

Abstract
The histone H3 N‐terminal protein domain (N‐tail) is regulated by multiple posttranslational modifications, including methylation, acetylation, phosphorylation, and by proteolytic cleavage. However, the mechanism underlying H3 N‐tail proteolytic cleavage is largely elusive. Here, we report that JMJD5, a Jumonji C (JmjC) domain‐containing protein, is a Cathepsin L‐type protease that mediates histone H3 N‐tail proteolytic cleavage under stress conditions that cause a DNA damage response. JMJD5 clips the H3 N‐tail at the carboxyl side of monomethyl‐lysine (Kme1) residues. In vitro H3 peptide digestion reveals that JMJD5 exclusively cleaves Kme1 H3 peptides, while little or no cleavage effect of JMJD5 on dimethyl‐lysine (Kme2), trimethyl‐lysine (Kme3), or unmethyl‐lysine (Kme0) H3 peptides is observed. Although H3 Kme1 peptides of K4, K9, K27, and K36 can all be cleaved by JMJD5 in vitro, K9 of H3 is the major cleavage site in vivo, and H3.3 is the major H3 target of JMJD5 cleavage. Cleavage is enhanced at gene promoters bound and repressed by JMJD5 suggesting a role for H3 N‐tail cleavage in gene expression regulation.
Keywords: H3K9me1, Histone H3, JMJD5, N‐tail cleavage
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Post-translational Modifications, Proteolysis & Proteomics
Introduction
In all eukaryotes, DNA is tightly associated with histone proteins in order to form chromatin, of which the fundamental subunit is the nucleosome 1. Histone proteins are subjected to a large number and variety of small chemical group modifications, such as methyl and acetyl groups and, together with DNA methylation, have a central role in regulating gene transcriptions 2.
A histone molecule can be divided into two distinct regions: a carboxy terminus that organizes DNA on the surface of each unit of the nucleosome, and a flexible amino terminus called the N‐tail, which spread out from the nucleosome. The histone H3 N‐terminal region is a flexible tail, which bears residues, with their side chains, that can be methylated, acetylated, and phosphorylated by different nuclear enzymes. Through these side‐chain posttranslational modifications, the H3 N‐tail contacts with other histones or transcription machinery to regulate chromatin folding, gene expression, DNA repair, and cell cycle regulation 3, 4.
There is evidence that histone turnover is regulated by proteolytic activities. An histone H2A‐specific protease activity during granulocyte differentiation removes a pentadecapeptide from H2A C‐terminus region, cutting between V114 and L115 5. The resulting H2A–H2B dimer has a reduced affinity for the H3–H4 tetramer, destabilizing the whole nucleosome. This function may contribute to a more “open” chromatin, facilitating gene transcription or DNA replication. Likewise, H3 N‐tail modifications also include cleavage of its peptide bond at different sites and emerging evidences reveal that H3 N‐tail cleavage differs from H3 protein turnover 6. Histone H3 N‐terminal region proteolytic cleavage by Cathepsin L has been shown to be required during early differentiation of mouse embryonic stem cell (mESC) 7, 8. Similar clipping events of H3 associated with other cellular processes including viral infection 9, 10, aging 11, 12, and yeast sporulation 13 have also been reported. Additionally, H3 protease activity was also found in chicken liver and Tetrahymena micronuclei 14, 15, 16, 17. Therefore, H3 N‐tail cleavage sweeps away various kinds of N‐tail histone modifications and serves as one of the posttranslational modifications in response to environmental changes 18.
Although the molecular consequences of any histone clipping event are yet to be defined, histone cleavage seem to suggest an evolutionary conserved process. So far, several cytoplasm or nuclear enzymes have been identified to contain histone H3 cleavage activity, such as Cathepsin L, which cleaves histone H3 between A21 and T22 to create a new N‐terminus starting at residue T22, Glutamate dehydrogenase and endopeptidase PRB1, which cleaves between K23 and A24 in vivo or in vitro to create a new N‐terminus starting at residue A24, and MMP‐9, which cleaves between K18 and Q19 to create a new N‐terminus starting at residue Q19 6, 7, 19, 20, 21, 22.
JMJD5, a Jumonji C (JmjC) domain‐containing protein, was reported to be responsible for gene transcription regulation through its histone H3 lysine 36 dimethylation (H3K36me2) demethylase activity 23, 24, 25 and to regulate osteoclastogenesis with its hydroxylase activity 26. Jmjd5 plays an important role in several biological processes, such as embryogenesis 23, 24, circadian rhythms 27, pluripotency of stem cells 28, cancer cell proliferation 25, and glucose metabolism 29.
We report here that change of cell stress conditions also leads to histone H3 N‐tail proteolytic cleavage at the carboxyl side of monomethylated lysine 9 (mMK9) by JMJD5. JMJD5 binds to gene promoters and cleaves H3 N‐tail; therefore, JMJD5 cleavage activity may play a role in gene transcription regulation.
Results
Appearance of a modified histone H3 is correlated with JMJD5 expression under the stressed conditions
Using antibody against H3 C‐terminal region (C‐terminal 10–20 residues) or antibodies against H3K27me1, H3K27me2, and H3K36me1, a weak faster‐migrating band was often detected in cells grown under the normal conditions, and expression of this protein band could be enhanced by serum starvation or TdR double block and release (Figs 1A and B, and EV1A–C). Interestingly, expression level of JMJD5, a Jumonji C (JmjC) domain‐containing protein responsible for H3K36me2 histone demethylation 24, 25, was elevated by serum starvation or TdR double block and release (Figs 1B and C, and EV1B and C), suggesting a correlation between induction of JMJD5 and appearance of fast‐migrating band. The faster‐migrating species was recognized by the antibodies recognizing H3K27me1, H3K27me2, or H3 C‐terminal region but not by the antibodies recognizing H3K9me1, H3K9me2, or H3K4me1 (Figs 1A and B, and EV1B), suggesting this species represents a form of modified histone H3 and probably lacks those common K9‐tail modifications. A similar pattern of H3 faster‐migrating species induction was detected in both acid‐extracted histones (Fig 1B) and whole cell extracts (Fig EV1B). Induction of this faster‐migrating species and expression of JMJD5 gradually decreased after cells were released from serum starvation and cultured in fresh medium for longer time (Fig EV1D). Moreover, UV treatment or addition of DNA damage reagents to cultured cells, such as Camptothecin (CPT) and Etoposide (ETO), which arrest cells at different cell cycle phase, also induced expression of JMJD5 and H3 faster‐migrating species (Fig EV1E and F). Note that induction of this H3 faster‐migrating species is independent of senescence, as only ETO treatment of cells induced senescence (Fig EV1E). Consistent with previous reports 30, 31, 32, serum starvation or TdR double block and release, but not G2 phase inhibitor RO‐3306, induced phosphorylation of H2AX at Ser 139 (γH2AX), a marker of DNA damage response 33 (Fig EV1G). These results suggested that DNA damage response, but not cell cycle arrest itself, induced expression of JMJD5 and H3 faster‐migrating species. Immunostaining revealed that, in cells expressing ectopic JMJD5, level of H3K9me1 or H3K9me2 to a lesser extent but not H3K9me3 and H3K27me1 was affected (Figs 1D and EV1H). The numbers of Myc‐JMJD5 transfected cells detected with H3K9me1 intensity reduction was significantly higher than those with other sites based upon three independent experiments (Fig 1E). These results indicated that N‐tail of histone H3 might undergo methylation‐dependent proteolysis, which correlated with the induction of JMJD5 expression under stressed conditions.
Figure 1. Induction of histone H3 N‐tail proteolysis correlates with JMJD5 expression under stressed conditions.
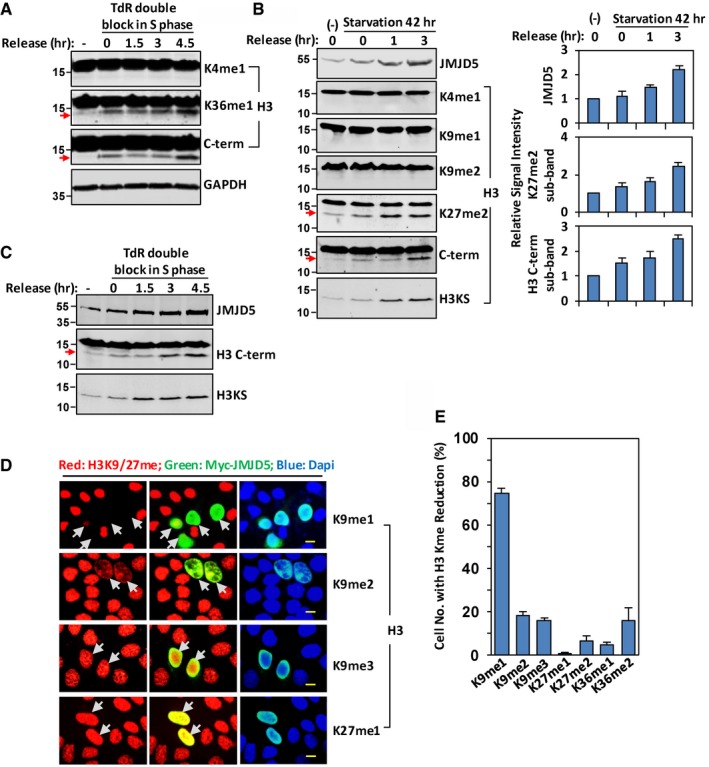
- HeLa cells were double‐blocked with TdR and released into fresh medium for indicated times. Western blots were performed with specific antibodies as indicated. GAPDH is used as protein loading control. Data shown are representative of three independent experiments.
- After serum starvation for 42 h, HeLa cells were released into fresh medium for indicated times. Acid‐extracted histones were analyzed with indicated H3 antibodies. JMJD5 induction was analyzed in whole cell extracts in Western blot (left panel). The bars and error bars shown in right panel represent mean ± SEM; n = 3 of densitometry analysis of three independent experiments for indicated induction of JMJD5 and faster‐migrating band detected with H3K27me2 or H3 C‐term antibody.
