

Abstract
The inaccuracy of 17-β estradiol (E2) measurements affects its use as a biomarker in patient care and research. Clinical and research communities called for accurate and standardized E2 measurements. Reference Measurement Procedures (RMPs), part of the CDC Hormone Standardization Program (HoSt), are essential in addressing this need and ensuring that methods are accurate and comparable across testing systems, laboratories, and over time. A candidate RMP (cRMP) was developed for the measurement of total E2 in serum using liquid chromatography-tandem mass spectrometry (LC-MS/MS) without derivatization. The cRMP meets suggested performance criteria for accuracy and precision through the use of isotope dilution, calibrator bracketing, and gravimetric measurements. The cRMP demonstrated high agreement with certified reference materials (no significant bias to BCR576, 577, and 578) and established RMPs (slope 1.00, 95% CI 1.00–1.01; intercept 0.02, 95% CI -0.01 to 0.06). The cRMP is highly precise with intra-assay, interassay, and total percent CVs of 2.7%, 1.3%, and 2.4%, respectively. A higher specificity was achieved by measuring E2 without derivatization, compared to methods using derivatization agents. The cRMP can serve as a higher-order standard for establishing measurement traceability and provides an accuracy base against which routine methods can be compared in HoSt.
Graphical Abstract
Estradiol (E2) measurements are used to monitor antiestrogen therapy (e.g., aromatase inhibitor therapy),1 assess fertility,2 monitor sex steroid metabolism disorders,3 identify E2 secreting tumors,4 and assess fracture risk.5 Several research studies and meta-analyses found that elevated E2 levels in postmenopausal women increase the risk for breast cancer with relative risk ratios similar to those observed for cholesterol levels and cardiovascular disease risk.6,7 Additional studies have reported associations between E2 and the risk of endometrial cancer,8,9 cardiovascular disease,10 and cognitive function.11
E2 measurements are reported to be inaccurate and highly variable, especially at the low concentrations observed in men, postmenopausal women, and women on aromatase inhibitor therapy.1,12–14 It has been suggested that the source of high variability at low concentrations is due to lack of method specificity,1,12,13,15 which can impact the diagnosis and treatment assessments of patients. For example, it has been suggested that in postmenopausal women on aromatase inhibitor therapy, inaccurate measurements can lead to incorrect assessments of treatment efficacy.15–17
A recent report from the clinical and research community and a position statement by The Endocrine Society recognized the problem with inaccurate E2 measurements and the need to improve steroid hormones measurements.1,12 To make improvements, clinical laboratories, proficiency testing (PT) providers, and assay manufacturers need access to highly accurate and precise reference measurement procedures (RMPs) to assess the trueness of analytical methods used in patient care and research. To address these needs, the CDC established the Hormone Standardization Program (HoSt).18,19 One key component of this program is an RMP for E2.
RMPs are analytically involved, are laborious, require larger sample volumes, and as a result are not practical for routine laboratories to adopt. Only three RMPs for E2 in serum are recognized by the Joint Committee for Traceability in Laboratory Medicine (JCTLM) as meeting criteria for higher-order reference method standards according to ISO 15193.20,21 Two of the methods use gas chromatography/mass spectrometry (GC/MS)22,23 and one uses liquid chromatography-tandem mass spectrometry (LC-MS/MS) methodology.24 The new proposed candidate RMP (cRMP) meets the accuracy and precision recommendations for measuring E2 with sufficient sensitivity for assigning target values in serum of men and women (pre- and postmenopausal) and is comparable to established RMPs. In contrast to the published LC-MS/MS RMP, this method uses optimized chromatography without requiring derivatization to improve specificity. In addition, no operational RMP exists in the United States to address the needs of the clinical laboratories, PT providers, and assay manufacturers. The cRMP presented here is suitable to meet these needs.
MATERIALS AND METHODS
Certified primary reference standard for 17-β estradiol NMIJ CRM 6004-a lot 009 from National Metrology Institute of Japan (Ibaraki, Japan) with a purity of 98.4% was used as calibrators. [2H5]-estradiol lot B0369 (Steraloids, Newport, RI), with an isotopic purity of 99.0%, was used as an internal standard (IS). HPLC-grade methanol and hexane, as well as reagent-grade ammonium carbonate and sodium acetate were obtained from Fisher Scientific (Suwanee, GA). Anhydrous ethanol was purchased from Sigma-Aldrich (St. Louis, MO). Ammonium fluoride was obtained from Acros (Pittsburgh, PA). Steroids analyzed for interference testing (Table 1) were obtained from Steraloids (Newport, RI), LGC Standards (Manchester, NH), Cerilliant (Round Rock, TX) and Sigma-Aldrich (St. Louis, MO). Three levels of serum-based certified reference materials (BCR 576, BCR 577, and BCR 578) were purchased from the Institute for Reference Materials and Measurements (IRMM, Geel, Belgium). Fresh-frozen, single-donor serum materials were obtained from Solomon Park Research Laboratories (Kirkland, WA). Other serum materials for QC preparation and matrix evaluations were obtained from BioreclamationIVT (Westbury, NY).
