

Abstract
SHP2 (Src homology 2 domain-containing protein tyrosine phosphatase 2; PTPN11) is a ubiquitous multidomain, nonreceptor protein tyrosine phosphatase (PTP) that plays an important role in diseases such as cancer, diabetes, and Noonan syndrome (NS). NS is one of the most common genetic disorders associated with congenital heart disease, and approximately half of the patients with Noonan syndrome have gain-of-function mutations in SHP2. One of the most common NS mutations is N308D. The activity of SHP2, like that of most PTPs, is reversibly inactivated by reactive oxygen species (ROS). However, the molecular basis of this inactivation and the consequences of NS-related mutations in PTPN11 on ROS-mediated inhibition are poorly understood. Here, we investigated the mechanistic and structural details of the reversible oxidation of the NS variant SHP2N308D. We show that SHP2N308D is more sensitive to oxidation when compared with wild-type SHP2. We also show that although the SHP2N308D catalytic domain can be reactivated by dithiothreitol as effectively as the wild-type, full-length SHP2N308D is only poorly reactivated by comparison. To understand the mechanism of oxidation at a molecular level, we determined the crystal structure of oxidized SHP2N308D. The structure shows that the catalytic Cys459 residue forms a disulfide bond with Cys367, which confirms that Cys367 functions as the “backdoor” cysteine in SHP2. Together, our data suggest that the reversible oxidation of SHP2 contributes negligibly, if at all, to the symptoms associated with NS.
Introduction
SHP2 (Src homology 2 domain-containing protein tyrosine phosphatase 2; PTPN11) is a ubiquitous multidomain, nonreceptor protein tyrosine phosphatase (PTP)1 that contains two regulatory SH2 domains [N-SH2 (residues 1–103) and C-SH2 (residues 112–216)] and a PTP domain (residues 221–524). The SHP2 PTP domain includes the structural features required for catalysis, including the PTP loop with the requisite catalytic cysteine residue, the WPD loop that is required for substrate hydrolysis, the Q-loop, the substrate-binding loop, and the E-loop. The activity of SHP2 is regulated by an intramolecular allosteric interaction between the SH2 and PTP domains.2,3 In the absence of phosphotyrosine (pTyr) docking sites created by receptor activation, SHP2 is not active. This is because the SH2 domains associate directly with the PTP domain and occlude the active site.4 However, receptor activation results in the generation of biphosphorylated tyrosine sequences that bind the SHP2 SH2 domains, which results in the dissociation of the SH2 domains from the PTP domain. This renders the SHP2 catalytic site accessible, resulting in substrate binding and dephosphorylation.
SHP2 has multiple biological functions, including the regulation of signaling pathways, especially the RAS/ERK signaling pathway that is downstream of most growth factors, cytokines, and integrins.5−7 Mutations in PTPN11 are correlated with approximately 50% of Noonan syndrome (NS) cases.8 NS is a congenital autosomal dominant disorder, affecting 1:1000 to 1:2500 live births, characterized by short stature, short neck, facial dysmorphia, pulmonary valve stenosis, congenital heart defects, variable coagulation defects, and lymphatic dysplasias.9 The most common NS variant is N308D, which leads to an increase in SHP2 activity (hyperactive SHP2),10 and is hypothesized to be mediated by a destabilization of the autoinhibited “closed” state. It has also been shown that reactive oxygen species (ROS), which are important mediators of cell growth, differentiation, and signaling, regulate SHP2 activity by reversible inactivation11−13 and that this is achieved through the formation of a disulfide bond between the catalytic cysteine (Cys459) and one of two potential “backdoor” cysteines (Cys333 or Cys367). Further, the same group identified the formation of a backdoor–backdoor disulfide following H2O2-mediated oxidation in the presence of Cys459, leading to a model in which the stably oxidized form of SHP2 consists of a reduced catalytic cysteine and a stable backdoor–backdoor disulfide. It is not known if or how this redox regulation is altered in PTPN11 variants correlated with NS.