- A549 cells were double‐blocked with TdR and released into fresh medium for indicated times. Western blots were performed with specific antibodies as indicated. Data shown are representative of three independent experiments.
- HeLa cells were transfected with Myc‐JMJD5, and immunostaining was performed to show H3K9me1, H3K9me2, H3K9me3, or H3K27me1 (red), and ectopic JMJD5 (green). Nuclei were stained with DAPI (blue). The scale bar represents 10 μm. Grey arrows indicate cells transfected with Myc‐JMJD5. Data shown are representative of three independent experiments.
- In HeLa cells transfected with JMJD5, the percentage of cells with the intensity reduction in H3 K9, K27, and K36 methylation were plotted as mean ± SEM; n = 3 of three independent experiments.
Figure EV1. H3 cleavage in HeLa cells under stressed conditions.
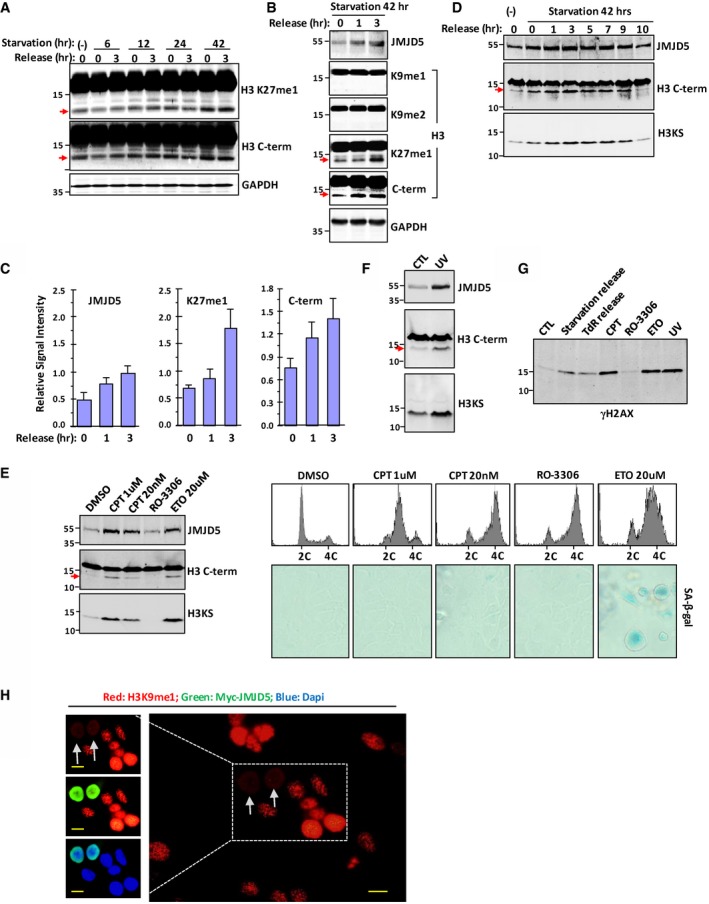
- HeLa cells were serum‐starved for 6, 12, 24, or 42 h, followed by releasing into fresh medium for 0 or 3 h. Processing of histone H3 was evidenced by the detection of fast‐migrating species in whole cell extracts with the antibodies against H3 K27me1 and H3 C‐terminal region. GAPDH was used as loading control. Data shown are representative of three independent experiments.
- After serum starvation for 42 h, HeLa cells were released into fresh medium for indicated times. Whole cell extracts were prepared, and Western blot was performed with indicated antibodies. Data shown are representative of three independent experiments.
- Densitometry of intensity of JMJD5 expression and H3 signal in (B). Data shown are represented as mean ± SEM; n = 3 of three independent experiments.
- HeLa cells were treated as in (B). Cells were released into fresh medium for indicated times. Whole cell extracts were prepared, and Western blot was performed with indicated antibodies. Data shown are representative of two independent experiments.
- A549 cells were treated with CPT, ETO, or RO‐3306 for 24 h, and then flow cytometry and cell senescence analysis were performed (right). The same cell lysates were subjected to Western blot with indicated antibodies (left). Data shown are representative of two independent experiments.
- A549 cells were exposed to UV (40 J/m2), and then cells were allowed to recover for 18 h. Cell lysates were subjected to Western blot with H3 C‐term antibody, and then the same membrane was stripped and reblotted with H3KS antibody.
- A549 cells were subjected to treatment as indicated. Cell lysates were used for Western blot with γH2AX antibody.
- HeLa cells were transfected with Myc‐JMJD5, and immunostaining was performed to show H3K9me1 (red) and ectopic JMJD5 (green). Nuclei were stained with DAPI (blue). The scale bars represent 10 μm. Grey arrows indicate cells transfected with Myc‐JMJD5. Data shown are representative of three independent experiments.
JmjC domain is for JMJD5 dimerization and modular H3 N‐tail docking
JMJD5 can be divided into the N‐terminal helical domain and a C‐terminal JmjC domain 34 (Fig 2A). The JmjC domain was previously demonstrated as the catalytic domain for H3 demethylation 34. To investigate whether JMJD5 was involved in H3 N‐tail manipulation, we constructed the N‐terminal domain (1–270), the JmjC domain (271–416), and full‐length JMJD5 (1–416) in glutathione S‐transferase (GST) recombinant form (Fig 2A). These GST‐JMJD5 protein variants prepared from bacteria were incubated with H3 proteins isolated from HeLa cells. As shown in Fig 2B, GST‐JMJD5 indeed interacted with histone H3. Intriguingly, the C‐terminal JmjC domain rather than the N‐terminal helical domain retained the major H3 binding activity (Fig 2B).
Figure 2. The JmjC domain is necessary for JMJD5 dimerization and modular H3 N‐tail docking.
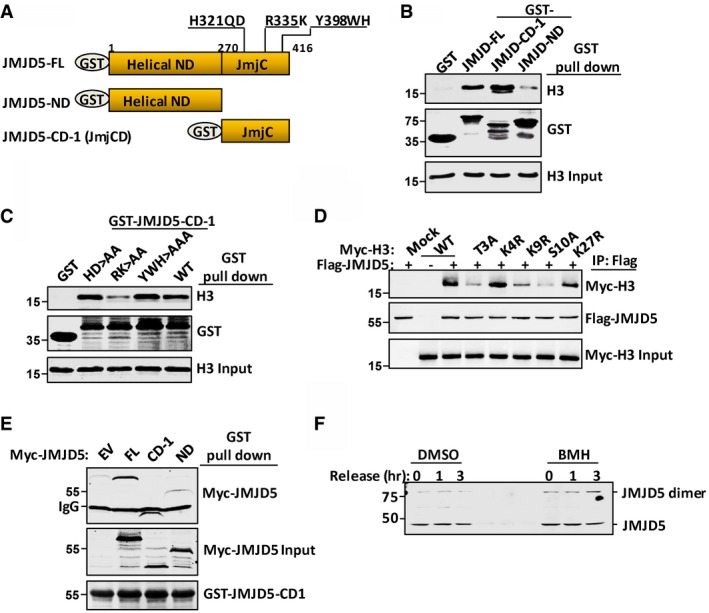
- Schematic representation of full length (FL), helical N‐terminal domain (ND), and C‐terminal JmjC domain (CD‐1) of JMJD5 tagged with GST.
- Bacterial purified GST control (mock), GST‐JMJD5‐FL, GST‐JMJD5‐ND, or GST‐JMJD5‐CD‐1 proteins were incubated with histone H3 prepared from HeLa cells. GST‐JMJD5 and GST‐JMJD5 precipitated H3 were analyzed with anti‐GST and anti‐H3 C‐term antibodies, respectively.
- GST‐JMJD5‐CD1, wild type (WT) or variants with different point mutations as indicated, was incubated with purified H3 histones. GST‐JMJD5‐CD1 precipitates were analyzed with anti‐GST and anti‐H3 C‐term antibodies, respectively.
- Different Myc‐H3 variants were cotransfected with Flag‐JMJD5. Anti‐Flag immunoprecipitates were analyzed for co‐immunoprecipitates with anti‐Myc antibody in Western blot.
- GST‐JMJD5‐CD‐1 was incubated with whole cell lysates prepared from 293T cells transfected with empty vector (EV) or Myc‐tagged JMJD5 (FL, CD‐1, and ND) as indicated. GST‐JMJD5‐CD‐1 was stained with Ponceau S. GST protein precipitates were subjected to anti‐Myc blotting.
- A549 cells were serum‐starved for 48 h followed by releasing for indicated times, and then cells were treated with DMSO or 1 mM BMH for 1 h. JMJD5 monomers and dimers were detected with anti‐JMJD5 antibody.
To identify the residue(s) responsible for histone H3 binding, we introduced mutations into the JmjC domain. JMJD5 with the putative α‐ketoglutarate (α‐KG) binding site mutation 34 (R335K→A335A) largely lost its interaction with H3, whereas JMJD5 with Fe2+ binding site mutation (H321QD→A321QA) or Cathepsin L light chain‐like positive motif mutation (KY398WH→KA398AA) retained the capability for H3 association (Fig 2C). Notably, histone H3 bears four “RK” motifs, three of which are RK9S, RK18Q, and RK27S located in the N‐tail. To test whether the histone H3 N‐tail provided the modular binding for JMJD5, Myc‐tagged histone H3 with S10→A substitution, as well as Myc‐tagged H3 with T3→A, K9→R, or K27→R substitution, was cotransfected with Flag‐JMJD5 in 293T cells. Immunoprecipitation with Flag‐JMJD5 revealed that histone H3 with the S10→A or T3→A, or K9→R mutation greatly reduced its binding with JMJD5 (Fig 2D), suggesting that residues T3, S10, and K9 in histone H3 are important for interaction between H3 and JMJD5.