Table 1.
List of Steroid Hormones Used for Interference Analysis Including the Molecular Weight, Concentration Used during Testing, and Source of Material Used
| steroid | MW (g/mol) | concn nM (ng/mL) | vendor |
|---|---|---|---|
| 4,16-Androstadien-3β-ol | 272.43 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| 5,16-Androstadien-3β-ol | 272.43 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| Androstenediol | 290.44 | 0.38 (0.11) | Steraloids, Inc. (Newport, RI) |
| Androstenedione | 290.44 | 0.38 (0.11) | LGC Standards (Manchester, NH) |
| 16,(5α)-Androsten-3-one | 272.43 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| 2,(5α)-Androsten-17-one | 272.43 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| Androsterone | 290.40 | 0.38 (0.11) | Steraloids, Inc. (Newport, RI) |
| 5α-Cholestane | 372.67 | 0.30 (0.11) | Steraloids, Inc. (Newport, RI) |
| 6-Dehydronandrolone | 272.38 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| Dehydroepiandrosterone (DHEA) | 288.42 | 0.39 (0.11) | Cerilliant (Round Rock, TX) |
| Dehydroepiandrosterone sulfate (DHEAS) | 368.49 | 0.30 (0.11) | Cerilliant (Round Rock, TX) |
| 1-Dehydrotestosterone | 286.41 | 0.39 (0.11) | Steraloids, Inc. (Newport, RI) |
| 3-Deoxydehydroepiandrosterone (3-deoxy DHEA) | 272.43 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| 17-Desoxyestradiol | 256.40 | 0.43 (0.11) | Steraloids, Inc. (Newport, RI) |
| 5a-Dihydrotestosterone (DHT) | 290.44 | 0.38 (0.11) | Cerilliant (Round Rock, TX) |
| Epiandrosterone | 290.44 | 0.38 (0.11) | Sigma-Aldrich (St. Louis, MO) |
| 17α-Estradiol (epiestradiol) | 272.38 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| Epitestosterone | 288.42 | 0.39 (0.11) | Cerilliant (Round Rock, TX) |
| 1,3,5(10)-Estratrien-16β, 17β-epoxy-3-ol | 272.38 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| 5(10)-Estren-3,17-dione | 272.40 | 0.41 (0.11) | Steraloids, Inc. (Newport, RI) |
| Estriol | 288.38 | 0.39 (0.11) | Cerilliant (Round Rock, TX) |
| Estrone | 270.37 | 0.41 (0.11) | Cerilliant (Round Rock, TX) |
| 17α-Ethynylestradiol | 296.40 | 0.37 (0.11) | Cerilliant (Round Rock, TX) |
| Etiocholanolone | 290.40 | 0.38 (0.11) | Cerilliant (Round Rock, TX) |
| 2-Methoxyestradiol | 302.41 | 0.37 (0.11) | Steraloids, Inc. (Newport, RI) |
| 17α-Methyl-5α-androstane-3α,17β-diol | 306.49 | 0.36 (0.11) | Cerilliant (Round Rock, TX) |
| 17-α Methyltestosterone | 302.46 | 0.37 (0.11) | Cerilliant (Round Rock, TX) |
| 19-Norethindrone | 298.42 | 0.37 (0.11) | Steraloids, Inc. (Newport, RI) |
| D(−)-Norgestrel | 312.45 | 0.36 (0.11) | Sigma-Aldrich (St. Louis, MO) |
Calibrator Preparation
All volumetric steps for the calibrator and sample preparation were controlled by gravimetric measurements. Mass fractions (mg/g) were calculated for all calibrator stock solutions and calibrators. For the purpose of this manuscript, mass fractions (mg/g) were converted into volume concentrations using the specific density determined with a DMA 500 density meter (Anton Paar, Ashland, VA). A primary stock solution (PSS) was prepared by dissolving 300 mg of E2 in 100 mL of anhydrous ethanol and further diluting was achieved through addition of 200 μL of this solution in 100 mL of anhydrous ethanol (Stock B). Stock C was created by diluting 100 μL of Stock B solution in 100 mL of anhydrous ethanol. Three working standard solutions were prepared by diluting 5.0 mL, 1.0 mL, and 100 μL of Stock C in 100 mL of anhydrous ethanol: WS1 (1.10 nM [300 pg/mL]), WS2 (220 pM [60.0 pg/mL]) and WS3 (22.0 pM [6.00 pg/ mL]). Internal standard (IS) PSS and working standard solutions ISWS1, ISWS2, and ISWS3 were prepared using [2H5]-estradiol in the same manner as unlabeled E2 to obtain similar final concentrations. WS1 and ISWS1 (high) solutions were used to quantitate values typically reported in premenopausal women (>257 pM [70.0 pg/mL]), WS2 and ISWS2 (medium) for men and some postmenopausal women (36.7–257 pM [10.0–70.0 pg/mL]), and WS3 and ISWS3 (low) for postmenopausal women (<36.7 pM [10.0 pg/mL]).
