that stably maintain HPV31 or HPV16 episomes. Since it was important to determine whether the effects varied during the various phases of the viral life cycle, both undifferentiated monolayer cultures and cells induced to differentiate using high-calcium media were examined for miR-424 expression. Reverse transcription-PCR (RT-PCR) analysis showed that cells that stably maintain complete HPV genomes have decreased levels of miR-424 compared to matched HFKs (Fig. 1A). Upon differentiation, the levels of miR-424 were observed to increase in normal keratinocytes. Although the levels of miR-424 also increased in HPV31-positive cells after differentiation, these were still substantially reduced from the levels seen in HFKs (Fig. 1B). Figure 1C shows that both HFKs and HPV-positive keratinocytes differentiate to comparable levels in high-calcium media, using loricrin and involucrin as differentiation markers (Fig. 1C). We have previously shown that upon differentiation most HPV-positive and -negative cells express markers of differentiation (9, 27) and that these are only modestly delayed in HPV-positive cells (9). A similar decrease of ∼5-fold in miR-424 levels is also seen in monolayer culture of CIN612 cells, a cell line isolated from a low-grade cervical biopsy specimen that stably maintains HPV31 episomes (Fig. 1D). Importantly, a 5-fold reduction in the levels of miR-424 was seen in organotypic raft cultures of in HPV31-positive cells compared to HFK rafts (28). We consistently find similar effects on the HPV life cycle in organotypic rafts as we do following calcium-induced differentiation (9, 29). In addition, several clinical studies have shown the low expression of miR-424 in cervical cancer tissues or high-grade cervical intraepithelial neoplasia (23, 25).
FIG 1.
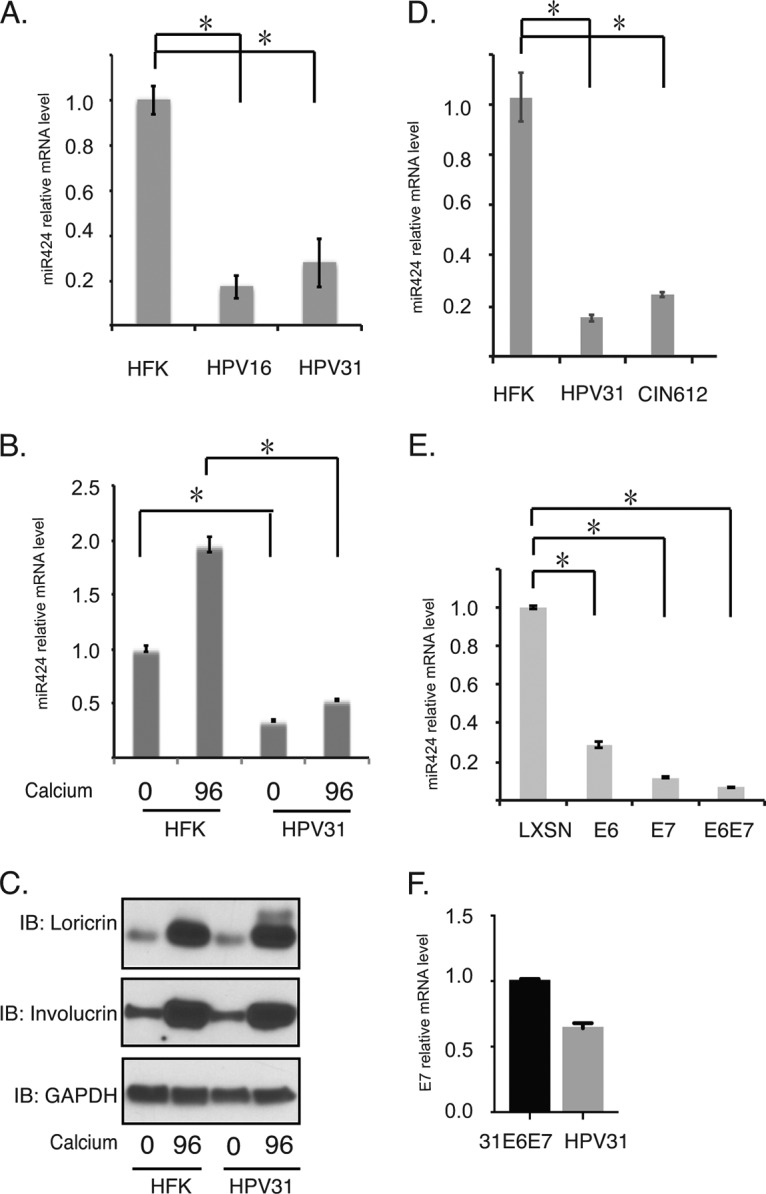
The expression of miR-424 is decreased in HPV-positive cells. (A) RT-PCR analysis of miR-424 in undifferentiated HFKs and HPV16- and HPV31-positive cells. (B) RT-PCR analysis of miR-424 in undifferentiated and differentiated HFKs or HPV31-positive cells. Differentiation was induced by addition of high calcium media for 96 h. (C) Western blot analysis of loricrin, involucrin, and GAPDH proteins in HFKs and HPV31-positive cells upon differentiation. (D) RT-PCR analysis of miR-424 in HFKs versus HPV31-positive or CIN612 cells. (E) RT-PCR analysis of miR-424 in HFKs expressing LXSN, 31E6, 31E7, and 31E6/E7. (F) RT-PCR analysis of E7 in HFKs expressing HPV31 E6/E7, and HPV31 genomes. The results were normalized to GAPDH. All results are representative of observations from three independent experiments. *, P < 0.05.
