

Abstract
Gene expression regulation is essential for cells to adapt to changes in their environment. Co‐activator complexes have well‐established roles in transcriptional regulation, but less is known about how they sense and respond to signaling cues. We have previously shown that, in fission yeast, one such co‐activator, the SAGA complex, controls gene expression and the switch from proliferation to differentiation in response to nutrient availability. Here, using a combination of genetic, biochemical, and proteomic approaches, we show that SAGA responds to nutrients through the differential phosphorylation of its Taf12 component, downstream of both the TORC1 and TORC2 pathways. Taf12 phosphorylation increases early upon starvation and is controlled by the opposing activities of the PP2A phosphatase, which is activated by TORC1, and the TORC2‐activated Gad8AKT kinase. Mutational analyses suggest that Taf12 phosphorylation prevents cells from committing to differentiation until starvation reaches a critical level. Overall, our work reveals that SAGA is a direct target of nutrient‐sensing pathways and has uncovered a mechanism by which TORC1 and TORC2 converge to control gene expression and cell fate decisions.
Keywords: differentiation, fission yeast, SAGA, signal transduction, TOR, transcription
Subject Categories: Metabolism, Signal Transduction, Transcription
Introduction
Gene regulation plays a fundamental role in the ability of cells to adapt to fluctuations in their environment. For example, nutrient availability controls whether cells proliferate or not, through a network of signaling kinases that drive specific gene expression programs 1. Nutrient sensing is mediated by several distinct kinases that are part of highly conserved signaling pathways 2. Of these, the target of rapamycin (TOR) atypical serine/threonine kinase plays a central role in coordinating cell growth with nutrient levels. TOR assembles into two distinct, highly conserved complexes, TORC1 and TORC2, which differ in their composition, regulation, and functions (reviewed in 3, 4). In the fission yeast Schizosaccharomyces pombe, TORC1 promotes cell growth and inhibits sexual differentiation when nutrients are present. In contrast, TORC2 induces cell cycle exit and differentiation upon starvation (reviewed in 5, 6, 7). Accordingly, TORC1 and TORC2 oppositely control the expression of ste11 + and mei2 +, which encodes the master regulators of mating and meiosis, respectively 8, 9, 10, 11, 12. However, the mechanism by which TORC1 and TORC2 regulate gene expression in this context is not well understood.
One critical step of gene expression is transcription initiation, which involves many different types of factors, including co‐activators 13. Co‐activators are typically large multi‐subunit complexes that possess multiple distinct activities, such as histone modification, nucleosome remodeling, and recruitment of general transcription factors. One such co‐activator is the SAGA complex (Spt‐Ada‐Gcn5 acetyltransferase). SAGA is essential for the transcription of many inducible genes and has key roles in cell cycle progression, stress responses, or during development (reviewed in 14, 15). SAGA is composed of 19 conserved subunits that are organized into functional modules with separate activities. For example, the Gcn5 subunit is a nucleosomal histone acetyltransferase (HAT) 16, 17, whereas the Spt8 subunit modulates the recruitment of the TATA box‐binding protein (TBP) to certain promoters 18, 19. Other subunits, including Spt7 16, Ada1 20, and five TBP‐associated factors (TAF5, 6, 9, 10, and 12) 21, are required for the integrity of the complex 18, 21, 22 and serve as structural scaffolds 23, 24, 25, 26. Interestingly, distinct SAGA activities function independently of each other and can have opposing roles in transcriptional regulation 27. For example, we have previously shown that, in S. pombe, SAGA uses distinct activities to either repress or induce the expression of differentiation genes, depending on nutrient levels 28, 29. However, it is not known how SAGA senses nutrient availability and, more generally, whether signaling pathways directly control the regulatory activities of co‐activator complexes.
Here, we have addressed these issues in the context of cell fate control by nutrient availability in S. pombe. Epistasis analyses established that the SAGA co‐activator regulates the expression of differentiation genes downstream of both the TORC1 and TORC2 signaling pathways. Quantitative proteomic and biochemical analyses revealed that the Taf12 subunit is phosphorylated early upon nutrient starvation. We then showed that TORC1 activates the PP2A phosphatase, via the Greatwall (Gwl) kinase, to de‐phosphorylate Taf12, whereas the TORC2‐Gad8AKT kinase pathway phosphorylates Taf12. Unexpectedly, we found that Taf12 phosphorylation inhibits sexual differentiation. We propose a model in which, upon nutrient starvation, the simultaneous inhibition of TORC1 and activation of TORC2‐Gad8AKT induces Taf12 phosphorylation, which modulates the timing and the amplitude of the differentiation response. Overall, our work reveals that Taf12 is a direct target of nutrient‐sensing pathways and that the TORC1 and TORC2 pathways converge to control a common effector, allowing the versatile expression of the master regulators of a differentiation program.
Results
The SAGA subunit Gcn5 regulates sexual differentiation downstream of both the TORC1 and TORC2‐Gad8AKT pathways
Schizosaccharomyces pombe is an ideal model to address how cells sense nutrients and coordinately regulate gene expression to control cell fate decisions. In the presence of nutrients, S. pombe grows and proliferates. Conversely, upon starvation, particularly of nitrogen, cells exit the cell cycle at the G1 phase and commit to sexual differentiation, which sequentially involves mating, meiosis, and sporulation. The Ste11 transcription factor is the master regulator of sexual differentiation and activates the expression of many genes, including mei2 +, which functions as the decisive trigger of meiosis (reviewed in 30). We have previously identified dual, opposite roles for SAGA in this process 28. In rich conditions, the SAGA HAT, Gcn5, directly represses the expression of ste11 + and mei2 +. In contrast, the Spt8 subunit is required for the induction of these genes upon starvation. We reasoned that, if SAGA is able to switch from a repressor to an activator in response to nutrients, it must be under the regulation of nutrient‐sensing signaling pathways.
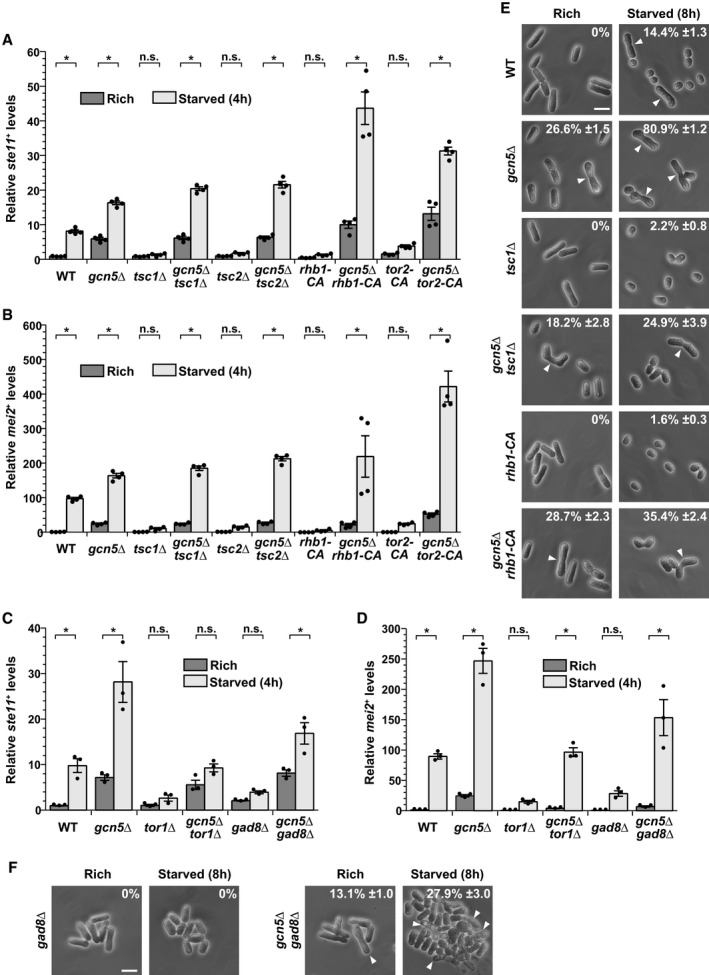
In rich conditions, the highly conserved TORC1 kinase and cyclic adenosine monophosphate (cAMP)‐activated protein kinase A, Pka1, are the two major pathways that both promote growth and inhibit differentiation 7. We first sought to identify which of these two pathways requires Gcn5 to inhibit differentiation. To do this, we quantified ste11 + and mei2 + expression in mutants that constitutively activate (CA) TORC1 signaling. These included point mutations in the TORC1‐specific kinase tor2 + (tor2‐CA) and in its activating GTPase Rheb, rhb1 + (rhb1‐CA), as well as deletion mutants of the tuberous sclerosis complex proteins Tsc1 and Tsc2. We observed that ste11 + and mei2 + expression (Fig 1A and B), as well as differentiation (Fig 1E), are weakly induced upon starvation of all mutants, tsc1Δ, tsc2Δ, rhb1‐CA, and tor2‐CA, as compared to wild‐type (WT) controls. Therefore, constitutive activation of TORC1 prevents the induction of differentiation upon starvation, as anticipated from previous work 9, 10, 31, 32, 33, 34. In marked contrast, in gcn5Δ mutants, ste11 + and mei2 + expression and differentiation are de‐repressed in rich conditions and further induced upon starvation, as compared to WT strains (Fig 1A, B and E). Strikingly, we observed that the expression profiles of ste11 + and mei2 + are indistinguishable between gcn5Δ single mutants and gcn5Δ tsc1Δ or gcn5Δ tsc2Δ double mutants (Fig 1A and B). Similar results are observed in gcn5Δ rhb1‐CA or gcn5Δ tor2‐CA double mutants. Accordingly, gcn5Δ mutants suppress the sterility of tsc1Δ and rhb1‐CA mutants (Fig 1E). We noticed that Gcn5 only partially rescues the differentiation phenotype of constitutively active TORC1 mutants, suggesting that TORC1 controls other processes that are important for sexual differentiation, independently of Gcn5. To conclude, the absence of Gcn5 rescues the inability of constitutively active TORC1 mutants to differentiate, indicating that TORC1 requires Gcn5 to inhibit differentiation.
Figure 1. SAGA is epistatic to the TORC1 and TORC2 pathways in the regulation of differentiation in response to nutrient availability.
-
A–DExpression of ste11 + (A, C) and mei2 + (B, D) using quantitative RT–PCR of RNA extracted from cells grown either in nutrient rich medium (dark gray) or shifted for 4 h to starvation medium (light gray). Cells of the following genotypes were analyzed: wild‐type isogenic controls (WT), gcn5Δ, tsc1Δ, gcn5Δ tsc1Δ, tsc2Δ, gcn5Δ tsc2Δ, rhb1‐DA4—a constitutively active (CA) rhb1 mutant 34, gcn5Δ rhb1‐DA4, tor2‐L1310P—a CA tor2 mutant 33, gcn5Δ tor2‐L1310P, tor1Δ, gcn5Δ tor1Δ, gad8Δ, and gcn5Δ gad8Δ. act1 + served as a control for normalization across samples. Values from a WT strain grown in rich medium were set at 1 to allow comparisons across culture conditions and mutant strains. Each column represents the mean value of 4 (A, B) or 3 (C, D) independent experiments, overlaid with individual data points and error bars showing the standard error of the mean (SEM). Statistical significance was determined by two‐way ANOVA followed by Bonferroni's multiple comparison tests (n = 4 for A, B; n = 3 for C, D); *P ≤ 0.01.
-
E, FCells were grown to mid‐log phase either in rich medium or shifted for 8 h to starvation medium. Zygotes and tetrads, which correspond to differentiated cells, were counted under a light microscope. Cells of the following genotypes were analyzed: WT isogenic controls, gcn5Δ, tsc1Δ, gcn5Δ tsc1Δ, rhb1‐DA4, gcn5Δ rhb1‐DA4, gad8Δ, and gcn5Δ gad8Δ. Each value represents the mean percentage and SEM of differentiating cells to the total number of cells, averaged from four independent experiments. At least 200 cells from the indicated genotypes were counted in each experiment. White arrowheads indicate zygotes. Scale bar, 10 μm.
We tested whether Gcn5 is also epistatic to the cAMP‐Pka1 pathway, which synergizes with TORC1 to control ste11 + expression 11. Activation of Pka1 either by adding cAMP or by deleting the cAMP‐specific phosphodiesterase gene, cgs2 +, markedly reduces ste11 + and mei2 + expression, both in rich and starved conditions (Appendix Fig S1A–D), as shown previously 35, 36, 37. In contrast to what we observed with TORC1, gcn5Δ mutants do not suppress this phenotype (Appendix Fig S1A–D), suggesting that Pka1 does not require Gcn5 to inhibit differentiation. Altogether, our genetic analyses indicate that the SAGA subunit Gcn5 inhibits differentiation downstream of TORC1.
Upon nutrient starvation, several signaling pathways are activated and required for sexual differentiation in S. pombe. Each is defined by a highly conserved kinase, including orthologs of the p38 MAPK, Sty1, and of the TORC2 complex, defined by the Tor1 kinase 7. Similar to other eukaryotes, S. pombe TORC2 mediates many of its functions through the activation of an AKT kinase, Gad8AKT 38, 39, 40, 41. Additionally, one S. pombe ortholog of the adenosine monophosphate (AMP)‐activated protein kinase, Ssp2, has important roles in starvation responses, notably through the inhibition of TORC1 42, 43.
We determined whether any of these kinases also functionally interacts with Gcn5 to regulate differentiation genes. To do this, we quantified their expression in mutants of each kinase, grown either in rich medium or shifted to starvation conditions. As compared to WT controls, we observed little, if any, induction of both ste11 + and mei2 + upon starvation of tor1Δ (Fig 1C and D), gad8Δ (Fig 1C and D), sty1Δ (Appendix Fig S1E and F), or ssp2Δ mutants (Appendix Fig S1G and H), as expected from previous work 38, 42, 44, 45. In contrast, expression of mei2 +, and to a lesser extent of ste11 +, is induced upon starvation of gcn5Δ gad8Δ or gcn5Δ tor1Δ double mutants (Fig 1C and D). In marked contrast, gcn5Δ mutants do not suppress the lack of induction observed in sty1Δ or ssp2Δ mutants (Appendix Fig S1E–H). Finally, gcn5Δ mutants partially suppress the sterility of gad8Δ mutants (Fig 1F) but, surprisingly, not of tor1Δ mutants (data not shown). We observed that gcn5Δ tor1Δ double mutants exhibit severe cell cycle progression defects, which likely interfere with their ability to exit from G1 and commit to sexual differentiation. To conclude, our genetic analyses suggest that Gcn5 regulates sexual differentiation downstream of the TORC2‐Gad8AKT pathway, but not of p38Sty1 or Ssp2AMPK. However, the partial suppression of the differentiation phenotypes of gad8Δ and tor1Δ mutants by gcn5Δ mutants suggests that the TORC2‐Gad8AKT pathway requires additional effectors to induce sexual differentiation upon starvation.