Results and Discussion
We used biochemistry and structural biology to determine how the NS variant SHP2N308D is differentially regulated by ROS. To understand how the presence of the SH2 domains affects oxidation and reactivation susceptibility, we examined both the catalytic domain in isolation (SHP2cat; aa 237–529; Figure 1A) and within the context of both SH2 domains (SHP21–526; Figure 1B). To determine the relative activities of the wild-type (WT) SHP2 and the SHP2N308D variants, we first measured their catalytic activities using para-nitrophenylphosphate (pNPP) as a substrate. Consistent with previous data, the presence of the SH2 domains, which block the SHP2 active site in the absence of phosphorylated peptides, dramatically reduces the activity of SHP2, as SHP21–526 is ∼70-fold less active than the catalytic domain alone (Table 1). However, although the catalytic efficiency of the NS variant, SHP2cat,N308D, is about 40% lower than that of WT SHP2cat, this difference is reversed for the SH2-containing constructs, i.e., the SHP21–526,N308D variant is about 4-fold more active than WT (SHP21–526). This increase in catalytic activity of the N308D mutant is consistent with what has been observed in vivo;14,15 namely, that the SHP2 NS variant N308D results in enhanced enzymatic activity, leading to the increased activation in the RAS/ERK pathway that is associated with the pathogenesis of NS.14
Figure 1.

Inhibition and reactivation of the steady-state activity of SHP2 variants. (A) Domain cartoon of the SHP2cat construct. (B) Domain cartoon of the SHP21–526 construct (SH2N, cyan; SH2C, blue; CAT, orange). (C, D) The steady-state activity of SHP2 variants (C, SHP2237-529; D, SHP21–526; WT, black squares, N308D variant, red diamonds) were measured after incubating the samples with the indicated concentrations of H2O2 for 15 min. The % remaining activity was calculated by normalizing the measured activity to the sample incubated without H2O2. (E, F) The ability of oxidized SHP2 variants (E, SHP2237-529; F, SHP21–526) to be subsequently reactivated was determined by measuring the activities of reduced (no H2O2) and oxidized (15 min incubation with 500 μM H2O2 prior to activity measurements) SHP2 samples in the presence of the indicated concentrations of DTT. The % reactivation was calculated by normalizing the activities of the oxidized samples to the corresponding reduced samples. ±SE, n = 3–4. Two-way ANOVA test, with ****p < 0.0001 (C, D) or *p < 0.05 (F) between WT and mutant.
Table 1. Catalytic Activities of SHP2cat and SHP21–526 Variants (WT and N308D) Using pNPP as a Substrate.
| PTP variant | kcat (s–1) | catalytic efficiency (kcat/Km; s–1 mM–1) | fold changea | IC50 H2O2 (μM) |
|---|---|---|---|---|
| SHP2cat | 3.7 ± 0.1 | 1.3 ± 0.1 | 72 | 148 ± 14 |
| SHP2cat,N308D | 3.0 ± 0.1 | 0.6 ± 0.04 | 33 | 38 ± 4b |
| SHP21–526 | 0.04 ± 0.01 | 0.018 ± 0.01 | 1 | 193 ± 7 |
| SHP21–526,N308D | 0.3 ± 0.02 | 0.078 ± 0.003 | 4 | 108 ± 3b |
Relative to Shp21–526 catalytic efficiency.
Unpaired t-test, p < 0.003, relative to WT.
