

Abstract
The site-specific recombinase encoded by bacteriophage λ (Int) is responsible for integrating and excising the viral chromosome into and out of the chromosome of its Escherichia coli host. Int carries out a reaction that is highly directional, tightly regulated, and depends upon an ensemble of accessory DNA bending proteins acting on 240 bp of DNA encoding 16 protein binding sites. This additional complexity enables two pathways, integrative and excisive recombination, whose opposite, and effectively irreversible, directions are dictated by different physiological and environmental signals. Int recombinase is a heterobivalent DNA binding protein and each of the four Int protomers, within a multiprotein 400 kDa recombinogenic complex, is thought to bind and, with the aid of DNA bending proteins, bridge one arm- and one core-type DNA site. In the 12 years since the publication of the last review focused solely on the λ site-specific recombination pathway in Mobile DNA II, there has been a great deal of progress in elucidating the molecular details of this pathway. The most dramatic advances in our understanding of the reaction have been in the area of X-ray crystallography where protein-DNA structures have now been determined for of all of the DNA-protein interfaces driving the Int pathway. Building on this foundation of structures, it has been possible to derive models for the assembly of components that determine the regulatory apparatus in the P-arm, and for the overall architectures that define excisive and integrative recombinogenic complexes. The most fundamental additional mechanistic insights derive from the application of hexapeptide inhibitors and single molecule kinetics.
INTRODUCTION
The λ site-specific recombination pathway has enjoyed the sequential attentions of geneticists, biochemists, and structural biologists for more than 50 years. It has proven to be a rewarding model system of sufficient simplicity to yield a gratifying level of understanding within a single (fortuitously timed) professional career, and of sufficient complexity to engage a small cadre of scientists motivated to peal this onion. The initiating highlight of the genetics phase was the insightful proposal by Allan Campbell for the pathway by which the λ chromosome integrates into, and excises from, the Escherichia coli host chromosome (1). The breakthrough for the biochemical phase was the purification of λ integrase (Int) and the integration host factor (IHF) by Howard Nash (2, 3). The first major step in the structural phase was the cocrystal structure of IHF bound to its DNA target site by Phoebe Rice and Howard Nash (4). Although the crystal structure of naked Fis protein had been determined earlier (5, 6), the full impact of Fis on understanding the fundamentals of the Int reaction did not come until much later (7, 8).
λ Integrase is generally regarded as the founding member of what is now called the tyrosine recombinase family, even though many family members are not strictly recombinases. Family membership is defined by the creation of novel DNA junctions via an active site tyrosine that cleaves and reseals DNA through the formation of a covalent 3′-phospho-tyrosine high-energy intermediate without the requirement for any high-energy cofactors. Other important, well studied, and highly exploited family members each have their own chapter in this volume of Mobile DNA III. Limitations on space prevent the inclusion in this chapter of the many other interesting family members, which comprise a wide range of biological functions and interesting variations on the themes discussed here, including other well-studied members of the heterobivalent subfamily, such as Tn916 (9), HP1 (10), and L5 (11). For previous reviews that include sections on the tyrosine recombinase family and λ Int see references 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, and 27. In this review, I will try to emphasize as much as possible those features of the λ Int pathway that have been reported since, or were not the focus of, earlier reviews, an intention that will consequently highlight recent advances in structural aspects of the pathway.
OVERVIEW OF THE REACTION
The λ Int recombination pathway has evolved to provide a conditional, effectively irreversible, DNA switch in the life cycle of the virus. The “cost” (complexity) associated with regulated directionality in the λ Int pathway is what distinguishes it from its Cre and Flp siblings (Fig. 1). As in most of the family members, each recombining partner DNA contains a pair of inverted repeat recombinase binding sites (called core-type sites) that flank a 7 bp over lap region (O) (6 to 8 bp in other systems) that is identical in both DNAs. (Evolution of new core-type and overlap DNA sequences has been proposed to proceed by low frequency λ phage insertions at sites other than the canonical attB [28].) DNA cleavage and exchange of the top strands on one side of the overlap region by two active Ints creates a four-way DNA junction [Holliday junction (HJ)] that is then resolved to recombinant products by the remaining pair of Ints cleaving and exchanging the bottom strands on the other side of the overlap region (Fig. 2). Appended to two of the four core-type sites are additional DNA sequences that encode binding sites for the second (NTD) DNA binding domain of Int and the accessory DNA bending proteins, IHF, Xis, and Fis. As indicated by color coding in Fig. 1, some sites are required only for integrative recombination between attP (on the phage chromosome) and attB (on the bacterial chromosome), some are required only for excisive recombination between the attL and attR sites (flanking the integrated prophage), and some sites are required for both reactions. For more detail, see reference 27. It has been suggested that the additional complexity of the λ pathway evolved to regulate the directionality of recombination in response to the physiological state of the host cell (29), a notion that is now well documented in latent human viruses, such as the ubiquitous herpes virus, cf., “… the [herpes] viral genome evolved to sense the infection status of the host… through highly evolved pathogen genomes with the capacity to sense host cytokines…” (30).
