

Abstract
MAVS signalosome plays an important role in RIG‐I‐like receptor (RLR)‐induced antiviral signaling. Upon the recognition of viral RNAs, RLRs activate MAVS, which further recruits TRAF6 and other signaling proteins to initiate type I interferon (IFN) activation. MAVS signalosome also regulates virus‐induced apoptosis to limit viral replication. However, the mechanisms that control the activity of MAVS signalosome are still poorly defined. Here, we report NLRP11, a Nod‐like receptor, is induced by type I IFN and translocates to mitochondria to interact with MAVS upon viral infection. Using MAVS as a platform, NLRP11 degrades TRAF6 to attenuate the production of type I IFNs as well as virus‐induced apoptosis. Our findings reveal the regulatory role of NLRP11 in antiviral immunity by disrupting MAVS signalosome.
Keywords: apoptosis, MAVS, NLRP11, TRAF6, type I IFNs
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Introduction
Innate immune responses against viral infection start with the recognition of pathogen‐associated molecular patterns (PAMPs) by pattern‐recognition receptors (PRRs), leading to the production of type I interferons (IFNs) 1. The RIG‐I‐like receptor (RLR) family, including RIG‐I, MDA5, and LGP2, are the primary PRRs to recognize viral RNAs and initiate antiviral responses 2. Upon binding to viral RNA, RIG‐I undergoes conformation change which subsequently leads to its interaction with MAVS 3. MAVS then aggregates and serves as a platform for recruiting downstream proteins, including TRAF3, TRAF5, and TRAF6, to form a large signalosome 4, 5. Subsequently, the MAVS signalosome activates IRF3/7 and NF‐κB pathway, leading to the production of type I IFNs (IFNα/β) and pro‐inflammatory cytokines 5, 6.
Sustained RLR activation results in extensive cell damage as well as the apoptosis via mitochondria‐dependent mechanism 7, 8. It is generally accepted that apoptosis of infected cells is critical in suppression of viral replication and production of progeny viruses 9. Recently, studies have clarified the essential roles of MAVS in the initiation of virus‐induced apoptosis 7, 10. Thus, MAVS signalosome is also known as an important mediator in antiviral responses, due to its dual functions in virus‐induced type I IFNs and apoptosis.
Nod‐like receptors (NLRs) are a large family of cytosolic proteins activated by intracellular PAMPs and danger‐associated molecular patterns (DAMPs) 11, 12. The best‐characterized NLRs are NOD1 and NOD2, which initiate innate immune signaling by activating RIP2 via their CARD domains upon binding to PAMPs, leading to the activation of MAPK and NF‐κB signaling pathways 13. Unlike NOD1 and NOD2, NLRP1, NLRP3, and NLRC4 form the large protein complexes called “inflammasome” with procaspase‐1 to mediate the mature of IL‐1β and IL‐18 14. In addition, recent studies have identified several NLRs functioned as negative regulators in innate immune responses, including NLRC5, NLRP4, NLRX1, and NLRC3, through diverse mechanisms 15, 16, 17, 18, 19. However, the regulatory roles of several other NLRs in antiviral responses are still need to be characterized. In this study, we identified NLRP11 as a negative regulator in antiviral responses. Upon viral infection, NLRP11 is upregulated and translocates to mitochondria, and subsequently attenuating the activation of MAVS signalosome by promoting the degradation of TRAF6. Besides inhibiting IRF3 activation, NLRP11 also suppresses virus‐induced apoptosis in a MAVS‐dependent manner, which serves as a dual mediator to maintain homeostasis of innate antiviral responses.
Results
NLRP11 is a negative regulator of type I IFN signaling induced by RNA viruses
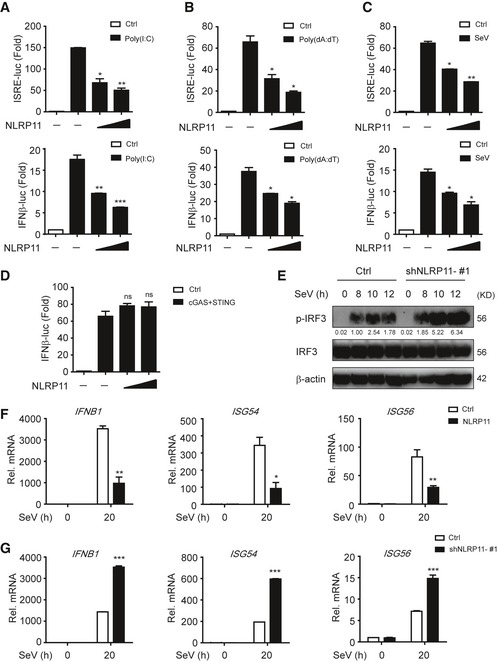
NLRP11 is a NLR protein which specifically exists in primates 20, and the biological function of NLRP11 in innate antiviral responses remains unclear. We found poly(I:C)‐ and poly(dA:dT)‐induced IFN‐stimulated response element (ISRE) or IFN‐β activation was remarkably attenuated by NLRP11 (Fig 1A and B). Similarly, overexpression of NLRP11 inhibited Sendai virus (SeV, a RNA virus)‐induced ISRE or IFN‐β activation (Fig 1C). Since both RNA and DNA viral infection activated the productions of type I IFNs, we also examined the functions of NLRP11 in IFN pathway induced by cGAS, which has been identified as a cytosolic sensor for DNA viruses through the adaptor STING 21. However, NLRP11 barely affected cGAS‐induced IFN‐β promoter activation via STING‐dependent pathway (Fig 1D). These results suggested NLRP11 specifically suppressed RLR‐mediated type I IFN signaling. To confirm the antiviral function of NLRP11, we constructed NLRP11 overexpressing and knockdown (shNLRP11) THP‐1 cell lines, respectively (Fig EV1A and B). Knockdown of NLRP11 enhanced IRF3 phosphorylation upon SeV, but not Herpes simplex virus type 1 (HSV‐1, a DNA virus) infection (Figs 1E and EV1C). In addition, the mRNA levels of IFNB1, IFN‐stimulated gene 54 (ISG54), and IFN‐stimulated gene 56 (ISG56) were inversely associated with the amount of NLRP11 after SeV infection (Figs 1F and G, and EV1D). In order to investigate the function of NLRP11 in primary cells, we knocked down endogenous NLRP11 in human peripheral blood mononuclear cells (PBMCs) by NLRP11‐specific siRNAs, and found that the expressions of IFNB1 and its downstream molecules ISG54 and ISG56 were enhanced, but SeV phosphoprotein expression was decreased in NLRP11‐knockdown PBMCs (Fig EV1E). Moreover, IFN‐β protein secretion was also increased in NLRP11‐knockdown PBMCs upon SeV infection (Fig EV1F). Collectively, our finding suggested that knockdown of NLRP11 enhanced type I IFN signaling induced by RLRs.
Figure 1. NLRP11 inhibits the activation of type I IFN signaling.
-
A–C293T cells were transfected with an ISRE or IFN‐β promoter reporter plasmid and pRL‐TK plasmid, together with an empty vector (EV) or NLRP11 construct for 24 h, and then transfected with poly(I:C) (5 μg/ml) (A), poly(dA:dT) (5 μg/ml) (B), or infected with Sendai virus (SeV) (MOI = 0.1) for 20 h (C), followed by ISRE‐ or IFN‐β‐dependent luciferase activity (fold induction) analysis. The data were normalized by using the values of ISRE‐luc or IFN‐β‐luc divided by the values of TK‐luc, and then, the results of each group were analyzed to compare with the control group.
-
D293T cells were transfected with the IFN‐β promoter reporter plasmid and pRL‐TK plasmid, together with an empty vector or cGAS and STING plasmids and increasing amount of NLRP11 for 24 h, and analyzed for IFN‐β‐dependent luciferase activity (fold induction).