Similar to procedures previously described, five point calibration curves were prepared by adding varying amounts of ISWS solution to WS solution to obtain mass ratios of unlabeled to labeled E2 of 0.5, 0.8, 1.0, 1.2, and 1.5.25,26 The procedure was applied to establish calibration curves for a concentration range of high 440–734 pM (120–200 pg/mL), medium 88.1–147 pM (24.0–40.0 pg/mL), and low 8.81–14.7 pM (2.40–4.00 pg/mL). Calibration curves were prepared in triplicate.
Sample Preparation
Determination of E2 concentration in a sample was performed using bracketing calibration as described previously.25,26 In brief, the amount of ISWS solution was adjusted to achieve an approximate mass ratio of one to the endogenous E2. For that, a routine method measured the approximate concentration of E2 in the sample. The result from this measurement was used to determine the amount of ISWS solution needed to achieve the ratio of one. The ISWS solution was transferred to a glass vial, and the solvent was evaporated under vacuum prior to addition of serum to avoid protein precipitation. For analysis of high concentration samples, 0.5 mL of serum was used; for medium concentration samples 1.0 mL was used; and for low concentration samples 3.0–5.0 mL was used, depending upon the approximate concentration of the sample. The samples and IS were allowed to equilibrate at room temperature for 30 min while gently shaking as previously described to improve accuracy and reproducibility.22
E2 was released from binding proteins with the addition of sodium acetate (0.5 M, pH 5.5) at a sample-to-buffer ratio of 1:2 (v/v) and equilibrated at room temperature (20 °C) for 2 h. Liquid/liquid extraction (LLE) was performed in duplicate using 2.5 mL of an ethyl acetate-hexane (3:2, v/v) solution. The two organic extracts were combined, evaporated to dryness under vacuum, and reconstituted in 0.5 mL of ammonium carbonate (0.2 M, pH 8.2). To remove polar lipids, the solution was extracted using 3 mL of ethyl acetate-hexane (3:2, v/v) in duplicate. The organic layers were dried under vacuum and reconstituted with 120 μL of a water-methanol mixture (80:20, v/v) for LC-MS/MS analysis. Three quality control (QC) materials from unaltered, pooled serum (QCLow, QCMedium, and QCHigh) and three IRMM certified reference materials (BCR 576, BCR 577, and BCR 578) were prepared by reconstitution according to the certificates of analysis and analyzed in duplicate in each run.
LC-MS/MS Conditions
Two injections for each preparation were performed on an AB/Sciex API 5500 tandem mass spectrometer (Foster City, CA) equipped with a Shimadzu LC-10AD/SIL-30AC Nexera system (Columbia, MD). A Phenyl/ Hexyl Kinetex, 50 mm × 4.6 mm, 2.6 μm analytical column with a guard column (Phenomenex, Torrance, CA) heated to 40 °C was used with 0.2 mM ammonium fluoride in water (Buffer A) and methanol only (Buffer B). The HPLC separation was achieved with a 9 min gradient from 48% to 62% Buffer B at a flow rate of 750 μL/min. The mass spectrometer was operated with electrospray ionization (ESI) in the negative ion mode with voltage set to −4 000 V, collision energy set to −50 eV, and Turbo Ion Spray Source temperature set at 650 °C. Selected reaction monitoring (SRM) was used with the following transitions: m/z 271 → 145 (quantitation ion, QI) and m/z 271 → 183 (confirmation ion, CI) for E2 and with 276 → 147 (QI) and m/z 276 → 187 (CI) for the IS.
Data Analysis
Analyst 1.6.2 software (AB/Sciex, Framingham, MA) was used to integrate selected peaks. Quantitation was performed with the peak area count ratio of the E2 QI to the IS QI. The mean area count ratios from the two injections were used for data analysis. To convert the mass fraction to the commonly used volume concentration, the density of the serum samples was used. For value assignment, duplicate preparations of serum were made in two independent runs (n = 4). The mean value from four measurements was used to assign an E2 concentration to the sample. Statistical evaluations for Deming regression and difference plots were performed with Analyze-It Software, Ltd. version 2.26 (Leeds, U.K.) using t test, α = 0.05.