SAGA is differentially phosphorylated in response to a change in nutrient levels
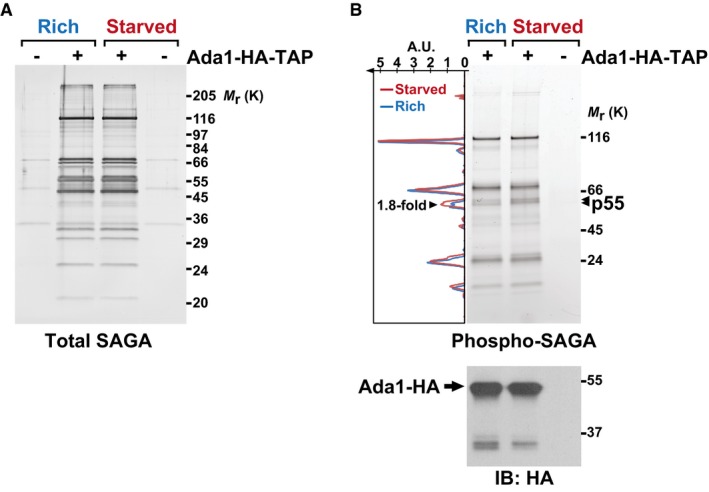
These genetic analyses prompted us to determine how TORC1 and TORC2 control Gcn5 in response to nutrients. To do this, we used the tandem affinity purification (TAP) procedure to purify Gcn5, as part of SAGA, from cells grown under nutrient rich conditions or shifted to starvation medium for 45 min. Both total protein staining and quantitative mass spectrometry (MS) analysis revealed that SAGA subunit composition does not change upon shifting cells from rich to starved conditions (Fig 2A, Appendix Table S1). To test whether SAGA is differentially phosphorylated, we re‐analyzed these purifications using an in‐gel fluorescent stain, Pro‐Q Diamond, which is specific for phosphorylated proteins (Fig 2B). Quantification of each lane indicates that one band, migrating at about 55 kDa and coined p55, is about 1.8‐fold more intense in SAGA purified from starved cells, as compared to rich conditions (Fig 2B). The predicted molecular weight of eight out of the 19 subunits of S. pombe SAGA falls between 50 and 60 kDa, including Gcn5 28.
Figure 2. SAGA is phosphorylated in response to nutrient starvation.
-
A, B4–20% gradient SDS–polyacrylamide gel electrophoresis analysis of SAGA purified from cells grown either in rich medium (R) or starved for 45 min (S). SAGA was purified using endogenously TAP‐tagged Ada1. A fraction of each eluate was loaded and either stained with silver, to detect all proteins (A), or with Pro‐Q® Diamond, which stains phosphorylated proteins (B). A strain without any TAP tag is shown as a negative control for the purification. The graph to the left of the gel in (B) shows the fluorescence intensity of the phospho‐specific stain, which was quantified along the left lane in blue (rich) and the middle lane in red (starved), using ImageJ. The area of one peak, which corresponds to the bands marked with arrowheads and was coined p55, is 1.8‐fold larger in SAGA purified from starved cells, as compared to rich conditions. Below is an anti‐HA immunoblot (IB) of each eluate, to reveal the amount of Ada1‐HA bait recovered. Shown are gels that are representative of three independent experiments. A.U., arbitrary units.
To test whether p55 corresponds to Gcn5, we purified SAGA using a strain in which Gcn5 is tagged with MYC epitopes, such as to slow its migration to a portion of the gel where no other SAGA subunits are detected (Appendix Fig S2A). Although Gcn5 is clearly phosphorylated (Appendix Fig S2B), its normalized levels are similar between rich and starved conditions (Appendix Fig S2C). Importantly, p55 is still detectable in SAGA purified from Gcn5‐MYC cells and its phosphorylation increases upon starvation (Appendix Fig S2A–C). Therefore, one SAGA component, but not Gcn5, is differentially phosphorylated in response to nutrients.
Taf12 is phosphorylated in response to nutrient availability
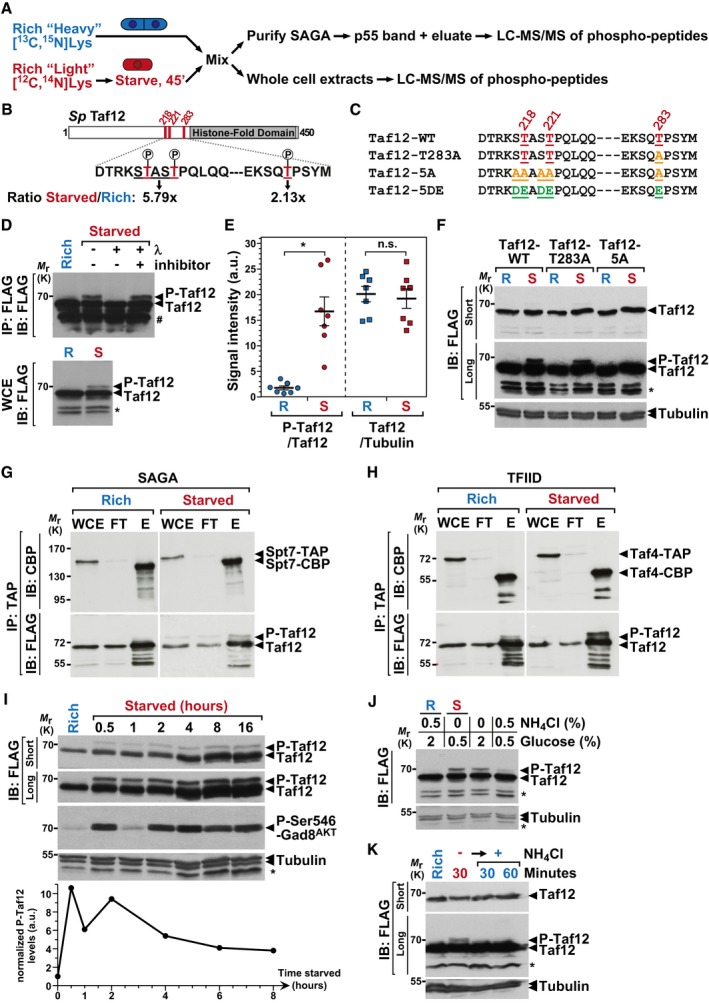
To identify p55, we performed a quantitative MS analysis of phospho‐peptides within SAGA, using a SILAC procedure (Fig 3A). We analyzed either whole cell extracts or purified SAGA complexes, comparing cells that were differentially labeled with lysine and arginine isotopes, grown either in rich conditions or shifted to starvation medium for 45 min. Strikingly, the core SAGA subunit Taf12 appeared differentially phosphorylated on several residues in all four independent phospho‐SILAC experiments, including when the p55 band was cut and directly analyzed by MS. Specifically, Taf12 shows increased phosphorylation of threonines (Thr) 218, 221, and 283 upon starvation (Fig 3B, Appendix Figs S3 and S4).
Figure 3. The SAGA and TFIID subunit Taf12 is phosphorylated early upon starvation.
-
AOverview of the quantitative proteomic approaches used to identify differentially phosphorylated peptides, either in crude extracts or in SAGA purifications. Cells were metabolically labeled using a SILAC procedure and grown either in rich medium or shifted to nutrient starvation conditions. Several independent experiments were carried out with forward and reverse labeling schemes (see Materials and Methods for details).
-
BSchematic view of the S. pombe (Sp) Taf12 protein sequence, including, in red, the three differentially phosphorylated Thr, at positions 218, 221, and 283 and, in gray, the histone‐fold domain. Shown below are the starved‐to‐rich SILAC ratios of the signal intensities observed for the Thr218‐Thr221 peptide (Appendix Fig S3) and for the Thr283 peptide (Appendix Fig S4).
-
CSummary of the different taf12 + point mutants that were constructed and analyzed.
-
DFLAG‐tagged Taf12 was purified from cells grown in rich medium (R) or shifted for 45 min to starvation medium (S). Anti‐FLAG immuno‐precipitations (IP) were incubated with and without λ‐phosphatase or its inhibitor and immunoblotted (IB), together with 1% of whole cell extracts (WCE), using an anti‐FLAG antibody.
-
EPhospho‐Taf12 (P‐Taf12), total Taf12, and tubulin levels were quantified from cells grown in rich medium (R) or starved for 45 min (S). P‐Taf12 levels were normalized to those of total Taf12, while total Taf12 levels were normalized to those of tubulin. Data points were individually plotted on the graph and overlaid with the mean and SEM. Signal intensities were quantified from IBs of seven independent experiments. Statistical significance was determined using a t‐test (n = 7, unpaired, two‐tailed); *P ≤ 0.01.
-
FP‐Taf12 and total Taf12 were followed in WT, taf12‐T283A and taf12‐5A mutants, grown in rich medium (R) or starved for 45 min (S).
-
G, HSAGA and TFIID were tandem affinity‐purified using endogenously TAP‐tagged Spt7 (G) or Taf4 (H), from strains containing FLAG‐tagged Taf12, grown in rich medium or starved for 45 min. TAP‐tagged Spt7 and Taf4 were eluted using the TEV protease, releasing a shorter form of each bait (Spt7‐CBP or Taf4‐CBP). Eluates were loaded and immunoblotted (IB) using anti‐FLAG or anti‐CBP antibodies, together with 1% of either whole cell extracts (WCE) or IgG‐Sepharose flow‐through (FT). Shown are IBs that are representative of two independent experiments.
-
IP‐Taf12, total Taf12, and Ser546‐phosphorylated Gad8AKT were followed in WT cells, grown in rich conditions or over a time‐course after a shift to starvation medium. Gad8AKT phosphorylation at Ser546 is a proxy of TORC2 activity in S. pombe.
-
JP‐Taf12 and total Taf12 were followed in WT cells, grown in rich conditions or shifted to different starvation media for 30 min. These include both the removal of the nitrogen source, ammonium chloride (NH4Cl), and the reduction in the carbon source, glucose, from a concentration of 2–0.5%. Alternatively, cells were either only deprived of NH4Cl or only exposed to reduced glucose levels (2–0.5%).
-
KP‐Taf12 and total Taf12 were followed in WT cells, grown in rich conditions or deprived of NH4Cl for 1 h. Then, NH4Cl was added back to the medium for 30 or 60 min.
To confirm and further characterize this observation, we examined the migration of endogenously FLAG‐tagged Taf12 by Western blot analysis of extracts from cells grown in rich medium or shifted to starved conditions. We detected an electrophoretic mobility shift of Taf12 following nutrient starvation, either in crude extracts (Fig 3D, lower panel) or after its immuno‐purification (Fig 3D, upper panel). We quantified the relative intensity of the phosphorylated form of Taf12, named hereafter P‐Taf12, and of total Taf12 from seven independent biological replicates and confirmed that P‐Taf12 increases reproducibly upon starvation, whereas total Taf12 levels do not change (Fig 3E). We then demonstrated that this shift is indeed caused by phosphorylation. First, this isoform of Taf12 disappears upon λ‐phosphatase treatment of FLAG purified Taf12 (Fig 3D, upper panel). Second, we constructed strains in which either one (Taf12‐T283A) or all three Thr were mutated to alanines (Ala) (Taf12‐5A). We also mutated the serines (Ser) that are adjacent to Thr218 and Thr221, in order to prevent their possible phosphorylation in the Thr‐to‐Ala mutant (Fig 3C). Western blotting confirmed that, at most, all five Ser/Thr residues contribute to this mobility shift, without affecting Taf12 steady‐state levels (Fig 3F). Altogether, these results indicate that Taf12 becomes phosphorylated upon nutrient starvation in S. pombe.
Next, we verified that Taf12 is phosphorylated within SAGA, as suggested by our phospho‐SILAC analyses. Similarly, we asked whether Taf12 is phosphorylated as part of the TFIID general transcriptional factor complex, of which S. pombe Taf12 is also a core component 46, similar to other eukaryotes. We used either Spt7 or Taf4 to purify SAGA or TFIID, respectively. Anti‐FLAG blotting of each eluate indicated that starvation induces Taf12 phosphorylation both within SAGA (Fig 3G) and TFIID (Fig 3H). Total protein staining of purified TFIID showed that its subunit composition does not change upon shifting cells from rich to starved conditions (Appendix Fig S5), similar to SAGA (Fig 2A). To conclude, Taf12 becomes phosphorylated upon nutrient starvation, as part of both the SAGA and TFIID complexes.
Dynamics of Taf12 phosphorylation
To further characterize Taf12 phosphorylation, we monitored its kinetics upon starvation and detected P‐Taf12 already 30 min after shifting cells to starved medium (Fig 3I). This observation suggests that Taf12 is directly controlled by nutrient‐sensing pathways, which respond to nutrient starvation within this timeframe in S. pombe. For example, TORC1 activity is fully inhibited within 30 min of nitrogen starvation 47. Likewise, we observed that phosphorylation of Ser546 of the AKT kinase, Gad8AKT, is maximal 30 min after removing nutrients (Fig 3I), consistent with previous observations 41. This modification is a hallmark of TORC2 activation both in S. pombe 38, 40 and in mammals 48. Interestingly, only a small fraction of total Taf12 is phosphorylated, about 7%, without further increasing with the duration of starvation, as more cells commit to differentiation (Fig 3I). Rather, we observed that Taf12 phosphorylation proportionally decreases about threefold at later time points of starvation (Fig 3I). To conclude, phosphorylation of Taf12 appears restricted to a small fraction of all Taf12 and peaks early upon nutrient starvation.
Our procedure for starving proliferating cells consists in both completely removing ammonium, a preferred nitrogen source, and lowering glucose levels fourfold. In S. pombe, TORC1 mostly responds to the availability of nitrogen. We thus tested which of nitrogen or carbon starvation induces Taf12 phosphorylation. Western blot analysis from cells shifted to various media showed that P‐Taf12 levels increase upon nitrogen starvation, whereas lowering glucose levels has a much weaker effect (Fig 3J). Additionally, P‐Taf12 levels rapidly decrease when nitrogen is added back to starved cells (Fig 3K). We thus conclude that Taf12 phosphorylation is dynamically regulated by nitrogen availability.
