

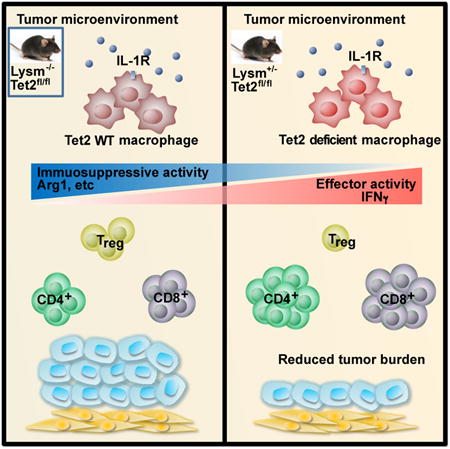
Summary
Ten-Eleven-Translocation-2 (Tet2) is a DNA methylcytosine dioxygenase that functions as a tumor suppressor in hematopoietic malignancies. In this study, we revealed a role for Tet2 in sustaining the immunosuppressive function of tumor-tissue myeloid cells. We found that Tet2 expression is increased in intratumoral myeloid cells both in mouse models of melanoma and in melanoma patients, and that this increased expression is dependent on an IL-1R-MyD88 pathway. Ablation of Tet2 in myeloid cells suppressed melanoma growth in vivo, and shifted the immunosuppressive gene expression program in tumor-associated macrophages to a proinflammatory one, with a concomitant reduction of the immunosuppressive function. This resulted in increased numbers of effector T cells in the tumor, and T cell depletion abolished the reduced tumor growth observed upon myeloid-specific deletion of Tet2. Our findings reveal a non-cell-intrinsic, tumor-promoting function for Tet2, and suggest that Tet2 may present a therapeutic target for the treatment of non-hematologic malignancies.
Graphical abstract
Introduction
Tumor-tissue myeloid cells have important roles in anti-tumor immunity and tumor progression. Monocytes, macrophages and granulocytes are actively recruited to tumors where they interact with the tumor microenvironment, often to accelerate tumor progression. Tumor associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are often regarded as having immunosuppressive functions that promote tumor growth, in part by suppressing adaptive immune responses to tumor cells (Marvel and Gabrilovich, 2015; Ostuni et al., 2015; Qian and Pollard, 2010; Yadav and Delamarre, 2016). TAMs are phenotypically and functionally distinct from tissue resident macrophages (Franklin et al., 2014). The tumor microenvironment acts on the gene expression program of TAMs to establish and maintain immunosuppressive functions (Amit et al., 2016; Church and Galon, 2015; Colegio et al., 2014; Noy and Pollard, 2014). Factors responsible for the establishment and maintenance of TAM functions are beginning to emerge (Colegio et al., 2014). In particular, proteins associated with the regulation of DNA methylation, and thereby gene expression, present interesting candidates.
Ten-Eleven-Translocation-2 (Tet2) is an important tumor suppressor within the hematopoietic system. Loss-of-function mutations in TET2, such as mutations resulting in reading-frame shifts and early stop codons, are frequently found in myeloid malignancies (Delhommeau et al., 2009; Figueroa et al., 2010; Ko et al., 2010; Langemeijer et al., 2009). Deletion of Tet2 in hematopoietic stem cells (HSCs) leads to increased self-renewal of HSCs and the expansion of the myeloid compartment, particularly the myelomonocytic lineage (Ko et al., 2011a; Li et al., 2011b; Moran-Crusio et al., 2011; Quivoron et al., 2011b), further supporting a tumor suppressive role for Tet2. Tet2, together with Tet1 and Tet3, form the Tet family, which biochemically catalyzes 5-methylcytosine (5mC) conversion to 5-hydroxymethylcytosine (5hmC) and its derivatives to mediate DNA demethylation (He et al., 2011; Huang and Rao, 2014; Ito et al., 2011; Tahiliani et al., 2009; Wu and Zhang, 2011). Recent studies have also revealed non-catalytic functions of Tet2 (Chen et al., 2013; Zhang et al., 2015), underscoring diverse mechanisms by which Tet2 regulates gene expression.
Although the function of Tet2 as a hematopoietic tumor suppressor is well established, it is unclear whether Tet2 activity within hematopoietic cells could impact solid tumors. In particular, the expansion of myelomonocytic lineages upon Tet2 deletion in HSCs raises the question of whether distinct functions of Tet2 may exist in these cells. Furthermore, recent findings of somatic TET2 mutations in peripheral blood cells, present in both healthy human individuals and solid cancer patients (Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014), raise the possibility that TET2-mutant myeloid cells may be “genetically reprogrammed” to elicit altered functions in solid cancer. Roles for Tet2 in T cells (Ichiyama et al., 2015; Tsagaratou et al., 2014; Yang et al., 2015) and dendritic cells in response to pathogens (Zhang et al., 2015) have been recently described. Whether Tet2 could impact gene expression and thereby function of TAMs, and how Tet2 might be regulated by the tumor environment remains to be investigated.
Successes of immune-checkpoint inhibitors in melanoma patients highlight the potential of immune cells in regulating melanoma biology (Bhatia and Thompson, 2014; Callahan, 2016). Human melanomas harbor activation mutations in BRAFV600E in ∼65% of cases (Chin, 2003; Davies et al., 2002), which often co-occur with loss-of-function mutations in tumor suppressors such as PTEN and CDKN2A. A mouse model of melanoma bearing the BrafV600E and Ptennull mutations recapitulates key features of human melanoma (Dankort et al., 2009b). Treatment of these mice with an inhibitor of the macrophage colony-stimulating factor receptor (Csf1r, important for macrophage differentiation, proliferation and survival) delayed tumorigenesis, suggesting the importance of TAMs in promoting tumor development in this model (Ngiow et al., 2016).
Here we explored the impact of myeloid-specific deletion of Tet2 on tumor growth using two murine melanoma models. Contrary to the recognized role of Tet2 as a tumor suppressor, we found that Tet2 maintains the immunosuppressive functions of tumor-tissue macrophages to promote tumor growth. Tet2 expression in TAMs was regulated via an interleukin-1 receptor (IL-1R)-Myd88 pathway, and deletion of Tet2 resulted in changes in gene expression and associated functional polarization of TAMs. Thus, Tet2, a protein regulating the DNA methylation landscape, mediates myeloid immunosuppression and melanoma tumor progression.
Results
Increased expression of Tet2 in TAMs and MDSCs during melanoma progression
As a main model in our study, we used the YUMM1.7 murine melanoma cell line, which was derived from the BrafV600EPten-/-Cdkn2a-/- mouse model (Dankort et al., 2009a). YUMM1.7 robustly gives rise to melanoma in syngeneic wildtype host mice with a substantial contribution of TAMs to tumor mass (Ho et al., 2015; Meeth et al., 2016), similar to what is often observed in human melanoma.
We first evaluated the RNA expression levels of Tet family members in myeloid cells after injecting YUMM1.7 cells subcutaneously into wildtype mice (Figure 1A). We found that TAMs isolated from tumor tissue had significantly higher Tet2 mRNA expression than macrophages isolated from peritoneum or bone marrow of control tumor-free mice (Figure 1B). In contrast, Tet3 mRNA expression levels were similar between these macrophage populations (Figure 1B), whereas Tet1 transcripts were barely detectable. We next harvested TAMs at two different time points during tumor progression (early and late stages) and determined that the amounts of Tet2 transcripts in TAMs increased during melanoma progression, whereas no change in Tet3 expression was observed (Figure 1C). Consistent with the increase of Tet2 mRNA expression, global 5hmC levels in TAM genomic DNA were increased by ∼2-fold during melanoma progression (Figure 1D). To further characterize Tet2 gene expression, we examined Tet2 RNA levels in TAMs, intratumoral MDSCs (CD11b+Gr1+), as well as splenic macrophages, splenic monocytic MDSCs (M-MDSCs; CD11b+Ly6ChiLy6G-) and splenic granulocytic MDSCs (G-MDSC; CD11b+Ly6CloLy6G+) from tumor-bearing mice (see Figure S1A for sorting scheme). Overall, intratumoral myeloid cells had ∼2-fold higher Tet2 mRNA levels than the corresponding splenic populations from tumor-bearing mice, which in turn were ∼2-fold higher than corresponding splenic populations from non-tumor-bearing mice (Figure 1E). We also observed that granulocytic populations had 2-3 fold lower Tet2 mRNA amounts than macrophage and monocytic populations from the same tissue, while Tet3 RNA levels were similar across all these subsets (Figure 1E). Similar results were observed when another melanoma model was used, induced by injecting B16-OVA cells (Figure S1B, S1C). To determine whether increased Tet2 expression in intratumoral myeloid cells is also seen in human melanoma, we isolated intratumoral and circulating CD11b+ myeloid cells from six melanoma patients. Intratumoral myeloid cells exhibited higher amounts of Tet2 mRNA than peripheral myeloid cells, whereas no appreciable difference was noted in Tet3 RNA amounts (Figures 1F). Taken together, the data above indicate that Tet2 expression is increased in TAMs and MDSCs during tumor progression, suggesting regulation by the tumor microenvironment.