The JmjC domains of JMJD6 interacted with each other during JMJD6 homodimerization 35. We incubated GST‐JMJD5 JmjC domain with Myc‐tagged full‐length JMJD5, JmjC domain, or N‐terminal helical domain. A strong interaction between two JmjC domains with different tags was detected, suggesting JMJD5 preferentially underwent a cis‐homodimerization between two monomers through its C‐terminal JmjC domain (Fig 2E). In addition, endogenous JMJD5 dimerization was stabilized in A549 cells treated with bis‐maleimidohexane (BMH) a cross‐linking reagent (Fig 2F).
JMJD5 is a Cathepsin L‐type protease for H3 N‐tail cleavage
The histone H3 N‐tail can be hydrolyzed by lysosomal cysteine protease Cathepsin L, which cleaves dibasic and monobasic processing sites 7, 36. Protein 3D structural alignment revealed that JmjC domain of JMJD5 and Cathepsin L1 catalytic domain shared a similar structural orientation (Fig 3A). Importantly, the active site H276 of Cathepsin L1 locates spatially near the active site H321 of JMJD5 (Fig 3A) 25, 37. For both JMJD5 and Cathepsin L, their N‐helical regions represent the regulatory domains and their C‐terminal enzymatic regions extend into a β‐sheet conformation similar to trypsin 1, which was reported to cleave histone tails at the carboxyl side of lysine or arginine residue, that is, “K‐X”, or “R‐X” motifs (X can be any residues; Fig EV2A) 38. A similar pattern of histone H3 cleavage was clearly induced by transient transfection of either JMJD5 or Cathepsin L1 (Fig 3B). Pretreatment of the transfectants with cysteine protease inhibitor E64 blocked H3 cleavage by JMJD5 or by Cathepsin L1 transfection (Fig 3B). While JMJD5 was exclusively nuclear localized (Fig 1D), Cathepsin L, however, was overall a cytoplasmic protein (Fig EV2B), despite that an isoform of Cathepsin L devoid of the signal peptide was capable to locate in nuclei 36, 39. Serum starvation/releasing induced histone H3 cleavage was dramatically reduced or even completely blocked by JMJD5 depletion with JMJD5‐specific siRNA but not with the control siRNA in A549 cells (Fig 3C) or HeLa cells (Fig EV2C). In contrast, depletion of Cathepsin L or trypsin showed no apparent effect on H3 cleavage in A549 cells (Fig 3D). Therefore, JMJD5 is apparently the nuclear protease responsible for histone H3 N‐tail cleavage under stressed conditions.
Figure 3. JMJD5 is a Cathepsin L‐type protease for H3 N‐tail cleavage.
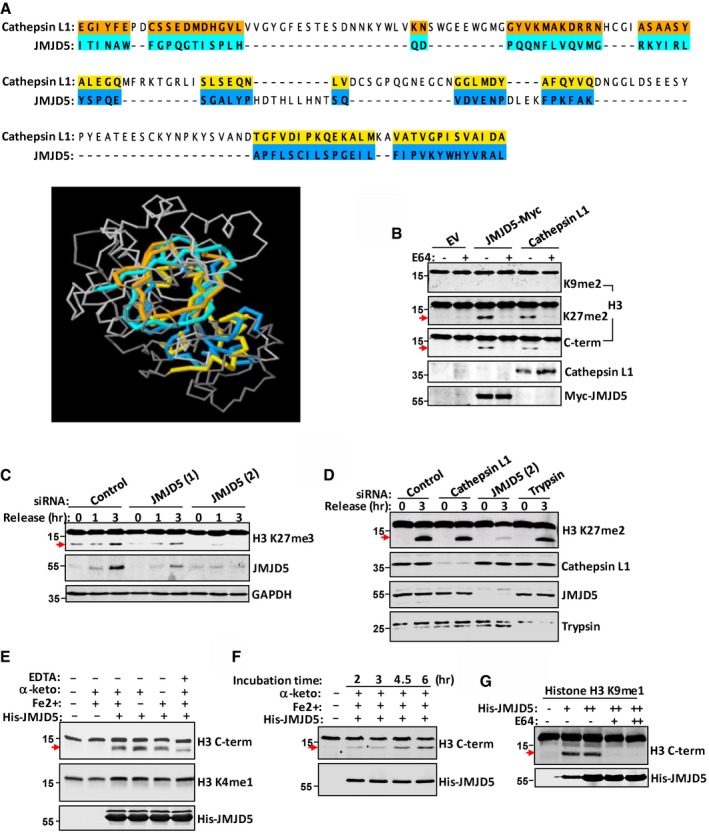
- PDB file for CTSL (2xu3) and JMJD5 (4gjz) was downloaded from RCSB protein data bank. The structure alignment of these two proteins was performed using RCSB PDB protein comparison tool “Calculate Structure Alignment.” Partial catalytic domain of CTSL: 146–330aa and partial JmjC domain of JMJD5: 305–405aa were chosen for structural comparison. Amino acids marked by different colors means they are aligned with each other in 3D structure.
- In A549 cells transfected with JMJD5 or Cathepsin L1, H3 N‐tail cleavage was blocked by treating the cells with E64. The faster‐migrating H3 was detected with indicated antibodies.
- A549 cells were transfected with control (CTL) siRNA or JMJD5 siRNA‐1 or JMJD5 siRNA‐2. After serum starvation for 24 h, the cells were released into fresh medium for indicated times. Western blot was performed with indicated antibodies.
- A549 cells were transfected with control siRNA, or siRNA against Cathepsin L, JMJD5, or trypsin. Western blot was performed with indicated antibodies.
- JMJD5 proteolytically processed H3 in vitro. Bacterial purified His‐JMJD5 was incubated with histone H3 purified from HeLa cells in the buffer with different chemicals as indicated. Full‐length H3 and H3 cleavage species were analyzed in Western blot with indicated antibodies. “α‐keto” represents “α‐ketoglutarate”.
- Bacterial purified His‐JMJD5 was incubated with histone H3 purified from HeLa cells as in (E) for different time. Full‐length H3 and H3 cleavage species were analyzed in Western blot with indicated antibodies.
- K9me1‐H3 was incubated with bacterial purified His‐JMJD5 in reaction buffer in the presence of absence of E64. Full‐length H3 and H3 cleavage species were analyzed with anti‐H3 antibody (C‐terminal) in Western blot.
Figure EV2. Histone H3 is a substrate of JMJD5 clipping activity.

- Alignment of trypsin‐1 and JmjC domain of JMJD5. Yellow color indicates β‐sheets, and blue color indicates the α‐helical structure. Red letters are identical amino acids in sequence alignment. Underlined amino acids H and D are Fe+ binding sites; R and K are α‐ketoglutarate binding site; K, Y and W are Cathepsin L light chain‐like positive motif.
- In HeLa cells, endogenous Cathepsin L1 was predominantly immunostained in cytoplasm. The scale bar represents 10 μm. Data shown are representative of three independent experiments.
- HeLa cells were transfected with JMJD5 siRNA or control siRNA; cell lysates were blotted with indicated antibodies. Data shown are representative of three independent experiments.
- JMJD5 proteolytically processed H3 in vitro. Flag‐JMJD5 purified from 293T cells was incubated with histone H3 purified from HeLa cells in the buffer with different chemicals as indicated. Full‐length H3 and H3 cleavage species were analyzed in Western blot with indicated antibodies. Data shown are representative of three independent experiments.
- Coomassie staining of bacterially purified His‐JMJD5.
- Bacterial purified His‐JMJD5 was incubated with various core histones purified from HeLa cells as in Fig 3F for different times. Western blot was performed with indicated antibodies.
- HeLa cells were transfected with Myc‐JMJD5, and cell lysates were subjected to Western blot with indicated antibodies targeting C‐terminal region of indicated histones. Data shown are representative of three independent experiments.
- HeLa cells were cotransfected with indicated plasmids, and cell lysates were subjected to Western blot with indicated antibodies. Data shown are representative of two independent experiments.
- Chromatin extracted from Myc‐JMJD5‐transfected A549 cells was subjected to Western blot with indicated antibodies. Data shown are representative of three independent experiments.
- HeLa cells were transfected with various Myc‐tagged plasmids, and cell lysates were subjected to Western blot with indicated antibodies. Data shown are representative of two independent experiments.
Total histone proteins purified from HeLa cells were then incubated with or without His‐JMJD5 prepared from bacteria under different in vitro cleavage conditions. The faster‐migrating H3 species was detected only when bacterial His‐JMJD5 from bacteria (Figs 3E and EV2E) or Flag‐JMJD5 from 293T cells (Fig EV2D) was presented. Level of faster‐migrating H3 species gradually increased after incubation with His‐JMJD5 for longer time (Fig 3F). We did not detect any faster‐migrating species from other core histones under same experimental condition (Fig EV2F). Although exogenous addition of α‐ketoglutarate and/or Fe2+ had a modest effect on H3 digestion, the overt negative effect of EDTA on H3 digestion (Fig 3E) supported an indispensable cofactor role of Fe2+ in JMJD5 enzymatic activity during H3 N‐tail cleavage. H3 cleavage by JMJD5 in vitro was further confirmed by using K9‐monomethylated H3 as the substrate and this JMJD5 cleavage activity was completely abolished by treating with E64 (Fig 3G). To test whether other core histones, such as H2A, H2B, and H4, are substrates of JMJD5 in vivo, cell lysates transfected with Myc‐JMJD5 were blotted with antibodies that targeting C‐terminal region of those histones and result showed that only endogenous histone H3 is targeted by JMJD5 (Fig EV2G). Of three histone H3 variants, H3.3 is most efficiently cleaved by JMJD5 (Fig EV2H). Next, chromatin was extracted from JMJD5 transfected cells and subjected to blot with H3 C‐terminal antibody, and result showed that chromatin‐bound histone H3 is also cleaved by JMJD5 (Fig EV2I). Interestingly, of six JmjC‐containing proteins, JMJD4 also showed a much weaker H3 N‐tail cleavage activity compared to JMJD5 (Fig EV2J).