The PP2A phosphatase represses differentiation downstream of TORC1
We next sought to identify which signaling pathways control Taf12 phosphorylation in response to nutrients. In rich conditions, TORC1 activity is high whereas Taf12 phosphorylation levels are low. We therefore hypothesized that TORC1 activates a phosphatase to prevent Taf12 phosphorylation in rich conditions. Functionally, this phosphatase should inhibit differentiation downstream of TORC1. Little is known about which phosphatases function in nutrient sensing and sexual differentiation in S. pombe. However, in S. cerevisiae, TORC1 regulates the type 2A and 2A‐related protein phosphatases (PP2A) to control nutrient stress responses 3, 49, including gametogenesis 50. PP2A is a conserved heterotrimeric complex formed by catalytic (C), structural (A), and regulatory (B55 or B56) subunits. The latter components are mutually exclusive and dictate substrate specificity 51. Schizosaccharomyces pombe PP2A has important roles in morphogenesis and mitosis 52, 53, 54, 55, but it is not known whether it also controls sexual differentiation.
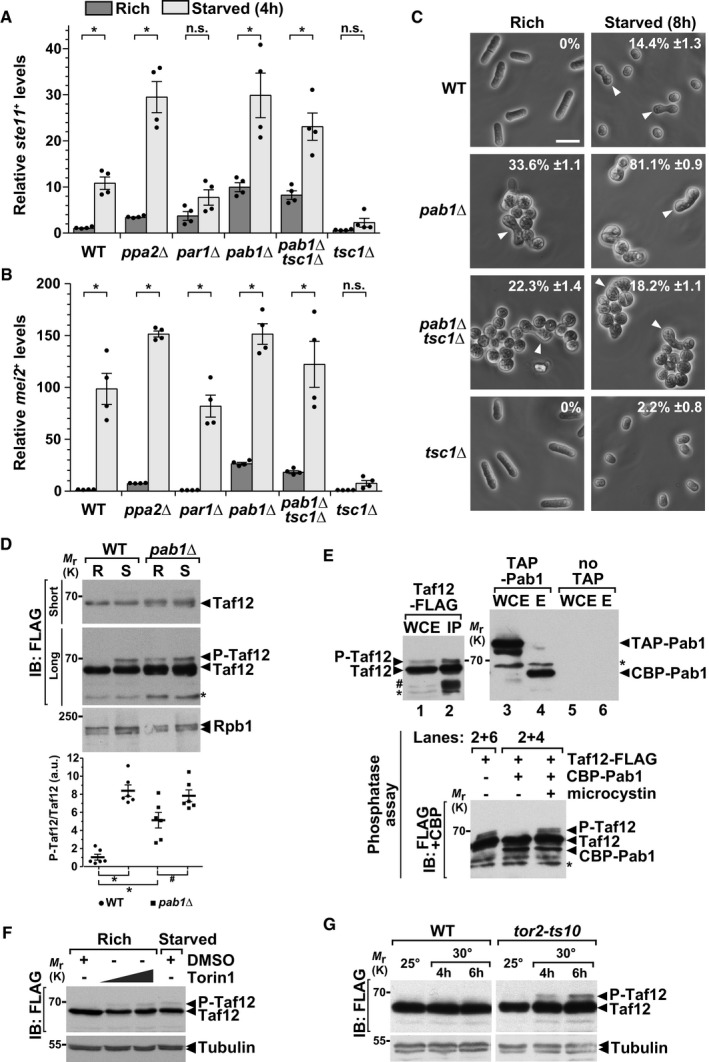
To answer this question, we quantified the expression of differentiation genes and the number of cells undergoing sexual differentiation in PP2A deletion mutants, grown either in rich conditions or shifted to starvation medium. We observed that the expression of both ste11 + and mei2 + is de‐repressed in a catalytic subunit mutant, ppa2Δ, as compared to WT controls (Fig 4A and B). Interestingly, both genes are de‐repressed in a mutant of the B55 regulatory subunit, pab1Δ, but not in a mutant of the B56 regulatory subunit, par1Δ (Fig 4A and B). In agreement, we counted a higher number of differentiated cells in pab1Δ mutants, in both rich and starved conditions, as compared to WT controls (Fig 4C). Therefore, similar to TORC1 and Gcn5, PP2A inhibits sexual differentiation when cells are grown in rich conditions and this function is specifically mediated by the B55 regulatory subunit Pab1B55.
Figure 4. The PP2A phosphatase inhibits differentiation and de‐phosphorylates Taf12.
-
A, BExpression of ste11 + (A) and mei2 + (B) using quantitative RT–PCR of RNA extracted from cells grown either in rich medium (dark gray) or starved for 4 h (light gray). Cells of the following genotypes were analyzed: wild‐type isogenic controls (WT), ppa2Δ, par1Δ, pab1Δ, tsc1Δ, and pab1Δ tsc1Δ. act1 + served as a control for normalization across samples. Values from a WT strain grown in rich medium were set at 1 to allow comparisons across culture conditions and mutant strains. Each column represents the mean value of four independent experiments, overlaid with individual data points and SEM. Statistical significance was determined by two‐way ANOVA followed by Bonferroni's multiple comparison tests (n = 4); *P ≤ 0.01.
-
CCells were grown to mid‐log phase either in rich medium or starved for 8 h. Zygotes and tetrads, which correspond to differentiated cells, were counted under a light microscope. Each value represents the mean percentage and SEM of differentiating cells to the total number of cells, averaged from four independent experiments. At least 200 cells from the indicated genotypes were counted in each experiment. White arrowheads indicate zygotes. Scale bar, 10 μm.
-
DP‐Taf12 was followed by anti‐FLAG IB of protein extracts from WT and pab1Δ strains, grown in rich conditions (R) or starved for 45 min (S). An anti‐Rpb1 IB is shown as a control for loading. The signal intensities of P‐Taf12 and total Taf12 were quantified in each strain and condition. Ratios of P‐Taf12 to total Taf12 were calculated from six independent experiments and individually plotted in a graph below the IBs, together with the mean and SEM. Averaged values from all WT controls grown in rich medium were set at 1 to allow comparisons across culture conditions and strains. Statistical significance was determined by two‐way ANOVA followed by Bonferroni's multiple comparison tests (n = 6; *P < 0.01; # P < 0.05). A short exposure and a long exposure of the FLAG IB are shown to detect total Taf12 and P‐Taf12, respectively, within the linear range of the chemiluminescence signal.
-
EFLAG‐tagged Taf12 and TAP‐tagged Pab1 were affinity‐purified separately from cells grown in rich conditions (upper panels). TAP‐tagged Pab1 was cleaved off from beads using the tobacco etch virus (TEV) protease, releasing a shorter form of Pab1 (CBP‐Pab1) in the eluate (E). CBP‐Pab1 was then mixed with beads containing FLAG‐Taf12 IPs and incubated in a phosphatase buffer, with and without 0.5 μM microcystin. Each reaction was then analyzed by IB and simultaneously probed with anti‐FLAG or anti‐CBP antibodies (lower panel).
-
FExponentially growing cells were treated for 1 h with Torin‐1, which was added at increasing concentrations, 7 and 21 μM, to rich medium. Dimethylsulfoxide (DMSO) was used as the vehicle and added as a negative control. Anti‐tubulin IBs are shown as a control for loading between samples.
-
GP‐Taf12 was followed by anti‐FLAG IB of protein extracts from WT and tor2‐ts10 strains, grown in rich conditions at 25°C or shifted to 30°C for 4 and 6 h. Anti‐tubulin IBs are shown as a control for loading between samples.
We then determined whether PP2A‐Pab1B55 functions downstream of TORC1 in this process, by testing whether a pab1Δ mutant suppresses the lack of differentiation observed in a constitutively active TORC1 mutant. We found that the expression profiles of ste11 + and mei2 + are similar between a pab1Δ single mutant and a pab1Δ tsc1Δ double mutant (Fig 4A and B). In agreement, the absence of Pab1B55 partially rescues the lack of differentiation of a tsc1Δ mutant (Fig 4C). As reported previously 53, pab1Δ mutants display severe morphological defects that likely interfere with sexual differentiation. In conclusion, the B regulatory subunit of PP2A, Pab1B55, functions downstream of Tsc1, and likely of TORC1, to inhibit differentiation when cells proliferate. Importantly, a very recent study has shown that TORC1 controls PP2A‐Pab1B55 to inhibit sexual differentiation in S. pombe 56.
The PP2A phosphatase de‐phosphorylates Taf12
These results prompted us to test whether PP2A regulates Taf12 phosphorylation in nutrient rich conditions. First, we observed that P‐Taf12 levels reproducibly increase in pab1Δ mutants, as compared to WT controls, whereas total Taf12 levels do not change (Fig 4D). Interestingly, P‐Taf12 levels appear to further increase in starved pab1Δ cells, suggesting that, in parallel to inhibiting PP2A, starvation activates a kinase which phosphorylates Taf12. Second, we tested whether PP2A is able to directly de‐phosphorylate Taf12. We separately purified Taf12 and Pab1B55, which were endogenously tagged with a FLAG and a TAP epitope, respectively (Fig 4E, upper panels). Incubation of the eluates together in a standard phosphatase buffer revealed that Pab1B55 de‐phosphorylates Taf12 in vitro (Fig 4E, lower panel). To control for the specificity of this assay, we verified that Taf12 is not de‐phosphorylated when a PP2A‐specific inhibitor, microcystin, is added, or when it is incubated with a control “no TAP” purification. Altogether, these experiments indicate that Taf12 is a substrate of the PP2A‐Pab1B55 phosphatase, both in vitro and in vivo.
To determine whether TORC1 modulates Taf12 phosphorylation, we followed P‐Taf12 levels upon addition of Torin‐1, which inhibits TOR kinase activity. Western blot analyses revealed that P‐Taf12 levels increase with the amount of Torin‐1 added to cells grown in rich medium (Fig 4F). We noticed that total Taf12 levels decreased upon Torin‐1 addition, possibly because TORC1 inhibition reduces translation. However, Torin‐1 is an ATP‐analogue inhibitor of TOR and therefore inhibits both TORC1 and TORC2 57, 58. We thus examined Taf12 phosphorylation in a temperature‐sensitive mutant of the TORC1‐specific kinase, tor2‐ts10 10. Western blot analyses showed that P‐Taf12 levels increase upon shifting tor2‐ts10 mutants to the restrictive temperature, as compared to WT controls (Fig 4G), indicating that Taf12 phosphorylation is specifically inhibited by TORC1.
It is possible that higher P‐Taf12 levels are an indirect consequence of the de‐repression of differentiation which is observed in both PP2A‐Pab1B55 and TORC1 mutants (Fig 4A–C) 10. However, Taf12 phosphorylation profile is normal in gcn5Δ cells (Appendix Fig S6A), even though a gcn5Δ mutant phenocopies a pab1Δ mutant (Figs 1 and 4A–C). Furthermore, constitutive activation of Pka1 does not affect Taf12 phosphorylation in both rich and starved conditions (Appendix Fig S6B and C), consistent with our genetic analyses, which indicated that SAGA does not function downstream of the cAMP‐Pka1 pathway (Appendix Fig S1A–D). In conclusion, we show that PP2A‐Pab1B55 is a crucial effector of TORC1 to prevent both differentiation and Taf12 phosphorylation.
The Ppk18Gwl‐Igo1 pathway promotes differentiation through PP2A and SAGA
Our data indicate that P‐Taf12 levels inversely correlate with TORC1 and PP2A‐Pab1B55 activities. We next investigated the mechanism by which TORC1 inhibits PP2A to allow Taf12 phosphorylation upon starvation. PP2A activity and substrate specificity are controlled by its interaction with distinct regulatory proteins. Of these, the essential PP2A‐associated protein Tap42 is a major effector of TORC1 signaling in S. cerevisiae 3, 49. BLAST searches identified one S. pombe protein, SPCC63.05, as the bona fide ortholog of S. cerevisiae Tap42 (E‐value = 2.3−27). However, conditional depletion of Tap42 had no effect on Taf12 phosphorylation (Appendix Fig S7).
In S. cerevisiae, another major effector of TORC1 signaling is the S6 kinase ortholog, Sch9, which regulates PP2A through the Greatwall (Gwl) kinase ortholog, Rim15. As in other eukaryotes, Rim15Gwl inhibits PP2A activity by phosphorylating proteins of the α‐endosulfine family 59, 60. Remarkably, this regulatory cascade is essential to induce quiescence upon starvation in S. cerevisiae 59. A recent study has identified a similar pathway in S. pombe, in which the Ppk18Gwl kinase phosphorylates Ser64 of the Igo1 α‐endosulfine to inhibit PP2A‐Pab1 and coordinate cell growth with mitosis 61.
To characterize the role of this pathway upon starvation in S. pombe, we first monitored the migration pattern of endogenously MYC‐tagged Igo1 in response to nutrient availability. Western blotting of a Phos‐tag‐containing acrylamide gel revealed several bands that migrated slower in extracts from starved cells (Fig 5A). This starvation‐induced shift is not observed in ppk18Δ mutants or in Ser64‐to‐Ala igo1 knock‐in mutants. We then quantified ste11 + and mei2 + expression and the number of differentiated cells in ppk18Δ, igo1Δ and igo1‐S64A mutants, grown either in rich conditions or shifted to starvation medium. We observed that the expression of both genes (Fig 5B and C) and sexual differentiation (Appendix Fig S8) are weakly induced upon starvation of all three mutants, as compared to wild‐type (WT) controls. Therefore, starvation induces the phosphorylation of Igo1‐Ser64, most likely through the Ppk18Gwl kinase, and this pathway is required to promote differentiation, analogous to its role in S. cerevisiae.
Figure 5. PP2A activity is regulated by the Ppk18Gwl‐Igo1 pathway to control differentiation and Taf12 phosphorylation, in response to nutrient availability.