FIGURE 1.
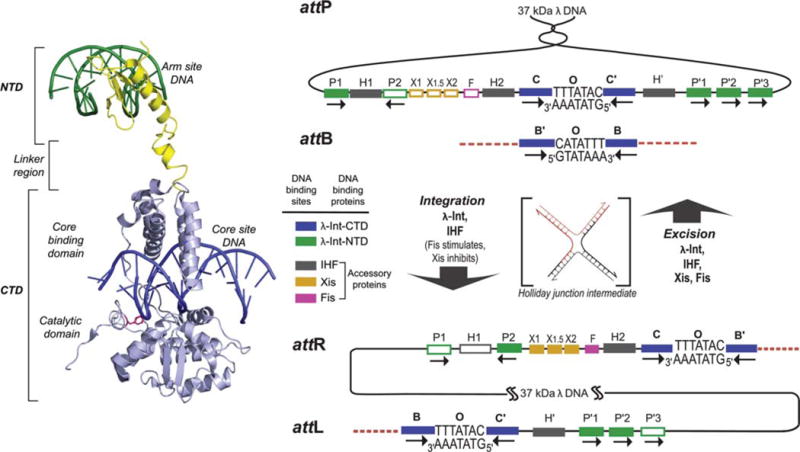
λ Integrase and the overlapping ensembles of protein binding sites that comprise att site DNA. The left panel shows the structure of a single λ Int protomer bound via its NTD to an arm site DNA and via its CTD to a core site DNA (adapted from the Int tetrameric structure determined by Biswas et al. [44], PDB code 1Z1G). The right panel shows the recombination reactions. Integrative recombination between supercoiled attP and linear attB requires the virally encoded integrase (Int) (2) and the host-encoded accessory DNA bending protein integration host factor (IHF) (4,177) and gives rise to an integrated phage chromosome bounded by attL and attR. Excisive recombination between attL and attR to regenerate attP and attB additionally requires the phage-encoded Xis protein (which inhibits integrative recombination) (140) and is stimulated by the host-encoded Fis protein (8). Both reactions proceed through a Holliday junction intermediate that is first generated and then resolved by single strand exchanges on the left and right side of the 7 bp overlap region, respectively. The two reactions proceed with the same order of sequential strand exchanges (not the reverse order) and use different subsets of protein binding sites in the P and P′ arms, as indicated by the filled boxes: Int arm-type P1, P2, P′1, P′2, and P′3 (green); integration host factor (IHF), H1, H2, and H′ (gray); Xis, X1, X1.5, and X2 (gold); and Fis (pink). The four core-type Int binding sites, C, C′, B, and B′ (blue boxes) are each bound in a C-clamp fashion by the CB and CAT domains, referred to here as the CTD. This is where Int executes isoenergetic DNA strand cleavages and ligations via a high-energy covalent 3′-phospho-tyrosine intermediate. The CTD of Int and the tetrameric Int complex surrounding the two overlap regions are functionally and structurally similar to the Cre, Flp, and XerC/D proteins. Reprinted with permission from reference 36. doi:10.1128/microbiolspec.MDNA3-0051-2014.f1
FIGURE 2.