JMJD5 preferentially cleaves monomethyl‐lysine H3 peptides in vitro
To verify that histone H3 N‐tail cleavage was methylation‐dependent and identify its cleavage site, synthetic methyl‐peptides of H3‐K4, H3‐K9, H3‐K27, and H3‐K36 sequences with N‐terminal cysteine were used as substrates for JMJD5 in vitro digestion analysis (Figs 4A–D and EV3A–C). Mass spectrometric analysis of in vitro peptide cleavages revealed that JMJD5 clipped all monomethyl‐peptides at the carboxyl side of K4me1, K9me1, K27me1, and K36me1 residues (Figs 4B and EV3A–C). Monomer and/or dimer cleavage products were observed. Cleavage efficiency by JMJD5 on K9me1 was not affected by increasing the length of synthetic peptides (Fig EV4H). However, JMJD5 failed to cleave unmethyl‐K‐, Kme2‐, or Kme3 peptide with the exception of a modest cleavage effect on K9me2 peptide (Figs 4A, C and D, and EV4A–F). Although R335K336 sites were critical for JMJD5 to interact with H3 as shown in Fig 2C, JMJD5‐R335K336A variant could still catalyze K27me1 peptide digestion with reduced efficiency (Fig EV3C). JMJD5 showed no cleavage effect on R2‐monomethylated H3 (H3 mMR2) peptide (Fig EV4G). Another His‐tagged recombinant protein ribonucleoside‐diphosphate reductase M2 subunit (RRM2) prepared under the same conditions failed to cleave monomethylated H3 peptides (Fig EV4I). It is worth to emphasize that no any JMJD5‐inducible monomethyl (‐14 Dalton), dimethyl (‐28 Dalton), or trimethyl (‐42 Dalton) mass change of H3 peptides was detected in our extensive mass spectrometric analysis. Thus, the results of in vitro peptide digestion further support the trypsin‐like proteolytic activity of JMJD5 in clipping specifically the carboxyl side of monomethylated lysine residues of histone H3.
Figure 4. JMJD5 predominantly cleaves H3K9me1/2 peptides.

-
A–DMALDI‐TOF mass spectra of H3 peptides cleaved by JMJD5 in vitro. Data shown are representative of three independent experiments.
Figure EV3. JMJD5 cleaves H3Kme1 peptides.
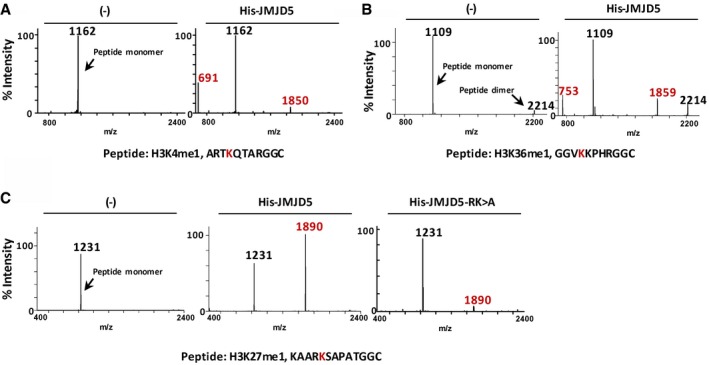
-
A–CMALDI‐TOF mass spectra of indicated H3 peptides cleaved by JMJD5 in vitro. Data shown are representative of three independent experiments.
Figure EV4. Mass spectra of H3 peptides digested with JMJD5 in vitro .
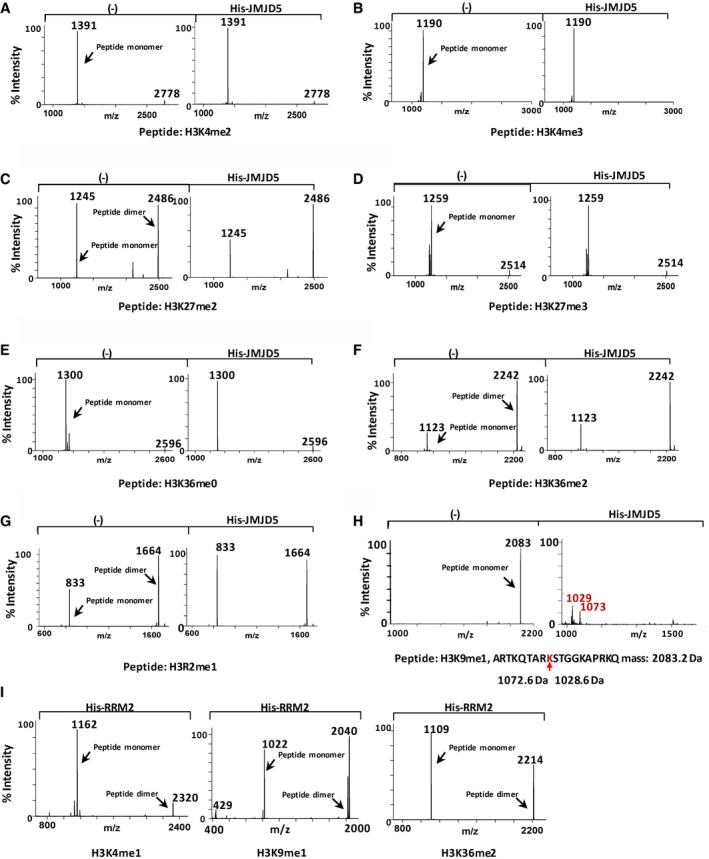
-
A–IMALDI‐TOF mass spectra of indicated H3 peptides cleaved by JMJD5 in vitro. Data shown are representative of three independent experiments.
JMJD5 catalytic domain is responsible for H3 cleavage between monomethyl‐K9 and S10
We then examined JMJD5 on histone H3 N‐terminal digestion in cells. Although a basal level of H3 cleavage was sometime observed with anti‐H3 C‐terminal antibody (Fig 5A), overexpression of JMJD5 increased H3 N‐terminal cleavage in either whole cell extracts (Fig 5A) or in acid‐extracted H3 proteins (Fig EV5A). Next, histone H3 N‐terminal cleavage in HeLa cells was systemically analyzed with an array of antibodies against different sites and posttranslational modifications in the H3. The fast‐migrating H3 species was detected by anti‐pS10‐H3 and all antibodies for those sites between S10 and K79 in addition to anti‐C‐terminal H3, which binds to last 10–20 amino acids of C‐terminus of H3 (Figs 5A and EV5A). In contrast, none of the antibodies against those different types of posttranslational modifications between N‐terminal K4 and K9 detected the fast‐migrating H3 (Fig 5A). A similar H3 cleavage pattern by overexpression of JMJD5 was obtained in A549 cells (Fig EV5B). H3 faster‐migrating species from either HeLa cells transfected with JMJD5 or incubation of purified H3 with JMJD5 were separated in a 15% gel and trypsinized for subsequent mass spectrometry analysis. The peptides recovered from the mass spectrometry supported the notion that JMJD5 only cleaved histone H3 within the N‐tail upstream of K27, most likely between K9 and S10 (Table EV1). When histone H3 N‐tail mutants were tested for digestion by JMJD5 in 293T cells, both H3 wild type and H3 K4R mutant could be effectively digested by transient transfection of JMJD5 (Fig 5B). In contrast, both H3 K9R and to a lesser extent H3 S10A mutants were resistant to JMJD5 digestion (Fig 5B). To better verify H3 N‐tail cleavage site by JMJD5, an antibody H3KS was generated commercially to recognize the primary site of H3 N‐tail cleavage between residue K9 and S10 with high specificity (Fig EV5C). We then used this antibody H3KS to immunoblot samples in previous experiments (Figs 1B and C, 5A, EV1D, E and F, and EV2J). Results showed that those faster‐migrating H3 sub‐band can also be recognized by H3KS antibody. Collectively, these results indicated that JMJD5 cleaved H3 N‐tail between K9 and S10 residues in cells.
Figure 5. JMJD5 cleaves H3 between K9 and S10.
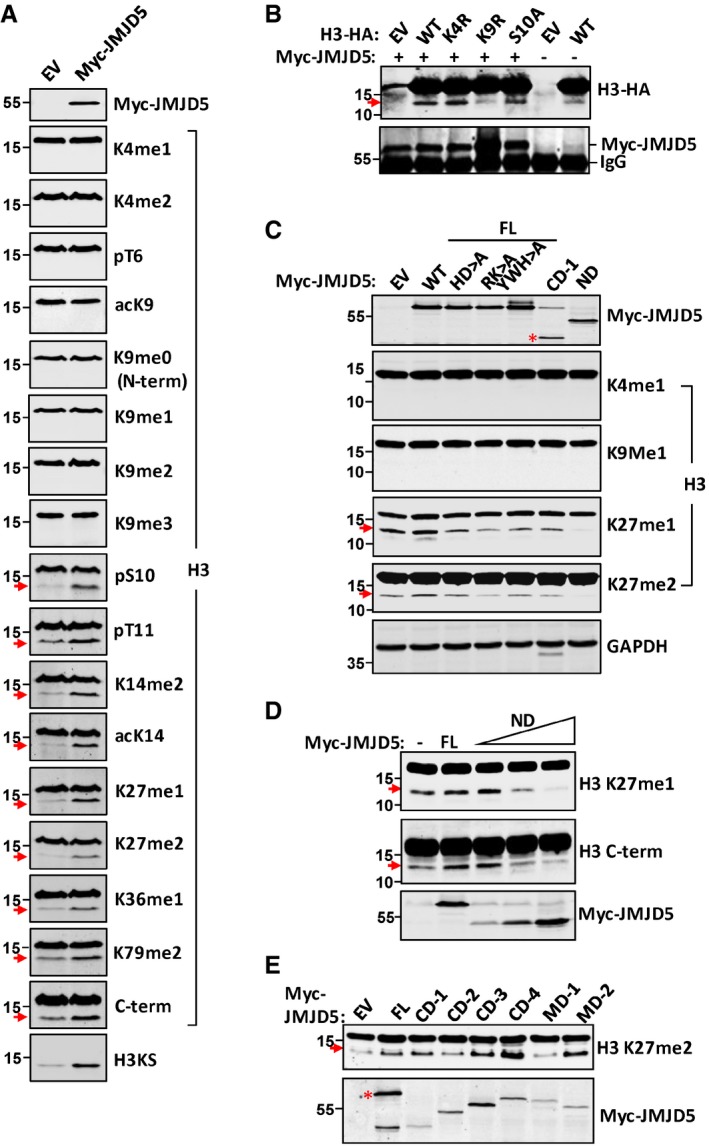
- EV or Myc‐JMJD5 was transiently transfected in HeLa cells, and H3 obtained from whole cell lysates was analyzed in Western blot with specific H3 antibodies as indicated.