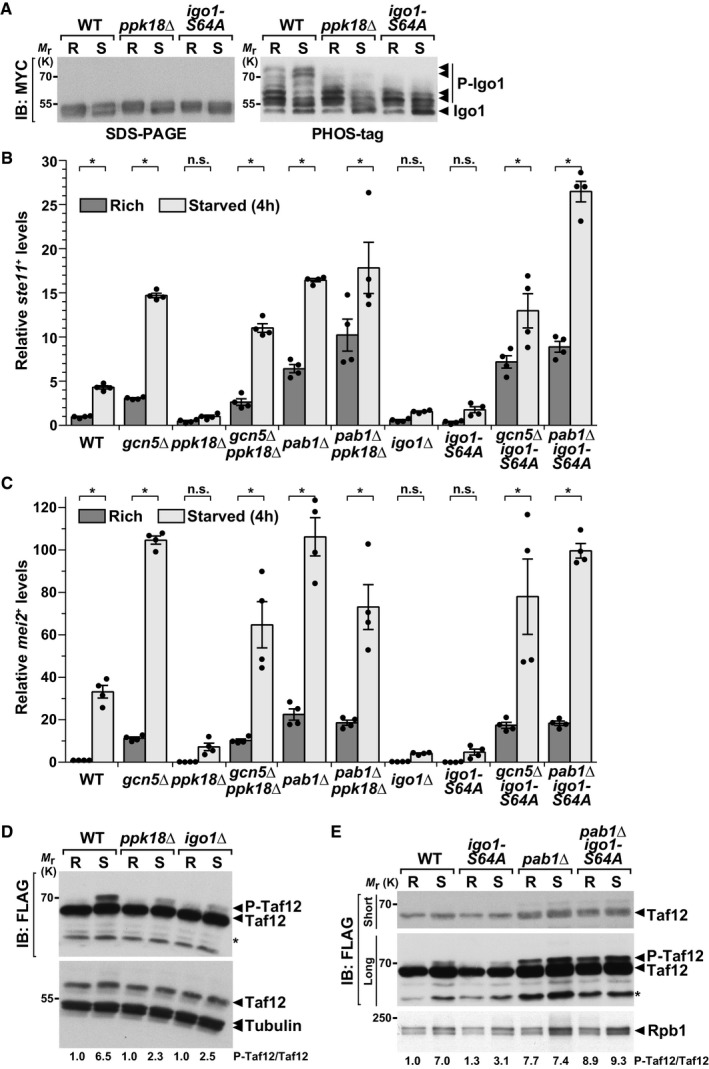
-
AIgo1 phosphorylation (P‐Igo1) was followed by anti‐MYC IB of protein extracts from WT, ppk18Δ, and igo1‐S64A strains, grown in rich conditions (R) or starved for 45 min (S). Igo1 migration was analyzed by electrophoresis of both a 12% SDS–polyacrylamide gel containing the Phos‐tag™ molecule (right panel) and a standard 10% SDS–polyacrylamide gel (left panel). Arrowheads indicate the various phosphorylated isoforms of Igo1‐MYC in each strain and condition. Shown is an IB representative of two independent experiments.
-
B, CExpression of ste11 + (C) and mei2 + (D) using quantitative RT–PCR of RNA extracted from cells grown either in rich medium (dark gray) or starved for 4 h (light gray). Cells of the following genotypes were analyzed: wild‐type isogenic controls (WT), gcn5Δ, ppk18Δ, gcn5Δ ppk18Δ, pab1Δ, pab1Δ ppk18Δ, igo1Δ, igo1‐S64A, gcn5Δ igo1‐S64A, and pab1Δ igo1‐S64A. act1 + served as a control for normalization across samples. Values from a WT strain grown in rich medium were set at 1 to allow comparisons across culture conditions and mutant strains. Each column represents the mean value of four independent experiments, overlaid with individual data points and SEM. Statistical significance was determined by two‐way ANOVA followed by Bonferroni's multiple comparison tests (n = 4); *P ≤ 0.01.
-
DP‐Taf12 was followed by anti‐FLAG IB of protein extracts from WT, ppk18Δ, and igo1Δ strains, grown in rich conditions (R) or starved for 45 min (S). An anti‐tubulin IB is shown as a control for loading.
-
EP‐Taf12 was followed in WT, igoS64A, pab1Δ, and pab1Δ igoS64A strains, grown in rich conditions (R) or starved for 45 min (S). An anti‐Rpb1 IB is shown as a control for loading.
This function of Ppk18Gwl‐Igo1 opposes that of PP2A‐Pab1B55 and of SAGA‐Gcn5, which both inhibit differentiation when cells proliferate. We thus tested whether Ppk18Gwl and Igo1 genetically interact with Pab1B55 and Gcn5 to control differentiation. We observed that ste11 + and mei2 + expression profiles are similar between gcn5Δ single mutants and gcn5Δ ppk18Δ or gcn5Δ igo1‐S64A double mutants (Fig 5B and C). Likewise, the expression profiles of pab1Δ single mutant are similar to those of pab1Δ ppk18Δ and pab1Δ igo1‐S64A double mutants (Fig 5B and C). Accordingly, gcn5Δ and pab1Δ mutants suppress the sterility of ppk18Δ and igo1‐S64A mutants (Appendix Fig S8). Thus, the absence of either Pab1B55 or Gcn5 suppresses the inability of ppk18Δ or igo1‐S64A mutants to differentiate upon starvation. We conclude that Ppk18Gwl‐Igo1 inhibits PP2A‐Pab1B55 to allow sexual differentiation in response to starvation and that Gcn5 functions downstream of this pathway.
The Ppk18Gwl‐Igo1 pathway inhibits Taf12 de‐phosphorylation by PP2A‐Pab1B55
We next determined whether Taf12 phosphorylation is modulated by the Ppk18Gwl‐Igo1‐PP2A cascade, by analyzing P‐Taf12 levels in ppk18Δ and igo1Δ mutants, grown either in rich conditions or shifted to starvation medium. Western blot analyses and quantifications revealed that Taf12 phosphorylation is reduced in starved ppk18Δ, igo1Δ, and igo1‐S64A mutants, as compared to WT controls (Fig 5D and E). Therefore, Ppk18Gwl‐Igo1 contributes to Taf12 phosphorylation upon starvation. Our genetic analyses (Fig 5B and C) predicted that this function of Ppk18Gwl‐Igo1 would require PP2A‐Pab1B55 activity. Indeed, we observed that the pattern of Taf12 phosphorylation is similar between pab1Δ single mutants and pab1Δ igo1‐S64A double mutants (Fig 5E). Therefore, the absence of PP2A‐Pab1B55 rescues the inability of igo1‐S64A mutants to induce Taf12 phosphorylation. These observations indicate that, upon starvation, Ppk18Gwl‐Igo1 is important to inhibit PP2A‐Pab1B55 activity toward Taf12 and allow its phosphorylation.
The TORC2‐Gad8AKT pathway induces Taf12 phosphorylation upon starvation
Our results show that, in rich conditions, Taf12 is de‐phosphorylated by PP2A‐Pab1B55 downstream of TORC1. Upon starvation, inhibition of PP2A‐Pab1B55 by Ppk18Gwl‐Igo1 prevents Taf12 de‐phosphorylation. However, Taf12 phosphorylation is expected to result from the opposing activities of the PP2A‐Pab1B55 phosphatase, whose activity decreases upon starvation, and a kinase, whose activity should increase concomitantly. Indeed, we noticed that P‐Taf12 levels still increase upon starvation of both starved PP2A‐Pab1B55 and Ppk18Gwl‐Igo1 mutants (Figs 4D and 5D and E).
Our genetic analyses suggested that the TORC2‐Gad8AKT functionally interacts with SAGA to regulate differentiation (Fig 1C,D and F). We thus tested whether the TORC2‐Gad8AKT pathway controls Taf12 phosphorylation upon starvation. Western blot analyses and quantifications revealed that, upon shifting cells to starvation conditions, Taf12 phosphorylation is reduced both in tor1Δ and in gad8Δ mutants, as compared to WT controls, whereas total Taf12 levels do not change (Fig 6A). We verified that this reduction is not an indirect consequence of the defects in G1 arrest and sexual differentiation of tor1Δ and gad8Δ mutants 38, 62, 63. Both sty1Δ and ssp2Δ mutants are also unable to arrest in G1 and to differentiate upon starvation 42, 45. However, Taf12 phosphorylation remains unchanged in either sty1Δ or ssp2Δ mutants (Fig 6B), in agreement with our genetic analyses (Appendix Fig S1E–H).
Figure 6. The TORC2‐Gad8AKT pathway induces Taf12 phosphorylation upon starvation.
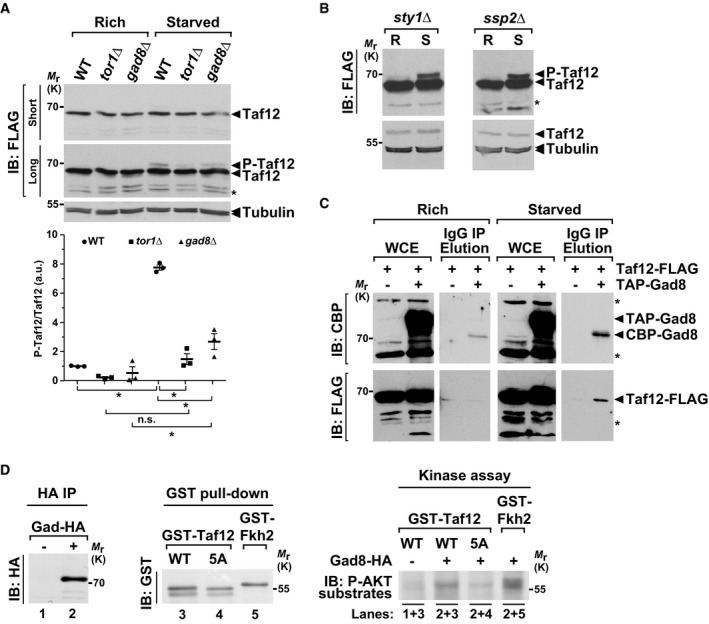
- P‐Taf12 was followed by anti‐FLAG IB of protein extracts from WT, tor1Δ, and gad8Δ strains, grown in rich conditions (R) or starved for 45 min (S). An anti‐tubulin IB is shown as a control for loading. The signal intensities of P‐Taf12 and total Taf12 were quantified in each strain and condition. P‐Taf12 to Taf2 ratios were calculated from three independent experiments and individually plotted in a graph below the IBs, together with the mean and SEM. Averaged values from all WT controls grown in rich medium were set at 1 to allow comparisons across culture conditions and mutant strains. Statistical significance was determined by two‐way ANOVA followed by Bonferroni's multiple comparison tests (n = 3); *P ≤ 0.01.
- P‐Taf12 was followed by anti‐FLAG IB of protein extracts from sty1Δ cells (left panel) or ssp2Δ cells (right panel), grown in rich conditions (R) or starved for 45 min (S). An anti‐tubulin IB is shown as a control for loading. Shown are IBs that are representative of two independent experiments.
- TAP‐tagged Gad8 was immuno‐precipitated using IgG‐Sepharose beads (IP) from strains containing FLAG‐tagged Taf12, grown either in rich medium (left panels) or starved for 45 min (right panels). TAP‐tagged Gad8 was eluted from beads using the TEV protease, releasing a shorter form of Gad8 (CBP‐Gad8). Eluates were loaded and immunoblotted (IB) using anti‐FLAG or anti‐CBP antibodies, together with 1% of whole cell extracts (WCE), to detect Taf12 co‐precipitation with Gad8. A strain which does not contain any TAP tag is shown as a negative control for the IP. Shown is an IB representative of two independent experiments.
- Endogenous Gad8‐HA was affinity‐purified from starved cells, in order to activate its kinase activity (left and middle panels), and mixed with recombinant, purified GST‐Taf12, GST‐Taf12‐5A, or GST‐Fkh2 proteins (right panels) in a kinase assay buffer. Each assay was then analyzed by IB and probed with an anti‐phospho‐AKT substrate antibody.
We then determined whether Gad8AKT interacts with Taf12, using a strain in which Taf12 and Gad8AKT were tagged with a FLAG and a TAP epitope, respectively. Affinity purification and elution of TAP‐tagged Gad8AKT specifically recover FLAG‐tagged Taf12, only in extracts from starved cells, suggesting that nutrient starvation promotes the interaction between Taf12 and Gad8AKT in vivo (Fig 6C). We then tested whether Gad8AKT is able to directly phosphorylate Taf12, by incubating affinity‐purified endogenous Gad8AKT with a recombinant fragment of Taf12, as well as a fragment of its well‐characterized substrate Fkh2 as a positive control (Fig 6D, left and middle panels). Kinase assays revealed that Gad8AKT phosphorylates Taf12 in vitro, as compared to a control “no tag” purification (Fig 6D, right panel). Importantly, using a Taf12‐5A mutant substrate, we found that Gad8AKT targets the same Ser/Thr residues that are phosphorylated upon starvation and de‐phosphorylated by PP2A‐Pab1B55. We conclude that Taf12 is a substrate of the Gad8AKT kinase, both in vitro and in vivo. Altogether, these results indicate that the TORC2‐Gad8AKT pathway mediates Taf12 phosphorylation in response to nutrient starvation.
Role of Taf12 phosphorylation during sexual differentiation
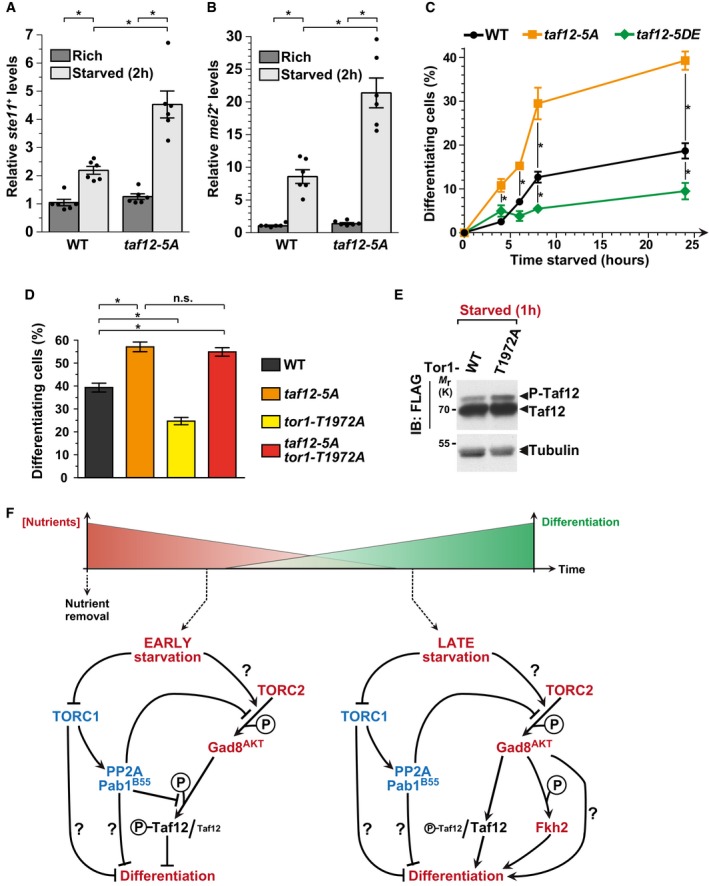
We next addressed the role of Taf12 phosphorylation in vivo. To do this, we quantified ste11 + and mei2 + expression and the number of differentiated cells in non‐phosphorylated taf12‐5A mutants. Surprisingly, both genes are more strongly induced upon starvation of taf12‐5A mutants, as compared to WT controls (Fig 7A and B). In agreement with Gad8AKT controlling Taf12 phosphorylation, the absence of Gad8AKT does not suppress the increased expression of ste11 + and mei2 + in starved taf12‐5A mutants (Appendix Fig S9). We then quantified the differentiation response of taf12‐5A mutants, at various time points upon nutrient starvation. In agreement with our previous observations, taf12‐5A mutants commit to differentiation more efficiently than WT cells (Fig 7C). To confirm this inhibitory role of Taf12 phosphorylation, we repeated this analysis using a strain in which all 5 Ser/Thr were mutated into aspartates (Asp) and glutamates (Glu) (taf12‐5DE), to mimic the negative charge of phosphorylation (Fig 3C). Indeed, in contrast to taf12‐5A mutants, we observed less differentiating cells in starved taf12‐5DE mutants, as compared to WT controls (Fig 7C). We noticed, however, that Taf12 phosphorylation represses differentiation genes only in the early time points of starvation (Appendix Fig S9 and data not shown), consistent with P‐Taf12 levels peaking early after starving cells (Fig 3I).