We next investigated whether E6 or E7 is responsible for the suppression of miR-424. For this analysis, HFKs were transduced with retroviruses expressing HPV31 E6, HPV31 E7, or HPV31 E6/E7, as well as the LXSN vector control. Interestingly, miR-424 levels were decreased in either E6- or E7-expressing cells by about 5- to 7-fold compared to those seen in LXSN-expressing HFKs. Furthermore, a reduction in miR-424 levels of ∼10-fold was seen in cells expressing both E6 and E7. This indicates that the most efficient reductions were seen when both viral oncoproteins are expressed (Fig. 1E). Figure 1F shows comparable levels of E7 expression in E6/E7 cells and HFKs stably expressing the HPV31 genome. Similar levels of E7 transcripts are also seen in HFKs that maintain HPV16 genomes and those expressing only HPV16 E6/E7.
Since the miR-424 levels are decreased in HPV16- and HPV31-positive cells, we next tested whether suppression of miR-424 is required either for the maintenance of HPV genomes in undifferentiated cells or for genome amplification upon differentiation. For this analysis, we infected HPV31-positive CIN612 cells with lentiviruses expressing miR-424 and screened for effects on stable maintenance and on amplification. The levels of miR-424 were increased by ∼10-fold in CIN612 cells stably expressing miR-424, as indicated by RT-PCR analysis (Fig. 2A). The miR-424-overexpressing cells were also induced to differentiate in high-calcium media for 72 h. Total DNA was harvested at 0 and 72 h for Southern analysis to screen for the effects on HPV genome amplification. Previous studies have shown that viral genome amplification increases steadily 48 h after a calcium switch (9, 29). Figure 2B shows that the CIN612 cells infected with miR-424 lentiviral vectors exhibit reduced levels of viral episomes in undifferentiated cells compared to cells infected with control lentiviruses. Furthermore, although the control cells amplified HPV31 episomes within 72 h of differentiation, miR-424-expressing CIN612 cells exhibited an impaired ability to amplify HPV31 episomes upon differentiation. The levels of HPV episome forms were quantified using ImageJ software (Fig. 2C). Our analysis indicates that reduced levels of miR-424 are important for efficient HPV genome amplification. To eliminate the possibility that loss of HPV episomes in miR-424-expressing cells in undifferentiated cells is due to cellular senescence, we examined the growth rates of miR-424-expressing cells and compared the findings to the control cells. Figure 2D indicates that there is no significant growth difference between the two sets of cells. We conclude that the levels of miR-424 do not correlate with altered rates of cellular growth. The levels of involucrin in control and miR-424-overexpressing cells were examined by Western analysis and found to be comparable (Fig. 3E).
FIG 2.
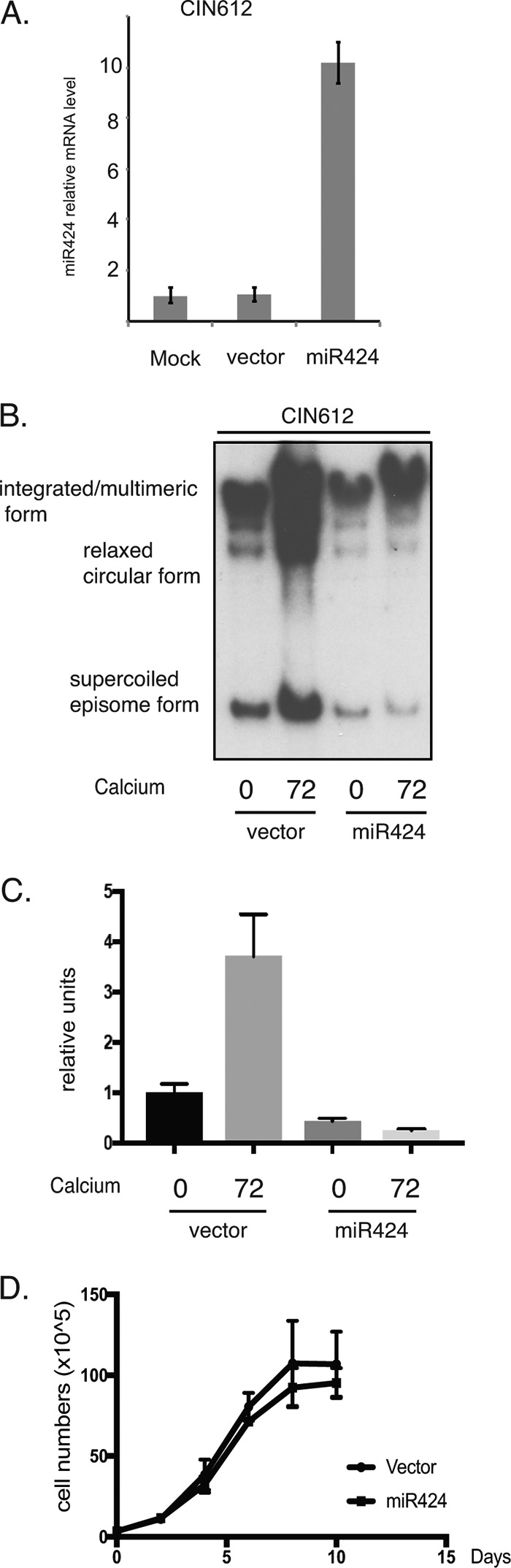
Overexpression of miR-424 suppresses HPV genome amplification upon keratinocyte differentiation. HPV31-positive CIN612 cells stably expressing a scrambled vector control or miR-424 were incubated for 72 h of differentiation in high-calcium media. (A) RT-PCR analysis of miR-424 in miR-424-overexpressed CIN612 cells grown in an undifferentiated monolayer culture. (B) Southern blot analysis of HPV31 episomes in CIN612 cells stably expressing miR-424 with differentiation in high-calcium media for the indicated times (in hours). (C) The levels of HPV episome shown in Fig. 2B were quantified using the ImageJ software and are presented graphically. (D) The growth rate of miR424-expressing cells was compared to the one of vector control cells. All results are representative of observations from two or more independent experiments.