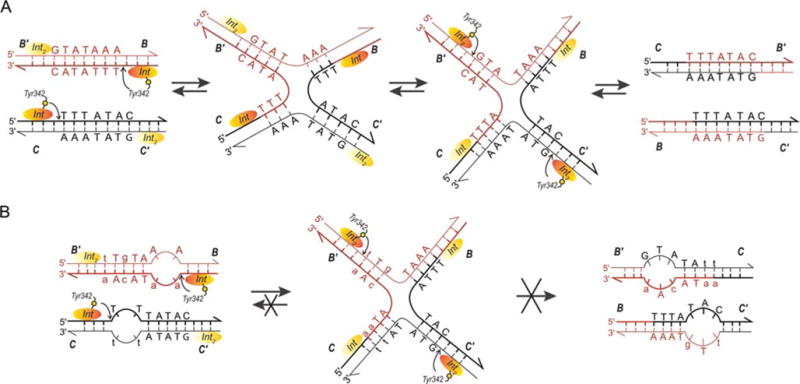
Formation, resolution, and trapping of Holliday junctions (HJ). (A) The top strand of each att site is cleaved via formation of a high-energy phosphotyrosine intermediate and the strands are exchanged (three bases are “swapped”) to form the HJ, thus, creating a branch point close to the center of the overlap regions. A conformational change of the complex that slightly repositions the branch point and more extremely repositions the Int protomers leads to the second swap of DNA strands and resolution of the HJ to helical products (44, 178). These features of the reaction suggested the mechanism-based method of trapping HJ complexes shown in (B). The left panel shows the DNA sequence changes made in the 7 bp overlap regions to trap HJ intermediates (lower case letters). Following the first pair of Int cleavages (via the active site Tyr) on one side of the overlap regions (arranged here in antiparallel orientation), the “top” strands are swapped to form the HJ; this simultaneously converts the unpaired (bubble) bases to duplex DNA. On the other side, the sequence differences between the two overlap regions strongly disfavor the second (“bottom”) strand swap that would resolve the HJ, because this would generate unpaired bubbles in the product complex (36, 37, 38, 39). This diagram applies to both integrative and excisive recombination (even though the labels refer to integrative recombination). Adapted in part, with permission, from reference 36. doi:10.1128/microbiolspec.MDNA3-0051-2014.f2
HOLLIDAY JUNCTION INTERMEDIATES
A hallmark of the tyrosine recombinase family, discussed here in terms of the λ pathway, is the formation of a four-way DNA junction (HJ) intermediate. For a long time, it was thought that this was a very unstable intermediate because it was difficult to identify without designing elaborate substrates (31, 32, 33). Only many years later was it discovered that the standard sodium dodecyl sulfate (SDS) treatment employed for visualizing naked HJ DNA gave misleading results because SDS fails to quench Int ligation activity fast enough to prevent reformation of the initial phosphodiester bonds (see also below) (34).
A striking feature of HJ formation in the Int reaction is that it is always initiated by cleavage and exchange of the same (“top”) strands in both integrative and excisive recombination (one of the facts indicating that the two reactions are not the reverse of one another) (Fig. 2) (31, 32, 33, 35). These features of the reaction suggested a mechanism-based method of trapping HJ complexes (outlined in Fig. 2) that would prove useful in studies of the complete higher-order complexes (described below) (36, 37, 38, 39).
Based upon structural snapshots from X-ray crystallographic models, patterns of amino acid residues in the active sites, mutational studies, and biochemical analyses, it is supposed there are only small differences between the pathways of λ Int, Cre, and Flp at the level of HJ formation, structure, and resolution, and likely only minor differences in their respective chemistries of DNA cleavage and ligation. One exception to this generalization is the manner in which the active site tyrosine nucleophile is delivered to the active site. In the case of Flp it is delivered in trans, that is, the tyrosine of one protomer in the tetramer is activated as a nucleophile in the active site of its adjacent neighbor (40, 41). While in both Int and Cre the tyrosine nucleophile is in cis, its proper positioning within the active site depends upon the nature of an interprotomer interaction between adjacent protomers within the tetrameric complex (42, 43, 44, 45), as discussed further below. In the absence of the NTD DNA binding domain Int can efficiently resolve HJs but it cannot carry out a recombination reaction (discussed further below) (46, 47, 48, 49). It is also clear from mutational analyses that there are Int residues that are specifically critical for HJ resolution but not DNA cleavage (50).
HEXAPEPTIDE INHIBITORS
In a bold and formidable effort to find recombination inhibitors that would trap the HJ intermediate, Anca Segall and her collaborators used deconvolution of synthetic hexapeptide libraries to search for hexapeptides that would block recombination subsequent to the first HJ-forming strand exchange(51, 52). Their most potent peptide inhibitor, WRWYCR, whose active form is a dimer assembled via a disulfide bridge between two peptide monomers, stably traps HJ complexes in all pathways mediated by Int as well as Cre (53, 54). Using this inhibitor, they were able to study the kinetics of HJ resolution under several different conditions and in several different Int-mediated pathways (55, 56). One of their conclusions from these studies was that spermidine stabilizes the “second” HJ isomeric form (the precursor to product formation) (57). Application of a hexapeptide inhibitor to studying the Bacteriodes NBU1 recombination pathway revealed that IntN1 recombinase is surprisingly more efficient when it forms HJs in the presence of mismatches, although their resolution to products does require homology (58).