- HA‐tagged WT, K4R, K9R, and S10A mutation form of H3 were cotransfected with or without Myc‐JMJD5 in 293T cells. H3‐HA was detected with anti‐HA antibody in Western blot. Myc‐immunoprecipitates were blotted with anti‐Myc antibody to show Myc‐JMJD5 expression.
- HeLa cells were transiently transfected with different domains or mutants of JMJD5 as indicated. Whole cell lysates were subjected to Western blotting analysis with indicated H3 antibodies. Red asterisk indicates corresponding protein expressed by vector.
- The dominant negative effect of JMJD5‐ND form on H3 cleavage was examined in HeLa cells with increasing amount of Myc‐JMJD5‐ND. Proteins extracted from these cells were analyzed with indicated H3 antibodies in Western blot.
- Full‐length JMJD5 and a series of domain deletion constructs of JMJD5 were transiently transfected in A549 cells. Histone proteins were prepared as above and blotted with anti‐H3K27me2 antibody. JMDJ5 was blotted with anti‐Myc antibody. Red asterisk indicates corresponding protein expressed by vector.
Figure EV5. JMJD5 induced H3 N‐tail cleavage in cultured cells.
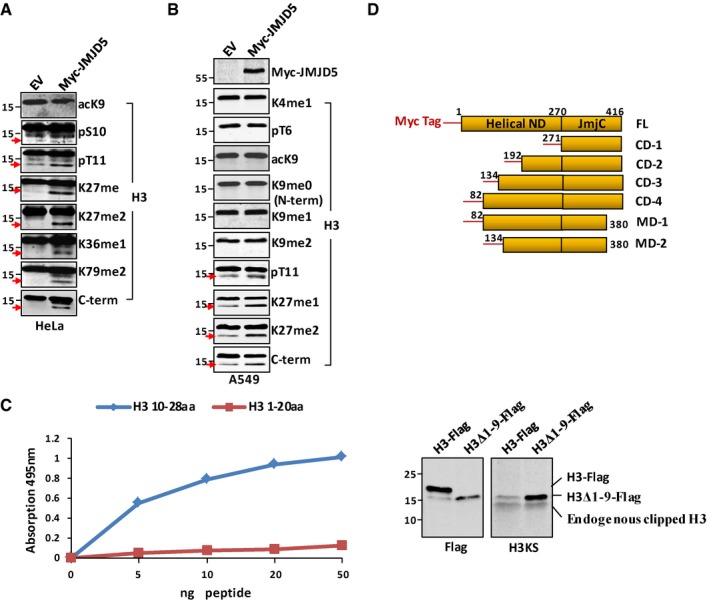
- In HeLa cells transiently transfected with EV (mock) or Myc‐JMJD5, acid‐extracted H3 and H3 fast‐migrating species were analyzed by Western blot with indicated antibodies. Data shown are representative of three independent experiments.
- In lung cancer cell line A549 transiently transfected with or without Myc‐JMJD5, whole cell lysates were prepared for Western blot analysis with indicated antibodies. Data shown are representative of three independent experiments.
- Determination of H3KS antibody specificity by ELISA using H3 peptides corresponding to either H3 N‐terminal 1–20aa or 10–28aa (left panel). Expression vector for C‐terminal Flag‐tagged full‐length H3 or H3Δ1–9 (amino acids 1–9 were deleted) was transfected into 293T cells. Cell lysates were blotted with either Flag or H3KS antibody (right panel).
- Schematic illustration of a series of Myc‐tagged JMJD5 domain deletion constructs.
Compared with wild‐type JMJD5, JMJD5 mutants (H321QD→AQA, Y398WH→AAA, and R335K→AA) all failed to increase H3 cleavage when they were overexpressed in HeLa cells (Fig 5C), even though they may have the potential to digest methyl‐peptide in vitro (Fig EV3C). The JMJD5 N‐terminal domain alone functioned dominant negatively on histone H3 N‐tail cleavage for overexpression of this domain dramatically reduced the basal H3 cleavage level in HeLa cells (Fig 5C). This dominant negative effect in H3 N‐tail cleavage was N‐terminal domain level‐dependent (Fig 5D), suggesting its interaction with the C‐terminal JmjC domain, though weak (Fig 2E), inhibited the proteolytic activity of JMJD5. Sharing structural homology with Cathepsin L1 light chain (critical for its proteolytic activity) as well as trypsin to certain degree (Figs 3A and EV2A), the JmjC domain of JMJD5 (CD‐1) was most likely for histone H3 N‐tail cleavage. In order to further confirm the catalytic domain of JMJD5 responsible for H3 digestion, a series of domain deletion constructs in addition to the CD‐1 (JmjC) and ND were prepared (Fig EV5D). In HeLa cells, the digestive effect of CD‐1 on H3 N‐tail was not conclusive presumably due to a relatively high H3 cleavage background in HeLa cells (Fig 5C). In A549 cells that bear a relatively low H3 cleavage background, increased H3 faster migration species was detected with CD‐1 transient transfection (Fig 5E). This indicates that the JmjC domain of JMJD5 is the catalytic domain responsible for histone H3 N‐tail digestion.
Regulation of H3 N‐tail dependent transcription by JMJD5 cleavage activity
Histone H3 N‐tail methylation and demethylation are involved in gene activation and silencing regulation 3. High levels of H3K4me1 and H3K9me1 were detected in active promoters surrounding transcription start sites, suggesting their roles in transcriptional activation 40. JMJD5 was reported to be a putative tumor suppressor and reduced expression of JMJD5 was noted in lung cancer tissues 41. In A549 cells, JMJD5 overexpression suppressed those genes including stat1 and stat4 with H3K4me1 or H3K9me1 in their promoters for expression (Fig 6A) 40. In contrast, JMJD5‐ND, the dominant negative form, failed to inhibit H3K4me1‐ and H3K9me1‐dependent gene regulation (Fig 6A). Both full‐length JMJD5 and JMJD5‐ND were expressed at similar level (Fig 6A, lower panel). To confirm above results, we performed chromatin immunoprecipitation (ChIP) assay. Full‐length JMJD5 but not JMJD5‐ND greatly reduced histone H3K9me1 association with promoters of those genes involved in cell cycle or growth/survival (Fig 6B, upper panel). In contrast, JMJD5 overexpression showed no effect on H3K9me3 association with the same promoters (Fig 6B, lower panel). Importantly, ChIP assay by using H3KS antibody and Flag antibody showed that level of N‐terminal cleaved H3 was enhanced at those gene promoters, where exogenous Flag‐tagged JMJD5 was also enriched, suggesting a positive correlation between occupation of JMJD5 and cleavage of H3 N‐tail at specific gene promoters (Fig 6C). These results have thus established the role of JMJD5 as a specific histone H3 protease in H3 monomethyl‐N‐tail activity regulation involved in gene expression.
Figure 6. Cleavage activity of JMJD5 modulates N‐tail‐dependent gene transcription.
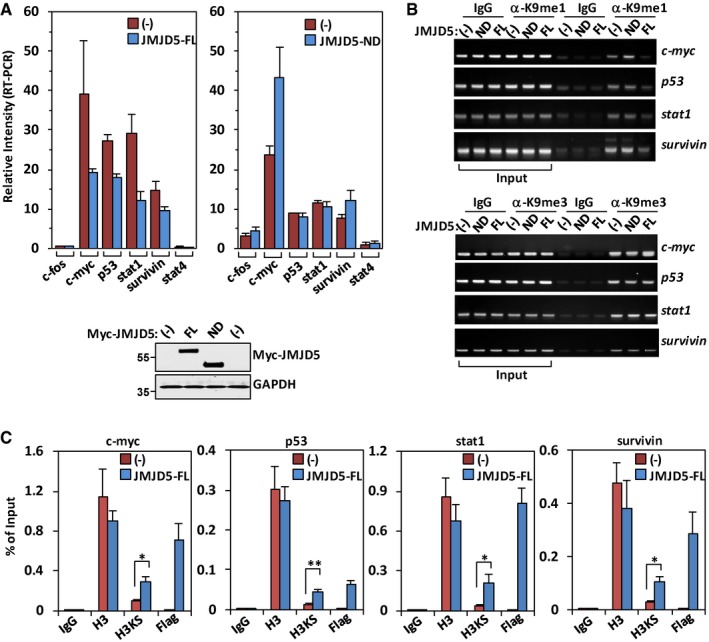
- In A549 cells transiently transfected with JMJD5‐FL (upper left panel) or JMJD5‐ND (upper right panel), the indicated genes were analyzed for their mRNA expression by real‐time PCR. The values for individual genes were normalized to beta‐actin. Data shown represent the mean ± SEM; n = 3 of three independent experiments. One representative full‐length JMJD5 or JMJD5‐ND protein expression in above samples was analyzed with anti‐Myc antibody (lower panel).