The prevailing hypothesis is that the N308D mutation destabilizes the interaction between the SH2 and catalytic domains (N308 is located at the interface of these two domains), leading to enhanced activity due to increased access to the SHP2 active site. We therefore asked if this putative increased access to the active site also confers differential susceptibility of SHP2N308D to reversible oxidation. To test this, we determined the oxidation profile of both WT SHP2 and the NS variant SHP2 N308D upon exposure to H2O2. As can be seen in Figure 1C,D, both the N308D variants (SHP2cat,N308D and SHP21–526,N308D) are more sensitive to oxidation than their WT counterparts. Namely, H2O2 inhibits SHP2cat,N308D with an IC50 of 38 μM, whereas the same H2O2 concentration has essentially no effect on its WT counterpart (Figure 1C, Table 1). A similar difference in H2O2 sensitivity is observed for the SH2-containing constructs, with H2O2 inhibiting the activity of SHP21–526,N308D with an IC50 of 108 μM, whereas the same H2O2 concentration has no effect on its WT counterpart (Figure 1D, Table 1). Thus, both SHP21–526,N308D and SHP2cat,N308D are more sensitive to oxidation than their WT SHP2 counterparts, with the catalytic domain construct (SHP2cat,N308D) being the most sensitive.
We then tested the ability of the N308D variants to be reactivated with DTT after a 15 min oxidation with 500 μM H2O2. We observed a striking difference in the ability of the NS variants to be reactivated upon the addition of reductant; namely, although the activity of SHP2cat,N308D recovers its activity as effectively as its WT counterpart (SHP2cat; Figure 1E), the SH2-containing variant does not and instead achieves only a 66% recovery in activity (Figure 1F). Thus, although both SHP21–526,N308D and SHP2cat,N308D are more sensitive to oxidation by H2O2 compared to their WT counterparts, only the catalytic domain is able to achieve WT-levels of reactivation; SHP21–526,N308D is not. This suggests that the N308D variant of SHP2 is not only more sensitive to oxidation by ROS, but it is also less effectively reactivated, at least by DTT. Thus, under conditions which lead to high concentrations of ROS, SHP2N308D would be expected to be less active and its activity recovered less effectively than its WT counterpart.
To gain further insights into the molecular basis of the reversible oxidation, we determined the crystal structure of oxidized SHP21–526,N308D to 2.5 Å. As SHP21–526,N308D is highly susceptible to oxidation, oxidized SHP21–526,N308D was formed by omitting reducing agents in the protein buffer prior to crystal formation. The electron density maps were well defined for the entirety of the protein molecule with the exception of five short loops. Like all previous structures of SHP2, oxidized SHP21–526,N308D is in the closed state, with the SH2 domains directly occluding the catalytic site (Figure 2A,B). The oxidized SHP21–526,N308D structure also contains three ordered phosphate molecules, two of which bind the phosphotyrosine recognition pockets in the SH2 domains. Unexpectedly, the third phosphate binds a pocket near the PTP active site, at a position that is very nearly continuous with the hydroxyl of Tyr279 (this residue defines the depth of the PTP active site; Figure 2C). Previous studies have shown that Tyr279 is phosphorylated in an Abl-kinase-dependent manner and functions to downregulate SHP2-dependent ERK signaling.16 Our structure shows that Tyr279 phosphorylation may function to stabilize the closed state, which would lead to a decrease in SHP2 activity and SHP2-dependent ERK signaling, as the bound phosphate ion is coordinated by residues from both the N-terminal SH2 domain (Asp61, Tyr62) and the catalytic PTP domain (Tyr279, Arg278, and Ser460) (Figure 2C).
Figure 2.
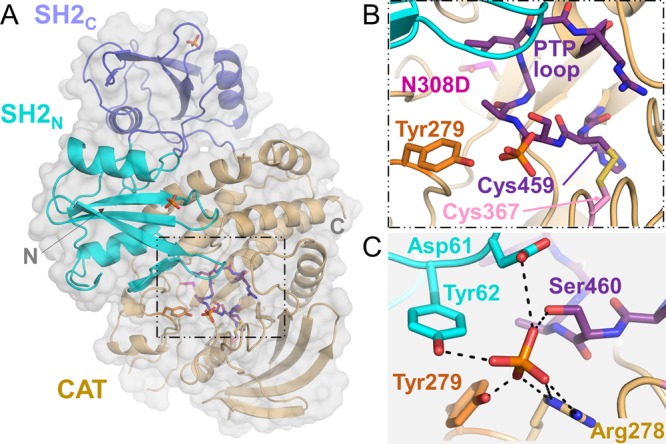
Oxidized SHP21–526,N308D results in the formation of a disulfide bond between the catalytic cysteine, Cys459, and the backdoor cysteine, Cys367. (A) Structure of oxidized SHP21–526 (SH2N, cyan; SH2C, blue; CAT, orange). The N- and C-termini are labeled. Three bound phosphate ions are shown as sticks and key residues discussed in the manuscript are shown as sticks and boxed. (B) Expanded view of the area boxed in (A). PTP loop (purple), N308D (magenta), Tyr279 (orange), Cys367 (pink), and catalytic cysteine Cys459 (purple) are shown as sticks. (C) View similar to (B), but highlighting the residues that coordinate the bound phosphate ion (shown as sticks and labeled; residues in cyan belong to the SH2N domain, whereas the rest are from the catalytic domain).