FIG 3.
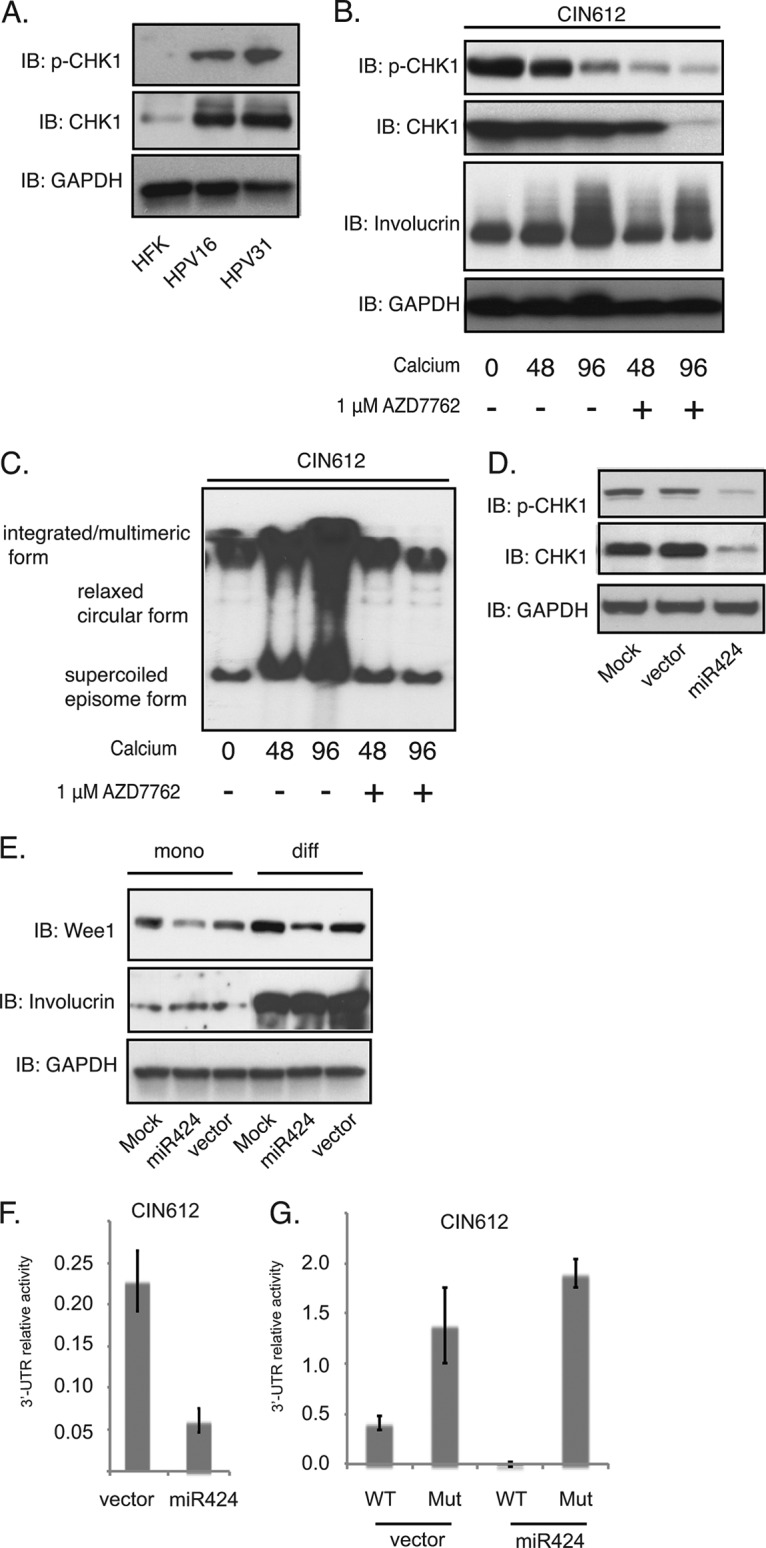
CHK1 is a miR-424 downstream target and is necessary for HPV31 genome amplification. (A) Western blot analysis of p-CHK1, CHK1, and GAPDH proteins in HFKs and in HPV16- and HPV31-positive cells. (B) Western blot analysis of p-CHK1, CHK1, involucrin, and GAPDH proteins in CIN612 cells in the presence or absence of AZD7762 (1 μM) after differentiation in high-calcium media for the indicated times. (C) Southern blot analysis for HPV31 episomes in CIN612 cells after AZD7762 treatment and differentiation in high-calcium media for the indicated times (in hours). (D) Western blot analysis of p-CHK1, CHK1, and GAPDH proteins in CIN612 cells stably expressing either vector alone or miR-424. (E) Western blot analysis of Wee1, involucrin, and GAPDH proteins in undifferentiated or differentiated (“mono” versus “diff”) CIN612 cells stably expressing either vector alone or miR-424. (F) The luciferase reporter vector containing wild-type CHK1 3′ UTR was transfected into CIN612 cells stably expressing either vector alone or miR-424. The firefly luciferase activities were determined 48 h after transfection and normalized to Renilla luciferase. The relative luciferase activities were normalized to the control pmiRGLO plasmid. Each column stands for the mean value plus the standard deviation of three independent experiments. (G) The luciferase reporter vector containing wild-type CHK1 3′ UTR or a mutant CHK1 3′ UTR was transfected into CIN612 cells transduced with miR424-expressing lentiviruses. The luciferase activities were determined as in panel F. All results are representative of observations from three independent experiments.
To investigate the mechanism by which miR-424 regulates HPV genome amplification, we tested whether miR-424 acts on the ATR DNA damage pathway to modulate HPV life cycle. One putative target of miR-424 is CHK1, a serine/threonine protein kinase that coordinates the ATR DNA damage response and regulates cell cycle check point arrest. Our previous studies using a general inhibitor suggested CHK1 activation was critical for both genome maintenance and amplification (10). We first observed that CHK1 levels and activation are increased in HPV16- and HPV31-positive cells compared to HFKs by Western blot analysis (Fig. 3A), findings consistent with our previous study (10). We next wanted to use a more specific and less toxic inhibitor than the one previously used to confirm the effect of CHK1 on HPV replication. For this analysis, CIN612 cells were induced to differentiation in high-calcium media in the presence or absence of the CHK1 phosphorylation-specific inhibitor AZD7762. Treatment of CIN612 cells with AZD7762 led to a reduction of phosphorylated CHK1 levels within 48 h. Interestingly, we also observed that the total levels of CHK1 were dampened