-
EImmunoblot analysis of the total and phosphorylated (p‐) IRF3 in THP‐1 cells stably transduced with recombinant lentivirus expressing empty vector or shNLRP11‐#1, which were left untreated or infected with SeV (MOI = 1) for indicated time points. Numbers between two blots indicate densitometry of phosphorylated proteins relative to that of total proteins, respectively.
-
F, GExpression of IFNB1, ISG54, and ISG56 mRNA in NLRP11 overexpressing THP‐1 cells (F) or NLRP11‐knockdown THP‐1 cells (G) infected with SeV (MOI = 1) for indicated time points.
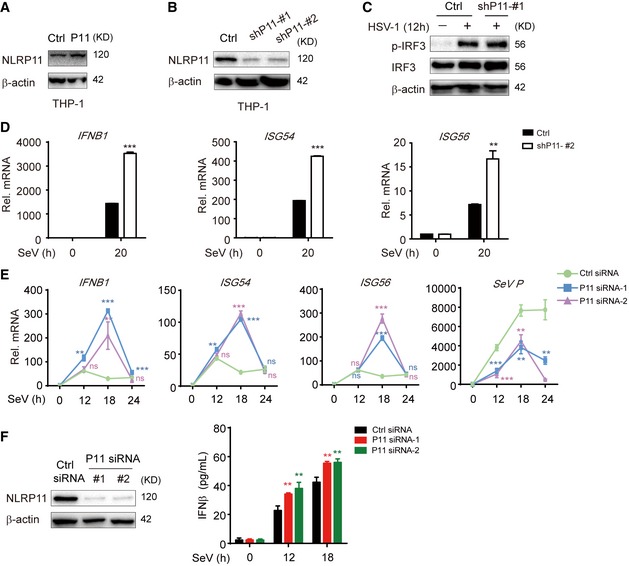
Figure EV1. Generation of NLRP11 overexpression or knockdown THP‐1 cell lines and knockdown of NLRP11 enhanced type I IFN signaling.
-
A, BThe lentivirus‐based NLRP11 (A) or NLRP11‐specific shRNAs (GIPZ shRNA set) (B) (puromycin resistance) were infected into THP‐1 cells. Next, these cells were selected using puromycin for at least 2 weeks. Immunoblot analysis of NLRP11 overexpression (A) or knockdown (B) in THP‐1 cells with anti‐NLRP11 antibodies.
-
CImmunoblot analysis of the total and phosphorylated (p‐) IRF3 in THP‐1 cells stably transduced with recombinant lentivirus expressing empty vector or shNLRP11‐#1, which were left untreated or were infected with HSV‐1 (MOI = 1) for 12 h.
-
DExpression of IFNB1, ISG54, and ISG56 mRNAs in NLRP11‐knockdown THP‐1 cells infected with Sendai virus (SeV) (MOI = 1) for indicated time points.
-
EExpression of IFNB1, ISG54, and ISG56 and SeV phosphoprotein mRNAs in control or NLRP11‐knockdown PBMCs upon SeV (MOI = 1) infection were determined by real‐time PCR.
-
FThe knockdown efficiency of NLRP11‐specific siRNAs in PBMCs was tested by immunoblot analysis (left). IFN‐β protein secretion was detected by ELISA in WT or NLRP11‐knockdown PBMCs upon SeV (MOI = 1) infection (right).
NLRP11 deficiency enhances IFN‐β expression as well as antiviral responses
To further confirm the negative role of NLRP11 in RLR‐induced antiviral responses, we constructed NLRP11 knockout (KO) 293T and THP‐1 cells, respectively, by the clustered regulatory interspersed short palindromic repeat (CRISPR)/CRISPR‐associated protein (Cas) system 22. The KO efficiency of NLRP11 was confirmed by immunoblot analysis and DNA sequencing (Fig EV2A and B). ISRE or IFN‐β activation was enhanced in NLRP11 KO cells after poly(I:C), poly(dA:dT) treatment, or SeV infection (Fig 2A and B). Next, we expressed a sgRNA‐resistant version of NLRP11 in NLRP11 KO cells and found it can reverse the enhancement of type I IFN activation caused by NLRP11 deficiency (Fig EV2C). In NLRP11 KO THP‐1 cells, the phosphorylation of IRF3 was enhanced compared to wild‐type (WT) cells upon SeV infection (Fig 2C). Consistently, the mRNA levels of IFNB1, ISG54, and ISG56 in NLRP11 KO THP‐1 cells were significantly increased after SeV, but not HSV‐1 infection (Figs 2D and EV2D). Moreover, pro‐inflammatory cytokines, such as IL6 and TNFA, were also upregulated in NLRP11 KO THP‐1 cells upon SeV infection (Fig EV2E). As expected, we found that NLRP11 deficiency reduced the number of GFP‐positive cells compared with WT THP‐1 cells upon vesicular stomatitis virus tagged with enhanced green fluorescent protein (VSV‐eGFP) infection (Fig 2E and F). Taking together, these data suggested that NLRP11 was a specific negative regulator in RLR pathway and limited the production of antiviral cytokines during antiviral immunity.
Figure EV2. Construction of NLRP11 knockout cell lines and function of NLRP11 in HSV‐1‐mediated IFNs or SeV‐mediated NF‐κB activation.
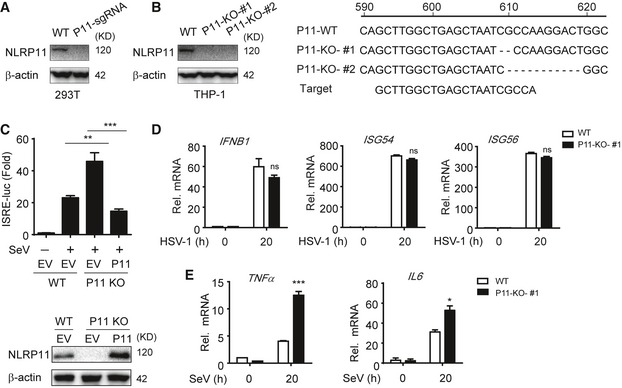
- 293T cells were transfected with NLRP11 sgRNA which contains anti‐puromycin gene, and then screened for three passages using puromycin and subjected to immunoblot analysis.
- Sequence alignment and immunoblot analysis of the targeted DNAs in WT and NLRP11 KO THP‐1 cells. THP‐1 cells were infected by lentivirus‐based Cas9‐ and NLRP11‐specific sgRNA (puromycin resistance). Next, these cells were selected using puromycin for at least 2 weeks. Finally, the cells were diluted to be cultured in 96‐well plates with one‐third cell per well, and individual cell clones were evaluated by immunoblotting and sequencing.
- 293T cells were transfected with NLRP11 sgRNA, and then screened for three passages using puromycin. Next, 293T‐NLRP11 sgRNA or WT cells were transfected with an ISRE promoter reporter plasmid and pRL‐TK plasmid, along with empty vector, or a sgRNA‐resistant NLRP11 plasmid for 24 h, and analyzed for ISRE‐dependent luciferase activity (fold induction) after infected with SeV (MOI = 0.1) for 20 h (top). NLRP11 was detected by immunoblotting (bottom).
- WT and NLRP11 KO THP‐1 cells were infected with HSV‐1 (MOI = 1) for 20 h, and then, IFNB1, ISG54, and ISG56 induction were measured by real‐time PCR.
- Expression of TNFA, IL6 mRNAs in NLRP11 knockout THP‐1 cells infected with SeV (MOI = 1) for indicated time points.