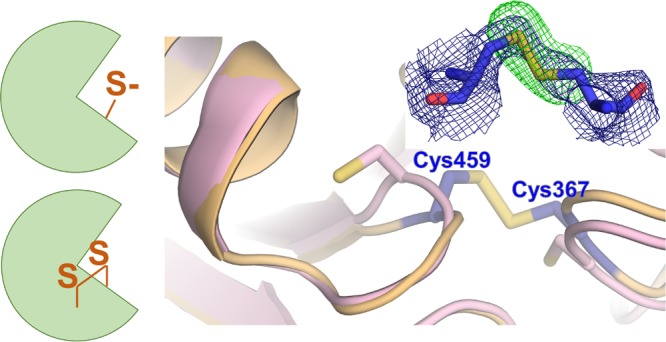
The structure of oxidized SHP21–526,N308D closely resembles that obtained under reducing conditions (PDB ID 4NWF; root-mean-square deviation of 0.58 Å over 471 residues; Figure 3A).17 However, the structures are not identical. In oxidized SHP2N308D, the catalytic Cys459 side chain rotates out of the PTP pocket by nearly 90° to form an intramolecular disulfide bond with Cys367 (Figure 3B). Thus, this structure confirms that the mechanism by which SHP2 achieves reversible oxidation is via the formation of a disulfide bond between its catalytic cysteine and a backdoor cysteine (and not by forming a cyclic sulphenamide with the immediate C-terminal Ser residue, Ser460, which has been observed in other PTPs, such as PTP1B18,19). Further, the structure reveals that the identity of the backdoor cysteine in SHP2 is Cys367. Although Chen et al. observed the existence of a reduced catalytic cysteine (Cys459) and the formation of a backdoor–backdoor disulfide bond following treatment with H2O2, we see no evidence of such a backdoor–backdoor (Cys333–Cys367) disulfide. Rather, oxidized SHP21–526,N308D is defined by a disulfide bond formed between the catalytic Cys459 and the backdoor Cys367 cysteines.
Figure 3.
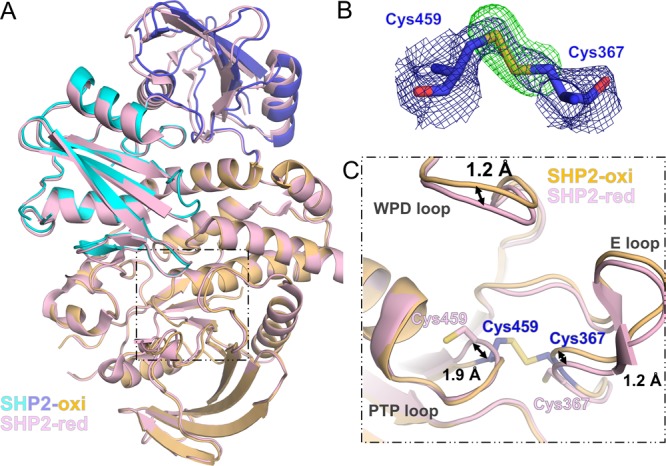
Oxidation of SHP2 results in only small, local conformational changes compared with the reduced state. (A) Overlay of oxidized SHP2 N308D (individual domains colored as in Figure 2) and reduced SHP2 N308D (pink; PDB ID 4NWF(20)). (B) FOM-weighted 2mFO–DFC electron density map (blue mesh; contoured at 1.0 σ to 2.50 Å) and mFO–DFC electron density map (green mesh; contoured at 3.0 σ to 2.50 Å) of residues Cys459 and Cys367 refined at an occupancy of 1.0, with γS atoms omitted. (C) Close-up of the residues that mediate SHP2 reversible oxidation: Cys459 and Cys367. Arrows highlight the small conformational changes observed in the PTP loop, the E-loop, and the WPD loop (labeled).