at late times of differentiation (96 h; Fig. 3B); this may be due to reduced numbers of amplifying cells at the very late stages of differentiation. Total DNA was also extracted from the differentiated cells with or without AZD7762 treatment and examined for HPV episomes by Southern blotting. Our data confirmed that inhibition of CHK1 suppresses HPV genome amplification and indicates that CHK1 is an important regulator of viral replication (Fig. 3C).
The studies described above indicate that the total levels of CHK1 proteins are increased in HPV-positive cells compared to normal cells, and we next wanted to demonstrate that the suppression of miR-424 was responsible for this increase. To confirm that high levels of miR-424 leads to suppression of CHK1 levels, we first examined the total levels of CHK1 and p-CHK1 in cells transduced with miR-424 lentiviral expression vectors. As seen in Fig. 3D, HPV31-positive cells were infected with miR-424 lentiviruses and selected to generate a stable cell line. The CIN612 cells stably expressing miR-424 exhibited reduced levels of both total CHK1 and p-CHK1. We also examined another miR-424 target, Wee1 (26, 30), which controls the G2 checkpoint, and found that increased expression of miR-424 similarly reduced Wee1 levels (Fig. 3E). The levels of Wee1 modestly increases upon 72 h of calcium differentiation in both vector control cells and miR424-expressing HPV31-positive cells; however, the presence of miR424 results in a lower level of Wee1 expression in miR424-expressing cells than in vector control cells.
It was next important to demonstrate that suppression of CHK1 levels occurred through miR-424 seed sequences located in the 3′ UTR of CHK1. For this analysis, a luciferase reporter was first constructed that contained sequences encompassing the putative miR-424-binding sites cloned into the 3′ region of the pmiRGLO dual-luciferase reporter vector. CIN612 cells were then transduced with the lentivirus expression vectors encoding miR-424, and stable cell lines were selected. The CHK1 3′ UTR reporter was transfected into miR-424-transduced cells, and the reporter activity was examined by luciferase reporter assay in comparison to the activity seen in the control transduced cells. We observed a significant decrease in the activities of the luciferase reporter containing CHK1 3′ UTR in cells expressing miR-424 (Fig. 3F). No such decrease was observed when the luciferase reporter vector carrying a mutation of the predicted binding site of miR-424 on CHK1 3′ UTR region was examined in these assays (Fig. 3G). This mutation has been previously characterized as representative of other mutations in the miR-424 recognition sequence (25). This analysis confirms that this region is a target of miR-424 activity and that it regulates CHK1 levels in HPV-positive cells.