Figure 1. Increased amounts of Tet2 transcripts in TAMs and MDSCs during melanoma progression.
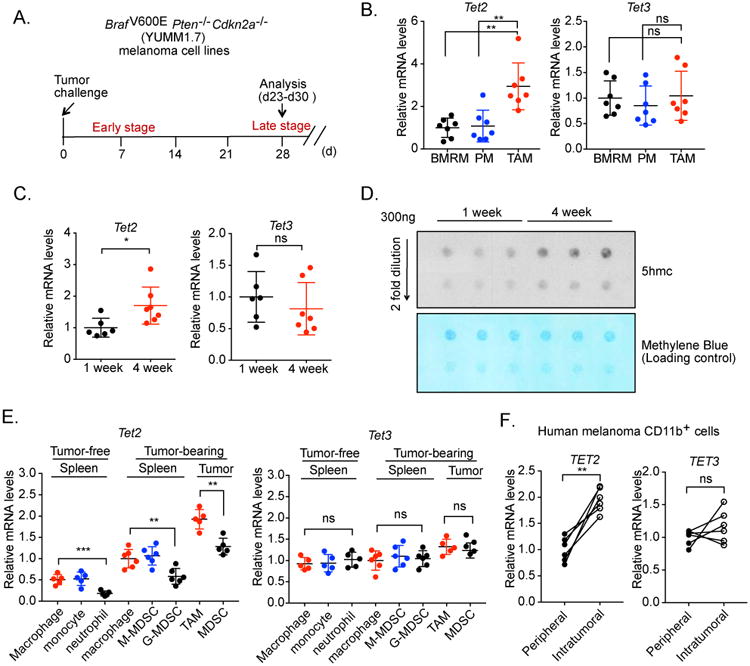
(A) Schematic illustration of the syngeneic tumor model in which BrafV600EPten-/- Cdkn2a-/-(YUMM1.7) melanoma cells were injected subcutaneously. Tumor-associated macrophages (TAMs) were harvested at one and four weeks. (B) Tet2 and Tet3 transcript amounts were determined in peritoneal macrophages (PM) and bone marrow resident macrophages (BMRM) from tumor-free mice and TAMs (four weeks after tumor cell injection) from tumor-bearing wildtype (WT) mice, using qRT-PCR. n=7. Normalized RNA expression is shown with each dot reflecting one mouse. (C) TAMs were harvested from WT mice at one week (n=6) and four weeks (n=7) post tumor cell injection. Tet2 and Tet3 transcript amounts were determined by qRT-PCR. Normalized RNA expression is shown with each dot reflecting one mouse. (D) TAMs were harvested from WT mice at the indicated time points after tumor cell injection. 5hmc levels in genomic DNA were determined by dot blot analysis, with methylene blue staining used as a loading control. (E) Tet2 and Tet3 transcript levels were determined in TAMs and MDSCs purified from tumor mass, splenic macrophages, monocytic MDSCs (M-MDSCs) and granulocytic MDSCs (G-MDSCs) purified from tumor-bearing WT mice, as well as splenic macrophages, monocytes and granulocytes purified from WT mice without tumor. (F) Tet2 and Tet3 transcript levels were determined in peripheral and intratumoral CD11b+ myeloid cells isolated from six human melanoma patients. Each line connects samples from the same patient. For all panels, *: P< 0.05; **: P < 0.01; ns: not significant. Data are representative of three (B-E) or two (D) independent experiments. Please also see Figure S1.
Myeloid-specific deletion of Tet2 suppresses melanoma growth
We next asked whether Tet2 in myeloid cells regulates tumor progression. We crossed Tet2fl/fl mice with LysM-cre mice to ablate Tet2 in the myeloid compartment, including macrophages (referred to as “Mye-Tet2 null”). Macrophages from Mye-Tet2 null mice had ∼80% reduction in Tet2 expression, which is consistent with the reported ∼70%-80% deletion efficiency in the LysM-cre model (Abram et al., 2014; Ye et al., 2003). We first determined that Mye-Tet2 null mice did not affect steady state myeloid cell lineage distribution. Indeed, similar numbers and percentages of myeloid populations, including macrophage and monocytic cells, were observed in spleen or bone marrow from 6 to 8-week-old Lysm+/wtTet2fl/fl mice, compared to wildtype control (WT, Lysmwt/wtTet2fl/fl) mice (Figure S1D, S1E). We next asked whether myeloid-specific deletion of Tet2 affected melanoma growth. We injected YUMM1.7 melanoma cells into syngeneic Mye-Tet2 null or WT host mice. Mye-Tet2 null mice exhibited significantly slower tumor growth (Figure 2A) and bore substantially smaller tumors than WT mice (Figure 2B, 2C). Similar results were observed when the B16-OVA melanoma model was used (Figure 2D, 2E). Taken together, the data above indicate that deletion of Tet2 in myeloid cells leads to the suppression of tumor growth.
Figure 2. Myeloid-specific deletion of Tet2 inhibits melanoma growth.
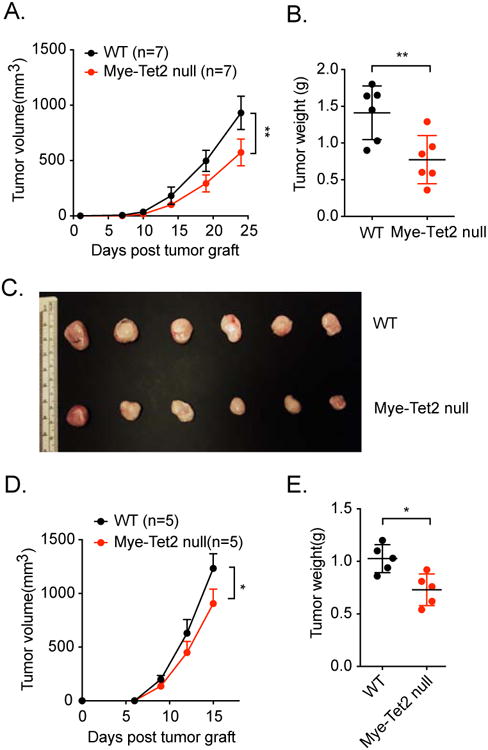
(A-C) YUMM1.7 melanoma cells were injected into WT and Mye-Tet2-null mice. (A) Mean tumor volume in WT and Mye-Tet2 null mice. n=7. (B) Tumor weights four weeks after tumor cell injection. n=6. (C) Pictures of tumors for (B). (D, E) B16-OVA melanoma cells were injected into WT and Mye-Tet2 null mice. (D) Mean tumor volume in WT and Mye-Tet2-null mice. n=5. (E) Tumor weights at the endpoint for (D). For all panels, *: P< 0.05; **: P < 0.01; ns: not significant. Data are representative of three (A-C) or two (D-E) independent experiments.
Tet2 expression is increased by IL-1 receptor signaling and dependent on the MyD88 pathway
The increase of Tet2 mRNA amounts in TAMs during tumor progression suggests that Tet2 is regulated by the tumor microenvironment. To investigate possible pathways controlling Tet2 expression, we hypothesized that IL-1 receptor signaling may regulate Tet2 expression. Several previous studies have demonstrated IL-1 cytokines as important factors in tumor regulation (Carmi et al., 2009; Cataisson et al., 2012; Lewis et al., 2006; Voronov et al., 2014), and that IL-1 cytokines are elevated in primary melanoma tissues (Voronov et al., 2003). We first performed a series of in vitro assays on bone marrow derived macrophages (BMDMs) and found that IL-1R signaling strongly increases Tet2 expression. Treatment of BMDMs with IL-1β markedly increased Tet2 RNA and Tet2 protein levels in BMDM cells within hours, while Tet3 mRNA amounts were not changed (Figure 3A, 3B). Consistent with the increase of Tet2 mRNA, IL-1β significantly increased global 5hmC levels in the macrophage genome by ∼2-fold within 9 hours (Figure 3C). These data indicate that an IL-1R ligand is sufficient to dynamically increase Tet2 expression and modulate 5hmC levels in the genome. To determine whether IL-1R signaling is required for sustaining elevated Tet2 expression in TAMs, we injected YUMM1.7 melanoma cells into Il1r1-/- or WT littermate control mice, and harvested TAMs from the resultant tumors. Indeed, Tet2 transcripts, as well as those of the previously established IL-1R downstream genes Vegfa and Mmp9, were significantly reduced in Il1r1-/- TAMs (Figure 3D). As negative controls, the mRNA levels of Ifnb were unchanged (Figure 3D). We also observed that tumor burden was largely reduced in Il1r1-/- mice compared to WT mice (Figure S2A). To determine the effect of acute loss of IL-1R signaling on Tet2 expression in TAMs and on tumor progression, we established tumor development in WT mice and acutely blocked IL-1R signaling afterwards (from day 9 post tumor cell injection) by administrating an IL-1R antagonist IL-1Ra in vivo (Figure 3E). IL-1Ra treatment led to a gradual reduction in tumor size (Figure 3F), and reduced Vegfa and Mmp9 expression in TAMs, both at an early time point before significant tumor size reduction was observed (day 12) and at a later time point when tumor size reduction became obvious (day 18) (Figure 3G). At both time points, we observed significant reductions in Tet2 RNA amounts in TAMs (Figure 3G). These data support that Tet2 expression in TAMs is positively regulated by IL-1R signaling within the tumor microenvironment.