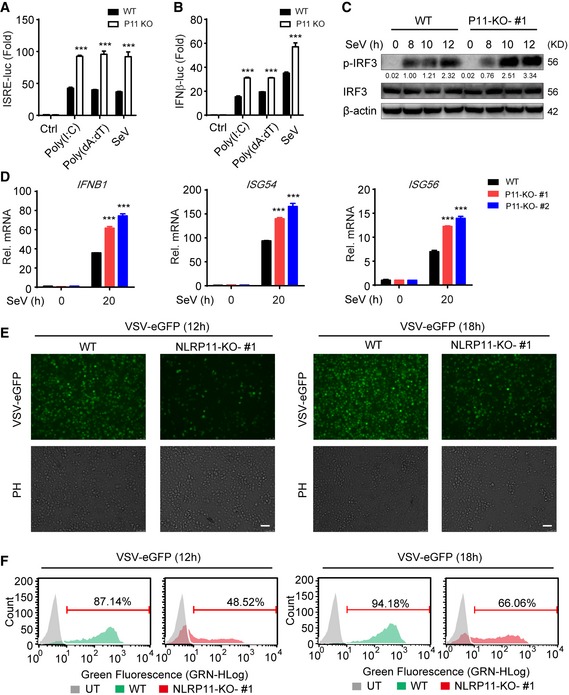
Figure 2. Knockout of NLRP11 enhances the production of IFN‐β and antiviral responses.
-
A, BWild‐type (WT) and NLRP11 knockout (KO) 293T cells were transfected with an ISRE (A) or IFN‐β (B) promoter reporter plasmid and pRL‐TK plasmid for 24 h, and then transfected with poly(I:C) (5 μg/ml) (A), poly(dA:dT) (5 μg/ml), or infected with Sendai virus (SeV) (MOI = 0.1) for 20 h, followed by ISRE‐ or IFN‐β‐dependent luciferase activity (fold induction) analysis.
-
CWT and NLRP11 KO THP‐1 cells were infected with SeV (MOI = 1) for indicated time points, and total and phosphorylated (p‐) IRF3 were analyzed by immunoblot analysis. Numbers between two blots indicate densitometry of phosphorylated proteins relative to that of total proteins, respectively.
-
DWT and NLRP11 KO THP‐1 cells were infected with SeV (MOI = 1) for 20 h, and then, IFNB1, ISG54, and ISG56 induction were measured by real‐time PCR.
-
E, FWT and NLRP11 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 10) for 12 h or 18 h, followed by phase‐contrast (PH) and fluorescence microscopy analysis (E) and flow cytometric analysis (F). Numbers above bracketed lines indicated the percentage of VSV‐eGFP‐infected cells. Scale bar, 100 μm.
NLRP11 inhibits IRF3 activation by targeting MAVS
To determine the molecular mechanisms by which NLRP11 inhibits type I IFN signaling, we co‐transfected 293T cells with expression vectors encoding RIG‐I (CARD) (an active domain of RIG‐I), MDA5, MAVS, TBK1, or IRF3 (5D) (a constitutively active mutant of IRF3) together with the ISRE luciferase reporter and the increasing amounts of NLRP11. We found that NLRP11 inhibited ISRE reporter activity induced by RIG‐I (CARD), MDA5 and MAVS, but not TBK1 or IRF3 (5D) (Figs 3A and EV3A). We also found NLRP11 inhibited IRF3 dimerization induced by RIG‐I (CARD) and MAVS, but not TBK1 (Fig EV3B). These results suggested that NLRP11 markedly inhibited type I IFN signaling at MAVS level. Next, we sought to determine whether NLRP11 could directly interact with MAVS or other signaling proteins within the type I IFN pathway. Co‐immunoprecipitation (Co‐IP) experiments revealed that NLRP11 strongly interacted with MAVS (Fig 3B). Moreover, endogenous NLRP11 weakly interacted with MAVS in THP‐1 cells, while the interaction between NLRP11 and MAVS was notably increased upon SeV infection (Fig 3C). Since NLRP11 expression was upregulated after SeV infection (Fig 3C), we overexpressed NLRP11 in 293T cells to eliminate the expression differences of NLRP11 during viral infection and found that the interaction between NLRP11 and MAVS was consistently enhanced during SeV infection (Fig EV3C). These results indicated that NLRP11 associated with MAVS during viral infection. Next, we investigated which domain of MAVS was responsible for its interaction with NLRP11. Since the CARD domain of MAVS is essential for RIG‐I‐MAVS interaction and transmembrane (TM) domain is critical for MAVS's mitochondria localization 23, we generated two MAVS deletion mutants, MAVS‐∆CARD and MAVS‐∆TM, respectively (Fig 3D). We found that the CARD deletion mutant markedly reduced the interaction between NLRP11 and MAVS (Fig 3E). In addition, the TM domain of MAVS was also essential for its interaction with NLRP11, since the deletion of TM domain in MAVS (MAVS‐∆TM) completely abolished NLRP11‐MAVS interaction (Fig 3E). These results indicated that both TM domain and CARD domain of MAVS were important for its interaction with NLRP11. TM domain guaranteed the mitochondria localization of MAVS to allow NLRP11 to approach it upon viral infection, while MAVS might directly interact with NLRP11 through its CARD domain. To identify the functional domains of NLRP11, we generated three domain constructs of NLRP11: NLRP11‐PYD, NLRP11‐NOD, and NLRP11‐LRR (Fig 3F). NOD and LRR, but not PYD, could interact with the full‐length MAVS protein (Fig 3G), as well as inhibit the ISRE activity induced by MAVS (Fig 3H). These results suggested that NLRP11‐LRR and NLRP11‐NOD were required for its binding ability with MAVS.
Figure 3. NLRP11 associates with MAVS to inhibit IRF3 activation.
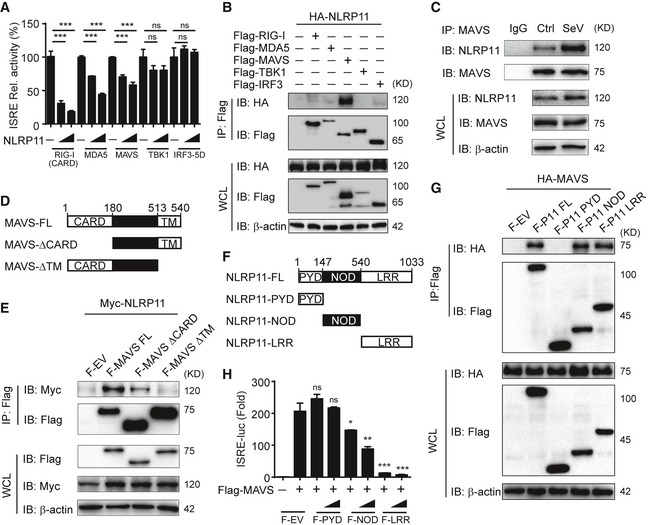
- Luciferase activity in 293T cells transfected with the ISRE promoter reporter plasmid and pRL‐TK plasmid for 24 h, together with expression plasmids encoding RIG‐I‐CARD, MDA5, MAVS, TBK1, and IRF3 (5D), and increasing amount of NLRP11.
- 293T cells were transfected with HA‐NLRP11 and Flag‐RIG‐I, Flag‐MDA5, Flag‐MAVS, Flag‐TBK1, and Flag‐IRF3 for 24 h, and cell lysates were subjected to immunoprecipitation with anti‐Flag beads, followed by immunoblot analysis with indicated antibodies.
- NLRP11 and MAVS interaction was analyzed by co‐immunoprecipitation assay in THP‐1 monocytes upon Sendai virus (SeV) (MOI = 1) infection for 20 h.
- The domain structure of MAVS. Numbers in parentheses indicate amino acid position in the construct.
- 293T cells were transfected with Myc‐NLRP11 and Flag‐MAVS, Flag‐MAVS‐ΔCARD, and Flag‐MAVS‐ΔTM for 24 h, and cell lysates were subjected to immunoprecipitation with anti‐Flag beads, followed by immunoblot analysis with indicated antibodies.
- The domain structure of NLRP11. Numbers in parentheses indicate amino acid position in the construct.
- 293T cells were transfected with HA‐MAVS and Flag‐NLRP11, Flag‐NLRP11‐PYD, Flag‐NLRP11‐NOD, and Flag‐NLRP11‐LRR for 24 h, and cell lysates were subjected to immunoprecipitation with anti‐Flag beads, followed by immunoblot analysis with indicated antibodies.