Figure 7. Taf12 phosphorylation inhibits sexual differentiation downstream of TORC2.
-
A, BExpression of ste11 + (A) and mei2 + (B) using quantitative RT–PCR of RNA extracted from cells grown either in rich medium (dark gray) or starved for 2 h (light gray). Cells of the following genotypes were analyzed: wild‐type isogenic controls (WT) and taf12‐5A mutants. act1 + served as a control for normalization across samples. Values from a WT strain grown in rich medium were set at 1 to allow comparisons across culture conditions and mutant strains. Each column represents the mean value of six independent experiments, overlaid with individual data points and SEM. Statistical significance was determined by two‐way ANOVA followed by Bonferroni's multiple comparison tests (n = 6); *P ≤ 0.01.
-
C, DZygotes and tetrads, which correspond to differentiated cells, were counted from cultures of homothallic cells grown to mid‐log phase in rich medium (t0) and shifted to starvation medium for up to 24 h (C) or from heterothallic cells mixed and incubated for 2 days on mating medium (D). Cells of the following genotypes were analyzed: wild‐type isogenic controls (WT), taf12‐5A, taf12‐5DE, tor1‐T1972A, and taf12‐5A tor1‐T1972A. Each value represents the mean percentage and SEM of differentiating cells to the total number of cells, averaged from six independent experiments. At least 200 cells from the indicated genotypes were counted in each experiment. Statistical significance was determined by two‐way ANOVA followed by Tukey's multiple comparison tests (n = 6); *P ≤ 0.01.
-
EP‐Taf12 was followed by anti‐FLAG IB of protein extracts from WT and tor1‐T1972A strains, grown in rich medium and shifted to starved conditions for 60 min. An anti‐tubulin IB is shown as a control for loading. Shown is an IB which is representative of two independent experiments.
-
FProposed model for the regulation and role of Taf12 phosphorylation during the early and late steps following nutrient starvation in S. pombe. Question marks represent unknown regulatory pathways. See Discussion for details.
We did not expect Taf12 phosphorylation to inhibit differentiation because we showed that it is induced upon starvation, by TORC2‐Gad8AKT, which promotes differentiation. However, a previous study has shown that constitutive activation of the TORC2 kinase Tor1 inhibits differentiation, similarly to a loss of Tor1 function 41. Therefore, although a basal level of TORC2‐Gad8AKT activity is required for cells to differentiate, it appears that TORC2‐Gad8AKT also buffers the differentiation response, for example to prevent too many cells from exiting the cell cycle immediately upon starvation. To address whether this function of TORC2 requires Taf12 phosphorylation, we determined the levels of P‐Taf12 in a constitutively active mutant of Tor1, tor1‐T1972A 41. Western blotting of FLAG‐tagged Taf12 revealed that its phosphorylation increases in starved tor1‐T1972A mutants, as compared to WT controls (Fig 7E). Altogether, our results show that TORC2 activity positively correlates with Taf12 phosphorylation (Figs 6A and 7E). We then asked whether tor1‐T1972A mutants differentiate less because of this increase in Taf12 phosphorylation. For this, we quantified sexual differentiation upon starvation of taf12‐5A tor1‐T1972A double mutants, as compared to single mutants and WT controls. We counted a similar number of differentiated cells between taf12‐5A tor1‐T1972A double mutants and taf12‐5A single mutants, indicating that taf12‐5A mutants rescue the reduced differentiation phenotype of tor1‐T1972A single mutants (Fig 7D). To conclude, upon nutrient starvation, TORC2 functions as both an activator and an inhibitor of sexual differentiation, the latter being mediated by Taf12 phosphorylation.
Importantly, we did not find any evidence of a loss of Taf12 function in either taf12‐5A or taf12‐5DE mutants. First, although S. pombe Taf12 is essential for viability 29, both mutants are viable and grow normally. Second, total protein staining of SAGA and TFIID complexes purified from either taf12‐5A or taf12‐5DE mutants, grown either in rich or starved conditions, revealed that the subunit composition of all mutant complexes is similar to that of WT cells, regardless of nutrient levels (Appendix Fig S10). Third, we did not observe any changes in the HAT activity of SAGA purified from either taf12‐5A or taf12‐5DE mutants, as compared to WT cells (Appendix Fig S11). Altogether, our results suggest that TORC2‐Gad8AKT‐mediated phosphorylation of Taf12 is important to modulate the timing and the amplitude of the sexual differentiation response upon nutrient starvation.
Discussion
Although it is well established that transcriptional co‐activator complexes regulate inducible genes, less is known about how they sense and respond to signaling cues. Here, we show that the SAGA and TFIID component Taf12 is differentially phosphorylated, depending on nutrient levels. Genetic and biochemical evidence support a model by which the opposing activities of the TORC1‐PP2AB55 and TORC2‐Gad8AKT pathways control the level of Taf12 phosphorylation, differentiation gene expression, and the switch from proliferation to mating and meiosis, in response to nutrient availability (Fig 7F).
Convergence of the TORC1 and TORC2 pathways on a transcriptional regulator
Nutrient availability is sensed by a network of regulatory pathways, many of which control proliferation through the TOR kinase. TOR forms two distinct complexes, TORC1 and TORC2, which differ in their composition, regulation, and function 3, 4. Here, we uncovered a previously unknown feature of the TORC1 and TORC2 pathways, which activate distinct effectors, the PP2A phosphatase, and the Gad8AKT kinase, respectively, to control the phosphorylation of the same target, Taf12. Taf12 is therefore an effector of the known, opposing regulatory roles of TORC1 and TORC2 in the control of gene expression and sexual differentiation in response to nutrients in S. pombe 7. However, our genetic analyses also indicate that TORC1 and TORC2 regulate sexual differentiation through other effectors than Taf12 and SAGA. For example, the absence of Gcn5 cannot completely rescue the lack of differentiation observed when TORC1 is constitutively activated or upon the loss of TORC2 (Fig 1). Future work will determine which other transcription factors and chromatin regulators contribute to the regulation of differentiation gene expression downstream of TORC1 and TORC2 in S. pombe.
Studies in various organisms have demonstrated that TORC1 promotes growth both by stimulating anabolic processes and by inhibiting stress responses, including through the regulation of RNA polymerase I‐, II‐, and III‐dependent transcription 3. For example, S. cerevisiae TORC1 represses nutrient stress‐responsive genes by sequestering transcription factors in the cytoplasm, primarily through the Tap42‐dependent inhibition of PP2A‐related phosphatases. Likewise, when nutrients are present, S. pombe TORC1 represses the expression of the master regulators of sexual differentiation, but the mechanism of this regulation is less well characterized 8, 9, 10, 11, 12. A previous study suggested that TORC1 inhibits meiosis through the phosphorylation‐dependent degradation of Mei2 12. Our work establishes that SAGA, a co‐activator of RNA polymerase II, is another important effector of TORC1 to regulate differentiation genes.
We found that TORC1 controls SAGA through Ppk18Gwl‐mediated inhibition of the PP2AB55 phosphatase. A very recent study proposed that a similar TORC1‐activated signaling cascade inhibits sexual differentiation, through the de‐phosphorylation of Gad8AKT by PP2AB55, thereby counteracting TORC2 activity 56. Our work reveals that an additional substrate of the TORC1‐PP2AB55 pathway contributes to control this process. However, we observed that Taf12 phosphorylation does not modulate sexual differentiation when cells are grown in nutrient rich conditions, indicating that PP2AB55 requires other effectors, possibly additional SAGA subunits, to inhibit differentiation when nutrients are present (Fig 7F).
TORC2 is also known to regulate RNA polymerase II‐dependent transcription, through the activation of the AKT kinase, which phosphorylates and inhibits FOXO transcription factors in mammals 4. In S. pombe, TORC2 and Gad8AKT are activated upon starvation and required for cells to arrest in G1 and initiate sexual differentiation (Fig 1) 38, 41, 62, 63. Interestingly, an S. pombe FOXO transcription factor, Fkh2, is also required for ste11 + induction and sexual differentiation upon nitrogen starvation 64 and is phosphorylated by Gad8AKT in vitro 39. Recently, a mutational analysis revealed that Gad8AKT‐dependent phosphorylation of Fkh2 is critical for sexual differentiation 56. Our results show that Taf12 phosphorylation is induced early upon starvation, positively correlates with TORC2 activity, but not any other nutrient‐sensing pathway, and negatively correlates with sexual differentiation. We therefore propose that the TORC2‐Gad8AKT pathway coordinates the opposing activities of Fkh2 and Taf12 at the promoters of differentiation genes to synchronize their expression with starvation response and allow the sequential induction of cell cycle exit, conjugation, and meiosis (Fig 7F).
Role of Taf12 phosphorylation in sexual differentiation
Intriguingly, phenotypic analyses revealed that Taf12 phosphorylation inhibits sexual differentiation upon starvation (Figs 7A–E and Appendix Fig S9). This function appears contradictory with our observation that Taf12 becomes phosphorylated upon starvation, by TORC2‐Gad8AKT. However, hyper‐activation of TORC2 upon starvation inhibits differentiation, similar to a loss of TORC2 41. Here, we show that this inhibitory role of TORC2 is mediated, at least in part, by Taf12 phosphorylation (Fig 7D and E).
One possible interpretation is that the phosphorylated form of Taf12 buffers the amplitude of the sexual differentiation response until cells reach a critical level of starvation. Similar functions have been described for two RNA‐binding proteins in S. pombe 65, 66. We propose that the ability of the TORC2‐Gad8AKT pathway to activate both a positive and a negative regulator of differentiation, Fkh2 and Taf12, respectively, contributes to buffering the differentiation response. Such a regulatory motif is reminiscent of an incoherent feedforward loop (Fig 7F), which can indeed establish pulse‐like dynamics 67. Schizosaccharomyces pombe may have evolved such regulation to ensure that not all cells in a population commit to meiosis and sporulation, particularly during a transient starvation event, because these processes are irreversible.
Another non‐mutually exclusive possibility is that Taf12 phosphorylation sequesters it in the cytoplasm, therefore preventing it from inducing differentiation genes early upon starvation. Then, Taf12 protein levels increase, reducing the proportion of phosphorylated Taf12 (Fig 3I) and, possibly, promoting its nuclear translocation. The TORC2‐Gad8AKT pathway might be required for this increase in Taf12 levels. De‐phosphorylated, nuclear Taf12 would induce the transcription of differentiating genes at later time points of starvation, to promote sexual differentiation once starvation has reached a critical level (Fig 7F).
Finally, we found that ste11 + and mei2 + expression is affected only at early time points of starvation in Taf12 phosphorylation mutants. Although this result is in agreement with our observation that P‐Taf12 levels peak early after starvation, it is conceivable that Taf12 phosphorylation regulates the transcription of other important differentiation genes. Transcriptome analyses of differentiating S. pombe cells have indeed identified approximately 2,000 genes whose expression changes in successive waves that correlate with the timing of each step of sexual differentiation 68, 69, 70. Finally, as opposed to Taf12 phosphorylation, we noted that Gad8AKT does not modulate the expression of differentiation genes early upon starvation (Appendix Fig S9). This observation might reflect the fact that Gad8AKT phosphorylates other substrates with an opposite function, such as Fkh2. Alternatively, Taf12 phosphorylation may be catalyzed by another kinase, which would be only active during the early steps following starvation.
Regulation of SAGA by Taf12 phosphorylation in response to environmental cues
We have previously shown that SAGA regulates the expression of differentiation genes and cell fate decisions in response to nutrient availability 28. Here, we provide genetic and biochemical evidence that SAGA functions downstream of TORC1 and TORC2, but not of any other nutrient‐sensing kinases. Several signaling pathways have already been shown to involve SAGA, consistent with its prominent role in the expression of stress‐regulated genes. Proteomic analyses of SAGA purified from S. cerevisiae or human cells have identified several phosphorylation sites on distinct subunits, including Taf12 71, 72, 73. However, few studies, if any, have explored whether these modifications vary in response to specific cues or signaling pathways. Our work shows that Taf12, a core component of both SAGA and TFIID, is rapidly and dynamically phosphorylated in response to changes in nutrient levels.
It is well established that co‐activator complexes are primarily regulated through their recruitment by promoter‐bound transcription factors. However, we showed that S. pombe SAGA is recruited to the promoters of differentiation genes regardless of nutrient levels, suggesting a different mode of regulation 28. Importantly, Taf12 is also phosphorylated as part of the TFIID general transcriptional factor complex, which functions both redundantly and specifically with SAGA in the regulation of transcription initiation 74, 75. Future studies will elucidate the exact contribution of S. pombe TFIID to the regulation of differentiation genes and cell fate decisions in response to nutrient availability.
Regardless, we show here that nutrient availability dictates whether Taf12 is phosphorylated or not. However, we found that neither Taf12 phosphorylation nor nutrient availability affect SAGA or TFIID subunit composition. Similarly, SAGA HAT activity remains unchanged in Taf12 phosphorylation mutants. We therefore propose that Taf12 phosphorylation causes a conformational change within SAGA and/or TFIID, for example to coordinate their regulatory activities at specific promoters. Taf12 is indeed an integral component of the core TAF module of both SAGA and TFIID, in which it forms a histone‐like dimer either with Ada1 or with Taf4, respectively 23, 25, 76, 77, 78, 79. This interaction is mediated by the histone‐fold domain (HFD) of Taf12, which is highly conserved and supports its essential functions in S. cerevisiae 80. All three phosphorylated Thr are outside, N‐terminal to the HFD (Fig 3B). We therefore speculate that these modifications will not impact the overall assembly of the complex but, rather, cause a subtle structural reorganization of specific modules. High‐resolution structural studies will determine whether Taf12 phosphorylation induces such structural rearrangements and how it modulates SAGA and TFIID regulatory functions.