Figure 3. Tet2 transcripts are increased by IL-1 receptor signaling and dependent on the MyD88 pathway.
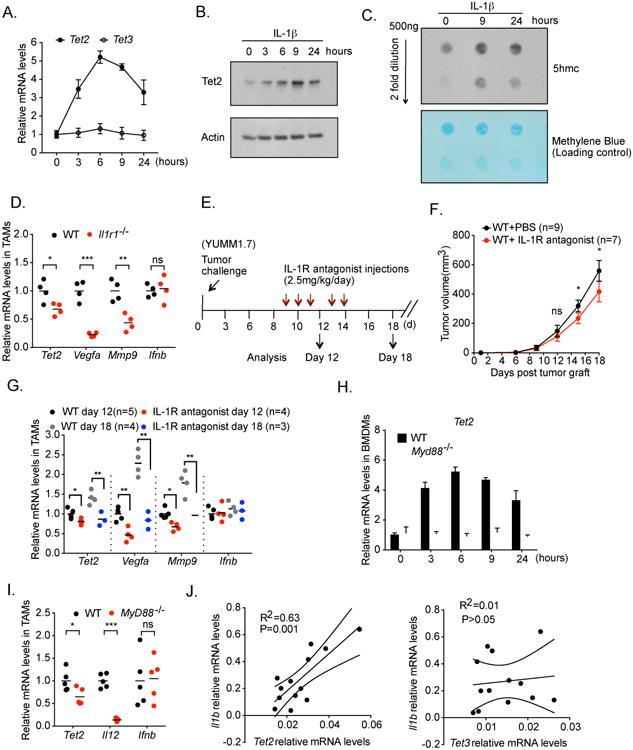
(A, B, C) Bone marrow derived macrophages (BMDM) were treated with 100 ng/ml IL-1β for the indicated hours. (A) Tet2 and Tet3 transcript levels were determined by qRT-PCR. (B) Representative western blot of Tet2 protein levels, with beta actin as a loading control. (C) Representative dot blot of 5hmc levels in genomic DNA for the indicated time points. Methylene blue staining was used as a loading control. (D) YUMM1.7 melanoma cells were injected into wildtype (WT) or Il1r1-/- mice. Tumor associated macrophages (TAMs) were harvested four weeks post injection. Transcript levels of Tet2, Vegfa, Mmp9 and Ifnb were determined by qRT-PCR, with each dot representing a mouse. n=4. (E-G) The effect of acute inhibition of IL-1R signaling by an IL-1R antagonist on Tet2 transcripts and tumor progression. (E) Experimental schematics. (F) Tumor volume. (G) TAMs were harvested at Day 12 and Day 18. Transcript levels of Tet2, Vegfa, Mmp9 and Ifnb were determined by qRT-PCR, with each dot representing a mouse. (H) WT or Myd88-/- BMDM cells were treated with 100 ng/ml IL-1β for the indicated hours. Tet2 transcript levels were determined by qRT-PCR. (I) YUMM1.7 melanoma cells were injected into wildtype (WT) or Myd88-/- mice. TAMs were harvested four weeks post injection. Transcript levels of Tet2, IL-12b and Ifnb were determined by qRT-PCR, with each dot representing a mouse. n=5. (J) Correlation analysis of the transcript levels of Il1b with those of Tet2 and Tet3 were determined in TAMs isolated from week 1 and week 4 YUMM1.7 tumors in WT mice. For all panels, *: P< 0.05; **: P < 0.01; ns: not significant. Error bars represent S.E.M. Data are representative of three independent experiments. Please also see Figure S2.
MyD88 is an important adaptor protein in the IL-1R signaling pathway (Muzio et al., 1997; Wesche et al., 1997). To determine the function of MyD88 in regulating Tet2 expression, we performed IL-1β treatment on BMDMs derived from WT or Myd88-/- mice. In Myd88-/- cells, we observed that the IL-1β-induced increase of Tet2 expression was abolished (Figure 3H), indicating that an IL-1R-MyD88 pathway regulates Tet2 expression in vitro. To determine the function of MyD88 in Tet2 regulation in vivo, we injected YUMM1.7 melanoma cells into WT or Myd88-/- mice. Consistent with the in vitro data, Tet2 mRNA levels, as well as those of the canonical MyD88 downstream gene Il12b, were reduced in TAMs in Myd88-/- mice (Figure 3I), while the levels of Ifnb were unchanged (Figure 3I). These results support a role for MyD88 in controlling Tet2 expression in TAMs.
IL-1R signaling has been shown to increase Il1b gene expression (Cataisson et al., 2012), Consistent with an IL-1R-MyD88 pathway regulating Tet2 expression, we observed a significant correlation between Tet2 and Il1b mRNA amounts when examining TAMs harvested from murine melanomas, but not between Tet3 and Il1b RNA levels (Figure 3J). Interestingly, when examining peripheral and intratumoral myeloid cells from human melanoma patients, we also observed a significant correlation between TET2 and IL1B transcripts, as well as between those of TET2 and IL1A (Figure S2B, S2C), suggesting that this regulatory pathway operates similarly in human cells. Taken together, the data above support an IL-1R-MyD88-Tet2 pathway in the tumor microenvironment regulating melanoma progression in vivo.
Tet2 sustains the expression of immunosuppressive genes in TAMs
To understand how Tet2 in myeloid cells regulates melanoma progression, we performed RNA-seq analysis on TAMs isolated from tumor-bearing mice of WT and Mye-Tet2 null genotypes. We observed that genes with reduced mRNA expression in Mye-Tet2 null TAMs were enriched for signatures of M2 immunosuppressive macrophages, whereas genes with increased expression in Mye-Tet2 null TAMs were enriched for signatures of M1 proinflammatory macrophages (Figure 4A). We further validated the reduced mRNA expression of a number of genes with immunosuppressive functions, including Arg1, Mgl2 and Il4 (Biswas et al., 2008; Rawal et al., 2011; Sharda et al., 2011a) (Figure 4B). At the same time, the mRNA amounts of a number of pro-inflammatory cytokines were increased, including Il6, Il12b, and Tnfa (Figure 4B). These data indicate that deletion of Tet2 shifted the balance between immunosuppressive and proinflammatory polarization of TAMs. The effect of Tet2 deletion on immunosuppressive gene expression was similarly observed when WT or Tet2 null BMDMs were polarized in vitro with IL-4 (Figure S3A). The mRNA expression levels of several immunosuppressive genes such as Arg1 and Mgl2 were reduced in both Mye-Tet2 null TAMs and IL-4 polarized Tet2 null BMDMs (Figure 4B, S3B). We further evaluated the protein activity of Arg1, and confirmed decreased Arg1 activity in Tet2 null TAMs (Figure 4C) and BMDMs (Figure S3C). These data establish that Tet2 is required to maintain the expression of multiple immunosuppressive genes in TAMs.
Figure 4. Tet2 sustains immunosuppressive function and gene expression in TAMs.

(A) Tumors were initiated by YUMM1.7 cell injection. RNA-seq experiments were performed on TAMs isolated from WT and Mye-Tet2 null mice ∼4 weeks post injection. n=4. A heat map of 214 differentially expressed genes are shown on the left. Right: two independent sets of M1 and M2 macrophage signatures were queried on the TAM gene expression using gene set enrichment analysis (GSEA) to reveal the decrease of M2 signature and the increase of M1 signature in Mye-Tet2 null TAMs. GSEA plots are shown. (B) qRT-PCR analysis of the mRNA levels of indicated genes in TAMs from WT or Mye-Tet2 null mice. n=5. (C) TAMs from WT or Mye-Tet2 null mice were analyzed for arginase activity in cell lysates. (D, E, F) TAMs purified from WT or Mye-Tet2 null mice were co-cultured with CD4+ T cells at the indicated cellular ratios for T cell activation assay. T cell proliferation was quantified by CFSE-labeling and dye dilution for three days in vitro. (D) Representative histograms of CFSE signals in the in vitro T cell activation assay. (E) Quantification of CD4+ T cell proliferation in (D). (F) Quantification of CD4+ T cell proliferation with recombinant human arginase 1 (1 μg/ml) or arginase inhibitor (100 μM) added into the co-culture medium. For all panels, *: P< 0.05; **: P < 0.01; ***: P< 0.001; ns: not significant. Error bars represent S.E.M. Data for all panels are representative of two or more independent experiments. Please also see Figure S3, S4.