In vitro, the hexapeptides inhibit a range of enzymes involving tyrosine-mediated transesterification, such as vaccinia virus topoisomerase and E. coli topoisomerase I (59). Subsequently, they were shown to be bacteriocidal to both Gram positive and Gram negative bacteria, presumably because they can interfere with DNA repair and chromosome dimer resolution by XerC/D. They were also shown to inhibit the excision of several different prophages in vivo (60). The in vivo successes of the hexapeptide inhibitors motivated the Segall group to search for therapeutically more useful small molecules with similar activities. Indeed, a search of over nine million compounds yielded one potentially interesting compound with properties that suggested the possible value of further searches for functional analogs of the hexapeptide inhibitors (61).
KINETICS
To overcome the difficulty of distinguishing kinetically relevant intermediates from off-pathway species, single molecule experiments were used to determine how binding energy from the multiple protein-DNA interactions is used to achieve efficiency and directionality in the overall Int recombination pathway (34). Protein binding (i.e., associated DNA bending), synapsis between attL and attR, HJ formation, and recombination were all monitored by changes in the length of a 1353 bp DNA that served as a diffusion-limiting tether of a microscopic bead to the flow chamber bed of a video-enhanced light microscope. In these experiments it was found that stable bent-DNA complexes containing Int, IHF, and Xis form rapidly (<20 s) and independently on attL and attR, and synapsis under these conditions is extremely rapid (1.0 min−1). These single molecule experiments strongly suggest there are no intrinsic mechanistic features of the pathway that make synapsis slow. While Int-mediated DNA cleavage, before or immediately after synapsis, is required to stabilize the synaptic complexes, those complexes that synapsed (~50% of the total) yield recombinant with an impressive ~100% efficiency. The rate-limiting step of excision occurs after synapsis, but closely precedes or is concomitant with the appearance of a stable HJ. This single molecule result is consistent with the observation that in solution rates of stable HJ formation are similar to the rates of excisive recombination (62).
Given the reversibility of the underlying chemistry of recombination, the apparent irreversibility observed in these experiments of each step of the reaction (except for synapsis) is notable. This result indicates that the overall directionality of excisive recombination is a direct consequence of the sequence of protein–protein and protein-DNA interactions that efficiently drive the reaction forward through nearly every step. It was proposed that the slow step in the reaction is some conformational change that stabilizes the HJ (34). Candidates for this rate-limiting step, such as the scissoring movement of the HJ arms, the shift in the localized bend of the HJ, or the reorientation of the active and inactive pairs of Int protomers, are suggested by comparison of the different X-ray crystal structures of tyrosine family recombinases complexed with their respective four-armed DNAs (15, 22, 43, 44, 63).
A totally different aspect of the kinetics of recombination concerns the process by which λ DNA, once inside the cell, finds its cognate attB site. Surprisingly, λ DNA does not carry out an active search but rather remains confined to the point where it entered the cell; it is the directed motion of the bacterial DNA during chromosome replication that delivers attB to a waiting, relatively stationary, attP (64).
STRUCTURE OF THE Int CTD
Among the most significant recent advances in our understanding of λ Int recombination were those emanating from the X-ray crystallographic studies by the Ellenberger laboratory (44, 45, 65). The second Int fragment to be used by the Ellenberger laboratory for X-ray crystallography, lacked the NTD (arm binding domain) and consisted of residues 75 to 356 (45). Referred to as C75 in the literature and here called the CTD, it corresponds to the two domains comprising the well-studied monovalent family members such as Cre, Flp, and XerC/D. The λ Int CTD is not competent for recombination but it is an efficient topoisomerase, binds weakly to single core-type DNA sites, and resolves λ att site HJs (48, 66, 67). The weak binding of the λ Int CTD to single core-type sites was circumvented by trapping covalent Int-att site complexes with a “flapped” suicide substrate containing a nick within the overlap region, three bases from the scissile phosphate (Fig. 3A).
FIGURE 3.