Method Validation
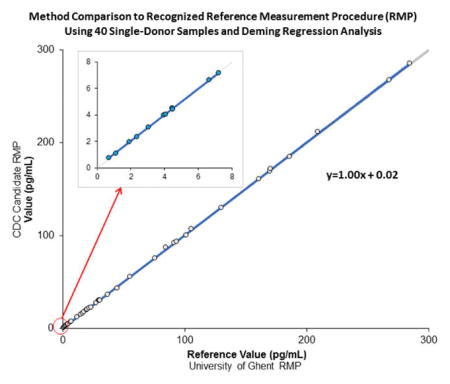
Accuracy was evaluated through a comparison with a JCTLM-recognized RMP at the University of Ghent (JCTLM code-NRMeth 10)23 and IRMM certified reference materials. The mean percent bias of the cRMP to the JCTLM-recognized RMP was determined following the CLSI document EP 9 A2 protocol comparing 40 single-donor serum samples.27 The cRMP was compared to the JCTLM-recognized RMP values using a Deming regression analysis. In addition, IRMM certified reference materials were analyzed, and the mean percent difference from the certified value was determined and evaluated for significance as described in NIST Special Publication 829.28
Imprecision was evaluated by analyzing 3 serum pools in duplicate over 5 days. CLSI EP 10-A3 was used to calculate the intra-assay, interassay, and total percent coefficients of variation (CVs).29 Standard uncertainty and expanded uncertainty of the cRMP following the ISO Guide to the Expression of Uncertainty in Measurement 2008 was also evaluated.30 The estimated variance of type A uncertainty was calculated from repeated measurements, and type B uncertainty was calculated using uncertainties in the purity of the reference compound, inaccuracy in the weighing of each component, and the measurement of the serum density. Calculated type A and B uncertainties were combined to determine the standard uncertainty and multiplied by a coverage factor, k = 2, to determine the expanded uncertainty using QCLow, QCMedium, and QCHigh.
In total, 29 structural analogues and other steroid hormones with relative molecular masses close to E2 and the IS were tested as potential interferences (Table 1). The absence of a peak with the mass transitions of E2 (m/z 271 → 145) or the IS (m/z 276 → 187) at the retention time of E2 at 11.3 min confirmed that the structural analogues did not interfere with the quantitation of E2. In addition, the QI/CI ratio of E2 was monitored in the 40 single-donor samples and compared to the QI/CI ratio of the calibrators. A difference of less than ±20% was used to confirm that no interfering compounds were present.31
The effect of different sample matrixes was determined following procedures previously described.25 Four sample matrixes (0.9% saline solution, charcoal stripped serum, male serum, and female serum) and one set of neat samples in ethanol were evaluated. A 7-point calibration curve ranging from 147 to 848 pM (40–231 pg/mL) was prepared in each matrix and spiked with 551 pM (150 pg/dL) IS solution. The calibrators in the matrixes were subjected to the sample preparation described above. The MS response (area count ratios of analyte to IS) was compared in all four matrixes to the MS response of the neat samples prepared in ethanol (matrix-free). The sample matrix effect (ME) was calculated with the following equation: ME % = B/A × 100, where B is the area count ratios of E2 to IS obtained from samples in matrix, and A is the area count ratios in matrix-free samples.
RESULTS AND DISCUSSION
The trueness and imprecision of an RMP needs to be smaller than that of routine measurement procedures to ensure target values assigned have a sufficiently low uncertainty so that any uncertainty added by a routine method can then still lead to clinically meaningful results. RMPs should have a limit that is half the limit for a routine method for imprecision and a limit that is one-third the maximum bias.32,33 The CDC HoSt Program for E2 certifies analytical performance criteria for a routine E2 assay with a difference of ±12.5% at concentrations <73.4 pmol/L (20.0 pg/mL) and ±2.5 pg at concentrations > 73.4 pmol/L (20.0 pg/mL) from the true value and includes a suggested imprecision of 11.3%.15 The described cRMP meets the suggested performance criteria for E2 with a bias within ±2.8% and imprecision of ≤5.7%. These characteristics are similar to those of established RMPs.22–24
A comparison of our cRMP against an established RMP using 40 samples over a concentration range of 2.61–1 050 pM (0.71–285 pg/mL) using Deming regression analysis with a variance ratio of 1 and t test, α = 0.05 found no statistically significant difference (Table 2). The agreement between the candidate and reference measurement procedure did not change when the samples were evaluated separately for low (typically found in postmenopausal women), medium (typically found in postmenopausal women and men), and high (typically found in premenopausal women) E2 concentrations. The mean percent bias of the cRMP to the established RMP for the 40 samples was 0.5% (95% CI, 0.1–0.9%; SE 0.21%), which is well within the suggested bias criterion of ±2.8%. All measurement results obtained with the IRMM serum-based certified reference materials were within ±0.9% of the assigned certified values for BCR 576, 577, and 578 and showed no significant bias when evaluated following the protocol described in NIST Special Publication 82928 (Table 3). Not only does the cRMP have an overall mean bias within suggested criteria, but each evaluated individual sample met this criteria, indicating no relevant sample specific bias.
Table 2.