We next asked whether the reduced expression of immunosuppressive genes can be explained by changes in local 5hmC alterations. Due to the difficulty of obtaining large numbers of TAMs, we performed genome-wide 5hmC DNA immunoprecipitation (5hmC-DIP) mapping in naïve (M0) and polarized (M2) BMDM cells from WT and Tet2 null mice. Overall, 5hmC levels around protein-coding genes were mildly decreased in Tet2 null cells (Figure S3D). Among the genes regulated by Tet2, Arg1 showed putative differential 5hmc peaks in gene proximal regions (Figure S3E). We observed that the upstream region of Arg1 contained a consistently reduced 5hmC peak in Tet2 null cells. The peak region was located close to a known enhancer region of Arg1 (Sharda et al., 2011b) (Figure S3F). Using DNA immunoprecipitation (DIP)-qPCR, we confirmed the decreased 5hmC levels at this region in Tet2 null BMDM cells, and observed a concomitant increase in 5mC levels (Figure S3G). Similar results were also observed in Mye-Tet2 null TAMs (Figure S3H). These data suggest that Tet2 is required to maintain a low 5mC level at the Arg1 gene locus by increasing 5hmC levels, implying regulation of the gene by DNA demethylation. However, other immunosuppression-related genes, such as Mgl2, Klf4 and Irf4, did not show major changes in 5hmC levels in gene proximal regions. Whether these genes could be regulated through a 5hmC-independent mechanism or through 5hmC-depdendent distal enhancers requires future analysis. Collectively, our data support that Tet2 maintains the expression of immunosuppressive genes in TAMs, likely through multiple mechanisms.
Reduced immunosuppressive functions of Tet2 null myeloid cells in vitro
Guided by the gene expression analysis, we tested whether Tet2 null TAMs and MDSCs have reduced immunosuppressive function, by an in vitro T cell activation assay. Specially, splenic CD4+ T cells were stimulated with anti-CD3 and anti-CD28 and T cell activation was determined by CFSE-based dye dilution (Doedens et al., 2010). When co-culturing CD4+ T cells with WT TAMs, strong suppression of T cell proliferation was observed (Figure 4D, 4E). In contrast, the suppressive capacity was significantly reduced when Mye-Tet2 null TAMs were used, under two different T cell to TAM ratios (Figure 4D, 4E). Similar results were observed when supernatants from IL-4-polarized WT or Tet2 null BDMDs were assayed (Figures S3I, 3J). Since MDSCs are also known to possess immunosuppressive functions and can regulate tumor development, we also examined intratumoral MDSCs and found that both Arg1 mRNA amounts and Arg1 protein activity were reduced in Tet2 null MDSCs, while reactive oxygen species (ROS) and iNos, Nox2, Ncf1 and Tgfb mRNA expression did not show significant changes (Figure S4A, S4B, S4C). We next measured the ability of intratumoral MDSCs to suppress T cells. While a decreased immunosuppressive function was observed for Tet2 null MDSCs at a T cell to MDSC ratio of 3:1 (Figure S4D, S4E), significant changes could not be detected when a different ratio was used (Figure S4E), unlike what we observed with TAMs (Figure 4E). These data support a reduction of immunosuppressive functions of Tet2 null TAMs and Tet2 null intratumoral MDSCs, with a stronger alteration in TAMs.
Since Arg1 gene expression was significantly reduced in Tet2 null TAMs, we asked whether Arg1 is an important mediator of Tet2 function in TAMs. In the TAM and CD4+ T cell co-culture, the addition of arginase inhibitor significantly reduced the immunosuppressive activity of WT TAMs, but not that of Mye-Tet2 null TAMs, leading to the abolishment of the difference on T cell suppression by these two groups of cells (Figure 4F). On the other hand, addition of recombinant Arg1 led to an almost complete blockage of T cell proliferation for both WT and Mye-Tet2 null TAMs, again abolishing the difference between these two TAM populations (Figure 4F). These data support that the reduction of Arg1 expression plays an important role in mediating the immunosuppressive function of Tet2 in TAMs.
Increased tumor-infiltrating T cells in mice with myeloid-specific deletion of Tet2
The reduction of the expression of immunosuppressive genes and the reduction of in vitro immunosuppressive functions of Mye-Tet2 null TAMs raised the possibility that the intratumoral immune cell repertoire was shifted to favor anti-tumor functions. We thus examined multiple immune subsets (Figure S5A) within the YUMM1.7-initiated tumors developed in WT or Mye-Tet2 null mice. We observed that the percentages of CD45+ cells were increased in tumors from Mye-Tet2 null mice (Figure 5A). Within CD45+ cells, the percentages of CD3+ T cells were significantly increased, whereas those of natural killer (NK), natural killer T (NKT) cells and macrophages were unchanged, and the percentages of granulocytes were mildly decreased in tumors (Figure 5B). When quantifying by cell numbers per tumor volume, the differences in granulocytes were no longer observed, with a substantial increase of CD3+ T cells and mild increases of TAMs, NK and NKT cells in Mye-Tet2 null tumors (Figure 5B). We further assessed the CD4+ and CD8+ T cell populations in tumor. Within the T cell compartment, the levels (both percentages within CD45+ cells and cell numbers per tumor volume) of both CD4+ and CD8+ T cells were increased in Mye-Tet2 null tumors, with a stronger elevation observed for CD8+ T cells (Figure 5C, 5D). In contrast, the percentages of CD4+ or CD8+ cells in the spleens of the tumor-bearing mice were not significantly altered in Mye-Tet2 null mice (Figure 5E). Among intratumoral CD4+ T cells, the percentages of T helper 1 (Th1) effector T cells were mildly increased whereas those of the immunosuppressive regulatory T cells (Treg) were mildly decreased (Figure 5F, 5J). In contrast, the corresponding populations in spleens were not changed (Figure 5G and 5K). Similar results were observed for CD8+ T cells (Figure 5H, 5I, 5L, 5M). This led to an overall significant elevation of the ratio of CD8+ T cells over Tregs, and a milder increase of the ratio of CD4+ T cells over Tregs (Figure 5N). The increase in the intratumoral T cell population in Mye-Tet2 null mice, particularly CD8+ T cells, could also be observed in the B16-OVA-initiated melanoma, indicating this effect was not dependent on a specific tumor model (Figure S5B-H). Overall, the data here suggest a shift of the intratumoral immune environment to favor anti-tumor activity in Mye-Tet2 null mice.
Figure 5. Myeloid-specific deletion of Tet2 leads to an increase of tumor-infiltrating T cells.
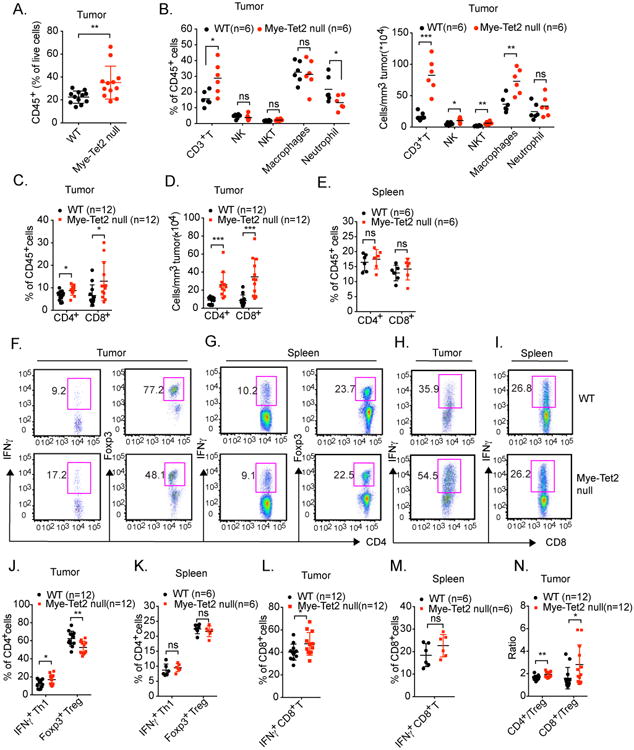
(A-N) YUMM1.7 melanoma cells were injected into WT or Mye-Tet2 null mice. Tumor infiltrating immune cells were examined by flow cytometry four weeks after injection. (A) The percentages of tumor-infiltrating CD45+ hematopoietic cells within all live cells from the tumor were quantified. Each dot represents one mouse. n=12. (B) For intratumoral cells, the percentages within total CD45+ cells (left) or numbers per tumor volume (right) of the indicated immune cell populations were quantified. Each dot represents one mouse. n=6. (C, D) Tumor-infiltrating CD4+ and CD8+ T cells were quantified either as (C) the percentages within total CD45+ cells, or (D) as absolute numbers per tumor volume. Each dot represents one mouse. n=12. (E) The percentages of splenic CD4+ and CD8+ T cells from tumor-bearing mice were quantified within total CD45+ splenocytes. n=6. (F-N) Intratumoral T cells or splenic T cells from tumor-bearing mice were further characterized by flow cytometry. (F, J) Statistical and representative flow cytometry plots showing the percentages of CD4+ IFNγ+ Th1 cells and CD4+ Foxp3+ Treg cells within total intratumoral CD4+ T cells. Each dot represents one mouse. n=12. (G, K) Similar analysis as (F, J) was performed on splenic T cells within tumor-bearing mice. n=6. (H, L) Statistical and representative flow cytometry plots showing the percentages of CD8+ IFNγ+ T cells within total intratumoral CD8+ T cells. Each dot represents one mouse. n=12. (I, M) Similar analysis as (H, L) was performed on splenic T cells within tumor-bearing mice. n=6. (N) Ratios of intratumoral CD4+ to Treg and CD8+ to Treg cells were quantified. Each dot represents one mouse. For all panels, *: P< 0.05; **: P < 0.01; ns: not significant. Data are representative of three independent experiments. Please also see Figure S5.