- Luciferase activity in 293T cells transfected with the ISRE promoter reporter, pRL‐TK plasmid, and MAVS or empty vector (EV), together with expression plasmids encoding Flag‐NLRP11‐PYD, Flag‐NLRP11‐NOD, or Flag‐NLRP11‐LRR for 24 h.
Figure EV3. NLRP11 suppresses MAVS activation, but does not affect the ubiquitination of MAVS or its interaction with RIG‐I.
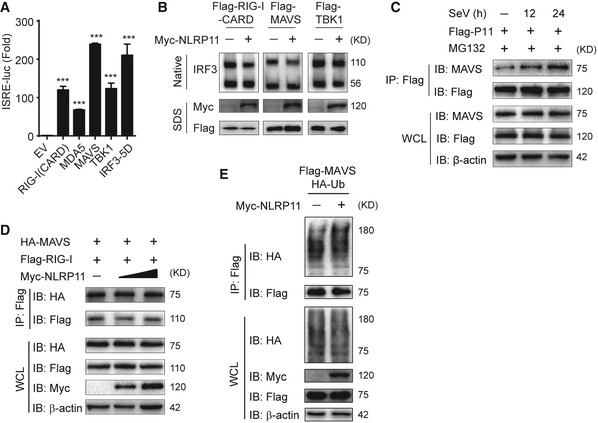
- Activation of RIG‐I (CARD)‐, MDA5‐, MAVS‐, TBK1‐, or IRF3 (5D)‐mediated ISRE activity in 293T cells. Data are expressed as means ± SEM of three independent experiments (***P < 0.001 versus cells transfected with EV with the same treatment, Student's t‐test).
- 293T cells were transfected with Flag‐RIG‐I‐CARD, Flag‐MAVS and Flag‐TBK1 together with empty vector or Myc‐NLRP11 for 24 h, and then, cell lysates were subjected to IRF3 dimer analysis by native PAGE (top) and RIG‐I‐CARD, MAVS, TBK1, and NLRP11 expression was detected by SDS–PAGE (bottom).
- 293T cells were transfected with Flag‐NLRP11, and then infected by SeV (MOI = 1) for indicated time points. MG132 was used to treat the cells for 6 h before harvesting. Whole‐cell extracts were immunoprecipitated with anti‐Flag beads, followed by IB analysis with anti‐MAVS antibody.
- 293T cells were transfected with Flag‐RIG‐I and HA‐MAVS together with increasing amount of NLRP11 for 24 h, and then, cell lysates were analyzed by co‐immunoprecipitation and immunoblot analysis with indicated antibodies.
- 293T cells were transfected with Flag‐MAVS and HA‐Ub along with empty vector or Myc‐NLRP11. Total ubiquitination of MAVS was analyzed by immunoblot.
Source data are available online for this figure.
NLRP11 targets TRAF6 for degradation in MAVS signalosome
Next, we investigated how NLRP11 negatively regulated MAVS‐mediated antiviral responses. Co‐IP assays showed NLRP11 did not disrupt the interaction between MAVS and its upstream molecule, RIG‐I (Fig EV3D). In addition, overexpression of NLRP11 barely affected the ubiquitination of MAVS (Fig EV3E), which is an important signal for MAVS activation in type I IFN signaling 5. It has been reported that MAVS polymers recruited multiple TRAF proteins to form MAVS signalosome, which finally led to activation of NF‐κB and IRF3 5, 6, 23. Then, we examined the interaction of NLRP11 and TRAFs. Co‐IP assays showed NLRP11 strongly interacted with TRAF6, rather than TRAF3 or TRAF5 (Fig 4A). We found TRAF6 is necessary for activating type I IFNs, and NLRP11 solely inhibited TRAF6‐induced ISRE activation in human cells (Fig EV4A and B). The interaction between NLRP11 and TRAF6 was further determined by endogenous co‐IP assay (Fig 4B). Our data revealed that the association between NLRP11 and TRAF6 was also markedly increased upon SeV infection (Fig EV4C). In TRAF6 KO 293T cells, NLRP11 failed to inhibit the ISRE promoter activity induced by SeV infection (Fig 4C). These findings suggested that TRAF6 was critical for NLRP11‐mediated inhibition of IFN activation.
Figure 4. NLRP11 promotes TRAF6 degradation in a MAVS‐dependent manner.
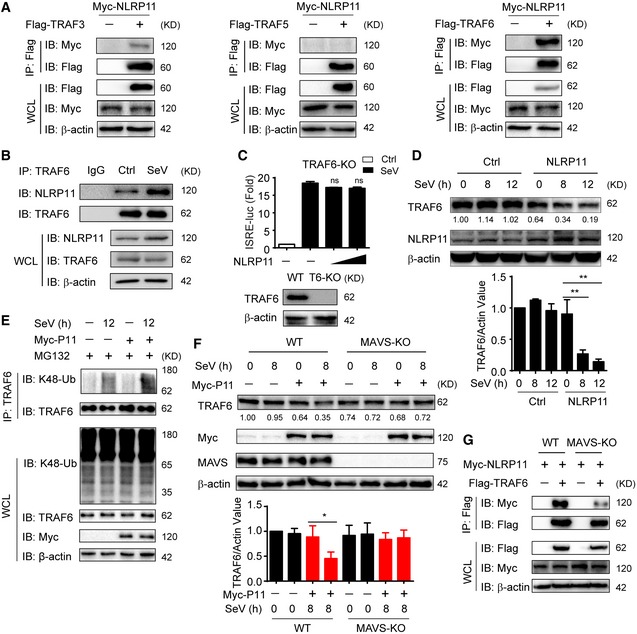
- 293T cells were transfected with Myc‐NLRP11 and Flag‐TRAF3, Flag‐TRAF5, or Flag‐TRAF6 for 24 h, and cell lysates were subjected to immunoprecipitation with anti‐Flag beads, followed by immunoblot analysis with indicated antibodies.
- NLRP11 and TRAF6 interaction was analyzed by co‐immunoprecipitation assay in THP‐1 monocytes upon Sendai virus (SeV) (MOI = 1) infection for 20 h.
- Luciferase activity in TRAF6 knockout (KO) 293T cells (bottom) transfected with ISRE promoter reporter and pRL‐TK plasmid and increasing amount of NLRP11 followed by SeV (MOI = 0.1) infection for 20 h. Data are expressed as means ± SEM of three independent experiments, versus cells transfected with EV with the same treatment, Student's t‐test, ns: no significant.
- Immunoblot analysis of indicated proteins in NLRP11 overexpressing THP‐1 cells followed with SeV (MOI = 1) infection for indicated time points (top). Numbers between two blots indicate densitometry of TRAF6 relative to that of β‐actin. Three independent experiments were quantified (bottom). Data are expressed as means ± SD of three independent experiments (**P < 0.01 versus uninfected cells, Student's t‐test).
- 293T cells were transfected with empty vector or Myc‐NLRP11 for 12 h, infected with SeV (MOI = 1) for 12 h, and then subjected to MG132 treatment for 6 h before harvesting. The cell lysates were subjected to immunoprecipitation with anti‐TRAF6 antibody followed by immunoblot analysis with indicated antibodies.
- Wild‐type (WT) and MAVS KO 293T cells were transfected with empty vector or Myc‐NLRP11 for 24 h, and the cells were then infected with SeV (MOI = 0.1) for indicated time points. Cell lysates were subjected to immunoblot analysis with indicated antibodies (top). Numbers between two blots indicate densitometry of TRAF6 relative to that of β‐actin. Three independent experiments were quantified (bottom). Data are expressed as means ± SD of three independent experiments (*P < 0.05 versus uninfected cells with the same treatment, Student's t‐test).
- WT and MAVS KO 293T cells were transfected with Myc‐NLRP11 and Flag‐TRAF6 for 24 h, and the cell lysates were analyzed by immunoprecipitation assay.