To test whether miR-424 acted in a similar manner in cells that maintained another high-risk type, we infected HPV16-positive SiHa cells with miR-424 lentivirus expression vectors. SiHa is an established cell line derived from a cervical cancer that contains integrated copies of HPV16 genomes and expresses only the E6 and E7 oncoproteins. We observed a 6-fold change for the mRNA levels of miR-424 in SiHa cells transduced by lentiviruses expressing miR-424 compared to the levels in SiHa cells transduced with empty vector (Fig. 4A). Similar to the experiments performed in CIN612 cells, the reporter vector containing CHK1 3′-UTR was transfected into SiHa cells that had been transduced with lentivirus expressing miR-424. As shown in Fig. 4B, the CHK1 3′-UTR activities were decreased in SiHa cells with miR-424 transduction compared to the vector control. Furthermore, the mutation of miR-424 binding site on CHK1 3′ UTR reversed the luciferase activity. Next, we examined the total CHK1, p-CHK1, and Wee1 levels in SiHa cells transduced with miR-424 expression vectors and found reductions similar to those seen in HPV31-positive cells (Fig. 4C and D). Finally, we infected keratinocytes that stably maintain HPV16 episomes with miR-424 lentiviral expression vectors and observed a reduction in the levels of stable viral episomes similar to the effects seen with HPV-31 episomes (Fig. 4E). We conclude that the suppression of miR-424 in HPV31- or HPV16-positive cells is important for the stable maintenance of episomes, as well as for amplification, and that this is mediated in part through control of the DNA damage kinase CHK1.
FIG 4.
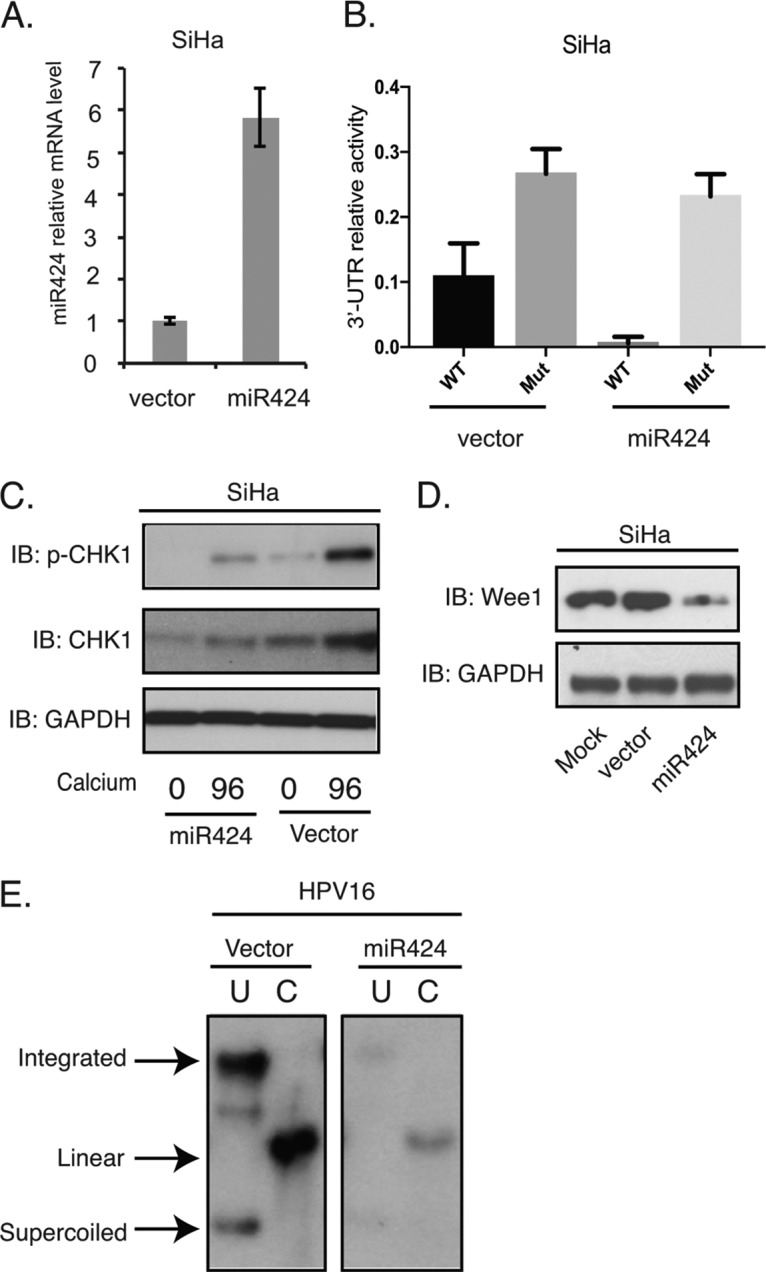
The suppression of miR-424 is required for HPV16 genome maintenance. (A) RT-PCR assay of miR-424 mRNA levels in SiHa cells transduced with lentiviruses expressing the control vector or miR-424 (P < 0.03). (B) The luciferase reporter vector containing wild-type CHK1 3′ UTR or a mutant CHK1 3′ UTR were transfected into SiHa cells transduced with miR424-expressing lentiviruses. The firefly luciferase activities were determined 48 h after transfection and normalized to Renilla luciferase. The relative luciferase activities were normalized to the control pmiRGLO plasmid. Each column stands for the mean value plus the standard deviation of three independent experiments. (C) Western blot analysis of p-CHK1, CHK1, and GAPDH proteins in SiHa cells transduced with the control and miR-424 lentiviruses, followed by differentiation in high-calcium media for 96 h. (D) Western blot analysis of Wee1 and GAPDH proteins in undifferentiated SiHa cells transduced with the control and miR-424 lentiviruses. (E) Southern blot analysis for HPV16 episomes in the control or miR-424-expressing HPV16 cells. The total DNAs from cells grown in monolayers were incubated with an enzyme that does not digest the HPV16 genome (lanes U) or cuts it once (lanes C). All results are representative of observations from three independent experiments.