Method Comparison to a Recognized RMP Using 40 Single-Donor Samples and Deming Regression Analysis
| sample group | n | range pM (pg/mL) |
slope (95% CI) [SE, p]a |
intercept (95% CI) [SE, p]a |
|---|---|---|---|---|
| 2.61–1 050 | 1.00 | 0.02 | ||
| all | 40 | (0.71–285) | (0.997–1.008) | (−0.02–0.05) |
| [0.003, 0.363] | [0.018, 0.311] | |||
| high (>257 pM)b | 277–1 050 | 1.00 | 1.38 | |
| 14 | (75.4–285) | (0.978–1.011) | (−1.17–3.93) | |
| [0.007, 0.499] | [1.170, 0.260] | |||
| medium (36.7–257 pM)c | 43.7–203 | 0.99 | 0.38 | |
| 15 | (11.9–55.3) | (0.962–1.010) | (−0.06–0.83) | |
| [0.011, 0.234] | [0.206, 0.085] | |||
| low (<36.7 pM)d | 2.61–26.5 | 0.99 | 0.03 | |
| 11 | (0.71–7.21) | (0.979–1.008) | (−0.02–0.08) | |
| [0.006, 0.365] | [0.021, 0.197] |
SE = standard error.
Concentrations typically found in premenopausal females.
Concentrations typically found in males and postmenopausal females.
Concentrations typically found in postmenopausal females.
Table 3.
Bias Assessment Using JCTLM-Recognized Reference Materials
| IRMM BCR material | reference target value, pM (pg/mL) | bias, % (95% CI) |
|---|---|---|
| BCR 576 | 114 (31.1) | 0.3 (0.2–0.5)a |
| BCR 577 | 690 (188) | −0.3 (−2.4–1.8)a |
| BCR 578 | 140 (365) | −0.9 (−4.0–2.2)a |
No significant bias when evaluated following the protocol described in NIST Special Publication 829.26
The intra-assay and interassay imprecision was less than 2.7% for all three levels of QC material tested (Table 4). The combined or total imprecision was less than 2.4%. Uncertainty was assessed at three levels as shown in Table 4 for both type A and type B factors. The relative expanded uncertainty was ≤ 4.8% at each level. Type B uncertainty was minimized through the use of primary standards with purity >98% and the use of gravimetric measurements in place of volumetric measurements in preparation of standards and samples. The imprecision and uncertainty were consistent and independent of concentrations of E2 in the cRMP, meeting performance goals across concentrations in premenopausal women, men, and postmenopausal women serum samples.
Table 4.
Inter-, Intra-, and Total Assay Imprecision and Estimation of Expanded Uncertainties of cRMP Measurements of Estradiol in Serum at Three Concentrationsa
| QCHigh | QCMedium | QCLow | |
|---|---|---|---|
| concn pM (pg/mL) | 455 (124) | 147 (40.0) | 19.2 (5.23) |
| interassay CV, % | 2.7 | 2.4 | 2.3 |
| intra-assay CV, % | 1.3 | 1.3 | 1.1 |
| total CV, % | 2.4 | 2.0 | 2.4 |
| type A (pM) | 10.92 | 2.94 | 0.38 |
| type B (pM) | 0.01 | 0.01 | 0.01 |
| combined standard uncertainty | 10.92 | 2.94 | 0.38 |
| coverage factor | 2 | 2 | 2 |
| expanded uncertainty (95% confidence interval) | 21.8 | 5.88 | 0.77 |
| relative expanded uncertainty (%) | 4.8 | 4.0 | 4.0 |
Samples were measured in triplicate over 5 days.
The cRMP provided high levels of accuracy and precision. This was achieved by gravimetric measurements, bracketing of calibrators, enhanced chromatographic resolution, and mass transitions specific to E2. Volumetric steps were replaced with gravimetric measurements to minimize the contributions of imprecision and inaccuracy from pipets and volume changes due to changes in temperature.34 Bracketing calibration around the target concentration, a common practice for reference methods, further increased precision and accuracy.35,36 Variability at the extremes of the large dynamic concentration range commonly observed in regular calibration curves (2.61-1 100 to pM) was minimized with bracketing into three different levels for quantitation. These adjustments were used so that the analyte and IS were close in concentration, which reduced the variability of signal intensities and measurement imprecision by reducing potential concentration-dependent ion suppression effects that would result from significantly different concentrations of IS and analyte entering the mass spectrometer.37
The structural analogues and potential interfering compounds of E2 listed in Table 1 either did not contain the same mass transitions or were chromatographically separated from E2 (11.3 min). When an IS-only sample was monitored, no additional peaks were observed in the analyte signal, confirming the absence of isotopic interferences from the use of the selected internal standard.
The mass transitions used for quantitation by the cRMP were highly specific to E2 and included the precursor ion for E2 (271 m/z) as well as a fragment ion (145 m/z). Several LC-MS/MS methods for E2, as well as the previously described LC-MS/ MS RMP, use dansyl chloride as a derivatizing agent to enhance the ionization of E2 in the source and to increase the method’s sensitivity. However, with these methods the dansyl fragment ion is used to quantitate E2, thereby limiting the specificity of the method. This limitation is overcome in our method by measuring E2 without derivatization.