Source data are available online for this figure.
Figure EV4. NLRP11 promotes the proteasomal degradation of TRAF6.
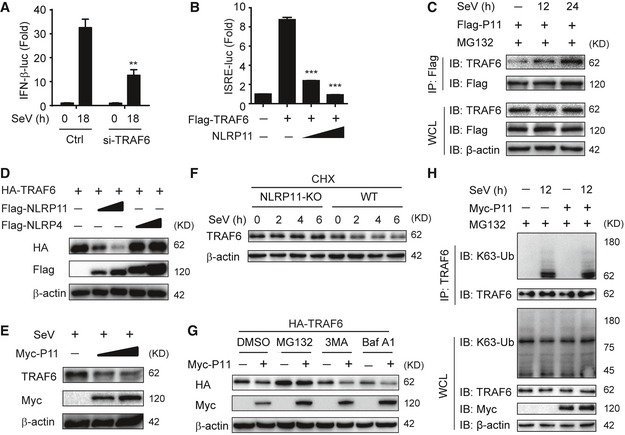
-
A293T cells were transfected with control or TRAF6 siRNA for 24 h, followed by transfection with an IFN‐β promoter reporter plasmid and pRL‐TK plasmid for 12 h, and analyzed for IFN‐β‐dependent luciferase activity (fold induction) after SeV (MOI = 0.1) infection for indicated time points.
-
BLuciferase activity in 293T cells transfected with the ISRE promoter reporter plasmid and pRL‐TK plasmid, together with expression plasmid encoding TRAF6 and increasing amount of NLRP11.
-
C293T cells were transfected with Flag‐NLRP11, and then infected by SeV (MOI = 1) for indicated time points. MG132 was used to treat the cells for 6 h before harvesting. Whole‐cell extracts were immunoprecipitated with anti‐Flag beads, followed by IB analysis with anti‐TRAF6 antibody.
-
D, EImmunoblot analysis of HA‐TRAF6 (D) or endogenous TRAF6 (E) in 293T cells transfected with Flag‐NLRP11 or Flag‐NLRP4 (D) or infected with Sendai virus (SeV) (MOI = 1) (E) for 24 h.
-
FWT or NLRP11 KO THP‐1 cells were pretreated with CHX for 2 h, and then infected with SeV (MOI = 1) for indicated time points, followed by immunoblot analysis.
-
G293T cells were transfected with Myc‐NLRP11 and HA‐TRAF6 for 24 h, and then treated with the indicated inhibitors for 6 h, followed by immunoblot analysis.
-
H293T cells were transfected with empty vector or Myc‐NLRP11 for 12 h, infected with SeV (MOI = 1) for 12 h, and then subjected to MG132 treatment for 6 h before harvesting. The cell lysates were subjected to immunoprecipitation with anti‐TRAF6 antibody followed by immunoblot analysis with indicated antibodies.
When TRAF6 and NLRP11 or NLRP4 were overexpressed in 293T cells, we observed that TRAF6 expression was lower in the presence of increasing amounts of NLRP11, but not NLRP4 (Fig EV4D). This finding prompted us to investigate the effect of NLRP11 on TRAF6 abundance. Next, we observed that endogenous TRAF6 protein level was decreased in NLRP11 overexpressing THP‐1 or 293T cells upon SeV infection (Figs 4D and EV4E). Consistently, TRAF6 was stabilized in NLRP11 KO cells by SeV infection in the presence of cycloheximide (CHX) (Fig EV4F). We also found that the proteasome inhibitor MG132, but not the autophagic sequestration inhibitor 3‐methyladenine (3‐MA) or the lysosomal acidification inhibitor bafilomycin A1 (Baf A1), inhibited the degradation of TRAF6 mediated by NLRP11 (Fig EV4G). Moreover, NLRP11 enhanced the K48‐, but not K63‐linked ubiquitination of endogenous TRAF6 upon SeV infection (Figs 4E and EV4H). These results suggested that NLRP11 induced the degradation of TRAF6 through enhancing its K48‐linked ubiquitination.
Since NLRP11 targets MAVS to inhibit IFN signaling (Fig 3B–E), we speculated whether MAVS plays as a platform for NLRP11 to degrade TRAF6. Indeed, MAVS deficiency abolished NLRP11‐induced TRAF6 degradation (Fig 4F). In addition, co‐IP assay showed that the interaction between NLRP11 and TRAF6 was remarkably suppressed in MAVS KO cells (Fig 4G). Taken together, these results indicated that NLRP11 regulated TRAF6 degradation in a MAVS‐dependent manner.
Virus infection induces NLRP11 expression and its mitochondria translocation
Immunoblot analysis showed that NLRP11 protein level is upregulated upon SeV infection (Figs 3C and 4B). To confirm it, we treated the THP‐1 and HeLa cells with poly(I:C) or SeV infection. We found that SeV infection and poly(I:C) stimulation increased NLRP11 mRNA and protein level in THP‐1 and HeLa cells (Figs 5A and B, and EV5A and B). As both poly(I:C) and SeV activate type I IFN signaling, we speculated that NLRP11 expression might rely on type I IFN secretion. Indeed, IFN‐β treatment increased NLRP11 expression in THP‐1 and HeLa cells (Figs 5C and EV5C), indicating that NLRP11 is an ISG gene, which can form a negative feedback loop to regulate type I IFN signaling.
Figure 5. NLRP11 is induced and aggregated to mitochondria by type I IFNs.
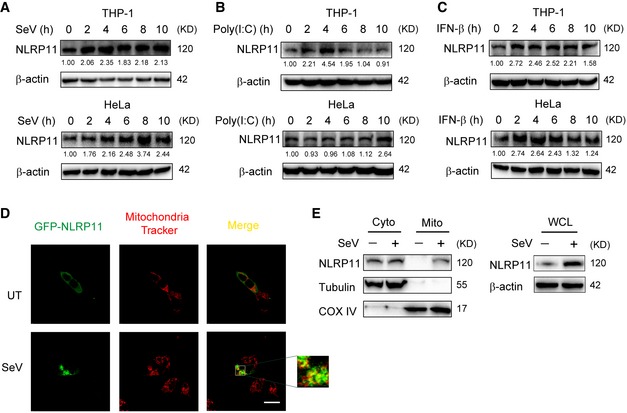
-
A–CImmunoblot analysis of NLRP11 protein expression in THP‐1 monocytes and HeLa cells infected with Sendai virus (SeV) (MOI = 1), transfected with poly(I:C) (10 μg/ml), or treated with IFN‐β (1,000 U/ml) for indicated time points. Numbers between two blots indicate densitometry of NLRP11 relative to that of β‐actin.
-
D293T cells were transfected with GFP‐NLRP11, and then were left untreated (UT) or infected with SeV (MOI = 1) for 12 h, followed by confocal fluorescence microscopy analysis. Mitochondria were detected with mitochondria tracker (red). Scale bar, 20 μm.
-
EHeLa cells were infected with SeV (MOI = 1) or not for 12 h, and then subjected to mitochondria isolation using cell mitochondria isolation kit (Beyotime Biotechnology) followed by immunoblot analysis. Tubulin and COX IV serve as cytosolic and mitochondrial markers, respectively.
Source data are available online for this figure.
Figure EV5. NLRP11 is induced and translocates to mitochondria by type I IFNs.