Disulfide bond formation results in distinct structural changes between the oxidized and reduced states (Figure 3C). First, in spite of disulfide bond formation, the structure of the PTP loop (458HCSAGIR465) is essentially unchanged in the oxidized versus reduced structure. The only significant conformational difference in this loop is the position of the Cα atom of Cys459 in the oxidized structure. Specifically, the Cα atom shifts ∼1.9 Å away from its corresponding position in the reduced structure to accommodate disulfide bond formation. Second, and in contrast to the PTP loop, whose changes are highly localized, both the E-loop (357TKEVERGKSKCVKY370, which includes the backdoor cysteine, Cys367) and to some extent the WPD loop (422TWPDHGVP429) shift away from the catalytic site as rigid bodies to accommodate disulfide bond formation. The maximum shift of both loops is ∼1.2 Å. Thus, disulfide bond formation is readily achieved in SHP2 with only minor changes in the overall conformation of the structure. Consistent with this, the position of the N308D mutation is identical between the oxidized and reduced structures.
Taken together, our data shows that both SHP2cat,N308D and SHP21–526,N308D are more susceptible to oxidation than WT SHP2 and furthermore that oxidized SHP21–526,N308D is comparatively resistant to reactivation by DTT. This suggests that under oxidizing conditions, the NS mutant is less active than WT and that its reactivation is less effective. This would result in an overall reduction in SHP2 activity for the N308D mutant compared to that in WT. Notably, this is the opposite of what is observed for NS mutants. Namely, NS variants exhibit increased SHP2 activities.17,20 Together, these data suggest that the reversible oxidation of SHP2 contributes negligibly, if at all, to the symptoms associated with NS. Our data also provide the first structural evidence for disulfide-mediated protection of SHP2 against irreversible oxidation. In our structure, Cys459 forms a disulfide bond with Cys367; no evidence is observed for a Cys333–Cys367 bond. Coupled with the observation that the γS atoms of C459 and C333 are separated by >8 Å, our data shows that Cys367 plays a dominant role over Cys333 in protecting Cys459 from irreversible oxidation.
Experimental Section
Expression and Purification
DNA coding the full length and the catalytic domain of SHP2 residues [1–526 and 237–526 (SHP2cat), respectively] were subcloned into RP1B and pNIC28, respectively, as previously described.21 Site-directed mutagenesis was used to create the N308D variant (Agilent Genomics). For protein expression, plasmid DNAs were transformed into Escherichia coli BL21 (DE3) RIL cells (Agilent). Cells were grown in Luria broth in the presence of selective antibiotics at 37 °C to an OD600 of ∼0.8, and the expression was induced by the addition of 1 mM isopropylthio-β-d-galactoside. Induction proceeded for ∼20 h at 18 °C prior to harvesting by centrifugation at 7647g (15 min, 4 °C). Cell pellets were stored at −80 °C until purification.
