

Abstract
In this issue of Cancer Cell, Hydbring and colleagues define a novel class of microRNAs (miRNAs), deemed “cell-cycle-targeting miRNAs,” that target several cyclins/CDKs, reduce tumor cell growth, and induce apoptosis. These miRNAs effectively suppressed chemoresistant patient-derived xenograft growth in vivo, and efficacy could be prospectively predicted with an expression-based algorithm.
One of the hallmarks of cancer is unbridled cell proliferation, often manifesting through alterations of the cell-cycle machinery, including cyclin-dependent kinases (CDKs) and their cognate catalytic partners, the cyclins (Hanahan and Weinberg, 2011). Cyclin/CDK complexes regulate cell-cycle phase transitions through selective phosphorylation of substrates that control all aspects of cell division, including commitment to completion of a mitotic cell cycle, achieved through phosphorylation and inactivation of the retinoblastoma (RB) tumor suppressor protein. These functions are counterbalanced by canonical endogenous CDK inhibitors and tumor suppressors. Loss of these cell-cycle-attenuating tumor suppressors, or, by contrast, deregulation of cyclins, CDKs, or CDK targets, can afford a mechanism for tumor cells to bypass the appropriate cell-cycle regulation and occur at high frequency in human malignancies (Otto and Sicinski, 2017). As such, developing effective therapeutic means to thwart cell-cycle dysregulation may lead to significant benefit for cancer patients.
The study by Hydbring et al. brings understanding to the role of miRNAs in controlling cell-cycle regulatory proteins and supports the concept that miRNA function could be harnessed as a means to counteract unchecked cell-cycle progression (summarized in Figure 1) (Hydbring et al., 2017). miRNAs are short, regulatory RNA species that reduce the expression of gene products by inducing mRNA degradation or inhibition of translation (Lagos-Quintana et al., 2001). Similar to the cell-cycle machinery, miRNAs show frequent alteration in human malignancy (Lu et al., 2005). Understanding of miRNA function is a fertile ground of current investigation, and a number of potential applications in both diagnostics and therapeutics have been realized (Adams et al., 2017; Hydbring and Badalian-Very, 2013). Hydbring et al. screened for miRNAs that target nine cyclins and CDKs of importance for cell division in cancer cells. The results of this screen revealed 30 miRNAs with the capacity to target five or more cyclins/CDKs and 14 miRNAs capable of targeting six or more cyclins/CDKs, thus demonstrating a novel class of miRNAs that regulate nearly all components of the cell-cycle machinery. The number of miRNAs that target multiple cyclins/CDKs is significantly higher than predicted by chance alone. Furthermore, prioritization studies identified anti-correlation of selected miRNAs with anticipated cyclin/CDKs in human tumors, and the miRNAs were found to be downregulated or deleted in several tumor types.
Figure 1. Summary of Hydbring et al.
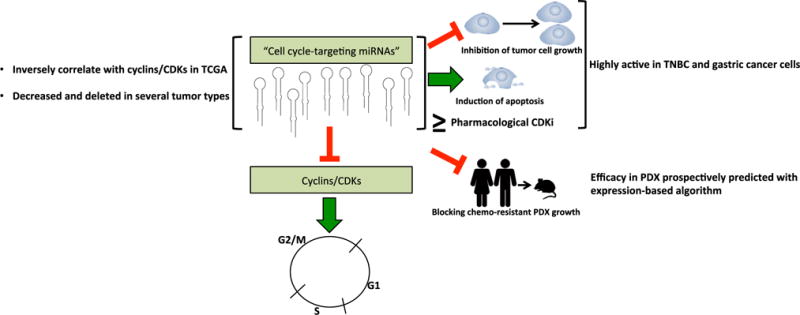
Through several screens, it was determined that a subset of miRNAs target cyclins and CDKs, which are positive drivers of the cell cycle. These “cell-cycle-targeting miRNAs” were found to be inversely correlated with cyclins/CDKs in The Cancer Genome Atlas (TCGA) data, as well as either reduced or deleted in several tumor types. Therapeutic efficacy of these cell-cycle-targeting miRNAs was enriched in TNBC and gastric cancer cell lines, which was validated in chemotherapy-resistant PDX models.
Building upon these initial findings, a second screen utilizing a 122-cancer-cell-line library comprised of 12 tumor types was employed to test the efficacy of these cell-cycle-targeting miRNAs. Four miRNAs identified in the initial screen demonstrated significant growth-inhibitory effects in the cell lines examined, albeit to differing magnitudes. Strikingly, cells derived from gastric cancers and triple-negative breast cancers (TNBCs) demonstrated a more robust response, potentially due to greater reliance on the targeted cyclins/CDKs. In these tissue types, it was further noted that cell-cycle-targeting miRNAs harbored the capacity to induce apoptosis, suggesting that the miRNAs elicit not only the expected cytostatic effects but also cytotoxic responses. Investigation of the underlying basis for enhanced responsiveness in TNBC and gastric cancer may therefore lay the foundation for determining which tumors may be most responsive to miR-based therapies targeting cell-cycle regulation.