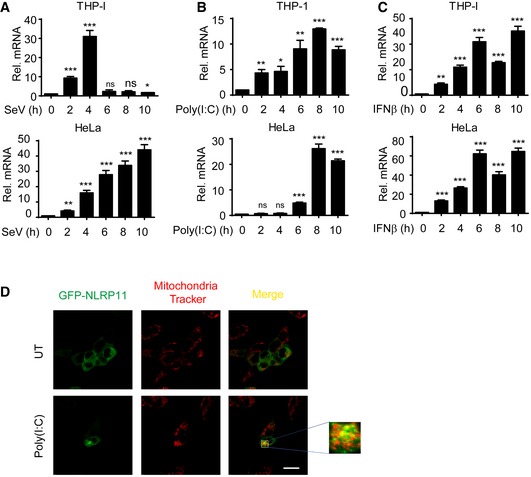
-
A–CNLRP11 mRNA expression in THP‐1 monocytes and HeLa cells infected with Sendai virus (SeV) (MOI = 1), or transfected with poly(I:C) (10 μg/ml) or treated with IFN‐β (1,000 U/ml) for indicated time points was analyzed by real‐time PCR. Data are expressed as means ± SEM of three independent experiments (*P < 0.05, **P < 0.01, and ***P < 0.001, versus untreated cells, Student's t‐test).
-
D293T cells were transfected with GFP‐NLRP11, and then, these cells were left untreated (UT) or were transfected with poly(I:C) (5 μg/ml) for 12 h, followed by confocal fluorescence microscopy analysis. Mitochondria were detected with mitochondria tracker (red). Scale bar, 20 μm.
Next, we transfected GFP‐NLRP11 into 293T cells to investigate the cellular localization of NLRP11. NLRP11 showed diffused expression in cytoplasm in resting cells (Figs 5D and EV5D). However, a proportion of NLRP11 aggregated after SeV infection or poly(I:C) treatment (Figs 5D and EV5D). It has been reported that RLR activation induced the prionlike polymerization of MAVS on mitochondria, which recruited downstream adaptors to amplify signaling 4, 23. We also found TM domain deletion of MAVS did not interact with NLRP11 anymore (Fig 3E). Based on these results, we reasoned that NLRP11 may aggregate on mitochondria. To test this hypothesis, we performed mitochondria isolation analysis and found that a fraction of NLRP11 translocated from cytosol to mitochondria upon SeV infection (Fig 5E).
NLRP11 suppresses virus‐induced apoptosis
Apoptosis is an important part of host defensing to limit virus replication and spreading 24. It has been reported that MAVS and TRAF6 have central roles in mediating the apoptosis of virus‐infected cells 25. Since we found NLRP11 disrupted MAVS signalosome by promoting TRAF6 degradation, we reasoned that NLRP11 may also regulate virus‐mediated apoptosis. To test this hypothesis, we infected WT or NLRP11 KO THP‐1 cells with VSV‐eGFP at a multiplicity of infection (MOI) of 10. We observed that trypan blue‐positive cells (dead cells) significantly increased in NLRP11 KO group (Fig 6A), indicating the potential inhibitory functions of NLRP11 on apoptotic cell death. Similarly, NLRP11 KO THP‐1 cells have a higher propidium iodide (PI) staining after VSV‐eGFP infection, as compared to WT cells (Fig 6B). In addition, a greater percentage of PI staining‐positive cells was observed in the absence of NLRP11 by flow cytometry analysis (Fig 6C). We next examined the effect of NLRP11 on poly‐ADP‐ribose polymerase (PARP) cleavage and observed that overexpression of NLRP11 reduced the cleavage of PARP upon VSV‐eGFP infection (Fig 6D). In addition, we found that the effect of NLRP11 on PARP cleavage is dependent on MAVS (Fig 6E). Consistently, an increasing cleavage of PARP was observed in NLRP11 KO cells after VSV‐eGFP infection, as compared to WT cells (Fig 6F). It has been reported that VSV M protein is a potent inhibitor of host gene expression, which could also trigger apoptosis of infected cells 26. In order to remove the apoptosis mediated by M protein, we transfected poly(I:C) to induce apoptosis. We observed more apoptotic cells by viral infection when NLRP11 was knocked down (Fig 6G and H). However, knockdown of TRAF6 abrogated the inhibition of poly(I:C)‐induced apoptosis mediated by NLRP11 (Fig 6I). Taken together, our finding indicated that NLRP11 attenuated virus or poly(I:C)‐induced apoptosis in a MAVS‐ and TRAF6‐dependent manner.
Figure 6. NLRP11 suppresses virus‐induced apoptosis.
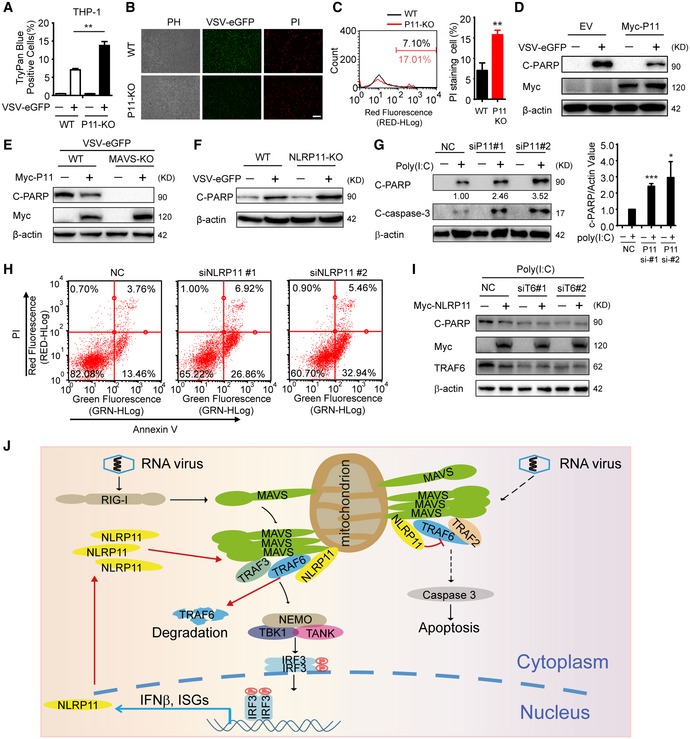
- Wild‐type (WT) and NLRP11 knockout (KO) THP‐1 cells were infected VSV‐eGFP (MOI = 10) for 24 h or not, and then stained with trypan blue.
- WT and NLRP11 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 10) for 12 h, and then stained with propidium iodide (PI), followed by fluorescence analysis. Scale bar, 100 μm.
- WT and NLRP11 KO THP‐1 cells were infected with VSV‐eGFP (MOI = 10) for 24 h, and then subjected to PI staining analysis by flow cytometry. The eGFP‐positive (infected) cells were gated to compare the cell death. Numbers indicated the percentage of PI‐stained cells (left). Three independent experiments were quantified (right).
- Cleaved PARP (C‐PARP) was analyzed by immunoblot analysis in 293T cells transfected with empty vector or Myc‐NLRP11 upon VSV‐eGFP (MOI = 10) infection for 18 h.
- Wild‐type (WT) and MAVS KO 293T cells were transfected with empty vector or Myc‐NLRP11, and then infected with VSV‐eGFP (MOI = 10) for 18 h. C‐PARP was analyzed by immunoblot analysis.
- Cleaved PARP (C‐PARP) was analyzed by immunoblot analysis in WT or NLRP11 KO THP‐1 cells upon VSV‐eGFP (MOI = 10) infection for 24 h.
- HT1080 cells were transfected with NLRP11 siRNAs or negative control (NC) siRNA for 48 h, and then transfected with poly(I:C) (5 μg/ml) for 24 h. Cells were harvested for immunoblot analysis (left). Three independent experiments were quantified (right). Numbers between two blots indicate densitometry of C‐PARP relative to that of β‐actin.
- HT1080 cells were transfected with NLRP11 siRNAs or negative control (NC) siRNA for 48 h, and then transfected with poly(I:C) (5 μg/ml) for 24 h. Cells were harvested for Annexin V and PI staining analysis by flow cytometry.
- HT1080 cells were transfected with Myc‐NLRP11 and TRAF6 siRNAs or negative control (NC) siRNA for 48 h, and then transfected with poly(I:C) (5 μg/ml) for 24 h were harvested for immunoblot analysis.
- A proposed model for the regulatory functions of NLRP11.