Materials and Methods
Schizosaccharomyces pombe procedures, growth conditions, and differentiation tests
Standard culture media and genetic manipulations were used, as described in 81. For all nutrient starvation experiments, prototrophic cells were inoculated in rich minimal medium (Edinburgh Minimal Medium, EMM + 5 g/l NH4Cl and 2% glucose) and grown at 30°C to mid‐log phase (~0.5 × 107 cells/ml). Cells were then pelleted at room temperature, washed once with starvation minimal medium (EMM without NH4Cl and 0.5% glucose), inoculated in either rich or starved medium, and incubated for various times at 30°C. In the text, “nutrient starvation” thus refers to the complete removal of the nitrogen source and to a fourfold decrease in glucose concentration in the medium. For mating assays, homothallic prototrophs were grown as described above and the number of zygotes and asci was counted under differential interference contrast microscopy. Alternatively, heterothallic prototrophs were grown as described above, mixed in equal proportion, plated onto mating medium (SPAS), and incubated for 2 days at 25°C. The proportion of differentiating cells (%) was calculated by dividing the sum of zygotes (one zygote counted as two cells) plus asci (one ascus counted as two cells) by the total number of cells (at least 200 cells). Torin‐1 (4247/10, Tocris Bioscience), cAMP (A9501, Sigma), or thiamine (T4625, Sigma) were added to the medium at the time points and concentrations indicated.
Strain and plasmid constructions
All S. pombe strains used are listed in Appendix Table S2. Genetic crosses were performed by mating strains at 25°C on SPAS medium. Strains with gene deletions or epitope‐tagged proteins were constructed by targeting the respective open reading frame (ORF) with kanMX6, natMX6, or hphMX6 cassettes, using PCR‐based gene targeting and lithium‐acetate transformation, as described in 82, 83, 84, 85. For epitope tagging, the corresponding tag was fused to either the 5′ or 3′ end of the targeted ORF, removing either the initiating ATG or the stop codon, respectively. In each case, PCR amplification was performed using primers of 100 bases, with 80 bases to direct homologous recombination. All primers used are listed in Appendix Table S3. Transformants were screened for correct integration by PCR and Western blot analyses. Point mutations were knocked within specific genes using a pop in, pop out allele replacement strategy, as described in 86. The integrating plasmids were constructed using the pKS‐ura4 vector, in which PCR products, restriction digest products, or synthesized DNA fragments were sub‐cloned using a Gibson assembly kit, according to the manufacturer's instructions (New England Biolabs). Plasmids and strains containing point mutations were eventually verified by Sanger sequencing.
We generated several plasmids to generate distinct Taf12 phospho‐mutant strains. First, DHB60 was synthesized with a fragment of the taf12 + gene, ranging from nucleotides (nt) +547 to +1,150 (+1 is defined as the A from the initiating codon) and comprising an A‐to‐T mutation, designed at nt +847 to replace Thr283 with an Ala283 residue. The DHO744‐745 primer pair was then used to amplify taf12 + from DHB60. This product was assembled into pKS‐ura4 together with three distinct PCR products, generated from S. pombe genomic DNA using DHO854‐855, DHO856‐857, and DHO858‐859, to create DHB62. These PCR products encompass the first 566 base pairs (bp) of taf12 +, the last 251 bp of taf12 + fused to an NdeI restriction site for genomic integration, and 590 bp of taf12 + 3′UTR, respectively. DHB62 was digested with NdeI and integrated into S. pombe using lithium‐acetate transformation to generate DHP1205.
We then introduced additional single base‐pair changes using the QuikChange Site‐Directed Mutagenesis kit, according to the manufacturer's instructions (Agilent Technologies). First, we used DHO909‐910 and the DHB62 plasmid template to mutate taf12 + codons so that the Ser217, Thr218, Ser220, and Thr221 residues are each replaced with Ala residues, creating DHB63. Second, we used DHO1035‐1036 and the DHB63 plasmids to replace these Ala residues with Asp217, Glu218, Asp220, and Glu221 residues, creating DHB64. Last, we used DHO1037‐1038 and the DHB64 plasmids to replace the Thr283 residue of taf12 + with a Glu283 residue, creating DHB65. The DHB63 and DHB65 plasmids were digested with NdeI and integrated into S. pombe using lithium‐acetate transformation to generate DHP1221 and DHP1222, respectively.
The igo1‐S64A knock‐in mutant strain was obtained using the two‐step delitto perfeto procedure 87. For this, a portion of the igo1 + gene, ranging from nt +29 to +571, was synthesized with a TCA‐to‐GCT mutation at nt +499 to +501 to replace Ser64 with an Ala residue, generating DHB61. The DHO1120‐1121 primer pair was then used to amplify igo1 + from DHB61, for S. pombe transformation and generation of DHP1210 and DHP1287.
RT–qPCR analysis
Quantitative PCR analyses of cDNA were performed using RNA extracted from 50 ml of exponentially growing cells. Total RNA was purified using hot, acidic phenol, and contaminating DNA was removed by DNase I digestion, using the TURBO DNA‐free™ kit (Ambion). 1 μg of RNA was then reverse‐transcribed (RT) at 55°C with random hexa‐nucleotide primers or an oligo‐dT primer, using the Invitrogen SuperScript III first‐strand synthesis kit (Invitrogen). Fluorescence‐based quantitative PCR was performed with SYBR Green, using Stratagene Mx3005P systems. The thermal cycling conditions comprised an initial denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for 30 s, 60°C for 1 min, and 72°C for 1 min. All analyses were performed according to the MIQE guidelines 88. Relative cDNA quantities were calculated using the Stratagene MxPro software, from the slope produced by standard curves. These were performed for each primer pair on at least one cDNA sample in each experiment. Standard curve slopes were comprised between −3.5 (90% efficiency) and −3.15 (110% efficiency), with an r 2 > 0.9. All primer sequences used are listed in Appendix Table S3.
Protein extractions
Protein extracts were prepared from 25 or 10 ml of exponentially growing cells, using either standard lysis buffer (LB: 40 mM HEPES‐NaOH pH 7.4, 350 mM NaCl, 0.1% NP‐40, and 10% glycerol) or trichloroacetic acid (TCA) precipitation, respectively. For standard lysis, LB was supplemented with protease inhibitors, including cOmplete EDTA‐free cocktails tablets (04693132001, Roche), 100 mM PMSF, 1 mg/ml Bestatin, and 1 mg/ml pepstatin A, as well as phosphatase inhibitors, including 50 mM β‐glycerophosphate, 1 mM NaF, and 1 mM Na3VO4. Glass beads were added to the cell suspension, and lysis was performed using FastPrep (MP Biomedicals). Beads were removed by centrifugation, and extracts were then cleared by centrifugation at 15,000 g, 4°C, for 10 min. For TCA extraction, cell pellets were washed once in 1 ml of 20% TCA and re‐suspended in 100 μl of 20% TCA. Glass beads were added, and cells were lysed using a FastPrep (MP Biomedicals). 400 μl of 5% TCA was added to the homogenates that were then vortexed and spun at 15,000 g for 10 min. The pellet was re‐suspended in 4× protein sample loading buffer, and the pH was adjusted with 1 M Tris base before boiling. 5 μl of each sample was loaded on a 10% SDS–acrylamide gel, which was stained with Coomassie blue or to normalize for protein amounts across samples.
Immuno‐precipitations
Of 50–100 ml of exponentially growing cells was harvested and re‐suspended in IP lysis buffer (IP‐LB: 50 mM HEPES‐NaOH pH 7.4, 150 mM NaCl, 0.5% NP‐40, 1 mM EDTA, 10% glycerol), supplemented with protease inhibitors and, when necessary, with phosphatase inhibitors, as described for protein extraction. Following lysis and extract clarification, 4–5 mg of total proteins was incubated first with 1–5 μg of anti‐FLAG antibody (M2, Sigma), for 2 h at 4°C, and then with 50 μl of a 1:1 protein G Sepharose 4 Fast Flow slurry (GE Healthcare Life Sciences), for 1 h at 4°C. Three washes were performed in IP‐LB without any protease or phosphatase inhibitors. Beads were eluted in 1× protein sample loading buffer and processed for Western blotting.
For TAP‐Gad8 or TAP‐Pab1 immuno‐precipitations, proteins extracts were incubated overnight with 50 μl of a 1:1 IgG‐Sepharose 6 Fast Flow slurry (GE Healthcare Life Sciences), for 2 h at 4°C. Three washes were performed in IP‐LB without any protease or phosphatase inhibitors, followed by one wash in TEV buffer (10 mM Tris pH 8.0, 150 mM NaCl, 0.1% NP‐40, 0.5 mM EDTA, 10% glycerol, 1 mM DTT). IgG beads were incubated for 1.5 h at room temperature in 100 μl of TEV buffer and 20 U of AcTEV protease (Invitrogen). IgG beads were then pelleted, and the supernatant was processed for Western blotting.
Tandem affinity purifications
Protein complexes were purified by the tandem affinity purification (TAP) method, as described previously 28, 89, with minor modifications. 2–4 l of exponentially growing cells was harvested, snap‐frozen, and ground in liquid nitrogen using a Freezer/Mill® (Spex SamplePrep). Following purifications, 20 mM EGTA eluates were concentrated and separated on 4–20% gradient SDS–polyacrylamide Tris–glycine gels (Bio‐Rad). Total protein content was visualized either by silver staining, using the SilverQuest kit (Invitrogen), or using the SYPRO Ruby/Red protein stains (Molecular Probes), following the manufacturer's instructions. Stained gels were scanned and imaged using either direct light (Amersham Imager 600, GE Healthcare Life Sciences) or a fluorescence imaging system (Typhoon Trio, GE Healthcare Life Sciences). For analysis of phospho‐protein content, EGTA eluates were delipidated and desalted using chloroform and methanol precipitation before loading. Phosphorylated proteins were visualized in gel by Pro‐Q® Diamond staining, according to the manufacturer's instructions (Molecular Probes). Stained gels were imaged using a Typhoon Trio imager (GE Healthcare Life Sciences) (532 nm laser excitation, 580 nm emission filter). Quantification of signal intensity was performed from scanned gels using ImageJ 90.
Western blots
Western blotting was performed using a peroxidase‐anti‐peroxidase (PAP) antibody for detection of the TAP tag (P1291, Sigma), an anti‐Calmodulin binding protein (CBP) antibody (RCBP‐45A‐Z, ICLab), an anti‐Rpb1 antibody (8WG16, Covance), an anti‐tubulin antibody (B‐5‐1‐2, Sigma), an anti‐FLAG antibody (M2, Sigma), an anti‐MYC antibody (9E10, Agro‐Bio), an anti‐HA antibody (12CA5, Agro‐Bio), an anti‐phospho‐AKT substrate (PAS, Cell Signaling Technology), and an anti‐phospho‐Ser546‐Gad8AKT antibody 40. Protein concentrations were measured by the Bradford method and used to load equal amounts of proteins across samples. Quantification of signal intensity was performed using film exposures that were within the linear range of ECL detection, as verified by loading serial dilutions of one sample, and analyzed with Image Studio™ Lite 4.0 (LI‐COR Biosciences). Typically, P‐Taf12 levels were measured from longer exposures, whereas total Taf12 levels were measured from shorter exposures.
For Phos‐tag gels, 12% resolving SDS–acrylamide gels were supplemented with 50 μM Phos‐tag™‐acrylamide (AAL‐107, Wako) and 100 μM MnCl2. TCA‐extracted samples were run at 30 mA for 2 h. Gels were washed twice in transfer buffer (25 mM Tris–HCl, 250 mM glycine) containing 1 mM EDTA and once without EDTA, before being processed for Western blotting.
Phosphatase assays
TAP‐Pab1 and Taf12‐FLAG were affinity‐purified separately, as described above, from 100 to 200 ml of exponentially growing cells. For TAP‐Pab1, IgG beads were washed three times in IP‐LB, once in phosphatase buffer (50 mM HEPES‐NaOH pH 7.4, 100 mM NaCl, 0.1% NP‐40, 2 mM MnCl2, 2 mM DTT), re‐suspended in 100 μl phosphatase buffer containing 10 U of AcTEV protease, and incubated at room temperature for 1.5 h. Equal amounts of the supernatant, normalized on the signal intensity of CBP‐Pab1, were mixed with Taf12‐FLAG‐bound protein G Sepharose beads, with and without 0.5 μM microcystin. Reactions were incubated for 40 min at 30°C, before processing for Western blotting. For λ‐phosphatase treatment of Taf12‐FLAG immuno‐precipitates, protein G Sepharose beads were re‐suspended in 100 μl 1× λ‐phosphatase buffer containing 1 mM MnCl2, with and without 1 μl of λ‐phosphatase enzyme, and incubated for 35′ at 30°C. As a control, 1 mM NaF and 1 mM Na3VO4 were added to inhibit phosphatase activity. Reactions were stopped by adding 4× protein sample loading buffer and boiling at 95°C for 10 min.
Kinase assays
PCR fragments ranging from nucleotides (nt) +439 to +1011, corresponding to residues Leu147 to Asp337, of the S. pombe taf12 gene were amplified from genomic DNA of either DHP148 (WT) or DHP1221 (taf12‐5A mutants), using DHO1393‐1394. These fragments were sub‐cloned into pGEX‐4T2 (GE Healthcare Life Sciences), 3′ and in frame to the GST coding sequence, using a Gibson assembly kit (New England Biolabs) to generate the DHB82 and DHB83 plasmids, respectively. Recombinant GST‐Taf12 and GST‐Taf12‐5A were produced by IPTG induction of transformed BL21 strains, purified on a column of 200 μl of glutathione Sepharose 4B (GE Healthcare Life Sciences) for 2 h at 4°C, and eluted using 10 mM reduced glutathione. As a positive control, a fragment corresponding to amino acid residues 209–411 of Fkh2 39 was cloned, expressed, and purified using an identical strategy. Endogenous Gad8‐HA was affinity‐purified, as described above, from 100 ml of S. pombe cells that were grown in rich medium and starved for nitrogen for 30 min, to activate Gad8AKT. 10 μg of recombinant GST‐Taf12 or GST‐Taf12‐5A was mixed with Gad8‐HA‐bound protein G Sepharose beads and incubated for 30 min at 30°C, in kinase buffer (25 mM Tris–HCl pH 8.0, 10 mM MgCl2, 100 μM ATP, 1 mM DTT). Reactions were stopped by adding 4× protein sample loading buffer and boiling at 95°C for 10 min.
HAT assays
Histone acetyltransferase (HAT) assays were performed with 10% of TEV eluates of purified SAGA complexes, using the EpiQuik™ HAT Activity/Inhibition Assay Kit (Epigentek), according to the manufacturer's instructions.