Cell pellets were resuspended in lysis buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5 mM imidazole, 0.1% Triton X-100) and lysed using high-pressure homogenization (Avestin C3 EmulsiFlex). The lysate was cleared by centrifugation (40905g, 45 min, 4 °C). The supernatant was filtered and loaded onto a Ni-NTA column equilibrated in buffer A (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5 mM imidazole). Protein was eluted using buffer B (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 300 mM imidazole). The elution was incubated with tobacco etch virus (TEV) protease overnight at 4 °C in dialysis buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl). The TEV protease, cleaved His6-tag, and uncleaved protein were removed using a second IMAC step. Final purification was achieved using size exclusion chromatography [SEC; Superdex 75 26/60 (GE Healthcare)] equilibrated in analysis buffer (50 mM Hepes pH 7.5, 150 mM NaCl). All experiments were performed in analysis buffer.
Determination of PTPase Catalytic Rates Using pNPP as a Substrate
The PTPase activity of SHP2cat, SHP2cat,N308D (0.25 μM), and SHP21–526,N308D (5 μM) was determined by first incubating protein with pNPP at different concentrations (0, 50, 100, 250, 500, 750, 1000, 2000, 3000, and 4000 μM) for 30 min at 30 °C. Next, the reactions were stopped with 100 μL of 1 M NaOH and the absorbance of pNPP 405 nm then measured using a microplate spectrophotometer (BioTek). The PTPase rates were calculated using the molar extinction coefficient of pNPP of 18 000 M–1 cm–1.
Inactivation of SHP2 Variants by H2O2
To measure the susceptibility of the SHP2 variants to oxidation, SHP21–526, SHP21–526,N308D, SHP2cat, and SHP2cat,N308D (20 μM) were incubated with different concentrations of H2O2 (0, 3.2, 8, 16, 32, 60, 80, 160, 320, 400, 1000, and 2000 μM) for 15 min at room temperature. Each reaction was incubated with analysis buffer for 10 min at room temperature. pNPP (2000 μM; 15 μL) was then added and the reactions incubated at 30 °C for an additional 15 min. The reactions were then stopped with 1 M NaOH and the absorbance of pNPP 405 nm measured using a microplate spectrophotometer (BioTek). The PTPase rates were calculated using the molar extinction coefficient of pNPP of 18 000 M–1 cm–1. The extent of remaining activity was normalized to the sample incubated without H2O2.
Reactivation Experiments
To analyze the ability of DTT to reverse the H2O2-mediated oxidation of SHP2 variants, 1.6 μM SHP21–526/SHP21–526,N308D and 0.6 μM SHP2cat/SHP2cat,N308D were incubated with or without 500 μM H2O2 for 15 min at room temperature. Five microliters (80 nM SHP2FL and 30 nM SHP2cat) of these reactions were then incubated in 95 μL of buffer containing 100 μM DiFMUP in the absence or presence of 0, 0.2, 0.5, 0.75, 1, 2, 4, and 6 mM DTT for 30 min at room temperature. The fluorescence intensity was then measured in the Synergy Epoch using an excitation filter of 340/40 and emission filter of 460/40. The recovered activities were determined by normalizing to the samples with 10 mM DTT and without H2O2.
Statistical Analysis
Data were analyzed using SigmaPlot 12.5 or Graph Pad Prism 6. The statistics for steady-state data was performed using unpaired t-test (SigmaPlot 13.0), with significance of p < 0.05 or by a two-way ANOVA analysis using Prism Graph Pad 6, with significance of p < 0.05.