Discussion
NOD‐like proteins are involved in the activation of diverse innate immune signaling pathways 11, 12. NOD1, NOD2, NLRP1, NLRP3, and NLRC4 have been extensively studied and shown to activate MAPK and NF‐ĸB pathways or form inflammasomes once they encounter relevant PAMPs 13, 14. Recently, accumulating evidence revealed the negative regulatory roles of NLRs in immune responses. NLRP12 has been reported to be involved in the regulation of inflammation 27, 28. We and others found that NLRX1 inhibits RNA virus‐induced type I IFN signaling and NF‐ĸB pathway by binding to MAVS and IKK complex, respectively 17, 29. We also found NLRC5 plays a critical role in the negative regulation of intracellular antiviral responses via interaction with RLRs 15, whereas NLRP4 reduces IFN production through promoting TBK1 degradation 16, 18. However, the role of NLRP11 in the regulation of antiviral immunity remains unknown. In this study, we report that NLRP11 serves as a negative regulator of type I IFN pathway and apoptosis by targeting MAVS signalosome for degradation of TRAF6 upon viral infection.
MAVS is a mitochondria adaptor protein, which acts as a major adaptor for RLRs 2. Upon viral infection, RLRs initiate the aggregation of MAVS 4. MAVS then recruits distinct TRAFs to form MAVS signalosome, which in turn activates IRF3/7 and NF‐κB pathway or apoptosis 5, 6, 7, 25. Up to now, the function of TRAF6 in type I IFN signaling is still under debate. Chattopadhyay et al 25 found TRAF6 was not necessary for IRF‐3‐mediated gene induction induced by poly(I:C) in MEFs. However, several other groups and our studies have clarified the essential roles of TRAF6 in virus‐induced IFN signaling (Fig EV4A) 23, 30. Chen et al 30 found that TRAF6 deficiency reduced IFN‐β expression in both mouse and human cells, and Liu et al 23 found TRAF6 was an IRF3 activator, and knockdown of TRAF6 reduced phosphorylation and dimerization of IRF3 in mouse cells. Based on these reports, we suggested that TRAF6 might function differently in different cell types or in response to different stimuli. Besides triggering apoptosis, virus could also activate pyroptosis in monocytes, macrophages, and dendritic cells 31. In our study, NLRP11 inhibits virus‐induced cell death and apoptosis in THP‐1 cells (Fig 6A–C and F), and whether NLRP11 affects pyroptosis needs further investigation.
In this study, we found NLRP11 played a dual regulatory role in virus‐induced IFN production and apoptosis in a MAVS‐dependent manner (Figs 4F and G, and 6E). Based on the experimental data, we proposed a working model to illustrate how NLRP11 could regulate virus‐triggered type I IFN and apoptosis signaling pathways (Fig 6J). After viral infection, NLRP11 expression is induced and translocates to mitochondria to target MAVS. Using MAVS as a platform, NLRP11 binds with TRAF6 to promote its degradation, and thus reduces type I IFN signaling. Along with the degradation of TRAF6, virus‐induced apoptosis is also decreased.
Multiple proteins, such as several E3 ubiquitin ligases, including TRIM38, WWP1, and STUB1, are identified to mediate the degradation of TRAF6 by proteasome‐dependent pathway 32, 33, 34. Since NLRP11 is not an E3 ubiquitin ligase, whether NLRP11 recruits other E3 ligases to degrade TRAF6 needs further investigation. In summary, our study identifies NLRP11 as a negative regulator of type I IFN and virus‐induced apoptosis via disrupting the activity of MAVS signalosome. NLRP11 might be used as a therapeutic target for inflammatory or autoimmune diseases, which were associated with aberrant RLR activation.
Materials and Methods
Antibodies
The following antibodies were used in this study: anti‐IRF3 (sc‐9082), donkey anti‐goat IgG‐HRP (sc‐2020), goat anti‐rabbit IgG‐HRP (sc‐2004), goat anti‐mouse IgG‐HRP (sc‐2005), tubulin (sc‐8035), MAVS (sc‐166583), TRAF6 (sc‐8409) (Santa Cruz Biotechnology); horseradish peroxidase (HRP)‐anti‐Flag (M2) (A8592), and anti‐β‐actin (A1978) (Sigma); HRP‐anti‐hemagglutinin (clone 3F10), anti‐Myc‐HRP (11814150001), unlabeled anti‐Myc (11667203001) (Roche Applied Science); NLRP11 (NBP‐1‐92186) (Novus Biologicals); anti‐p‐IRF3 (#4947S), c‐PARP (#5625), caspase‐3 (#9661), TRAF6 (#8028), MAVS (#3993), mouse anti‐rabbit IgG‐HRP (#5127), K48‐Ub‐HRP (#12805), K63‐Ub‐HRP (#12930) (CST); NLRP11 (ab88732), COX IV (ab16056) (Abcam).
Virus infection
VSV‐eGFP was kindly provided by Dr. Xiaofeng Qin (Suzhou Institute of Systems Medicine), and herpes simplex virus type 1 (HSV‐1, KOS strain) was kindly provided by Dr. Guoying Zhou (Guangzhou Medical University). Cells were infected at various MOI, as previously described 35.
Real‐time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) and reverse‐transcribed using oligo‐dT primers and reverse transcriptase (TAKARA). Real‐time quantitative PCR was performed using SYBR green qPCR Mix kit (Genstar) and specific primers using the Primer 5.0 analyzer (Applied Biosystems). Data were normalized to the Rpl13a gene, and the relative abundance of transcripts was calculated by the 2−ΔΔCt models. The sequences of primers are as follows:
IFNB1: Forward 5′‐TGATACTCCTGGCACAAAT‐3′
Reverse 5′‐TTGAGCCTTCTGGAACTGT‐3′
ISG54: Forward 5′‐GGAGGGAGAAAACTCCTTGGA‐3′
Reverse 5′‐GGCCAGTAGGTTGCACATTGT‐3′
ISG56: Forward 5′‐TCAGGTCAAGGATAGTCTGGAG‐3′
Reverse 5′‐AGGTTGTGTATTCCCACACTGTA‐3′
SeV phosphoprotein: Forward 5′‐GACGCGAGTTATGTGTTTGC‐3′
Reverse 5′‐TTCCACGCTCTCTTGGATCT‐3′
TNFA: Forward 5′‐CCAGACCAAGGTCAACCTCC‐3′
Reverse 5′‐CAGACTCGGCAAAGTCGAGA‐3′
IL6: Forward 5′‐AGAGGCACTGGCAGAAAACAAC‐3′
Reverse 5′‐AGGCAAGTCTCCTCATTGAATCC‐3′
Rpl13a: Forward 5′‐GCCATCGTGGCTAAACAGGTA‐3′
Reverse 5′‐GTTGGTGTTCATCCGCTTGC‐3′
Immunoprecipitation and immunoblot analysis
Chemiluminescent HRP substrate (MILLIPORE) was used for protein detection, and ChemiDoc™ XRS+ imaging system (BIO‐RAD) was used for immunoblot imaging. Procedures were done as previously described 36.
Luciferase reporter assays
293T (2 × 105) cells were seeded in 24‐well plates and transfected with plasmids encoding an IFN‐β or ISRE luciferase reporter (firefly luciferase; 100 ng) and pRL‐TK (renilla luciferase plasmid; 10 ng), together with various amounts of the appropriate control or protein‐expressing plasmid(s). An empty vector (pcDNA3.1) was used to maintain equal amounts of DNA among wells. Cells were collected at 24–36 h after transfection, and luciferase activity was measured with a dual‐luciferase assay (Promega) with a Luminoskan Ascent luminometer (Thermo Scientific) according to the manufacturer's protocol. Reporter gene activity was determined by normalizing to renilla luciferase activity as previously described 16.