SILAC and mass spectrometry analyses
SILAC procedure for SAGA purification analyses
For SILAC analyses of purified SAGA, lysine auxotrophs (DHP828: h − lys1‐131 spt7‐HA3‐TAP2::kanMX6) were pre‐cultured in minimal rich medium (EMM) supplemented with 225 mg/l of either “light” L‐lysine or “heavy” L‐lysine (13C6‐15N2‐L‐lysine, Cambridge Isotope Laboratories), for more than twelve doublings, to maximize metabolic labeling of proteins. Cells were grown to mid‐logarithmic phase, either in 2 l of EMM supplemented with 45 mg/l of either “light” L‐lysine or “heavy” L‐lysine. The “light” L‐lysine culture was then shifted to starved medium, without any L‐lysine, for 45 min. A reverse labeling scheme was used in an independent replicate of this experiment. Cells were harvested, snap‐frozen, and weighted. Equal quantities of dried cells were pooled before TAP purification, which was performed as described above, using Spt7‐TAP as the bait. EGTA eluates were processed for silver staining and for mass spectrometry analysis.
Mass spectrometry (MS) analysis
Peptides were re‐suspended in 5% formic acid, 5% acetonitrile and loaded onto a 100‐μm ID × 3‐cm precolumn packed with Maccel C18 1.9‐μm, 200‐Å particles (The Nest Group) for nRPLC‐MS/MS analysis in a Velos Orbitrap mass spectrometer (Thermo Fisher). Peptides were eluted over a 100‐μm ID × 30‐cm analytical column packed with the same material. Gradient conditions were tailored to the complexity and chemical properties of each sample; generally, the gradient was 9–32% acetonitrile in 0.15% formic acid over the course of 90 min. All MS spectra were collected with Orbitrap detection. MS/MS spectra were collected in the linear ion trap; the 20 most abundant ions were selected in a data‐dependent manner.
Identification and quantification of peptides, proteins, and modifications
MS/MS spectra were searched with Comet 91 against the S. pombe proteome (downloaded November 19, 2015). The precursor mass tolerance was set to 50 p.p.m., and the fragment ion tolerance was set to 0.36 Da with a 0.11‐Da offset. Variable modification of methionine oxidation (15.994914 Da), serine, threonine, and tyrosine phosphorylation (79.966331 Da) was used for all searches. For SILAC experiments, an additional variable modification of heavy lysine (8.014198 Da) was considered. Search results were filtered to a 1% FDR at PSM level using Percolator 92. Phosphorylation sites were localized using an in‐house implementation of the Ascore algorithm 93. Phosphorylation sites with an Ascore > 13 (P < 0.05) were considered confidently localized. Peptides were quantified using in‐house software measuring chromatographic peak maximum intensities. The peak maxima for peptides with the same combination of modification sites were summed before calculating a ratio. Proteins were quantified by first summing maximum peak intensities of all unmodified peptides for a given protein. The ratio was calculated between the sum of all peptides for a protein from heavy‐labeled cells and the sum from light‐labeled cells. In instances in which only the light or heavy form was identified, the noise signal level was used as the signal of the missing peptide.
SILAC and mass spectrometry analyses of total protein extracts
For SILAC analyses of total protein extracts, auxotrophic strains were cultured in minimal medium (EMM) supplemented with 225 mg/l of either “light” L‐lysine and L‐arginine (Lys‐0/Arg‐0) or “medium” L‐lysine and L‐arginine (2H4‐L‐lysine/13C6‐arginine, Lys4/Arg6). Supernatants derived from the “light”‐ and “medium”‐labeled cell cultures were combined, and proteins were precipitated at −20°C using ice‐cold acetone/methanol left on ice overnight. The proteins were pelleted by centrifugation (2,200 g, 20 min, 4°C) and washed with 80% ice‐cold acetone. Dried proteins were resolved in digestion buffer (6 M urea, 2 M thiourea, 10 mM Tris, pH 8.0) and mixed in 1:1 ratio according to measured protein amounts. The mixtures were digested in solution with trypsin as described previously 94. For proteome analyses, 100 μg of the mixtures was fractionated by isoelectric focusing on an OffGel 3100 Fractionator (Agilent) according to the manufacturer's instructions. Focusing was performed using 13‐cm (12‐well) Immobiline DryStrips pH 3–10 (Bio‐Rad) at a maximum current of 50 μA for 24 kVh. Peptide fractions were collected and desalted separately using C18 StageTips 95.
For phospho‐proteome analyses, 8 mg of each peptide mixture was subjected to phospho‐peptide enrichment as described previously 96, with minor modifications: Peptides were separated by strong cation‐exchange (SCX) chromatography with a gradient of 0–35% SCX solvent B resulting in seven fractions that were subjected to phospho‐peptide enrichment by TiO2 beads. Elution from the beads was performed three times with 100 μl of 40% ammonia hydroxide solution in 60% acetonitrile (pH > 10.5). Fractions rich in peptides were subjected to multiple TiO2 enrichment. Enrichment of phospho‐peptides from the SCX flow‐through was completed in five cycles.
LC‐MS/MS analyses were performed on an EasyLC nano‐HPLC (Proxeon Biosystems) coupled to an LTQ Orbitrap XL (Thermo Scientific) for phospho‐peptide analyses, or an LTQ Orbitrap Elite mass spectrometer (Thermo Scientific) for proteome analyses as described previously 97. The peptide mixtures were injected onto the column in HPLC solvent A (0.5% acetic acid) at a flow rate of 500 nl/min and subsequently eluted with a 87‐min (proteome) or a 127‐min (phospho‐proteome) segmented gradient of 5–33–90% HPLC solvent B (80% ACN in 0.5% acetic acid). During peptide elution, the flow rate was kept constant at 200 nl/min. For proteome analysis, the twenty most intense precursor ions were sequentially fragmented in each scan cycle. For the phospho‐proteome analysis, the five most intense precursor ions were fragmented by multistage activation of neutral loss ions at −98, −49, and −32.6 Th relative to the precursor ion 98. In all measurements, sequenced precursor masses were excluded from further selection for 90 s. Full scans were acquired at resolution of 60,000 (Orbitrap XL) or 120,000 (Orbitrap Elite). The target values were set to 5,000 charges for the LTQ (MS/MS) and 106 charges for the Orbitrap (MS), respectively; maximum allowed fill times were 150 ms (LTQ) and 1,000 ms (Orbitrap). The lock mass option was used for real‐time recalibration of MS spectra 96.
The MS data of all SILAC experiments were processed using default parameters of the MaxQuant software (v1.2.2.9) 99. Extracted peak lists were submitted to database search using the Andromeda search engine 100 to query a target‐decoy 101 database of S. pombe proteome (http://www.pombase.org/, Protein Dataset in FASTA format, downloaded on the April 6, 2011), containing 5,076 protein entries and 248 commonly observed contaminants.
Statistics
Statistical tests were performed using GraphPad Prism. t‐Tests were used when comparing two means. One‐way or two‐way analyses of variance (ANOVA) were performed for comparing more than two means, across one (for example “genotype”) or two distinct variables (for example “genotype” and “nutrients”). ANOVAs were followed by Bonferroni or Tukey post hoc pairwise comparisons. An α level of 0.01 was used a priori for all statistical tests, except otherwise indicated. Comparisons that are statistically significant (P ≤ 0.01) are marked with the star sign (*), whereas those that are statistically not significant (P > 0.01) are labeled n.s.
Author contributions
DH designed and supervised the project; TL, DD, CF, YR, and DH designed experiments, performed research, and analyzed data; RAR‐M performed the SILAC analyses of purified SAGA, supervised by JV; MF‐W and KK performed the SILAC analyses of total protein extracts, supervised by BM and JP; DH wrote the manuscript, and all authors reviewed and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank Sandra Lopez‐Aviles for sharing strains, protocols, and ideas, as well as Bérengère Pradet‐Balade and Fred Winston for critical reading of the manuscript. We thank an anonymous referee for contributing to the proposed model in Fig 7F. We also thank Gwendaline Lledo for invaluable technical assistance and all members of the Helmlinger laboratory for helpful suggestions and discussions. We are grateful to Fuyu Tamanoi (UCLA, USA), Tomohiro Matsumoto (Kyoto University, Japan), and Masayuki Yamamoto (University of Tokyo, Japan) for sharing strains. T.L. is a recipient of graduate fellowships from the Labex EpiGenMed and from the Fondation ARC pour la Recherche Contre le Cancer. D.D. is a recipient of a graduate fellowship from the Ligue Contre le Cancer. Y.R. was a recipient of post‐doctoral fellowships from the University of Montpellier and the Fondation pour la Recherche Médicale. This work has benefited from support by the Labex EpiGenMed, an “Investissements d'avenir” program (ANR‐10‐LABX‐12‐01). This work was supported by funds from the CNRS (ATIP‐Avenir), the FP7 Marie Curie Actions (FP7‐PEOPLE‐2012‐CIG/COACTIVATOR), the Fondation ARC (PJA‐20131200471), and the Agence Nationale de la Recherche (ANR‐15‐CE12‐0009‐01) to D.H.
EMBO Reports (2017) 18: 2197–2218
References
- 1. de Nadal E, Ammerer G, Posas F (2011) Controlling gene expression in response to stress. Nat Rev Genet 12: 833–845 [DOI] [PubMed] [Google Scholar]
- 2. Yuan H‐XX, Xiong Y, Guan K‐LL (2013) Nutrient sensing, metabolism, and cell growth control. Mol Cell 49: 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loewith R, Hall MN (2011) Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189: 1177–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149: 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Otsubo Y, Yamamato M (2008) TOR signaling in fission yeast. Crit Rev Biochem Mol Biol 43: 277–283 [DOI] [PubMed] [Google Scholar]
- 6. Davie E, Petersen J (2012) Environmental control of cell size at division. Curr Opin Cell Biol 24: 838–844 [DOI] [PubMed] [Google Scholar]
- 7. Otsubo Y, Yamamoto M (2012) Signaling pathways for fission yeast sexual differentiation at a glance. J Cell Sci 125: 2789–2793 [DOI] [PubMed] [Google Scholar]
- 8. Shinozaki‐Yabana S, Watanabe Y, Yamamoto M (2000) Novel WD‐repeat protein Mip1p facilitates function of the meiotic regulator Mei2p in fission yeast. Mol Cell Biol 20: 1234–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alvarez B, Moreno S (2006) Fission yeast Tor2 promotes cell growth and represses cell differentiation. J Cell Sci 119: 4475–4485 [DOI] [PubMed] [Google Scholar]
- 10. Matsuo T, Otsubo Y, Urano J, Tamanoi F, Yamamoto M (2007) Loss of the TOR kinase Tor2 mimics nitrogen starvation and activates the sexual development pathway in fission yeast. Mol Cell Biol 27: 3154–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valbuena N, Moreno S (2010) TOR and PKA pathways synergize at the level of the Ste11 transcription factor to prevent mating and meiosis in fission yeast. PLoS One 5: e11514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otsubo Y, Yamashita A, Ohno H, Yamamoto M (2014) Schizosaccharomyces pombe TORC1 activates the ubiquitin‐proteasomal degradation of the meiotic regulator Mei2 in cooperation with Pat1 kinase. J Cell Sci 127: 2639–2646 [DOI] [PubMed] [Google Scholar]
- 13. Rando OJ, Winston F (2012) Chromatin and transcription in yeast. Genetics 190: 351–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koutelou E, Hirsch CL, Dent SY (2010) Multiple faces of the SAGA complex. Curr Opin Cell Biol 22: 374–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weake VM, Workman JL (2012) SAGA function in tissue‐specific gene expression. Trends Cell Biol 22: 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grant PA, Duggan L, Côté J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen‐Hughes T, Allis CD, Winston F et al (1997) Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev 11: 1640–1650 [DOI] [PubMed] [Google Scholar]
- 17. Grant PA, Eberharter A, John S, Cook RG, Turner BM, Workman JL (1999) Expanded lysine acetylation specificity of Gcn5 in native complexes. J Biol Chem 274: 5895–5900 [DOI] [PubMed] [Google Scholar]
- 18. Sterner DE, Grant PA, Roberts SM, Duggan LJ, Belotserkovskaya R, Pacella LA, Winston F, Workman JL, Berger SL (1999) Functional organization of the yeast SAGA complex: distinct components involved in structural integrity, nucleosome acetylation, and TATA‐binding protein interaction. Mol Cell Biol 19: 86–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhaumik SR, Green MR (2002) Differential requirement of SAGA components for recruitment of TATA‐box‐binding protein to promoters in vivo . Mol Cell Biol 22: 7365–7371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horiuchi J, Silverman N, Piña B, Marcus GA, Guarente L (1997) ADA1, a novel component of the ADA/GCN5 complex, has broader effects than GCN5, ADA2, or ADA3. Mol Cell Biol 17: 3220–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grant PA, Schieltz D, Pray‐Grant MG, Steger DJ, Reese JC, Yates JR, Workman JL (1998) A subset of TAF(II)s are integral components of the SAGA complex required for nucleosome acetylation and transcriptional stimulation. Cell 94: 45–53 [DOI] [PubMed] [Google Scholar]
- 22. Wu P‐YJY, Winston F (2002) Analysis of Spt7 function in the Saccharomyces cerevisiae SAGA coactivator complex. Mol Cell Biol 22: 5367–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu P‐YJY, Ruhlmann C, Winston F, Schultz P (2004) Molecular architecture of the S. cerevisiae SAGA complex. Mol Cell 15: 199–208 [DOI] [PubMed] [Google Scholar]
- 24. Durand A, Bonnet J, Fournier M, Chavant V, Schultz P (2014) Mapping the deubiquitination module within the SAGA complex. Structure 22: 1553–1559 [DOI] [PubMed] [Google Scholar]
- 25. Han Y, Luo J, Ranish J, Hahn S (2014) Architecture of the Saccharomyces cerevisiae SAGA transcription coactivator complex. EMBO J 33: 2534–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Setiaputra D, Ross JD, Lu S, Cheng DT, Dong MQ, Yip CK (2015) Conformational flexibility and subunit arrangement of the modular yeast Spt‐Ada‐Gcn5 acetyltransferase complex. J Biol Chem 290: 10057–10070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu Y, Eriksson P, Bhoite LT, Stillman DJ (2003) Regulation of TATA‐binding protein binding by the SAGA complex and the Nhp6 high‐mobility group protein. Mol Cell Biol 23: 1910–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helmlinger D, Marguerat S, Villén J, Gygi SP, Bähler J, Winston F (2008) The S. pombe SAGA complex controls the switch from proliferation to sexual differentiation through the opposing roles of its subunits Gcn5 and Spt8. Genes Dev 22: 3184–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helmlinger D, Marguerat S, Villén J, Swaney DL, Gygi SP, Bähler J, Winston F (2011) Tra1 has specific regulatory roles, rather than global functions, within the SAGA co‐activator complex. EMBO J 30: 2843–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anandhakumar J, Fauquenoy S, Materne P, Migeot V, Hermand D (2013) Regulation of entry into gametogenesis by Ste11: the endless game. Biochem Soc Trans 41: 1673–1678 [DOI] [PubMed] [Google Scholar]