In addition the specificity of the mass transitions to E2 without derivatization for both the QI and CI allowed for the use of QI/CI ratios to rule out any potential interferences. For this method, the QI/CI ratios in 40 single-donor serum samples were compared to the neat calibrators. The mean QI/ CI ratio of 9 replicates of each calibrator was 1.12 (95% CI of 1.11–1.13) and mean ratio of 40 samples from 2.61 to 1 050 pM was 1.11 (95% CI of 1.11–1.12). The QI/CI ratios of all 40 serum samples were well within ±20% of the mean QI/CI ratio of calibrators, indicating that the cRMP is not affected by interfering compounds.
Chromatographic resolution was important for the cRMP to ensure any potential interfering compounds that could be identified by the QI/CI were properly resolved. An example is shown in Figure 1, where several additional peaks can be found resolved from the E2 peak at 11.3 min in the serum sample (Figure 1B) as compared to the neat sample (Figure 1A). In example, if the peaks in the 2 min window (9.3–11.3 min) in Figure 1B were not properly resolved, the concentration of the serum sample in Figure 1B, with a true E2 concentration of 26.5 pM (7.21 pg/mL), would increase by approximately 173% to 72.7 pM (19.8 pg/mL).
Figure 1.

Chromatograms of neat and serum sample for E2 (retention time, 11.3 min): (A) neat, E2 concentration of 20.3 pM (5.52 pg/mL), (B) native serum sample, E2 concentration of 26.5 pM (7.21 pg/mL) resolved from additional interference peaks.
A phenyl/hexyl column was used to improve the separation of aromatic compounds with a 9 min gradient from 48% to 62% of buffer B to further resolve E2. The change in percent buffer B (1.6% per min) is similar to the previously published LC-MS/MS RMP24 (1.4% per min) but was accomplished in less than half the time, thus increasing the throughput while still achieving the required resolution. This chromatographic approach allowed us to resolve several additional peaks that could potentially contribute to inaccurate E2 results.
Maximizing the recovery of E2 during sample preparation is important in ensuring adequate signal strength during LC-MS/MS analysis. Low recovery can affect the method’s LOD as well as precision if ample analyte and IS are not recovered, especially at the low concentrations of E2. The overall extraction efficiency of the procedure was assessed by spiking IS prior to sample preparation and injection onto the LC-MS/ MS in duplicate at a concentration of 1.02 nM (279 pg/mL) as described previously.38 An extraction recovery of 90.2% (7.7% CV) was achieved. To enhance ionization and sensitivity of the method without a derivatizing agent, 0.2 mM ammonium fluoride was used in the LC buffer to promote ionization of E2 under negative ionization mode.39 Sensitivity sufficient for assigning target values to sera from men and pre- and postmenopausal women was achieved and is the intended use of this method. Through this approach, the limit of detection (LOD) (at a signal-to-noise ratio (S/N) of 10) was 2.61 pM (0.71 pg/mL), which is a slight improvement over the previously published LC-MS/MS RMP and has additional specificity achieved by avoiding dansyl chloride derivatization.
For evaluation of ME, the slope and r2 values were determined for each matrix and the neat calibrators as well as the mean ME % for each matrix (Table 5). On the basis of the data presented, the sample matrix effect can be evaluated by two separate observations. First, as the slopes of all matrixes and matrix free curves are identical, the absence of any significant sample ME on quantification can be confirmed. Second, the sample ME % showed either no effect or a slight enhancement for the saline and charcoal stripped matrixes evaluated (>100%). The serum samples had a ME % of 96.2% and 92.2%, indicating slight ion suppression from the matrix, as expected with increased serum volume requirements for samples with lower concentrations of E2 such as male samples. However, our evaluations have shown no significant effect of this matrix effects on the accuracy and precision of our method in both male and female samples.
Table 5.
Matrix Effect Evaluation Using a 7-Point Calibration Curve (147 to 848 pM, 40–231 pg/mL) was Prepared in 4 Different Matrixes and Spiked with 551 pM (150 mg/mL) Internal Standard
| matrix
|
|||||||
|---|---|---|---|---|---|---|---|
| neat | saline | charcoal | male | female | mean | 95% CI | |
| slope | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 | 0.0060–0.0061 |
| R2 | 0.99 | 0.99 | 0.99 | 0.98 | 0.98 | 0.99 | 0.986–0.989 |
| mean ME% | 103.5 | 102.8 | 92.2 | 96.2 | 98.7 | 96.8–100.5 | |
Sample preparation of the cRMP plays a key role in ensuring that no interferences and no significant matrix effects exist. This is achieved in part through the selected conditions used during the isolation of the analyte from the matrix. The first LLE isolated the lipids from polar compounds. A solution of ethyl acetate/hexane at 3:2, v/v, was selected to maximize isolation of the analyte from the matrix. The addition of a basic solution (ammonium carbonate buffer, pH 8.2) to the sample extract and the second LLE allowed for the removal of acidic impurities, such as fatty acids and phospholipids. These lipids are a source of ion suppression and matrix effects, particularly in LC-MS/MS analyses.37 A higher pH for the ammonium carbonate buffer would increase the removal of impurities;25 however, the pH of 8.2 was selected to be two pH units below the pKA of E2 (10.5) to avoid deprotonation and subsequent decrease in recovery of E2.40
The cRMP has been automated using the Hamilton Starlet (Reno, NV) equipped with an analytical balance. The enhancement to the method doubled the sample throughput compared to the manual sample preparation which helps to meet the increasing demand for target value assignments.