- A549 cells were transiently transfected with JMJD5‐FL or JMJD5‐ND, and the levels of H3K9me1 or H3K9me3 on promoter regions of indicated genes were examined by ChIP assay. IgG was used as a negative control. Data shown are representative of three independent experiments.
- A549 cells were transiently transfected with Flag‐JMJD5. ChIP assays were performed using indicated antibodies and primers for promoter regions of indicated genes. IgG was used as a negative control. Data shown represent mean ± SEM; n = 3 of three independent experiments. *P < 0.05; **P < 0.01. P‐values were calculated by a one‐tailed paired Student's t‐test using Microsoft Excel.
Discussion
Limited proteolysis of nuclear proteins is an important means of regulating transcription and other cell processes 39, 42. Histone cleavage was first reported several decades ago 43, 44. However, histone cleavage was not come into epigenetic landscape until recently with the discovery of transient histone H3 clipping in differentiating mESC 19 and sporulating yeast 13. By histone H3 N‐tail clipping, multiple H3 posttranslational modification (PTM) marks including methylation, acetylation, and phosphorylation are simultaneously removed from the N‐tail of histone H3 in mouse and yeast cells, respectively. This mechanism involves enzymatic cleavage of the first 21 amino‐acid residues at the N‐terminus of H3, thus sweeping away en masse all modifications present in this region.
In mammalian cells, histone H3 N‐tail cleavage is initiated under various conditions, such as mouse ESC differentiation, osteoclastogenesis, and senescence 7, 8, 22; especially, histone H3 N‐tail cleavage between K18 and Q19 is greatly enhanced by K18 acetylation during mouse osteoclastogenesis 22. In oncogene‐induced senescent cells, however, one histone H3 N‐tail cleavage site lies between residue A21 and T22, and another one lies between K9 and K14, though exact clipping site for the latter is not determined 8. Those reports suggest that histone H3 N‐tail clipping can be initiated at different residues on a context dependent manner. We reports here that histone H3 N‐tail is also proteolytically cleaved between the site K9 and S10 by JMJD5, a Jumonji C (JmjC) domain‐containing protein, under the stressed conditions in the cells. This histone H3 N‐tail clipping event is a monomethylation‐dependent process, which may represents a novel mechanism by which JmjC domain proteins regulate histone activity in cells.
Currently, there are more than 30 known mammalian JmjC domain‐encoding genes, and some have been reported to be involved in cell proliferation by regulating the expression of cell cycle inhibitors 34. JMJD5 has been reported to be required for the cell cycle progression through transcriptional regulation of cell cycle‐related genes 24, 25, 28. JMJD5 is a nuclear protein, and the N‐terminal domain of JMJD5 is necessary for its nuclear localization 45. Though JMJD5 was originally identified as a histone demethylase for dimethylated lysine 36 of histone H3 25, the enzymatic activity of JMJD5 is still controversial. Crystal structure analysis has suggested that JMJD5 functions as a protein hydroxylase rather than a histone demethylase 46, 47. In addition, a recent study has indicated that Jmjd5 works as a protein recruiter to translocate the binding protein into the nucleus 29. Under the stressed conditions, such as thymidine block and release and DNA damage response, we found that histone H3 N‐tail is cleaved by JMJD5, which is independent of cell cycle and/or senescence, suggesting JMJD5 plays a role in DNA damage response. Interestingly, JMJD5 in C. elegans is important for DNA damage response and repair by demethylating H3K36me2 48. In mammalian cells, however, DNA damage‐induced dimethylation at H3K36 is counteracted by KDM2, suggesting JMJD5 is not a major demethylase for H3K36me2 under the DNA damage response in mammals 49. DNA damage response resulted in reduction in H3K9me1 at the sites of damaged chromosome loci, which is achieved by either degradation of methyltransferase G9a or induction of demethylase such as KDM4B and PHF8 50, 51, 52, 53. Because H3K9me1 is a preferred substrate for JMJD5 clipping activity in our study, it is possible that cleavage of H3K9me1 by JMJD5 may also contribute to reduction in H3K9me1 under DNA damage response. Furthermore, depletion of JMJD5 in mouse embryo results in upregulation of p53 gene expression, which is independent of H3K36me2 level 23. We observed that overexpression of JMJD5 downregulated p53 mRNA level, which may resulted from reduced H3K9me1 at p53 gene promoter, as active promoter marker H3K9me1 is cleaved by binding of JMJD5 to p53 promoter. Interestingly, we found that histone H3.3 is a major target of JMJD5 cleavage activity. H3.3 has been shown to be a clipping substrate of protease Cathepsin L under condition of oncogene‐induced senescence 8. Like senescence, DNA damage response leads to transcriptional inhibition at some gene promoters 8, 54. As H3.3 is closely associated with transcriptionally active foci in the cells 55, 56, it is possible that cleavage of H3.3 N‐tail at different amino acids by different protein enzymes may represent a common way of gene expression regulation under specific stressed conditions. However, precise molecular mechanisms of JMJD5 functions in gene expression regulation under specific conditions have not been fully characterized and are waiting for further investigations.
Our data suggest here that the JmjC domain proteins may have functions more diverse than previously thought including H3 dMR2 demethylation, U2AF65 lysyl hydroxylation, and recently discovered mRNA binding 57, 58, 59. Interestingly, JMJD4 also exhibits a much weaker H3 N‐tail cleavage activity compared to JMJD5. Both JMJD4 and JMJD5 are the only JmjC domain‐containing proteins that have a serine residue in place of the normal threonine in the first αKG‐binding site. As opposed to the less bulky serine residue, threonine is preferred in forming an enzymatically competent JmjC domain active site 34. Notably, the JmjC domain (101‐C) alone without the N‐terminal domain of JMJD5 has been recently reported as a potential H3K36me2 demethylase in breast cancer cells 60, suggesting that the N‐terminal domain of JMJD5 (ND) blocks the JmjC domain to function as a conventional H3 demethylase. We were also unable to detect any intrinsic demethylation activity of JMJD5 in full length. The JmjC domain is not only for histone H3 N‐tail modular interaction and JMJD5 homodimerization, but also for the H3 N‐tail cleavage. Moreover, H3 N‐tail cleavage by JMJD5 is specifically H3 Kme1 dependent. While Cathepsin L1 clips the H3 N‐tail between “A21‐T22” and between “K27‐S28” in stem cells 7, it was unclear whether K27 methylation would affect Cathepsin L proteolytic activity. In S. cerevisiae, however, a trimethylation mark, H3K4me3, which prevents clipping by the yeast endopeptidase in vitro, is maintained in chromatin at promoters during gene activation 61. This observation supports a mechanism that nucleosomes that do not contain K4me3 are marked for H3 cleavage and subsequent displacement. Our study showed that JMJD5 preferred to clip “K9‐S10” site over other Kme1 sites in vivo. Given that JMJD5 was able to cleave H3 peptides between “K27‐S28” and between “K36‐K37” in vitro, it did not rule out the possibility that JMJD5 might also cleave these sites of H3 either inefficiently in the cells we tested or under different yet unknown conditions in vivo.
In summary, histone H3 N‐tail cleavage is specifically designated to counteract H3 N‐tail monomethylation, as it eventually depends on disassembly of the nucleosome and replacement of the histone molecule to regenerate an intact unmodified nucleosome. This process causes loss of a portion of the histone monomethylation marks. Our data suggest that mammalian cells employ another regulated H3 proteolysis mechanism that may serve to alter epigenetic signatures made in cells during stress.
Materials and Methods
Antibodies, peptides, and other reagents
Antibodies: Antibody H3KS is commercially generated by Hangzhou Jucheng Bio‐Technology Co. Ltd (China). A 2× branched peptide corresponding to histone H3 sequence 10–14 was conjugated to KLH and injected into rabbits. Serum was collected, purified, and tested for specificity as described in Fig EV5C; anti‐K4me1‐H3 (Abcam #8895), anti‐K4me2‐H3 (Abcam #7766), anti‐pT6‐H3 (Abcam #14102), anti‐K9me0‐H3 (Abcam #61251), anti‐K9me1‐H3 (Abcam #8896), anti‐K9me2‐H3 (Abcam #1220), anti‐K9me3‐H3 (Abcam #8898), anti‐acK9‐H3 (Abcam #4441), anti‐pS10‐H3 (Abcam #5176), anti‐pT11‐H3 (Abcam #5168), anti‐K14me2‐H3 (Upstate #07‐427), anti‐acK14‐H3 (Abcam #52946), anti‐K27me1‐H3 (Upstate #07‐448), anti‐K27me2‐H3 (Abcam #24684), anti‐K36me1‐H3 (Abcam #9048), anti‐K79me2‐H3 (Abcam #3594), anti‐H3 C‐terminal region (Abcam #1791), anti‐JMJD5 (Abcam #28883), anti‐Myc (Santa Cruz Biotech #40), anti‐H2A (CST #12349), anti‐H2B (CST #2934), anti‐H4 (Abcam #ab10158), α‐GAPDH (MultiSciences #Mab5079), anti‐GST (GenScript #A00865), anti‐HA (Santa Cruz Biotech #7392), Alexa‐680 or IRDye‐800 goat anti‐mouse or ‐rabbit secondary antibody (Li‐COR).
Peptides: K4me1‐H3 peptide (Abcam #1340), K4me2‐H3 peptide (Abcam #7768), K4me3‐H3 peptide (Abcam #1342), K9me1‐H3 peptide (Abcam #1771), K9me2‐H3 peptide (Abcam #1772), K9me3‐H3 peptide (Abcam #1773), K27me1‐H3 peptide (Abcam #1780), K27me2‐H3 peptide (Abcam #1781), K27me3‐H3 peptide (Abcam #1782), K36me1‐H3 peptide (Abcam #1783), K36me2‐H3 peptide (Abcam #1784), K36me3‐H3 peptide (Abcam #1785), and R2me1‐H3 peptide (Abcam #1775).
Cathepsin L inhibitor E64 was purchased from Sigma, and Cathepsin L1 gene was obtained from Addgene (Boston, MA).