DISCUSSION
In this study, we identified miR-424 as a critical negative regulator of both stable HPV genome replication and amplification. The levels of miR-424 are decreased in undifferentiated keratinocytes that stably maintain HPV31 or HPV16 episomes, as well as in cervical cancer cell lines, compared to the levels detected in normal cells. Although the levels of miR-424 in HPV-positive cells increase upon differentiation, they are still substantially decreased from those seen in HFKs. Interestingly, both E6 and E7 oncoproteins act to suppress miR-424 levels with the most significant effects seen when both proteins are coexpressed. In exfoliated cells from high-grade cervical intraepithelial neoplasia (CIN) 2 and 3 cervical lesions, as well as from head and neck cancer tissues, miR-424 levels have been reported to be decreased (23, 31). However, prior to this study it was not clear whether miR-424 had any effect on the HPV life cycle or whether its effects were manifested only in high-grade lesions. Our work demonstrates that the suppression of miR-424 levels is important for HPV replication and that this occurs in part through the regulation of factors in the DNA damage repair pathways.
One downstream target of miR-424 is CHK1, a downstream effector kinase in the ATR DNA repair pathway. Our previous studies have shown that activation of both the ATM and the ATR DNA damage pathways by HPV proteins is required for HPV genome amplification (9, 10). Our data demonstrate an increase in the levels of CHK1, as well as of p-CHK1, in HPV-positive cells, which we show is due, at least in part, to decreased levels of miR-424. Our previous studies on the ATR/CHK1 pathway showed that inhibition of CHK1 phosphorylation with UNC01 inhibited amplification, and similar effects are seen with the specific p-CHK1 inhibitor AZD7762. Our data further confirm that CHK1 inhibition results in a decrease in HPV viral amplification (Fig. 3C), indicating that CHK1 activation is necessary for the HPV life cycle. Interestingly, the overexpression of miR-424 reduces stable HPV genome replication in undifferentiated cells and amplification upon differentiation. It has been reported that inhibition of the ATR/CHK1 pathway also reduces long-term stable maintenance of HPV episomes in established cell lines (32), and our preliminary results suggest that this is also seen with HPV31 episomes (S. Hong and L. A. Laimins, unpublished data). It is also possible that additional targets of miR-424 besides CHK1 contribute to regulating stable HPV genome replication. This is consistent with our observation that Wee1, a kinase that regulates G2, is also controlled by miR-424 (30). Since miRNAs target a large spectrum of cellular genes, it is likely other miR-424 targets are also involved in regulating HPV replication. Similarly, other miRNAs also may contribute to the regulation of CHK1 levels, and this is suggested by the finding that miR-424 is part of a family of related miRNAs, including miR-15/16/195/497, some of which could also regulate CHK1 (26). It is likely that HPVs regulate CHK1 through multiple pathways, and future studies will try to underline the mechanisms of how HPV oncoproteins mediate CHK1. Taken together, our study shows that HPV suppresses the levels of miR-424, resulting in increased levels of CHK1 that in turn controls viral replication.
The role of miR-424 in the development of cervical cancers has not been well studied. It is possible that miR-424 regulates other aspects besides the effects on HPV genome amplification via modulation of the ATR DNA damage pathway. Recent studies have shown that miR-424 also regulates BCL-2 and insulin-like growth factor 1, suggesting that it has tumor suppressor activities (33). In contrast to our observations with HPV, the levels of miR-424 are increased in EBV-positive, diffuse, large B cell lymphomas (34). These findings suggest miR-424 may act in different ways in cancers induced by other oncogenic viruses. Overall, our studies demonstrate that HPV proteins downregulate the levels of miR-424, and this is critical for HPV amplification through its effects on DNA damage factors.
MATERIALS AND METHODS
Cell culture.