T cell depletion abolished the reduced tumor growth observed upon myeloid-specific deletion of Tet2
We reasoned that if increased intratumoral T cells were functionally important for the reduced melanoma growth in Mye-Tet2 null mice, we would expect that depleting T cells could lead to a rescue of tumor size. We tested this possibility by antibody-based depletion of both CD8+ and CD4+ T cells, given the increase of both cell types observed in YUMM1.7-intiated tumors. After melanoma growth was initiated by injecting WT and Mye-Tet2 null mice with tumor cells, we treated mice with both anti-CD4 and anti-CD8 antibodies from day 7, with one dose per 7 days (Figure 6A). The antibody treatments effectively depleted T cells, as evidenced by the loss of CD4+ and CD8+ T cell populations in spleen, lymph node and tumor (Figures 6B). This treatment did not lead to any major noticeable changes in TAMs with regard to both immunostaining and gene expression (Figure S6A, S6B). Similar to the aforementioned phenotypes, mock-treated mice bore tumors with reduce size in Mye-Tet2 null mice compared to WT controls (Figure 6C). T cell depletion resulted in a mild increase (1.4 fold) in tumor size in WT mice (Figure 6C, 6E). In contrast, T cell depletion in Mye-Tet2 null mice led to a stronger (>2.2-fold) increase in tumor size. No significant changes were observed when comparing tumor sizes in Mye-Tet2 null mice to those from WT mice upon T cell depletion (Figure 6C). Indeed, statistical analyses confirmed that the reduction of tumor size in Mye-Tet2 null mice were largely abolished upon T cell depletion (Figure 6D), and that T cell depletion had a significantly stronger effect on tumor size in Mye-Tet2 null mice than in WT control mice (Figure 6E). When depleting CD8+ and CD4+ T cells separately, we observed a stronger effect by CD8+ T cell depletion (Figure S6C), consistent with a stronger increase in intratumoral CD8+ T cells in the Mye-Tet2 null tumors. Similar effects of CD8+ and CD4+ T cell depletion were also observed in the B16-OVA-initiated tumor model (Figure S6D, S6E). Taken together, these data indicate that T cell depletion rescued the reduced tumor growth phenotype in Mye-Tet2 null mice.
Figure 6. T cell depletion abolishes the reduced tumor size phenotype seen in Mye-Tet2 null mice.
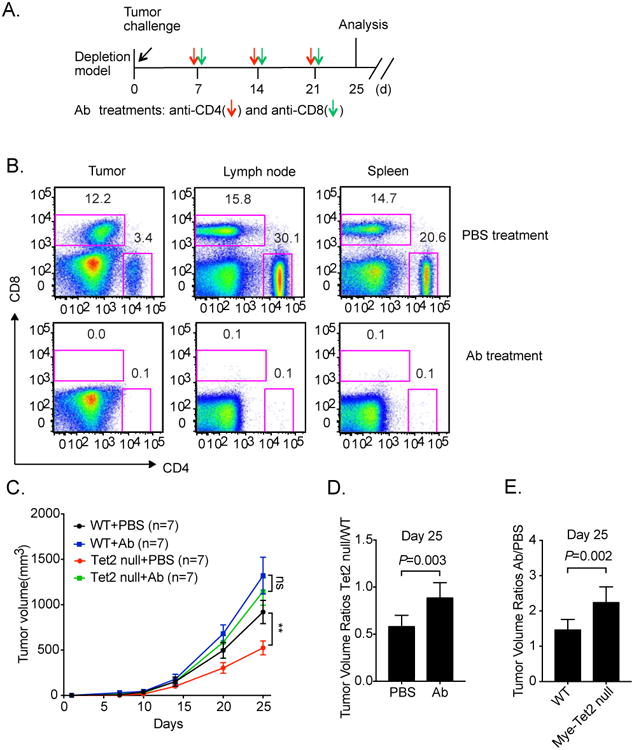
(A) Schematic illustration of the T cell depletion experiment. YUMM1.7 melanoma cells were injected into WT or Mye-Tet2 null mice. Anti-CD4 and anti-CD8 antibodies (Ab) were intraperitoneally injected every 7 days at the indicated time points. PBS was injected as a mock control. (B) T cell depletion efficiencies in spleen, lymph node, and tumor were determined after three antibody injections. Representative flow cytometry plots are shown. (C) Tumor growth curves in WT and Mye-Tet2 null mice either treated with PBS or with antibodies are shown. n=7. **, P < 0.01; ns: not significant. Error bars represent S.E.M. (D) Statistical analysis of tumor volume ratio between Mye-Tet2 null and WT mice at day 25, for PBS or Ab treatment groups. (E) Statistical analyses of tumor volume ratio between Ab and PBS treatment groups at day 25, for WT and Mye-Tet2 null groups. Error bars for (D, E) represent S.D. See methods for P value determination. Data represent three (B) and two (C-E) independent experiments. Please also see Figure S6.
Discussion
Tet2 is recognized as one of the major tumor suppressors within the hematopoietic system. It is often mutated somatically in hematopoietic cells in healthy individuals, as well as patients with myeloid malignancies or solid tumors (Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). This tumor suppressive function has been attributed to increased hematopoietic stem and progenitor cells in mice with Tet2 deletion (Ko et al., 2011b; Li et al., 2011a; Moran-Crusio et al., 2011; Quivoron et al., 2011a). While the expansion of myelomonocytic lineages have been observed when Tet2 is deleted in murine HSCs, it is unclear whether ablation of Tet2 in downstream myeloid cells may impact malignant processes. Here, we focus on the myeloid compartment and demonstrate that Tet2 plays a surprising tumor-promoting role in melanoma models by sustaining immunosuppressive functions of intratumoral myeloid cells. Whether this role also applies to other solid cancers awaits future exploration.
Since the cre stain we used cannot genetically distinguish the different myeloid subpopulations, it is likely that both TAMs and MDSCs contribute to the phenotype of reduced tumor growth in mice with myeloid-specific deletion of Tet2. Both TAMs and intratumoral MDSCs had higher levels of Tet2 mRNA amounts than similar splenic populations of tumor-bearing mice or tumor-free mice. The expression of Arg1, the signature gene for TAMs, M2 macrophages and M-MDSCs, was reduced in TAMs and intratumoral MDSCs. We did not detect a change in ROS level in the intratumoral MDSC population, suggesting that G-MDSCs may be less impacted. On the gene expression level, Mye-Tet2 null TAMs showed a significant increase in the M1 macrophage signature and a concurrent decrease in the M2 macrophage signature. Which of the differentially expressed gene(s) mediates the function of Tet2 on immunosuppression? Our data support that the reduction of Arg1 is an important event downstream of Tet2 ablation. These results are consistent with myeloid Arg1 promoting tumor growth in a lung tumor model in vivo (Colegio et al., 2014; Rodriguez et al., 2005) and that Arg1 catalyzes the degradation of L-arginine from the extracellular environment to decrease antigen-specific T cell proliferation (Huber et al., 2010; Pauleau et al., 2004). Our findings on Arg1, however, do not exclude the possibility that other Tet2-regulated genes, such as Mgl2, also contribute to the phenotype. Similarly, it is also possible that the increase of the M1 macrophage gene expression program in Tet2 null TAMs plays important functional roles.