CONCLUSION
The described ID LC-MS/MS cRMP for measuring total 17-β estradiol with sufficient sensitivity for assigning target values to sera from men, and pre- and postmenopausal women, has higher specificity at similar accuracy and precision performance compared to existing RMPs. High specificity is needed to ensure accurate target value assignment, especially in samples with low E2 concentrations, where small amounts of interfering compounds can have a high impact on the measurement result. The cRMP is suitable to address the current unmet needs of clinical laboratories, assay manufacturers, and PT providers by providing value assignments to serum for calibration and verification of method performance as part of the CDC HoSt Program.
Acknowledgments
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the views of the Centers for Disease Control and Prevention. The authors would like to acknowledge support from the Division of Laboratory Science at the CDC, the CDC Foundation, Drs. Linda Thienpont and Kathleen Van Uytfanghe from the University of Ghent, CDC Hormone Laboratory members Otoe Sugahara, Jacob Farris, Lumi Duke, Paul Kim, Brandon Laughlin, Susan Ma, Krista Poynter, Christopher Ghattas, Dr. Uliana Danilenko, and CDC PBL/LRL. In addition, the authors would like to acknowledge The Endocrine Society, AACC, and the Partnership for the Accurate Testing of Hormones (PATH) for their support.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Demers LM, Hankinson SE, Haymond S, Key T, Rosner W, Santen RJ, Stanczyk FZ, Vesper HW, Ziegler RG. J Clin Endocrinol Metab. 2015;100:2165–70. doi: 10.1210/jc.2015-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demers LM. Steroids. 2008;73:1333–8. doi: 10.1016/j.steroids.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Rahhal SN, Fuqua JS, Lee PA. Steroids. 2008;73:1322–7. doi: 10.1016/j.steroids.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Fossa SD, Klepp O, Barth E, Aakvaag A, Kaalhus O. Int J Androl. 1980;3:487–501. doi: 10.1111/j.1365-2605.1980.tb00137.x. [DOI] [PubMed] [Google Scholar]
- 5.Roddam AW, Appleby P, Neale R, Dowsett M, Folkerd E, Tipper S, Allen NE, Key TJ. J Bone Miner Metab. 2009;27:485–93. doi: 10.1007/s00774-009-0060-z. [DOI] [PubMed] [Google Scholar]
- 6.Key TJ, Appleby PN, Reeves GK, Travis RC. Steroids. 2015;99:49–55. doi: 10.1016/j.steroids.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Key TJ. Steroids. 2011;76:812–5. doi: 10.1016/j.steroids.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Allen NE, Key TJ, Dossus L, Rinaldi S, Cust A, Lukanova A, Peeters PH, Onland-Moret NC, Lahmann PH, Berrino F, et al. Endocr-Relat Cancer. 2008;15:485–97. doi: 10.1677/ERC-07-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukanova A, Lundin E, Micheli A, Arslan A, Ferrari P, Rinaldi S, Krogh V, Lenner P, Shore RE, Biessy C, et al. Int J Cancer. 2004;108:425–32. doi: 10.1002/ijc.11529. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Zeleniuch-Jacquotte A, Arslan AA, Wojcik O, Toniolo P, Shore RE, Levitz M, Koenig KL. Atherosclerosis. 2011;216:414–9. doi: 10.1016/j.atherosclerosis.2011.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boss L, Kang D, Marcus M, Bergstrom N. West J Nurs Res. 2014;36:388–426. doi: 10.1177/0193945913500566. [DOI] [PubMed] [Google Scholar]
- 12.Rosner W, Hankinson SE, Sluss PM. J Clin Endocrinol Metab. 2013;98:1376–1387. doi: 10.1210/jc.2012-3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vesper HW, Botelho JC, Wang Y. Asian J Androl. 2014;16:178–84. doi: 10.4103/1008-682X.122338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Handelsman DJ, Newman JD, Jimenez M, McLachlan R, Sartorius G, Jones GR. Clin Chem. 2014;60:510–7. doi: 10.1373/clinchem.2013.213363. [DOI] [PubMed] [Google Scholar]
- 15.Vesper HW, Botelho JC, Vidal ML, Rahmani Y, Thienpont LM, Caudill SP. Steroids. 2014;82:7–13. doi: 10.1016/j.steroids.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson RE, Grebe SK, Okane DJ, Singh RJ. Clin Chem. 2004;50:373–384. doi: 10.1373/clinchem.2003.025478. [DOI] [PubMed] [Google Scholar]
- 17.Jaque J, Macdonald H, Brueggmann D, Patel SK, Azen C, Clarke N, Stanczyk FZ. SpringerPlus. 2013;2:5. doi: 10.1186/2193-1801-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Centers for Disease Control and Prevention. [Accessed March 2012];Laboratory Quality Assurance and Standardization Programs: Hormone Standardization Program. http://www.cdc.gov/labstandards/hs.html.