Recent clinical successes targeting CDK activity have increased enthusiasm for refining the means to suppress cell-cycle control in a subset of tumor types, most particularly as related to CDK4/6 inhibitors (CDK4/6i). The CDK4/6i palbociclib is FDA approved for the treatment of estrogen receptor (ER)-positive, HER2-negative advanced breast cancer in combination with estrogen deprivation or ER-targeted therapies (letrozole or fulvestrant, respectively) (Finn et al., 2015; Turner et al., 2015), based in part on clinical trial data demonstrating that the combination resulted in improved progression-free survival (PFS) compared to hormone therapy alone. More recently, the CDK4/6i ribociclib was FDA approved for this same patient population in combination with letrozole, based on clinical data indicating that the combination lengthened PFS (Hortobagyi et al., 2016). In principle, the studies here have the potential to build on the success of CDK4/6i, as the miRNAs identified proved capable of simultaneously suppressing more than five to six CDKs or cyclins and therefore could provide a more potent mechanism to suppress the cancer cell cycle. Furthermore, whereas CDK4/6i are approved for ER-positive disease, the data from Hydbring et al. identified ER-negative tumor models (TNBC) as exceptional responders to miRNA-based cell-cycle suppression. Head-to-head comparison revealed that miR193a-3p is more effective than palbociclib in blocking TNBC cell growth, and miR193a-3p elicits anti-tumor effects even in the context of RB loss, which renders cells resistant to palbociclib. miR193a-3p was also more effective than lapatinib in HER2-positive cells and was comparable to the combination of lapatinib and palbociclib. While these findings require validation and development of translational potential, the study by Hydbring et al. provides robust evidence that cell-cycle-targeting miRNAs hold promise as anti-cancer agents that may hold advantages over CDK4/6 targeting alone in breast cancer.
Harnessing the miRNAs for translational potential yielded promising results in patient-derived xenograft (PDX) models of chemotherapy-resistant TNBC and colorectal cancer. In both systems, miRNA-containing nanoparticles were effective in eliciting anti-tumor responses, whereas normal tissues were spared any toxicity. In a doxorubicin- and taxane-resistant TNBC PDX, nanoparticles containing miR193a-3p reduced proliferation and induced apoptosis, demonstrating that cell-cycle-targeting miRNA therapy may be effective in chemotherapy-resistant tumors. Furthermore, in a KRAS mutant FOLFOX- and FOLFIRI-resistant colorectal tumor PDX, miR214-5p blocked tumor growth while sparing normal tissues and was more effective in KRAS mutant than wild-type cells. These data indicate that tumor phenotype and/or genotype may influence the utility of cell-cycle-targeting miRNAs. Given the demonstrated selectivity, it is intriguing to speculate that the heightened reliance of tumor cells on cyclins/CDKs can be effectively targeted in malignant tissues using the cell-cycle-targeting miRNA strategy.
Hydbring et al. also developed an expression-based algorithm to predict responses to cell-cycle-targeting miRNAs. As proof of principle, the authors transcriptionally profiled an aggressive dermatofibrosarcoma protuberans that was resistant to both Gleevec and PI3K inhibition. Utilizing the developed algorithm, it was predicted that the tumor would be responsive to miR-193a-3p. Subsequent administration of nanoparticles containing miR193a-3p to mice harboring a PDX of the dermatofibrosarcoma protuberans resulted in significant reduction of tumor growth, demonstrating the utility of the algorithm. Based on these exciting findings, future studies examining combinatorial strategies with cell-cycle-targeting miRNAs may lead to effective means of managing malignancy.
There are currently three clinical trials utilizing miRNA-based therapies listed on www.clinicaltrials.gov. Two (NCT01829971, NCT02862145) were recently terminated due to immune-related serious adverse events, suggesting that a proper nomination schema, as might be done with the expression-based algorithm described by the authors, may be needed to optimize miRNA-based therapies. The third therapeutic miRNA trial is a phase I trial utilizing a miR-16 mimic in progressive mesothelioma and non-small-cell lung cancer (NCT02369198), which is employing an epidermal growth factor receptor-targeting mechanism. A targeting strategy may be needed to circumvent the possible toxicity of miRNA-based cancer therapy, although the work by Hydbring et al. demonstrates that use of nanoparticles to deliver cell-cycle-targeting miRNAs does not elicit toxicity in immunocompromised mice. Importantly, while several studies that have examined the capacity of miRNA-based technology to be clinically useful report mixed results, the strategy defined by the authors provides hope for using cell-cycle-targeting miRNAs for cancer management in the near future.
References
- Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. J Clin Invest. 2017;127:761–771. doi: 10.1172/JCI84424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, André F, Winer EP, et al. N Engl J Med. 2016;375:1738–1748. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- Hydbring P, Badalian-Very G. F1000Res. 2013;2:136. doi: 10.12688/f1000research.2-136.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydbring P, Wang Y, Fassl A, Li X, Matia V, Otto T, Choi YJ, Sweeney KE, Suski JM, Yin H, et al. Cancer Cell. 2017;31:576–590. doi: 10.1016/j.ccell.2017.03.004. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Otto T, Sicinski P. Nat Rev Cancer. 2017;17:93–115. doi: 10.1038/nrc.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, et al. PALOMA3 Study Group N Engl J Med. 2015;373:209–219. [Google Scholar]