TAMs are heterogeneous in terms of function and phenotype (Okabe and Medzhitov, 2014). It is highly likely that the different activation states of TAMs reflect responses to dynamic local microenvironmental cues within the tumor (Amit et al., 2016). We examined this topic in our murine tumor models, and found an IL-1R-MyD88 pathway enhancing Tet2 expression both in vitro and in vivo. Our data on MDSCs and human melanoma specimens suggest that the regulation of Tet2 gene expression by tumor microenvironment could be extended to other myeloid populations and is conserved from mouse to human. IL-1R can be regulated by multiple ligands, including IL-1β, IL-1α and IL-1Ra (Voronov et al., 2014). Our work suggests that cytokines derived from tissue microenvironment can affect the DNA-methylation-related pathway in TAMs and control macrophage function. It is also interesting to note that chronic IL-1 exposure drives HSCs to precociously differentiate toward the myeloid lineage (Pietras et al., 2016). Whether a similar pathway regulates Tet2 during HSC differentiation could be examined in the future.
Our work suggests that the DNA-methylation regulatory pathway in myeloid cells could be further explored to elucidate immune regulation of solid tumors. Other than Tet2, Tet3 and Dnmt3a have been recently reported to modulate gene expression in macrophages and control antiviral responses (Li et al., 2016; Xue et al., 2016). DNMT3A is also frequently mutated in HSCs, whose ablation leads to HSC expansion and inhibition of differentiation (Cullen and Goodell, 2015). Although TET3 is not a major target of genetic mutations in human, murine studies have revealed that combined Tet2 and Tet3 ablation leads to accelerated development of a myeloid malignancy (An et al., 2015), suggesting functional cooperation between Tet2 and Tet3. Further mechanistic studies in this area may lead to new avenues of clinical intervention.
Star Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Jun Lu (jun.lu@yale.edu).
Experimental Model and Subject Details
Mice
All animal experiments were performed in accordance with the guidelines of Yale University's Institutional Animal Care and Use Committee and the guidelines of NIH. All mice were housed in facilities of the Yale Animal Resources Center (YARC) with husbandry service provided by YARC. Yale University is registered as a research facility with the United States Department of Agriculture, and is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. In addition, an Animal Welfare Assurance is on file with OLAW-NIH. Lysmcre/wt, Tet2fl/fl, Tet2-/- and C57BL/6J mice were purchased from the Jackson Laboratory. Mye-Tet2 null mice were generated by crossing Lysmcre/wt and Tet2fl/fl mice. Litter mate Lysmwt/wt,Tet2fl/fl mice were used as controls. All mice used were on a C57BL/6J background. Myd88-/- and Il1r1-/- mice were originated from the Jackson Laboratory. In all tumor experiments, 6-8 week old male mice were used due to YUMM1.7 cells being of male origin and the B16 tumor model having similar behavior in male and female hosts.
Human samples
All human samples were obtained with informed consent and approved by the Research Ethics Board of Sun Yat-Sen University affiliated Cancer Center. A total of six Chinese patients with melanoma were enrolled during 2017. Peripheral blood mononuclear cells were collected and enriched by density gradient centrifugation. Freshly resected melanoma tissue was dissociated and single cell suspension was obtained. CD11b+ myeloid cells from peripheral blood and melanoma tissue were further enriched by CD11b MicroBeads (Miltenyi Biotec).
Cell lines
YUMM1.7 melanoma cells (BrafV600EPten-/-Cdkn2a-/-) and B16-OVA cells were maintained in complete RPMI medium 1640 (Thermo Fisher), with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin/glutamine (PSG) at 37°C and 5% CO2. Raw264.7 cells were kept in complete Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS and 1% PSG at 37°C and 5% CO2. Raw264.7 cells were gently scraped off from culture dish using a sterile scraper when passage was needed.
Method Details
Murine tumor models and tumor harvest
YUMM1.7 melanoma cells and B16-OVA cells were injected subcutaneously into the flanks of mice (2×105 cells per injection in 100 μl PBS). Male C57BL/6J mice of 6 to 8 weeks of age were used. Tumor volume was determined by measuring the length, width and height of the tumor with a caliper, and calculated by (1/6) × π × (Length × Width × Height). The mice were euthanized on indicated days in the figures. Tumors were resected and transferred to 5 ml PBS on ice. Tumor weight was measured on a scale by transferring the specimen to a sterile Petri dish after removal of surface moisture with Kimwipes. The tumors from all experiments were then processed for flow cytometry analysis or FACS-sorting on the same day. The resected mouse tumors were mechanically dissociated with surgical scissors and digested with Collagenase IV (Worthington) and DNase I (Roche) in PBS for 30min in a 37 °C shaking incubator (150 r.p.m.). After enzymatic dissociation, the samples were transferred to ice to stop the reaction. The tumor suspension was then filtered using a 70 μm cell strainer (Becton Dickinson) and washed with the FACS buffer (0.5% FBS in PBS) and centrifuged at 1,300 rpm. at 4 °C in an Eppendorf 5810R centrifuge (similar centrifugation parameters were used throughout). Red blood cells were lysed with ACK lysis buffer (Thermo Fisher) followed by washing with the FACS buffer. The samples were then resuspended in the FACS buffer. The samples were kept on ice throughout the rest of the staining procedure.
Isolation of human melanoma CD11b+ cells from surgical tissues
The tumor tissue was placed on ice and a scalpel was used to trim off any evident stromal, fat, and necrotic portions of the tumor. The remaining tumor tissue was then finely minced with a razor blade on a petri dish to break up large pieces >1–2 mm in diameter. The melanoma tissue was then dissociated in a tube containing ∼10–30 ml of RPMI 1640 medium with Liberase Blendzyme (Roche) added at a final concentration of 60 μg/ml. Dissociation was performed in a 37°C incubator on a rocking platform with constant mixing for 45-60 minutes. The dissociated tissue was then filtered through a 70 μm nylon filter into a 50 ml conical tube. A 30 ml solution of PBS and 2% heat inactivated FBS was added to neutralize the dissociation enzyme. The cell suspension was centrifuged at 1500 rpm for 5 min at 4°C. The pellet was washed twice with 30 ml of PBS with 2% FBS If the initial surgical sample contained a significant amount of red blood cells, the cells pellet was resuspended in 2 ml of ACK lysis buffer (Thermo Fisher) and incubated for 1 min on ice, followed by the addition of 30 ml of PBS with 2% FBS, and centrifuged. The cells were subjected to the isolation of CD11b+ cells by using CD11b MicroBeads (Miltenyi Biotec). Peripheral blood samples were similarly enriched for CD11b+ cells after red cell lysis. Overall, paired peripheral blood samples and tumor tissues were obtained from six melanoma patients. Examination of mRNA expression by qRT-PCR was performed in triplicates for each sample.
Cell and tissue collection, and FACS analysis
Peritoneal macrophages were collected from the peritoneal cavity of untreated mice by injecting 10 ml of PBS followed by gentle abdominal massaging. Macrophage enrichment was performed by plating cells in RPMI with 10% FBS and 1% penicillin/streptomycin for 2 hours at 37 °C and 5% CO2. After 2 hours, non-adherent cells were removed with three PBS washes, and cells were harvested in TRIZol for RNA extraction and qPCR analysis. Bone marrow macrophages were isolated by FACS-sorting of CD11b+F4/80+ cells. Experiments were repeated twice or more times. In each experiment, cells were collected from ∼ 7 mice for each group. qRT-PCR analysis was performed with two replicates.
For flow cytometry analysis or FACS-sorting, single cell suspension form the tumor tissue was washed with the FACS buffer and stained with the following antibodies: anti-mouse CD45.2– Pacific blue antibody (Biolegend; clone, 30-F11; used at 1:100), anti-mouse CD11b–PE antibody (Biolegend; clone, M1/70; used at 1:200), anti-F4/80–APC antibody (eBioscience; clone BM8; used at 1:100), anti-Gr1 (Biolegend; clone RB6-8C5; used at 1:100), anti-Ly-6G-PE/Cy7 (Biolegend; clone 1A8; used at 1:400), anti-Ly-6C-APC (Biolegend; clone HK1.4; used at 1:200), anti-CD3-PE (Biolegend; clone 145-2C11; used at 1:200), anti-CD4–APC (Biolegend; clone GK1.5; used at 1:100), anti-mouse CD8a-FITC (Biolegend; clone 53-6.7; used at 1:100), anti-NK1.1-APC (Biolegend; clone PK136; used at 1:100) and anti-Ly6g-FITC (Biolegend; clone 1A8; used at 1:200) at 4 °C for 15 minutes in the dark. The samples were then washed twice and resuspended in FACS buffer for sorting. For FACS=-sorting, live cells were first selected on the basis of forward scatter (FSC) and side scatter (SSC), followed by excluding non-single-cell events with SSC-A, SSC-W and FSC-A and FSC-W. The indicated populations were selected and sorted. Cells from other tissues were similarly processed.