HFKs and HPV genome-expressing cells were prepared as previously described (35). E6- and E7-expressing cells were generated by retroviral infection as previously described (36). CIN612 cells that stably maintain HPV 31 episomes are derived from a CIN II biopsy specimen. To induce differentiation, HPV-positive cells were grown in keratinocyte basal medium without supplements, and 1.5 mM CaCl2 was added for up to 96 h (35).
RT-PCR.
Total small RNA populations were isolated from HPV-positive cells using the Roche miRNA isolation kit. The products were then transcribed using an Exiqon cDNA synthesis kit. The cDNAs were next mixed with LightCycler 480 SYBR green I master mix (Roche, Indianapolis, IN) in the presence of RT-PCR primers, and PCR was performed using a LightCycler 480. The specific miR-424 primers were purchased from Exiqon, Woburn, MA. The results are normalized to those for GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and the data shown is representative of observations from three independent experiments. Significance was determined using Student t test, and a P value of <0.05 was considered significant.
Inhibitors, antibodies, and Western blot analysis.
The inhibitor (1 μM AZD7762) used in this study was purchased from Selleckchem, Houston, TX. The antibodies used in this study were as follows: anti-Wee1, anti-involucrin, and anti-GAPDH (Santa Cruz, Santa Cruz, CA) and anti-CHK1 and anti-p-CHK1 (Cell Signaling, Danvers, MA). For Western blot analysis, cell lysates were processed as previously described (37). Briefly, keratinocytes were first separated from J2 feeders by Versene (phosphate-buffered saline [PBS] containing 0.5 mM EDTA) treatment and lysed in ice-cold radioimmunoprecipitation assay lysis buffer for 30 min. The protein samples were separated on SDS-page gels and then transferred to polyvinylidene difluoride membranes. The membranes were then developed using ECL Prime or ECL reagents (Amersham, Pittsburgh, PA). Chemiluminescence signals were detected using Eastman Kodak X-ray films.
Southern blot analysis.
Total DNA was exacted by phenol-chloroform, precipitated, and washed with 70% alcohol. The products were air dried for 10 min and then dissolved in water. The DNA sample concentrations were determined by multiple NanoDrop measurements, and equal amounts of samples were loaded on ethidium bromide-containing gels for electrophoresis. The gels were then processed by Southern blotting as previously described (38).
Lentiviral virion production and transduction.
plv-miR-424 lentiviral vectors were purchased from Biosettia, San Diego, CA. Lentiviral particles expressing miR-424 were prepared as previously described (38). CIN612 cells were incubated with 5 ml of fresh E media, including concentrated miR-424 or scramble lentiviral soup in the presence of 4 μg of hexadimethrine bromide (Polybrene; Sigma-Aldrich, St. Louis, MO)/ml overnight at 37°C. The next day, the transduced cells were washed once with PBS and then cultured in fresh E medium for an additional 48 h before analysis.
Luciferase reporter assay.
The sequences from the CHK1 3′ UTR containing the miR-424-binding sites were subcloned into the pmiRGLO dual-luciferase reporter vector. The vector carrying a mutation of the predicted binding site of miR-424 on CHK1 3′ UTR was a generous gift from Junfen Xu from Zhejiang University, China (39). The miR-424 transduced cells were washed with PBS and transfected with the pmiRGLO-CHK1 plasmid using polyethylenimine reagents. The following day, the transfected cells were transferred to fresh E medium, followed by incubation at 37°C for 24 h. The effects of the CHK1 3′ UTR on expression were examined by luciferase reporter assays by comparing the transduced cells to the control cells. The luciferase activity was measured using the Dual-Luciferase reporter assay system (Promega, Madison, WI), with Renilla luciferase as an internal control according to the manufacturer's instructions. Significance was determined using a Student t test.
ACKNOWLEDGMENTS
We thank Junfen Xu and Weiguo Lv from Zhejiang University (Hangzhou, China) for valuable reagents.
This study was supported by grants from the National Cancer Institute to L.A.L. (CA 59655 and CA 142861).