Protein purification
JMJD5 (NP_079049) was expressed as a GST‐tag fusion using the pGEX‐4T3 expression vector or as a His6‐tag fusion using the pET‐28b(+) expression vector. All protein coding sequences were verified by sequencing. Following expression in Rosetta Escherichia coli at 25°C induced by 1 mM IPTG when the OD reached 0.5–0.6, proteins were purified using glutathione agarose beads (Amersham Biosciences) or Ni‐NTA beads (Qiagen) according to the manufacturer's instructions.
In vitro H3 N‐tail cleavage assay
Acid‐extracted H3 from HeLa cells, K9me1 H3 purchased from Active Motif (Carlsbad, CA), or synthetic H3 peptides were incubated with purified His‐tagged JMJD5 in buffer (150 mM NaCl, 20 mM Tris–HCl pH 8.0, 2 mM ascorbic acid, with or without 1 mM α‐ketoglutarate, 20 mM EDTA, and 50 μM (NH4)2Fe(SO4)2) at 37°C for 2–3 h. A total of 20–50 g of full‐length His‐tagged JMJD5 was added to the reactions. The reactions were stopped by 2× SDS loading buffer or desalted using a μ‐C18 Ziptip. Reaction products were analyzed either by standard Western blot analysis or mass spectrometry.
Matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI‐TOF) mass spectrometry
Synthetic peptide substrates derived from H3 were incubated with purified recombinant JMJD5 protein in demethylation buffer at 37°C. Then, the demethylation products were enriched and desalted using a μ‐C18 Ziptip (Millipore) and eluted directly onto a MALDI plate containing 2 μl of a CHCA (α‐cyano‐4‐hydroxy cinnamic acid) saturated solution in 50% acetonitrile (ACN) and 0.1% trifluoroacetic acid (TFA). MALDI‐TOF spectra were acquired on a Voyager DE Pro mass spectrometer (Applied Biosystem, Foster City, CA). For histone digestion analysis with mass spectrometry, acid‐extracted H3 prepared from HeLa cells transfected with or without JMJD5 or in vitro digested K9me1‐H3 and H3 faster‐migrating species were separated in 12% gel. Selected Coomassie Blue‐stained H3 and H3 fast‐migrating species were excised from gels using a scalpel blade. Excised bands were subjected to tryptic digestion (trypsin or chymotrypsin). Mass–mass spectrometry was performed with an Applied Biosystems 4800 Proteomics analyzer.
Cell culture, transfection, and immunostaining experiments
HeLa cells, A549 cells, and human embryonic kidney (HEK) 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) or RPMI medium supplemented with 10% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C, 5% CO2. No mycoplasma contamination was found in cell cultures.
For microscopy, cells were grown to 50–70% confluence on 18 × 18 glass coverslips and transfected with the indicated expression constructs using Lipofectamine 2000 transfection reagent (Invitrogen) or Superfact reagent (Qiagen) according to the manufacturer's instructions. After 48 h, cells were washed with cold phosphate‐buffered saline (PBS) and fixed in 4% paraformaldehyde for 15 min. The cells were washed three times with PBS and then permeabilized for 15 min with 0.2% Triton X‐100. Then, the samples were blocked with 1% BSA, stained with antibodies, and visualized using a fluorescence microscope (OLYMPUS).
GST‐JMJD5 pull‐down and co‐immunoprecipitation assays
GST precipitation was carried out by incubating acid‐extracted histones from HeLa cells or whole cell extract from HEK293T cells expressing Myc‐JMJD5 with GST‐fusion proteins.
For co‐immunoprecipitation assays, HEK293T cells were transfected with Myc‐JMJD5 and HA‐H3 using Lipofectamine 2000.
3D protein alignment
PDB file for CTSL (2xu3) 62 and JMJD5 (4gjz) 46 were downloaded from RCSB protein data bank. The structure alignment of these two proteins was performed using RCSB PDB protein comparison tool “Calculate Structure Alignment” via Java webstart 63. Ranges of “Chain A: 1‐220” for 2xu3 and “Chain A: 183‐416” for 4gjz were chosen for structural comparison, and jCE Circular Permutation algorithm with default parameters (except that the penalties for gap open and extension were increased to 6.0 and 0.6) was applied in this alignment 64.
RNA interference, qRT–PCR, and ChIP assay
For RNA interference, siRNAs were provided by Sigma or Santa Cruz and transfected according to the manufacturer's instructions using Lipofectamine 2000 (Invitrogen). Small interfering RNA against luciferase was used as a control. The siRNA sequences were as follows: JMJD5 siRNA in sense strand, 5′‐GUGAUCCUGGGCUACUCCUTT‐3′; luciferase siRNA (control) in sense strand, 5′‐CGUACGCGGAAUACUUCGATT‐3′. Total RNA was prepared using the Qiagen RNeasy kit and reverse‐transcribed using Invitrogen's First Strand Synthesis kit.
Quantitative PCR was performed in accordance with standard procedures with SYBR Green mix (Takara). The primer sequences used for JMJD5 were 5′‐CGTGCATGCAGAAGTGGAGT‐3′ and 5′‐TACCCGACGTCCCTTGGCTC‐3′.
ChIP assay was performed by using Upstate kit (Catalog # 17‐295). JMJD5 transiently transfected A549 cells (1 × 106) were washed once with PBS and placed in PBS containing 1% formaldehyde at 37°C for 10 min to covalently cross‐link any DNA‐protein complexes. Cells were lysed in 200 μl of SDS lysis buffer for 10 min on ice and sonicated for 20 min. The samples were precleared with 75 μl protein‐A/G agarose slurry; 1% of the precleared chromatin sample was set aside as input DNA. Immunoprecipitations were prepared with ˜1 mg chromatin and 2 μg of antibody (i.e., control IgG, anti‐K9me1‐H3, or anti‐K9me3‐H3). 60 μl protein‐A/G agarose slurry was added to each sample and rotated at 4°C for 2 h. DNA was then purified with spin columns (QIAquick PCR purification kit). Primer sets used in subsequent qPCR for ChIP are survivin‐F: TGGCACCCTGTAAAGCTCTCC; survivin‐R: ATGCCTGTAATCCCAACTACTCG. p53‐F: GGGCGCAGCAGGTCTTG: p53‐R: TCAGTACATGGAAACGTAAGCCT. c‐myc‐F: GTGCGTTCTCGGTGTGGAG; c‐myc‐R: GCTCCCTCTCAAACCCTCTCC. stat1‐F: GTCAACTCTGCCCCATGCTT; stat1‐R: CCGAGAATCCAGGCAGAGG.
Statistical analysis
An one‐tailed paired Student's t‐test was used to calculate P‐values in statistical analysis.
Author contributions
YEC and CH designed the project. CH, JS, XX, LC, LW, YS, LM, XG, HL, and LW performed the experiments. HW performed protein 3D alignment. YEC, CH, JS, and Y‐nY analyzed the data. CH and YEC wrote and revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 3
Acknowledgements
This work was supported by the National Key Basic Research Program of China (2013CB910900), the Natural Science Foundation of China (81672804), and MOST (2015CB910402).
EMBO Reports (2017) 18: 2131–2143
Contributor Information
Jimin Shao, Email: shaojimin@zju.edu.cn.
Chao Huang, Email: c_huang_bio@msn.com.
Y Eugene Chin, Email: yechin@sibs.ac.cn.