Crystallization, Data Collection, and Structure Determination
SHP21–526,N308D was crystallized in 0.2 M sodium phosphate pH 9.1, 20% (w/v) poly(ethylene glycol) (PEG) 3350 using the sitting drop vapor diffusion method at 4 °C. Crystals were cryoprotected in 0.18 M sodium phosphate pH 9.1, 18% (w/v) PEG 3350, 20% (v/v) glycerol (direct soak, 1 min) prior to diffraction screening and data collection. Crystallographic data was collected at Brookhaven National Laboratory National Synchroton Light Source (BNL-NSLS) Beamline X25 at 100 K using an ADSC QUANTUM 315 CCD detector (Table 2). Crystallographic data were indexed, scaled, and merged using HKL2000 0.98.692i.22 The structure of SHP21–526,N308D was solved by molecular replacement using the program Phaser 1.3.223 and the structure of SHP21–527 T2K/F41L/F513S (PDB ID: 2SHP) as a search model, after omitting solvent molecules. The resulting rotation- and translation-function Z-scores were 24.3 and 19.7 (molecule A) and 31.2 and 61.2 (molecule B), respectively. The model was completed by cycles of manual building using the program Coot 6.0.224 coupled with structure refinement using RefMac 5.2.0019,25 with a final round of refinement using PHENIX.26 The structure of SHP21–526,N308D was determined to 2.50 Å resolution and refined to Rcryst = 19.5% and Rfree = 23.7% and contains 2 molecules of Shp2, 197 water molecules, and 4 phosphate molecules per asymmetric unit (SHP2 molecule A residues 1, 91–92, 157–162, 236–244, 298–299, 314–322, and 526, and SHP2 molecule B residues 1, 91–92, 156–162, 236–244, 298–299, 314–323, and 526, were not observed in the electron density map and so were not modeled). The stereochemical quality of the model was analyzed using MolProbity,27 which performs Ramachandran plot, Cβ deviation, and rotamer analyses. The agreement of the model to the diffraction data was analyzed using SFCheck 7.2.02.28 Atomic coordinates and structure factors for SHP21–526,N308D have been deposited with the Protein Data Bank as entry 6ATD.
Table 2. Data Collection and Refinement Statistics.
| SHP21–526,N308Da,b | |
|---|---|
| protein | |
| organism | Homo sapiens |
| PDB ID | 6ATD |
| data collection | |
| space group | P21 |
| cell dimensions | |
| a, b, c (Å) | 46.3, 216.1, 55.7 |
| α, β, γ (deg) | 90.0, 97.3, 90.0 |
| resolution (Å) | 50.0–2.50 (2.54–2. 50) |
| Rmerge | 10.3 (57.5) |
| I/σI | 18.5 (2.4) |
| completeness (%) | 98.3 (97.8) |
| redundancy | 3.7 (3.5) |
| refinement | |
| resolution (Å) | 38.7–2.50 (2.57–2.50) |
| no. reflections | 36 774 |
| Rwork/Rfree | 0.195 (0.245)/0.237 (0.285) |
| no. atoms | |
| protein | 7792 |
| ligand/ion | 20 |
| water | 197 |
| B-factors | |
| protein | 41.68 |
| ligand | 57.8 |
| water | 37.7 |
| rms deviations | |
| bond lengths (Å) | 0.004 |
| bond angles (deg) | 0.653 |
| Ramachandran | |
| outliers (%) | 0.5 |
| allowed (%) | 2.9 |
| favored (%) | 96.6 |
| clashscore | 3.5 |
Data was collected from a single crystal.
Values in parentheses are for the highest-resolution shell.
Acknowledgments
Crystallographic data was collected at NSLS beamline X25. Use of the NSLS at Brookhaven National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-98CH10886. This work was supported in part by grants from the National Institutes of Health (R01GM098482 to R.P.; R01NS091336 to W.P.) and the American Diabetes Association (Pathway to Stop Diabetes Grant 1-14-ACN-31 to W.P.).
Author Present Address
§ Department of Molecular Structure & Design, Discovery Chemistry & Molecular Technologies, Bristol-Myers Squibb, Princeton, New Jersey 08543, United States (D.A.C.).
The authors declare no competing financial interest.