For intracellular cytokine staining, cells obtained from the digested tumor mass or other sources were incubated in a tissue culture incubator for five hours at 37 °C with phorbol 12-myristate 13-acetate (PMA, 50 ng/ml; Sigma), ionomycin (750 ng/ml; Sigma) and brefeldin A (10 μg/ml; Sigma). Surface staining was performed as described above with anti–mouse CD4-APC (Biolegend; clone GK1.5; used at 1:100), or anti-mouse CD8a-FITC (Biolegend; clone 53-6.7; used at 1:100). After surface staining, the cells were resuspended in Fixation/Permeabilization solution (Cytofix/Cytoperm Kit; BD Biosciences) and intracellular cytokine staining was performed according to the manufacturer's protocol with anti-mouse IFN-γ-PE (Biolegend; clone XMG1.2; used at 1:100). For Foxp3 staining, the cells were not stimulated with PMA and ionomycin; instead, they were stained according to the manufacturer's protocol (Foxp3 Staining Buffer Set, eBioscience). For all flow cytometry data, each dot represents one mouse. Experiments were repeated twice or more times. For one batch of experiments, usually more than five mice were used for each group.
Differentiation and treatment of BMDMs
Total bone marrow cells were harvested from femur bones into the FACS buffer. Cell suspension was filtered through a 70 μm nylon strainer into a 50 ml tube. ACK lysis buffer (Thermo Fisher, 5 ml) was then added to lyse the red blood cells for 2 minutes at room temperature. To stop the lysis reaction, 20 ml RPMI with 2% FBS was added and the tube was centrifuged at 1500 rpm for 5 min at 4°C. The cell pellet was washed and resuspended in BMDM medium (30% L929-conditioned medium, 20% FBS, 10 ng/ml recombinant murine M-CSF (Biolegend, cat#576402) in RPMI 1640 medium, with 1% penicillin/streptomycin/glutamine). The cells were then plated at 2.5×106 cells per well in a sterile six-well tissue culture plate, and cultured for 7 days. To activate BMDM cells, BMDM medium was changed into DMEM medium (DMEM, 10% FBS, 1% PSG) the night before the stimulation. For IL-1β activation, 100 ng/ml IL-1β was added; for M2 activation, 20 ng/ml IL4 was added. The cells were then harvested at the indicated time points, and used in subsequent experiments. Two to three independent experiments were performed, with each containing three biological replicates or more (except for the RNA-seq samples with two replicates). qRT-PCR analysis was performed with two replicates for each sample.
RNA-Seq and data analysis
RNA-Seq library preparation and sequencing for BMDM samples were performed by Yale Center for Genomic Analysis, through ribosomal RNA depletion. Sequencing was performed on an Illumina 2500 machine. RNA-Seq library preparation and sequencing for TAM samples were conducted at Yale Stem Cell Center Genomics Core facility through poly A enrichment (Illumina TruSeq Stranded mRNA Library Prep Kit). RNA-Seq fastq files were processed in the GenePattern package, by first performing TopHat alignment to the mm9 mouse genome assembly, followed by quantification by CuffDiff. The resultant fpkm data were analyzed with custom matlab codes. For BMDM data, differentially expressed genes were identified by ANOVA analysis between the experimental groups and time points, and further filtered for at least 1.5 fold changes. Pathway analysis was performed using DAVID and Ingenuity Pathway Analysis. For TAM data, differentially expressed genes were identified by satisfying both student's t-test nominal p value of less than 0.05 and having mean log2 expression difference of at least 1. For analysis of M1 and M2 signatures, gene set enrichment analysis (GSEA) was performed by using the Broad Institute's GSEA program, using custom gene sets. Two independent sets of signatures were used (Table S2). M1 Signature 1 and M2 Signature 1 were derived ourselves by comparing transcriptomic data of M1 (LPS treated) and M2 (IL4 treated) BMDMs treated for 10 hours and 24 hours versus those from M0 BMDMs (not treated). M1 Signature 2 and M2 Signature 2 were copied from M1 distinct gene set and M2 distinct gene set in Jablonski et al. (Jablonski et al., 2015) with duplicate gene symbols removed. Our own M1 and M2 signatures were derived as follows. For either M1 or M2, treatment data were compared to untreated cells (M0) at 10 hour and 24 hours. Signature genes satisfied the following criteria: mean log2 fpkm expression of treated samples is more than 2; mean log2 expression of treated samples minus the mean log2 expression of M0 samples is more than log2(5); mean log2 expression of opposite polarization at 10 hour minus mean log2 expression of M0 samples is less than 0.2; signature genes satisfying the above criteria at either 10 hours or 24 hours of treatment.
Quantitative RT-PCR
Total RNA was extracted from the cells and mouse tissues with TRIzol reagent (Thermo Fisher) according to the manufacturer's protocol. cDNA was synthesized with SuperScript III reagent kit (Thermo Fisher). The expression of the genes encoding mouse Tet2, Tet3, Arg1, Mgl2, Il10, Irf4, and Klf4 etc. were quantified by real-time PCR using the SYBR Green PCR Master Mix (Thermo Fisher). All gene expression results were normalized to the expression of the housekeeping gene Rpl13a. Amplification of cDNA was performed on an ABI Prism 7900 HT cycler (Applied Biosystems) or a Biorad CFX96 machine. Gene expression data were analyzed by calculating the threshold values (Ct) and fold changes relative to an internal control. Primers used in this study are listed in Table S1. qRT-PCR analysis of gene expression was usually performed with more than two technical replicates.
5hmC DIP
For genome-wide analysis of 5hmC distributions, 5hmC DIP analysis was performed. Library preparation was performed according to the protocols in NEBNext DNA Library Prep Master Mix Set for Illumina (NEB, E6040). Briefly, genomic DNA was sonicated to ∼200bp fragments. Ten micrograms of sonicated genomic DNA were then used to ligate with the adaptor, provided in the kit, following the manufacturer's instructions. Sonicated and adaptor-ligated genomic DNA fragments were incubated with 5 μl of 5hmC antibody (Active Motif, 39791) at 4°C overnight in a final volume of 500 μl in DIP buffer (10 mM sodium phosphate, pH 7.0, 140 mM NaCl, 0.05% Triton X-100). The immunoprecipitated DNA fragments were enriched with Dynabeads Protein G (Thermo Fisher) and amplified with the primers provided in the NEBNext Multiplex Oligos for Illumina preparation kit (NEB, E7335). The prepared libraries were sequenced on an Illumina HiSeq2000 machine at Yale Stem Cell Center. One library was prepared for each condition indicated.
In vitro T-cell activation assay
Mouse splenic CD4+ T cells were isolated by MACS-based purification (Stem Cell Technologies). T cells were labeled with 1 mM CFSE (Invitrogen) in pre-warmed PBS for 10 min at 37 °C. The CFSE-labelled CD4+ T cells were then plated in complete RPMI media (with 10% FBS, 2mM glutamine, 1% penicillin/streptomycin) supplemented with 0.05 M β-mercaptoethanol in round bottom 96-well plates (2.5 × 104 cells per well). The plates were pre-coated with 1 μg ml-1 anti- CD3 (eBioscience clone 145-2C11). 5 μg ml-1 anti-CD28 (eBioscience clone 37.51) antibodies were directly added into the medium. Purified myeloid cells (TAMs or MDSCs) were added in indicated ratios and plates were incubated at 37 °C and in 5% CO2. In experiments with BMDMs, BMDM cells were cultured under M0 or M2 conditions for more than 24h, and then the culture supernatants (200 ul) were mixed with the CFSE-labeled T cells and were added into the 96-well assay plates pre-coated with 1 μg ml-1 anti-CD3 and 5μg ml-1 anti-CD28. After 3 days, cells were harvested and CFSE signal in the gated CD4+ T cells was measured by flow cytometry (LSRII Flow Cytometer, BD Biosciences). Two or more independent experiments were performed.
Arginase and ROS assays
Arginase inhibitor Nω-Hydroxy-nor-L-arginine was obtained from Sigma (#399275), and used at 100 μM. Recombinant human arginase 1 protein was obtained from Biolegend (#552502), and used at 1 μg/ml. To measure Arg1 activity, the Arginase Activity Assay Kit from Sigma (# MAK112) was used following manufacturer's instructions.
For ROS assay of MDSCs, the oxidation-sensitive dye Carboxy-H2DFFDA was obtained from Sigma (#C13293). Briefly, FACS-sorted MDSC cells were incubated at 37°C in RPMI medium in the presence of 2.5 μM DCFDA for 30 min. For induced activation, cells were simultaneously cultured, along with DCFDA, with 30ng/ml PMA. Analysis was then conducted by flow cytometry as described above. Data are representative of two independent experiments.
T cell depletion and IL-1R acute inhibition
T cell depletion was performed by intraperitoneal injection of anti-CD4 and anti-CD8 antibodies (400ug each for a single dose per mouse) every 7 days from day 7 post tumor cell injection. Antibodies were from BioXCell (clones GK1.5 and 53-6.72). Recombinant IL-1 receptor antagonist (IL-1Ra, anakinra) was from Biolegend (Cat#553908). IL1Ra was administered by i.v. injection at a dose of 2.5 mg/kg. Injections were performed daily for three consecutive days from day 9 after tumor cell injections. For experiments involving later time points, two additional injections were performed on day 13 and 14 after tumor cell injection. For each experiment, seven to nine mice were used for each group.