References
- 1. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260 [DOI] [PubMed] [Google Scholar]
- 2. Du J, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693–705 [DOI] [PubMed] [Google Scholar]
- 4. Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- 5. Elia MC, Moudrianakis EN (1988) Regulation of H2a‐specific proteolysis by the histone H3:H4 tetramer. J Biol Chem 263: 9958–9964 [PubMed] [Google Scholar]
- 6. Osley MA (2008) Epigenetics: how to lose a tail. Nature 456: 885–886 [DOI] [PubMed] [Google Scholar]
- 7. Duncan EM, Muratore‐Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, Allis CD (2008) Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135: 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duarte LF, Young ARJ, Wang ZC, Wu HA, Panda T, Kou Y, Kapoor A, Hasson D, Mills NR, Ma'ayan A et al (2014) Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat Commun 5: 5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Falk MM, Grigera PR, Bergmann IE, Zibert A, Multhaup G, Beck E (1990) Foot‐and‐mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J Virol 64: 748–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tesar M, Marquardt O (1990) Foot‐and‐mouth disease virus protease 3C inhibits cellular transcription and mediates cleavage of histone H3. Virology 174: 364–374 [DOI] [PubMed] [Google Scholar]
- 11. Gonzalo S (2010) Epigenetic alterations in aging. J Appl Physiol 109: 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mahendra G, Kanungo MS (2000) Age‐related and steroid induced changes in the histones of the quail liver. Arch Gerontol Geriatr 30: 109–114 [DOI] [PubMed] [Google Scholar]
- 13. Santos‐Rosa H, Kirmizis A, Nelson C, Bartke T, Saksouk N, Cote J, Kouzarides T (2009) Histone H3 tail clipping regulates gene expression. Nat Struct Mol Biol 16: 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Allis CD, Wiggins JC (1984) Proteolytic processing of micronuclear H3 and histone phosphorylation during conjugation in Tetrahymena thermophila . Exp Cell Res 153: 287–298 [DOI] [PubMed] [Google Scholar]
- 15. Allis CD, Bowen JK, Abraham GN, Glover CV, Gorovsky MA (1980) Proteolytic processing of histone H3 in chromatin: a physiologically regulated event in Tetrahymena micronuclei. Cell 20: 55–64 [DOI] [PubMed] [Google Scholar]
- 16. Mandal P, Azad GK, Tomar RS (2012) Identification of a novel histone H3 specific protease activity in nuclei of chicken liver. Biochem Biophys Res Comm 421: 261–267 [DOI] [PubMed] [Google Scholar]
- 17. Mandal P, Verma N, Chauhan S, Tomar RS (2013) Unexpected histone H3 tail‐clipping activity of glutamate dehydrogenase. J Biol Chem 288: 18743–18757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin R, Cook RG, Allis CD (1991) Proteolytic removal of core histone amino termini and dephosphorylation of histone H1 correlate with the formation of condensed chromatin and transcriptional silencing during Tetrahymena macronuclear development. Genes Dev 5: 1601–1610 [DOI] [PubMed] [Google Scholar]
- 19. Duncan EM, Allis CD (2011) Errors in erasure: links between histone lysine methylation removal and disease. Prog Drug Res 67: 69–90 [DOI] [PubMed] [Google Scholar]
- 20. Vossaert L, Meert P, Scheerlinck E, Glibert P, Van Roy N, Heindryckx B, De Sutter P, Dhaenens M, Deforce D (2014) Identification of histone H3 clipping activity in human embryonic stem cells. Stem Cell Res 13: 123–134 [DOI] [PubMed] [Google Scholar]
- 21. Xue Y, Vashisht AA, Tan Y, Su T, Wohlschlegel JA (2014) PRB1 is required for clipping of the histone H3 N terminal tail in Saccharomyces cerevisiae . PLoS One 9: e90496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim K, Punj V, Kim JM, Lee S, Ulmer TS, Lu WE, Rice JC, An W (2016) MMP‐9 facilitates selective proteolysis of the histone H3 tail at genes necessary for proficient osteoclastogenesis. Genes Dev 30: 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oh S, Janknecht R (2012) Histone demethylase JMJD5 is essential for embryonic development. Biochem Biophys Res Comm 420: 61–65 [DOI] [PubMed] [Google Scholar]
- 24. Ishimura A, Minehata K, Terashima M, Kondoh G, Hara T, Suzuki T (2012) Jmjd5, an H3K36me2 histone demethylase, modulates embryonic cell proliferation through the regulation of Cdkn1a expression. Development 139: 749–759 [DOI] [PubMed] [Google Scholar]
- 25. Hsia DA, Tepper CG, Pochampalli MR, Hsia EY, Izumiya C, Huerta SB, Wright ME, Chen HW, Kung HJ, Izumiya Y (2010) KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci USA 107: 9671–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Youn MY, Yokoyama A, Fujiyama‐Nakamura S, Ohtake F, Minehata K, Yasuda H, Suzuki T, Kato S, Imai Y (2012) JMJD5, a Jumonji C (JmjC) domain‐containing protein, negatively regulates osteoclastogenesis by facilitating NFATc1 protein degradation. J Biol Chem 287: 12994–13004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones MA, Covington MF, DiTacchio L, Vollmers C, Panda S, Harmer SL (2010) Jumonji domain protein JMJD5 functions in both the plant and human circadian systems. Proc Natl Acad Sci USA 107: 21623–21628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu H, Hu S, Baker J (2014) JMJD5 regulates cell cycle and pluripotency in human embryonic stem cells. Stem Cells 32: 2098–2110 [DOI] [PubMed] [Google Scholar]
- 29. Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF, Chen CC, Izumiya Y, Yu JS, Kung HJ et al (2014) JMJD5 regulates PKM2 nuclear translocation and reprograms HIF‐1alpha‐mediated glucose metabolism. Proc Natl Acad Sci USA 111: 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi YD, Felley‐Bosco E, Marti TM, Orlowski K, Pruschy M, Stahel RA (2012) Starvation‐induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer 12: 571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gagou ME, Zuazua‐Villar P, Meuth M (2010) Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 21: 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD et al (2011) Compromised CDK1 activity sensitizes BRCA‐proficient cancers to PARP inhibition. Nat Med 17: 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- 34. Klose RJ, Kallin EM, Zhang Y (2006) JmjC‐domain‐containing proteins and histone demethylation. Nat Rev Genet 7: 715–727 [DOI] [PubMed] [Google Scholar]
- 35. Mantri M, Krojer T, Bagg EA, Webby CA, Butler DS, Kochan G, Kavanagh KL, Oppermann U, McDonough MA, Schofield CJ (2005) Crystal structure of the 2‐oxoglutarate‐ and Fe(II)‐dependent lysyl hydroxylase JMJD6. J Mol Biol 401: 211–222 [PubMed] [Google Scholar]
- 36. Yasothornsrikul S, Greenbaum D, Medzihradszky KF, Toneff T, Bundey R, Miller R, Schilling B, Petermann I, Dehnert J, Logvinova A et al (2003) Cathepsin L in secretory vesicles functions as a prohormone‐processing enzyme for production of the enkephalin peptide neurotransmitter. Proc Natl Acad Sci USA 100: 9590–9595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berti PJ, Storer AC (1995) Alignment/phylogeny of the papain superfamily of cysteine proteases. J Mol Biol 246: 273–283 [DOI] [PubMed] [Google Scholar]
- 38. Harborne N, Allan J (1983) Modulation of the relative trypsin sensitivities of the core histone ‘tails’. FEBS Lett 155: 88–92 [DOI] [PubMed] [Google Scholar]
- 39. Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A (2004) A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell 14: 207–219 [DOI] [PubMed] [Google Scholar]
- 40. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (2007) High‐resolution profiling of histone methylations in the human genome. Cell 129: 823–837 [DOI] [PubMed] [Google Scholar]
- 41. Suzuki T, Minehata K, Akagi K, Jenkins NA, Copeland NG (2006) Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm‐deficient mice. EMBO J 25: 3422–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vogel JL, Kristie TM (2006) Site‐specific proteolysis of the transcriptional coactivator HCF‐1 can regulate its interaction with protein cofactors. Proc Natl Acad Sci USA 103: 6817–6822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johns EW (1964) Studies on histones. 7. Preparative methods for histone fractions from calf thymus. Biochem J 92: 55–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Phillips DM, Johns EW (1959) A study of the proteinase content and the chromatography of thymus histones. Biochem J 72: 538–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang X, Zhang L, Qi H, Shao J, Shen J (2013) Identification and functional implication of nuclear localization signals in the N‐terminal domain of JMJD5. Biochimie 95: 2114–2122 [DOI] [PubMed] [Google Scholar]
- 46. Del Rizzo PA, Krishnan S, Trievel RC (2012) Crystal structure and functional analysis of JMJD5 indicate an alternate specificity and function. Mol Cell Biol 32: 4044–4052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang H, Zhou X, Wu M, Wang C, Zhang X, Tao Y, Chen N, Zang J (2013) Structure of the JmjC‐domain‐containing protein JMJD5. Acta Crystallogr D Biol Crystallogr 69: 1911–1920 [DOI] [PubMed] [Google Scholar]
- 48. Amendola PG, Zaghet N, Ramalho JJ, Johansen JV, Boxem M, Salcini AE (2017) JMJD‐5/KDM8 regulates H3K36me2 and is required for late steps of homologous recombination and genome integrity. PLoS Genet 13: e1006632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang YH, Qian X, Shen JF, Wang YG, Li XJ, Liu R, Xia Y, Chen QM, Peng G, Lin SY et al (2015) Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol 17: 1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takahashi A, Imai Y, Yamakoshi K, Kuninaka S, Ohtani N, Yoshimoto S, Hori S, Tachibana M, Anderton E, Takeuchi T et al (2012) DNA damage signaling triggers degradation of histone methyltransferases through APC/C‐Cdh1 in senescent cells. Mol Cell 45: 123–131 [DOI] [PubMed] [Google Scholar]
- 51. Maroschik B, Gurtler A, Kramer A, Rossler U, Gomolka M, Hornhardt S, Mortl S, Friedl AA (2014) Radiation‐induced alterations of histone post‐translational modification levels in lymphoblastoid cell lines. Strahlenther Onkol 190: 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Q, Ma S, Song N, Li X, Liu L, Yang SD, Ding X, Shan L, Zhou X, Su DX et al (2016) Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J Clin Invest 126: 2205–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Palomera‐Sanchez Z, Bucio‐Mendez A, Valadez‐Graham V, Reynaud E, Zurita M (2010) Drosophila p53 Is required to increase the levels of the dKDM4B demethylase after UV‐induced DNA damage to demethylate histone H3 lysine 9. J Biol Chem 285: 31370–31379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Polo SE, Jackson SP (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25: 409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chow CM, Georgiou A, Szutorisz H, Maia e Silva A, Pombo A, Barahona I, Dargelos E, Canzonetta C, Dillon N (2005) Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep 6: 354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ahmad K, Henikoff S (2002) The histone variant H3.3 marks active chromatin by replication‐independent nucleosome assembly. Mol Cell 9: 1191–1200 [DOI] [PubMed] [Google Scholar]
- 57. Chang B, Chen Y, Zhao Y, Bruick RK (2007) JMJD6 is a histone arginine demethylase. Science 318: 444–447 [DOI] [PubMed] [Google Scholar]
- 58. Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR III et al (2009) Jmjd6 catalyses lysyl‐hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325: 90–93 [DOI] [PubMed] [Google Scholar]
- 59. Hong X, Zang J, White J, Wang C, Pan CH, Zhao R, Murphy RC, Dai S, Henson P, Kappler JW et al (2010) Interaction of JMJD6 with single‐stranded RNA. Proc Natl Acad Sci USA 107: 14568–14572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hsia DA, Tepper CG, Pochampalli MR, Hsia EY, Izumiya C, Huerta SB, Wright ME, Chen HW, Kung HJ, Izumiya Y (2010) KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci USA 107: 9671–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ (2005) Single‐nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol 3: e328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J et al (2011) Systematic investigation of halogen bonding in protein‐ligand interactions. Angew Chem 50: 314–318 [DOI] [PubMed] [Google Scholar]
- 63. Prlic A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE (2010) Pre‐calculated protein structure alignments at the RCSB PDB website. Bioinformatics 26: 2983–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shindyalov IN, Bourne PE (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11: 739–747 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 3