References
- Geiger T.; Velic A.; Macek B.; Lundberg E.; Kampf C.; Nagaraj N.; Uhlen M.; Cox J.; Mann M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteomics 2013, 12, 1709–1722. 10.1074/mcp.M112.024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechleider R. J.; Sugimoto S.; Bennett A. M.; Kashishian A. S.; Cooper J. A.; Shoelson S. E.; Walsh C. T.; Neel B. G. Activation of the SH2-containing phosphotyrosine phosphatase SH-PTP2 by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor. J. Biol. Chem. 1993, 268, 21478–21481. [PubMed] [Google Scholar]
- Sugimoto S.; Lechleider R. J.; Shoelson S. E.; Neel B. G.; Walsh C. T. Expression, purification, and characterization of SH2-containing protein tyrosine phosphatase, SH-PTP2. J. Biol. Chem. 1993, 268, 22771–22776. [PubMed] [Google Scholar]
- Hof P.; Pluskey S.; Dhe-Paganon S.; Eck M. J.; Shoelson S. E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. 10.1016/S0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Stein-Gerlach M.; Wallasch C.; Ullrich A. SHP-2, SH2-containing protein tyrosine phosphatase-2. Int. J. Biochem. Cell Biol. 1998, 30, 559–566. 10.1016/S1357-2725(98)00002-8. [DOI] [PubMed] [Google Scholar]
- Qu C.-K. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochim. Biophys. Acta 2002, 1592, 297–301. 10.1016/S0167-4889(02)00322-1. [DOI] [PubMed] [Google Scholar]
- Neel B. G.; Gu H.; Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Niemeyer C. M.; Fragale A.; Song X.; Buechner J.; Jung A.; Hählen K.; Hasle H.; Licht J. D.; Gelb B. D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Kalidas K.; Shaw A.; Song X.; Musat D. L.; van der Burgt I.; Brunner H. G.; Bertola D. R.; Crosby A.; Ion A.; Kucherlapati R. S.; Jeffery S.; Patton M. A.; Gelb B. D. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 2002, 70, 1555–1563. 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T.; Chan G.; Newbigging S.; Morikawa L.; Bronson R. T.; Neel B. G. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4736–4741. 10.1073/pnas.0810053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibrecht I.; Böhmer S.-A.; Dagnell M.; Kappert K.; Ostman A.; Böhmer F.-D. Oxidation sensitivity of the catalytic cysteine of the protein-tyrosine phosphatases SHP-1 and SHP-2. Free Radical Biol. Med. 2007, 43, 100–110. 10.1016/j.freeradbiomed.2007.03.021. [DOI] [PubMed] [Google Scholar]
- Chen C.-Y.; Willard D.; Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry 2009, 48, 1399–1409. 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- Heneberg P.; Dráber P. Regulation of cys-based protein tyrosine phosphatases via reactive oxygen and nitrogen species in mast cells and basophils. Curr. Med. Chem. 2005, 12, 1859–1871. 10.2174/0929867054546636. [DOI] [PubMed] [Google Scholar]
- Fragale A.; Tartaglia M.; Wu J.; Gelb B. D. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum. Mutat. 2004, 23, 267–277. 10.1002/humu.20005. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Niemeyer C. M.; Fragale A.; Song X.; Buechner J.; Jung A.; Hählen K.; Hasle H.; Licht J. D.; Gelb B. D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- Mitra S.; Beach C.; Feng G.-S.; Plattner R. SHP-2 is a novel target of Abl kinases during cell proliferation. J. Cell Sci. 2008, 121, 3335–3346. 10.1242/jcs.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W.; Wang X.; Romanov V.; Hutchinson A.; Lin A.; Ruzanov M.; Battaile K. P.; Pai E. F.; Neel B. G.; Chirgadze N. Y. Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11). BMC Struct. Biol. 2014, 14, 10. 10.1186/1472-6807-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeen A.; Andersen J. N.; Myers M. P.; Meng T.-C.; Hinks J. A.; Tonks N. K.; Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- van Montfort R. L. M.; Congreve M.; Tisi D.; Carr R.; Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 2003, 423, 773–777. 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- Keilhack H.; David F. S.; McGregor M.; Cantley L. C.; Neel B. G. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J. Biol. Chem. 2005, 280, 30984–30993. 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- Peti W.; Page R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expression Purif. 2007, 51, 1–10. 10.1016/j.pep.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Storoni L. C.; Read R. J. Likelihood-enhanced fast translation functions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005, 61, 458–464. 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B.; de Bakker P. I. W.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Vaguine A. A.; Richelle J.; Wodak S. J. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 191–205. 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]