Quantification and Statistical Analysis
Student's t-test (2-tailed) was applied to all analysis comparing between two groups of samples. For the T- cell depletion experiment, permutation-based tests were performed to evaluate statistical significance, using custom matlab codes. Specifically, we evaluated the probability to reject the null hypothesis that T-cell depletion does not alter reduced tumor size phenotype in Mye-Tet2 null mice in comparison with mock treatment. This was performed by taking all possible ratios of tumor sizes between Tet2 null and WT groups (7 tumors per group, and thus 49 total possible ratios) under a specific treatment condition. The 49 ratios in T-cell-depletion conditions were then compared to the 49 ratios in the mock group to derive a t-test score. Permutations were then performed for 10,000 times by randomizing the labels of WT and Tet2 null assignment, and for each permutation, a t-test score was derived by comparing the two sets of 49 ratios. Two-tailed p value was calculated as the probability of observing an equal or more extreme t-test score in the permutation cases than the score of the original data. Similar tests were performed to reject the null hypothesis that T-cell-depletion (in comparison to mock) has the same impact on Tet2 null groups as WT groups.
Data and Software Availability
Data have been deposited in Gene Expression Omnibus. The accession number for the sequencing data reported in this paper is GSE98964.
Supplementary Material
Acknowledgments
We thank Susan Kaech, Carla Rothlin, Shangqin Guo, Tae Kon Kim, Yi Yang and Anna Baccei for helpful discussions, technical expertise, or critical review of this manuscript. We thank Mei Zhong from Yale Stem Cell Center Genomics Core for assistance with deep sequencing, which was supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation. We thank the assistance from the cores of Yale Cooperative Center of Excellence in Hematology. This work is supported in part by NIH grant R01CA149109 (to J.Lu), Connecticut RMRF grant 15-RMB-YALE-06 (to J.Lu), a Leukemia and Lymphoma Society Fellowship (to W.P.), a fellowship from Helen Hay Whitney Foundation-Howard Hughes Medical Institute (to S.Z.), and a grant (No. XDPB03) from the Strategic Priority Research Program of the Chinese Academy of Sciences (to S.Z.).
Footnotes
Author Contributions: W.P. designed and conducted most of the experiments. S.Z., K.M. J.C., K.H., H.M., Z.W., C.R. J.Liu, Z.T., J.Z. and B.G. participated in the experiments. K.Q., J.Z. W.P. and J.Lu performed bioinformatics analysis. Y.L., X.W. collected and analyzed human samples. M.B., R.F. and S.K. assisted with reagents, experimental design and data interpretation. J.Lu supervised the study. J.Lu. and W.P. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abram CL, Roberge GL, Hu YM, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. Journal of immunological methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nature immunology. 2016;17:18–25. doi: 10.1038/ni.3325. [DOI] [PubMed] [Google Scholar]
- An J, Gonzalez-Avalos E, Chawla A, Jeong M, Lopez-Moyado IF, Li W, Goodell MA, Chavez L, Ko M, Rao A. Acute loss of TET function results in aggressive myeloid cancer in mice. Nature communications. 2015;6:10071. doi: 10.1038/ncomms10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S, Thompson JA. Melanoma: immune checkpoint blockade story gets better. Lancet. 2014;384:1078–1079. doi: 10.1016/S0140-6736(14)61140-5. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008;180:2011–2017. doi: 10.4049/jimmunol.180.4.2011. [DOI] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nature genetics. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK. Immune Checkpoint Therapy in Melanoma. Cancer journal. 2016;22:73–80. doi: 10.1097/PPO.0000000000000183. [DOI] [PubMed] [Google Scholar]
- Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, Huszar M, White MR, Dinarello CA, Apte RN. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. Journal of immunology. 2009;183:4705–4714. doi: 10.4049/jimmunol.0901511. [DOI] [PubMed] [Google Scholar]
- Cataisson C, Salcedo R, Hakim S, Moffitt BA, Wright L, Yi M, Stephens R, Dai RM, Lyakh L, Schenten D, et al. IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. The Journal of experimental medicine. 2012;209:1689–1702. doi: 10.1084/jem.20101355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nature reviews Cancer. 2003;3:559–570. doi: 10.1038/nrc1145. [DOI] [PubMed] [Google Scholar]
- Church SE, Galon J. Tumor Microenvironment and Immunotherapy: The Whole Picture Is Better Than a Glimpse. Immunity. 2015;43:631–633. doi: 10.1016/j.immuni.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen SM, Goodell MA. Dynamic DNA methylation discovered during HSC differentiation. Cell cycle. 2015;14:693–694. doi: 10.1080/15384101.2015.1006558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009a;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009b;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. Mutation in TET2 in myeloid cancers. The New England journal of medicine. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Doedens AL, Stockmann C, Rubinstein MP, Liao DB, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage Expression of Hypoxia-Inducible Factor-1 alpha Suppresses T-Cell Function and Promotes Tumor Progression. Cancer research. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England journal of medicine. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu XJ, Amezquita R, Tsui YC, Cui GL, Micevic G, Perales JC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends in Genetics. 2014;30:464–474. doi: 10.1016/j.tig.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. 2015;42:613–626. doi: 10.1016/j.immuni.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel Markers to Delineate Murine M1 and M2 Macrophages. Plos One. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. P Natl Acad Sci USA. 2011a;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011b;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nature genetics. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- Lewis AM, Varghese S, Xu H, Alexander HR. Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. Journal of translational medicine. 2006;4 doi: 10.1186/1479-5876-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, Shen Q, Liu X, Zhu X, Li N, et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nature immunology. 2016;17:806–815. doi: 10.1038/ni.3464. [DOI] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011a;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cai XQ, Cai CL, Wang JP, Zhang WY, Petersen BE, Yang FC, Xu MJ. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011b;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. The Journal of clinical investigation. 2015;125:3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeth K, Wang J, Micevic G, Damsky W, Bosenberg MW. The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment cell & melanoma research. 2016 doi: 10.1111/pcmr.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- Ngiow SF, Meeth KM, Stannard K, Barkauskas DS, Bollag G, Bosenberg M, Smyth MJ. Co-inhibition of colony stimulating factor-1 receptor and BRAF oncogene in mouse models of BRAFV600E melanoma. Oncoimmunology. 2016;5:e1089381. doi: 10.1080/2162402X.2015.1089381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends in immunology. 2015;36:229–239. doi: 10.1016/j.it.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. Journal of immunology. 2004;172:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nature cell biology. 2016;18:607–618. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer cell. 2011a;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis (vol 20, pg 25, 2011) Cancer cell. 2011b;20:276–276. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Rawal S, Park HJ, Chu FL, Zhang M, Nattamai D, Kannan SC, Sharma R, Delgado DA, Chou T, Davis RE, Neelapu SS. Role of IL-4 in Inducing Immunosuppressive Tumor Microenvironment in Follicular Lymphoma. Blood. 2011;118:349–350. [Google Scholar]
- Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. Journal of Experimental Medicine. 2005;202:931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM, Hankey PA. Regulation of Macrophage Arginase Expression and Tumor Growth by the Ron Receptor Tyrosine Kinase. J Immunol. 2011a;187:2181–2192. doi: 10.4049/jimmunol.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM, Jr, Hankey PA. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. Journal of immunology. 2011b;187:2181–2192. doi: 10.4049/jimmunol.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE, Lahdesmaki H, Rao A. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A. 2014;111:E3306–3315. doi: 10.1073/pnas.1412327111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronov E, Carmi Y, Apte RN. The role IL-1 in tumor-mediated angiogenesis. Frontiers in physiology. 2014;5:114. doi: 10.3389/fphys.2014.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, Apte RN. IL-1 is required for tumor invasiveness and angiogenesis. P Natl Acad Sci USA. 2003;100:2645–2650. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes & development. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Liu C, Sun X, Li W, Zhang C, Zhou X, Lu Y, Xiao J, Li C, Xu X, et al. TET3 Inhibits Type I IFN Production Independent of DNA Demethylation. Cell reports. 2016;16:1096–1105. doi: 10.1016/j.celrep.2016.06.068. [DOI] [PubMed] [Google Scholar]
- Yadav M, Delamarre L. IMMUNOTHERAPY. Outsourcing the immune response to cancer. Science. 2016;352:1275–1276. doi: 10.1126/science.aag1547. [DOI] [PubMed] [Google Scholar]
- Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, Chen C, Liu S, Liu D, Chen Y, et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity. 2015;43:251–263. doi: 10.1016/j.immuni.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Iwasaki H, Laiosa CV, Stadtfeld M, Xie H, Heck S, Clausen B, Akashi K, Graf T. Hematopoietic stem cells expressing the myeloid lysozyme gene retain long-term, multilineage repopulation potential. Immunity. 2003;19:689–699. doi: 10.1016/s1074-7613(03)00299-1. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.