RNA interference
LipoRNAiMAX (Invitrogen) was used for transfection of siRNAs into cells, according to the manufacturer's instructions. The sequences of siRNAs are as follows:
NLRP11‐siRNA‐1#:
Sense: GCGAUAUCUCUCAAUAUAUTT
Antisense: AUAUAUUGACAGAUAUCGCTT
NLRP11‐siRNA‐2#:
Sense: GCCAUGAGAACGUCAAAUATT
Antisense: UAUUUGACGUUCUCAUGGCTT
TRAF6‐siRNA‐1#:
Sense: GCGCUGUGCAAACUAUAUATT
Antisense: UAUAUAGUUUGCACAGCGCTT
TRAF6‐siRNA‐2#:
Sense: GCGCUUGCACCUUCAGUUATT
Antisense: UAACUGAAGGUGCAAGCGCTT
Negative control siRNA:
Sense: UUCUCCGAACGUGUCACGUTT
Antisense: ACGUGACACGUUCGGAGAATT
Generation of knockout cells by CRISPR/Cas9 technology
293T or THP‐1 knockout cells were generated by a CRISPR/Cas9 system, and the sequences of target sgRNAs are as follows:
NLRP11‐sgRNA: Sense: GCTTGGCTGAGCTAATCGCCA
Antisense: TGGCGATTAGCTCAGCCAAGC
TRAF6‐sgRNA: Sense: CGTCTCGGCGCGCAGTGTCT
Antisense: AGACACTGCGCGCCGAGACG
MAVS‐sgRNA: Sense: GATTGCGGCAGATATACTTAT
Antisense: ATAAGTATATCTGCCGCAATC
Statistical analysis
Data are represented as mean ± SEM or mean ± SD when indicated, and Student's t‐test was used for all statistical analyses. Differences between groups were considered significant when P‐value was < 0.05.
Author contributions
JC designed the research; YQ, ZS, YW, WX, and SJ performed the research; CW, WJ, and RZ provided technical help; and JC, YQ, YW, and ZS analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by National Natural Science Foundation of China (31370869, 31522018, and 31601135), National Key Basic Research Program of China (2014CB910800 and 2015CB859800). Yunfei Qin is partially supported by Outstanding Young Talent Research Fund of Zhengzhou University (F0000953), and the Startup Research Fund of Zhengzhou University (F0000922).
EMBO Reports (2017) 18: 2160–2171
References
- 1. Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124: 783–801 [DOI] [PubMed] [Google Scholar]
- 2. Yoneyama M, Fujita T (2009) RNA recognition and signal transduction by RIG‐I‐like receptors. Immunol Rev 227: 54–65 [DOI] [PubMed] [Google Scholar]
- 3. Wu B, Hur S (2015) How RIG‐I like receptors activate MAVS. Curr Opin Virol 12: 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (2011) MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell 146: 448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vazquez C, Horner SM (2015) MAVS coordination of antiviral innate immunity. J Virol 89: 6974–6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seth RB, Sun LJ, Ea CK, Chen ZJJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappa B and IRF3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- 7. Chattopadhyay S, Sen GC (2017) RIG‐I‐like receptor‐induced IRF3 mediated pathway of apoptosis (RIPA): a new antiviral pathway. Protein Cell 8: 165–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC (2016) Ubiquitination of the transcription factor IRF‐3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 44: 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chattopadhyay S, Yamashita M, Zhang Y, Sen GC (2011) The IRF‐3/Bax‐mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol 85: 3708–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lei Y, Moore CB, Liesman RM, O'Connor BP, Bergstralh DT, Chen ZJJ, Pickles RJ, Ting JPY (2009) MAVS‐mediated apoptosis and its inhibition by viral proteins. PLoS ONE 4: e5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen G, Shaw MH, Kim YG, Nunez G (2009) NOD‐like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol‐Mech 4: 365–398 [DOI] [PubMed] [Google Scholar]
- 12. Shaw MH, Reimer T, Kim YG, Nunez G (2008) NOD‐like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol 20: 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caruso R, Warner N, Inohara N, Nunez G (2014) NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41: 898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lechtenberg BC, Mace PD, Riedl SJ (2014) Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol 29: 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cui J, Zhu L, Xia XJ, Wang HY, Legras X, Hong J, Ji JB, Shen PP, Zheng S, Chen ZJJ et al (2010) NLRC5 negatively regulates the NF‐kappa B and type I interferon signaling pathways. Cell 141: 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, Wang RF (2012) NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol 13: 387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia XJ, Cui J, Wang HLY, Zhu L, Matsueda S, Wang QF, Yang XA, Hong J, Songyang Z, Chen ZJJ et al (2011) NLRX1 negatively regulates TLR‐induced NF‐kappa B signaling by targeting TRAF6 and IKK. Immunity 34: 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin M, Zhao ZY, Yang ZF, Meng QC, Tan P, Xie WH, Qin YF, Wang RF, Cui J (2016) USP38 inhibits type I interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome. Mol Cell 64: 267–281 [DOI] [PubMed] [Google Scholar]
- 19. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA et al (2014) NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 40: 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tian X, Pascal G, Monget P (2009) Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol 9: 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai X, Chiu YH, Chen ZJ (2014) The cGAS‐cGAMP‐STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54: 289–296 [DOI] [PubMed] [Google Scholar]
- 22. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, Chen ZJ (2013) MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2: e00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barber GN (2001) Host defense, viruses and apoptosis. Cell Death Differ 8: 113–126 [DOI] [PubMed] [Google Scholar]
- 25. Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BRG, Sen GC (2010) Viral apoptosis is induced by IRF‐3‐mediated activation of Bax. EMBO J 29: 1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kopecky SA, Willingham MC, Lyles DS (2001) Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J Virol 75: 12169–12181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RMT, Davis BK, Uronis JM, Herfarth HH et al (2012) NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF‐kappa B signaling. Immunity 36: 742–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lukens JR, Gurung P, Shaw PJ, Barr MJ, Zaki MH, Brown SA, Vogel P, Chi H, Kanneganti TD (2015) The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin‐4 production in T cells. Immunity 42: 654–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti‐Loper MA, Madden VJ, Sun LJ, Ye ZM et al (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451: 573–577 [DOI] [PubMed] [Google Scholar]
- 30. Chen H‐W, Yang Y‐K, Xu H, Yang WW, Zhai ZH, Chen DY (2015) Ring finger protein 166 potentiates RNA virus‐induced interferon‐β production via enhancing the ubiquitination of TRAF3 and TRAF6. Sci Rep 5: 14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Danthi P (2016) Viruses and the diversity of cell death. Annu Rev Virol 3: 533–553 [DOI] [PubMed] [Google Scholar]
- 32. Zhao W, Wang L, Zhang M, Yuan C, Gao C (2012) E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR‐mediated immune responses by proteasomal degradation of TNF receptor‐associated factor 6 in macrophages. J Immunol 188: 2567–2574 [DOI] [PubMed] [Google Scholar]
- 33. Lin XW, Xu WC, Luo JG, Guo XJ, Sun T, Zhao XL, Fu ZJ (2013) WW domain containing E3 ubiquitin protein ligase 1 (WWP1) negatively regulates TLR4‐mediated TNF‐alpha and IL‐6 production by proteasomal degradation of TNF receptor associated factor 6 (TRAF6). PLoS ONE 8: e67633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li S, Shu B, Zhang YQ, Li J, Guo JW, Wang YY, Ren FL, Xiao G, Chang ZJ, Chen D (2014) Carboxyl terminus of Hsp70‐interacting protein regulation of osteoclast formation in mice through promotion of tumor necrosis factor receptor‐associated factor 6 protein degradation. Arthritis Rheumatol 66: 1854–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang RF et al (2016) TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell 64: 105–119 [DOI] [PubMed] [Google Scholar]
- 36. Qin Y, Liu Q, Tian S, Xie W, Cui J, Wang RF (2016) TRIM9 short isoform preferentially promotes DNA and RNA virus‐induced production of type I interferon by recruiting GSK3beta to TBK1. Cell Res 26: 613–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6