- 19.Vesper HW, Botelho JC. J Steroid Biochem Mol Biol. 2010;121:513–9. doi: 10.1016/j.jsbmb.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 20.Joint Committee for Traceability of Laboratory Measurements (JCTLM) [accessed January 2011];Database of higher order reference materials; measurement methods/procedures and services. http://www.bipm.org/jctlm/
- 21.International Organization of Standardization (ISO) ISO 15193:2009(E): In vitro diagnostic medical devices–Measurement of quantities in samples of biological origin–Requirements for content and presentation of reference measurement procedures. International Organization of Standardization; Geneva, Switzerland: [Google Scholar]
- 22.Siekmann L, Siekmann A, Breuer H. Fresenius' Z Anal Chem. 1978;290:122–3. [Google Scholar]
- 23.Thienpont LM, De Leenheer AP. Clin Chem. 1998;44:671–674. [PubMed] [Google Scholar]
- 24.Tai SSC, Welch MJ. Anal Chem. 2005;77:6359–63. doi: 10.1021/ac050837i. [DOI] [PubMed] [Google Scholar]
- 25.Botelho JC, Shacklady C, Cooper HC, Tai SS, Van Uytfanghe K, Thienpont LM, Vesper HW. Clin Chem. 2013;59:372–380. doi: 10.1373/clinchem.2012.190934. [DOI] [PubMed] [Google Scholar]
- 26.Vesper HW, Botelho JC, Tai SSC, Van Uytfanghe K, Thienpont LM. Clin Chem. 2013;59:1130–1. doi: 10.1373/clinchem.2013.205856. [DOI] [PubMed] [Google Scholar]
- 27.CLSI. EP9-A2: Method comparison and bias estimation using patient samples; approved guideline. 2. Clinical and Laboratory Standards Institute; Wayne, PA: 2002. [Google Scholar]
- 28.Becker D, Christensen R, Currie L, Diamondstone B, Eberhardt K, Gills T, Hertz H, Klouda G, Moody J, Parris R, et al. NIST Special Publication 829. U.S. Department of Commerce, Technology Administration, National Institute of Standards and Technology; Gaithersburg, MD: 1992. Use of NIST standard reference materials for decisions on performance of analytical chemical methods and laboratories. [Google Scholar]
- 29.CLSI. EP10-A3: Preliminary evaluation of quantitative clinical laboratory measurement procedures: approved guideline. 3. Clinical and Laboratory Standards Institute; Wayne, PA: 2006. [Google Scholar]
- 30.ISO. 98-3:2008: Uncertainty of measurement–Part 3: Guide to the expression of uncertainty in measurement. International Organization of Standardization; Geneva, Switzerland: [Google Scholar]
- 31.CLSI. C-62A: Liquid chromatography-mass spectrometry methods: approved guideline. Clinical and Laboratory Standards Institute; Wayne, PA: 2014. [Google Scholar]
- 32.Stöckl D, Sluss PM, Thienpont LM. Clin Chim Acta. 2009;408:8–13. doi: 10.1016/j.cca.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 33.Petersen PH, Fraser CG, Kallner A, Kenny D. Scand J Clin Lab Invest. 1999;59:477–478. [PubMed] [Google Scholar]
- 34.Bowers GN, Fassett JD, White E. Anal Chem. 1993;65:475–9. doi: 10.1021/ac00060a620. [DOI] [PubMed] [Google Scholar]
- 35.Siekmann L. J Steroid Biochem. 1979;11:117–23. doi: 10.1016/0022-4731(79)90285-1. [DOI] [PubMed] [Google Scholar]
- 36.Thienpont LM, De Brabandere VI, Stöckl D, De Leenheer AP. Anal Chem. 1994;66:4116–4119. doi: 10.1021/ac00094a041. [DOI] [PubMed] [Google Scholar]
- 37.Sargent M, Harrington C, Harte R, editors. LGC Limited. Guidelines for achieving high accuracy in isotope dilution mass spectrometry (IDMS) Royal Society of Chemistry; Cambridge, U.K: 2002. [Google Scholar]
- 38.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal Chem. 2003;75:3019–30. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 39.Fiers T, Casetta B, Bernaert B, Vandersypt E, Debock M, Kaufman JM. J Chromatogr B: Anal Technol Biomed Life Sci. 2012;893–894:57–62. doi: 10.1016/j.jchromb.2012.02.034. [DOI] [PubMed] [Google Scholar]
- 40.Hurwitz AR, Liu ST. J Pharm Sci. 1977;66:624–627. doi: 10.1002/jps.2600660504. [DOI] [PubMed] [Google Scholar]