- 31. Mach KE, Furge KA, Albright CF (2000) Loss of Rhb1, a Rheb‐related GTPase in fission yeast, causes growth arrest with a terminal phenotype similar to that caused by nitrogen starvation. Genetics 155: 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakase Y, Fukuda K, Chikashige Y, Tsutsumi C, Morita D, Kawamoto S, Ohnuki M, Hiraoka Y, Matsumoto T (2006) A defect in protein farnesylation suppresses a loss of Schizosaccharomyces pombe tsc2+, a homolog of the human gene predisposing to tuberous sclerosis complex. Genetics 173: 569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Urano J, Sato T, Matsuo T, Otsubo Y, Yamamoto M, Tamanoi F (2007) Point mutations in TOR confer Rheb‐independent growth in fission yeast and nutrient‐independent mammalian TOR signaling in mammalian cells. Proc Natl Acad Sci USA 104: 3514–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murai T, Nakase Y, Fukuda K, Chikashige Y, Tsutsumi C, Hiraoka Y, Matsumoto T (2009) Distinctive responses to nitrogen starvation in the dominant active mutants of the fission yeast Rheb GTPase. Genetics 183: 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watanabe Y, Lino Y, Furuhata K, Shimoda C, Yamamoto M (1988) The S. pombe mei2 gene encoding a crucial molecule for commitment to meiosis is under the regulation of cAMP. EMBO J 7: 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sugimoto A, Iino Y, Maeda T, Watanabe Y, Yamamoto M (1991) Schizosaccharomyces pombe ste11+ encodes a transcription factor with an HMG motif that is a critical regulator of sexual development. Genes Dev 5: 1990–1999 [DOI] [PubMed] [Google Scholar]
- 37. Davidson MK, Shandilya HK, Hirota K, Ohta K, Wahls WP (2004) Atf1‐Pcr1‐M26 complex links stress‐activated MAPK and cAMP‐dependent protein kinase pathways via chromatin remodeling of cgs2+. J Biol Chem 279: 50857–50863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsuo T, Kubo Y, Watanabe Y, Yamamoto M (2003) Schizosaccharomyces pombe AGC family kinase Gad8p forms a conserved signaling module with TOR and PDK1‐like kinases. EMBO J 22: 3073–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ikeda K, Morigasaki S, Tatebe H, Tamanoi F, Shiozaki K (2008) Fission yeast TOR complex 2 activates the AGC‐family Gad8 kinase essential for stress resistance and cell cycle control. Cell Cycle 7: 358–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Du W, Hálová L, Kirkham S, Atkin J, Petersen J (2012) TORC2 and the AGC kinase Gad8 regulate phosphorylation of the ribosomal protein S6 in fission yeast. Biol Open 1: 884–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hálová L, Du W, Kirkham S, Smith DL, Petersen J (2013) Phosphorylation of the TOR ATP binding domain by AGC kinase constitutes a novel mode of TOR inhibition. J Cell Biol 203: 595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Valbuena N, Moreno S (2012) AMPK phosphorylation by Ssp1 is required for proper sexual differentiation in fission yeast. J Cell Sci 125: 2655–2664 [DOI] [PubMed] [Google Scholar]
- 43. Davie E, Forte GM, Petersen J (2015) Nitrogen regulates AMPK to control TORC1 signaling. Curr Biol 25: 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kato T, Okazaki K, Murakami H, Stettler S, Fantes PA, Okayama H (1996) Stress signal, mediated by a Hog1‐like MAP kinase, controls sexual development in fission yeast. FEBS Lett 378: 207–212 [DOI] [PubMed] [Google Scholar]
- 45. Shiozaki K, Russell P (1996) Conjugation, meiosis, and the osmotic stress response are regulated by Spc1 kinase through Atf1 transcription factor in fission yeast. Genes Dev 10: 2276–2288 [DOI] [PubMed] [Google Scholar]
- 46. Elmlund H, Baraznenok V, Linder T, Szilagyi Z, Rofougaran R, Hofer A, Hebert H, Lindahl M, Gustafsson CM (2009) Cryo‐EM reveals promoter DNA binding and conformational flexibility of the general transcription factor TFIID. Structure 17: 1442–1452 [DOI] [PubMed] [Google Scholar]
- 47. Nakashima A, Sato T, Tamanoi F (2010) Fission yeast TORC1 regulates phosphorylation of ribosomal S6 proteins in response to nutrients and its activity is inhibited by rapamycin. J Cell Sci 123: 777–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- 49. De Virgilio C, Loewith R (2006) Cell growth control: little eukaryotes make big contributions. Oncogene 25: 6392–6415 [DOI] [PubMed] [Google Scholar]
- 50. Sarkar S, Dalgaard JZ, Millar JB, Arumugam P (2014) The Rim15‐endosulfine‐PP2ACdc55 signalling module regulates entry into gametogenesis and quiescence via distinct mechanisms in budding yeast. PLoS Genet 10: e1004456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janssens V, Goris J (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353: 417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kinoshita N, Ohkura H, Yanagida M (1990) Distinct, essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell 63: 405–415 [DOI] [PubMed] [Google Scholar]
- 53. Kinoshita K, Nemoto T, Nabeshima K, Kondoh H, Niwa H, Yanagida M (1996) The regulatory subunits of fission yeast protein phosphatase 2A (PP2A) affect cell morphogenesis, cell wall synthesis and cytokinesis. Genes Cells 1: 29–45 [DOI] [PubMed] [Google Scholar]
- 54. Jiang W, Hallberg RL (2000) Isolation and characterization of par1(+) and par2(+): two Schizosaccharomyces pombe genes encoding B' subunits of protein phosphatase 2A. Genetics 154: 1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, Smith DL, Pines J, Hagan IM (2015) A PP1‐PP2A phosphatase relay controls mitotic progression. Nature 517: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martin R, Portantier M, Chica N, Nyquist‐Andersen M, Mata J, Lopez‐Aviles S (2017) A PP2A‐B55‐mediated crosstalk between TORC1 and TORC2 regulates the differentiation response in fission yeast. Curr Biol 27: 175–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP‐competitive mammalian target of rapamycin inhibitor reveals rapamycin‐resistant functions of mTORC1. J Biol Chem 284: 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Atkin J, Halova L, Ferguson J, Hitchin JR, Lichawska‐Cieslar A, Jordan AM, Pines J, Wellbrock C, Petersen J (2014) Torin1‐mediated TOR kinase inhibition reduces Wee1 levels and advances mitotic commitment in fission yeast and HeLa cells. J Cell Sci 127: 1346–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. De Virgilio C (2012) The essence of yeast quiescence. FEMS Microbiol Rev 36: 306–339 [DOI] [PubMed] [Google Scholar]
- 60. Lorca T, Castro A (2013) The Greatwall kinase: a new pathway in the control of the cell cycle. Oncogene 32: 537–543 [DOI] [PubMed] [Google Scholar]
- 61. Chica N, Rozalen AE, Perez‐Hidalgo L, Rubio A, Novak B, Moreno S (2016) Nutritional control of cell size by the greatwall‐endosulfine‐PP2A.B55 pathway. Curr Biol 26: 319–330 [DOI] [PubMed] [Google Scholar]
- 62. Kawai M, Nakashima A, Ueno M, Ushimaru T, Aiba K, Doi H, Uritani M, Not Available Not A (2001) Fission yeast tor1 functions in response to various stresses including nitrogen starvation, high osmolarity, and high temperature. Curr Genet 39: 166–174 [DOI] [PubMed] [Google Scholar]
- 63. Weisman R, Choder M (2001) The fission yeast TOR homolog, tor1+, is required for the response to starvation and other stresses via a conserved serine. J Biol Chem 276: 7027–7032 [DOI] [PubMed] [Google Scholar]
- 64. Shimada M, Yamada‐Namikawa C, Murakami‐Tonami Y, Yoshida T, Nakanishi M, Urano T, Murakami H (2008) Cdc2p controls the forkhead transcription factor Fkh2p by phosphorylation during sexual differentiation in fission yeast. EMBO J 27: 132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsukahara K, Yamamoto H, Okayama H (1998) An RNA binding protein negatively controlling differentiation in fission yeast. Mol Cell Biol 18: 4488–4498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jeong HT, Ozoe F, Tanaka K, Nakagawa T, Matsuda H, Kawamukai M (2004) A novel gene, msa1, inhibits sexual differentiation in Schizosaccharomyces pombe . Genetics 167: 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461 [DOI] [PubMed] [Google Scholar]
- 68. Mata J, Lyne R, Burns G, Bähler J (2002) The transcriptional program of meiosis and sporulation in fission yeast. Nat Genet 32: 143–147 [DOI] [PubMed] [Google Scholar]
- 69. Mata J, Bähler J (2006) Global roles of Ste11p, cell type, and pheromone in the control of gene expression during early sexual differentiation in fission yeast. Proc Natl Acad Sci USA 103: 15517–15522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Amorim MJ, Cotobal C, Duncan C, Mata J (2010) Global coordination of transcriptional control and mRNA decay during cellular differentiation. Mol Syst Biol 6: 380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mischerikow N, Spedale G, Altelaar AF, Timmers HT, Pijnappel WW, Heck AJ (2009) In‐depth profiling of post‐translational modifications on the related transcription factor complexes TFIID and SAGA. J Proteome Res 8: 5020–5030 [DOI] [PubMed] [Google Scholar]
- 72. Chen SH, Albuquerque CP, Liang J, Suhandynata RT, Zhou H (2010) A proteome‐wide analysis of kinase‐substrate network in the DNA damage response. J Biol Chem 285: 12803–12812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spedale G, Timmers HT, Pijnappel WW (2012) ATAC‐king the complexity of SAGA during evolution. Genes Dev 26: 527–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee TI, Causton HC, Holstege FC, Shen WC, Hannett N, Jennings EG, Winston F, Green MR, Young RA (2000) Redundant roles for the TFIID and SAGA complexes in global transcription. Nature 405: 701–704 [DOI] [PubMed] [Google Scholar]
- 75. Huisinga KL, Pugh BF (2004) A genome‐wide housekeeping role for TFIID and a highly regulated stress‐related role for SAGA in Saccharomyces cerevisiae . Mol Cell 13: 573–585 [DOI] [PubMed] [Google Scholar]
- 76. Gangloff YG, Werten S, Romier C, Carre L, Poch O, Moras D, Davidson I (2000) The human TFIID components TAF(II)135 and TAF(II)20 and the yeast SAGA components ADA1 and TAF(II)68 heterodimerize to form histone‐like pairs. Mol Cell Biol 20: 340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Selleck W, Howley R, Fang Q, Podolny V, Fried MG, Buratowski S, Tan S (2001) A histone fold TAF octamer within the yeast TFIID transcriptional coactivator. Nat Struct Biol 8: 695–700 [DOI] [PubMed] [Google Scholar]
- 78. Brand M, Leurent C, Mallouh V, Tora L, Schultz P (1999) Three‐dimensional structures of the TAFII‐containing complexes TFIID and TFTC. Science 286: 2151–2153 [DOI] [PubMed] [Google Scholar]
- 79. Bieniossek C, Papai G, Schaffitzel C, Garzoni F, Chaillet M, Scheer E, Papadopoulos P, Tora L, Schultz P, Berger I (2013) The architecture of human general transcription factor TFIID core complex. Nature 493: 699–702 [DOI] [PubMed] [Google Scholar]
- 80. Moqtaderi Z, Yale JD, Struhl K, Buratowski S (1996) Yeast homologues of higher eukaryotic TFIID subunits. Proc Natl Acad Sci USA 93: 14654–14658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Forsburg SL, Rhind N (2006) Basic methods for fission yeast. Yeast 23: 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR‐based gene targeting in Schizosaccharomyces pombe . Yeast 14: 943–951 [DOI] [PubMed] [Google Scholar]
- 83. Sato M, Dhut S, Toda T (2005) New drug‐resistant cassettes for gene disruption and epitope tagging in Schizosaccharomyces pombe . Yeast 22: 583–591 [DOI] [PubMed] [Google Scholar]
- 84. Hentges P, Van Driessche B, Tafforeau L, Vandenhaute J, Carr AM (2005) Three novel antibiotic marker cassettes for gene disruption and marker switching in Schizosaccharomyces pombe . Yeast 22: 1013–1019 [DOI] [PubMed] [Google Scholar]
- 85. Van Driessche B, Tafforeau L, Hentges P, Carr AM, Vandenhaute J (2005) Additional vectors for PCR‐based gene tagging in Saccharomyces cerevisiae and Schizosaccharomyces pombe using nourseothricin resistance. Yeast 22: 1061–1068 [DOI] [PubMed] [Google Scholar]
- 86. Gao J, Kan F, Wagnon JL, Storey AJ, Protacio RU, Davidson MK, Wahls WP (2013) Rapid, efficient and precise allele replacement in the fission yeast Schizosaccharomyces pombe . Curr Genet 60: 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Storici F, Lewis LK, Resnick MA (2001) In vivo site‐directed mutagenesis using oligonucleotides. Nat Biotechnol 19: 773–776 [DOI] [PubMed] [Google Scholar]
- 88. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL et al (2009) The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55: 611–622 [DOI] [PubMed] [Google Scholar]
- 89. Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- 90. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Eng JK, Jahan TA, Hoopmann MR (2013) Comet: an open‐source MS/MS sequence database search tool. Proteomics 13: 22–24 [DOI] [PubMed] [Google Scholar]
- 92. Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ (2007) Semi‐supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4: 923–925 [DOI] [PubMed] [Google Scholar]
- 93. Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP (2006) A probability‐based approach for high‐throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24: 1285–1292 [DOI] [PubMed] [Google Scholar]
- 94. Borchert N, Dieterich C, Krug K, Schutz W, Jung S, Nordheim A, Sommer RJ, Macek B (2010) Proteogenomics of Pristionchus pacificus reveals distinct proteome structure of nematode models. Genome Res 20: 837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906 [DOI] [PubMed] [Google Scholar]
- 96. Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C‐trap. Mol Cell Proteomics 4: 2010–2021 [DOI] [PubMed] [Google Scholar]
- 97. Koch A, Krug K, Pengelley S, Macek B, Hauf S (2011) Mitotic substrates of the kinase aurora with roles in chromatin regulation identified through quantitative phosphoproteomics of fission yeast. Sci Signal 4: rs6 [DOI] [PubMed] [Google Scholar]
- 98. Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ (2004) A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal Chem 76: 3590–3598 [DOI] [PubMed] [Google Scholar]
- 99. Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- 100. Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- 101. Elias JE, Gygi SP (2007) Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File