REFERENCES
- 1.zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 3.Bodily J, Laimins LA. 2011. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 19:33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huibregtse JM, Scheffner M, Howley PM. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 10:4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 6.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 7.Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 8.Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. 1989. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J 8:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong S, Cheng S, Iovane A, Laimins LA. 2015. STAT-5 regulates transcription of the topoisomerase IIβ-binding protein 1 (TopBP1) gene to activate the ATR pathway and promote human papillomavirus replication. mBio 6:e02006–02015. doi: 10.1128/mBio.02006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace NA, Galloway DA. 2014. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin Cancer Biol 26:30–42. doi: 10.1016/j.semcancer.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Songock WK, Kim SM, Bodily JM. 2017. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res 231:56–75. doi: 10.1016/j.virusres.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong S, Laimins LA. 2013. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol 8:1547–1557. doi: 10.2217/fmb.13.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat Methods 2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 16.Harwig A, Das AT, Berkhout B. 2014. Retroviral microRNAs. Curr Opin Virol 7:47–54. doi: 10.1016/j.coviro.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Cai X, Li G, Laimins LA, Cullen BR. 2006. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J Virol 80:10890–10893. doi: 10.1128/JVI.01175-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna DJ, McDade SS, Patel D, McCance DJ. 2010. MicroRNA 203 expression in keratinocytes is dependent on regulation of p53 levels by E6. J Virol 84:10644–10652. doi: 10.1128/JVI.00703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melar-New M, Laimins LA. 2010. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol 84:5212–5221. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harden ME, Prasad N, Griffiths A, Munger K. 2017. Modulation of microRNA-mRNA target pairs by human papillomavirus 16 oncoproteins. mBio 8:e02170-16. doi: 10.1128/mBio.02170-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunasekharan V, Laimins LA. 2013. Human papillomaviruses modulate miR-145 expression to directly control genome amplification. J Virol 87:6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yablonska S, Hoskins EE, Wells SI, Khan SA. 2013. Identification of miRNAs dysregulated in human foreskin keratinocytes (HFKs) expressing the human papillomavirus (HPV) type 16 E6 and E7 oncoproteins. Microrna 2:2–13. doi: 10.2174/2211536611302010002. [DOI] [PubMed] [Google Scholar]
- 23.Tian Q, Li Y, Wang F, Li Y, Xu J, Shen Y, Ye F, Wang X, Cheng X, Chen Y, Wan X, Lu W, Xie X. 2014. MicroRNA detection in cervical exfoliated cells as a triage for human papillomavirus-positive women. J Natl Cancer Inst 106:dju241. doi: 10.1093/jnci/dju241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Wang HK, Li Y, Hafner M, Banerjee NS, Tang S, Briskin D, Meyers C, Chow LT, Xie X, Tuschl T, Zheng ZM. 2014. microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc Natl Acad Sci U S A 111:4262–4267. doi: 10.1073/pnas.1401430111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Li Y, Wang F, Wang X, Cheng B, Ye F, Xie X, Zhou C, Lu W. 2012. Suppressed miR-424 expression via upregulation of target gene Chk1 contributes to the progression of cervical cancer. Oncogene 32:976–987. doi: 10.1038/onc.2012.121. [DOI] [PubMed] [Google Scholar]
- 26.Pouliot LM, Chen YC, Bai J, Guha R, Martin SE, Gottesman MM, Hall MD. 2012. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res 72:5945–5955. doi: 10.1158/0008-5472.CAN-12-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruesch MN, Stubenrauch F, Laimins LA. 1998. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J Virol 72:5016–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunasekharan V, Laimins LA. 2013. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol 87:6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langsfeld ES, Bodily JM, Laimins LA. 2015. The deacetylase Sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog 11:e1005181. doi: 10.1371/journal.ppat.1005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B, Duan L, Yin G, Tan J, Jiang X. 2013. Simultaneously expressed miR-424 and miR-381 synergistically suppress the proliferation and survival of renal cancer cells—Cdc2 activity is up-regulated by targeting WEE1. Clinics (Sao Paulo) 68:825–833. doi: 10.6061/clinics/2013(06)17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hui AB, Lenarduzzi M, Krushel T, Waldron L, Pintilie M, Shi W, Perez-Ordonez B, Jurisica I, O'Sullivan B, Waldron J, Gullane P, Cummings B, Liu FF. 2010. Comprehensive MicroRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res 16:1129–1139. doi: 10.1158/1078-0432.CCR-09-2166. [DOI] [PubMed] [Google Scholar]
- 32.Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. 2013. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J Virol 87:3979–3989. doi: 10.1128/JVI.03473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Barrueco R, Nekritz EA, Bertucci F, Yu J, Sanchez-Garcia F, Zeleke TZ, Gorbatenko A, Birnbaum D, Ezhkova E, Cordon-Cardo C, Finetti P, Llobet-Navas D, Silva JM. 2017. miR-424(322)/503 is a breast cancer tumor suppressor whose loss promotes resistance to chemotherapy. Genes Dev 31:553–566. doi: 10.1101/gad.292318.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imig J, Motsch N, Zhu JY, Barth S, Okoniewski M, Reineke T, Tinguely M, Faggioni A, Trivedi P, Meister G, Renner C, Grasser FA. 2011. microRNA profiling in Epstein-Barr virus-associated B-cell lymphoma. Nucleic Acids Res 39:1880–1893. doi: 10.1093/nar/gkq1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fehrmann F, Laimins LA. 2005. Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol 292:317–330. [DOI] [PubMed] [Google Scholar]
- 36.Hong S, Mehta KP, Laimins LA. 2011. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 85:9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong S, Brass A, Seman M, Haag F, Koch-Nolte F, Dubyak GR.. 2007. Lipopolysaccharide, IFN-γ, and IFN-β induce expression of the thiol-sensitive ART21 Ecto-ADP-ribosyltransferase in murine macrophages J Immunol 179:6215–6227. [DOI] [PubMed] [Google Scholar]
- 38.Hong S, Laimins LA. 2013. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog 9:e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Li Y, Wang F, Wang X, Cheng B, Ye F, Xie X, Zhou C, Lu W. 2013. Suppressed miR-424 expression via upregulation of target gene Chk1 contributes to the progression of cervical cancer. Oncogene 32:976–987. doi: 10.1038/onc.2012.121. [DOI] [PubMed] [Google Scholar]