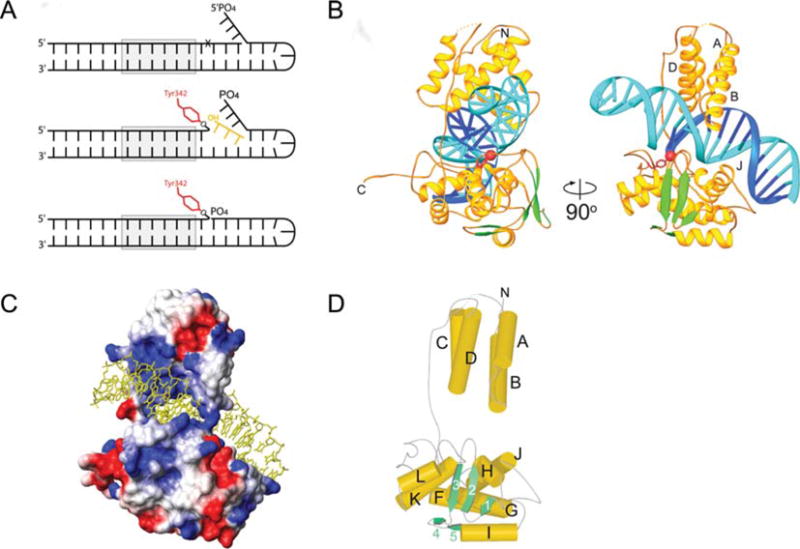
X-ray crystal structure of the Int CTD. (A) With this modified version of previously designed suicide recombination substrates (35, 47) covalently trapped CTD-DNA complexes were stable for weeks. Formation of the phosphotyrosine bond and diffusion of the three base oligonucleotide is followed by annealing of the three base flap to the three nucleotide gap, thus, positioning the 5′-phosphate such that it repels water and shields the phosphotyrosine linkage from hydrolysis. (B) Ribbon diagrams showing the central domain (residues 75 to 160; above the DNA) and the catalytic domain (residues 170 to 356; below the DNA) of λ Int, and their interactions with the major and minor grooves on the opposite sides of the DNA. A long, extended linker (residues I160 to R176) connects these domains. The scissile phosphate that is covalently linked to Y342 is shown as a red sphere. The central domain inserts into the major groove adjacent to the site of DNA cleavage. The catalytic domain makes interactions with the major and minor groove on the opposite side of the DNA, straddling the site of DNA cleavage. (C) The solvent accessible surface of the Int protein is shown, colored according to electrostatic potential. The DNA binding surface is highly positive (blue) and makes numerous interactions with the phosphates of the DNA (cf. Figure 3B). The polypeptide linker between domains joins the central and catalytic domains on one side of the DNA. A salt bridge between the Nζ of K93 and the carbonyl oxygen of S234 bridges between domains on the other side of the DNA, completing the ring-shaped structure that encircles the DNA. (D) The architecture of the λ Int C-75 protein is shown with cylinders and arrows representing helices and β strands, respectively. This view is oriented similarly to that in (A) (right side). The central domain of λ Int lacks helix E, corresponding to the fifth helix of Cre’s N-terminal domain, which is involved in subunit interactions. Reprinted with permission from reference 45. doi:10.1128/microbiolspec.MDNA3-0051-2014.f3
As shown in Fig. 3, the λ Int CTD consists of a catalytic domain that is joined to the central binding (CB) domain by a flexible, interdomain linker, residues I160-R176, that is extremely sensitive to proteolytic degradation (45, 48). The CB and catalytic domains of Int both contribute to recognition of the core site, although the former, whose structure has also been determined (68), confers most of the sequence specificity (69, 70). Only two residues from each domain (K95 and N99 in the CB domain and K235 and R287 in the catalytic domain) directly form hydrogen bonds with DNA bases. Interestingly, one of them, K95, interacts with a base, Gua30, that is absent in the B′ site, the weakest of the four core sites (71). The base-specific interactions are consistent with the effects of mutations of these and nearby residues that affect DNA binding specificity (72, 73, 74).
In comparison to the monovalent family members, Cre and Flp (41, 42, 43), the λ Int CTD displays fewer hydrogen bonds and total direct contacts to DNA bases in both its amino- and carboxy-terminal domains. Additionally, the extended unstructured interdomain linker of λ Int appears to be more flexible than the Cre linker (43), suggesting an increase in entropic cost of binding to DNA. Indeed, the helpful and informative int-h mutant (E174K), which substitutes a lysine in the middle of the interdomain linker adjacent to the site of DNA cleavage, increases the DNA binding affinity of λ Int and relaxes or eliminates the requirement for IHF during recombination (75, 76). It was proposed that the substituted lysine might enhance DNA binding affinity by contributing a stabilizing interaction with DNA, and/or by constraining the movement of the interdomain linker (45).
A comparison of the structure of the CTD Int covalently bound to DNA (45) with that of the unliganded catalytic domain (65), revealed that the tyrosine342 nucleophile had moved approximately 20 Å into the active site where it forms a 3′-phosphotyrosine linkage with the cleaved DNA (Fig. 4). Additionally, in the tetrameric complex, the eight carboxy-terminal residues (349 to 356) of a protomer extend away from the protein and pack against a neighboring protomer, contributing in trans an additional strand (β7) to the sheet formed by strands β1, β2, and β3 of the catalytic domain. This trans packing arrangement of β7 is required for appropriate placement of the Tyr342 nucleophile into the active site. This fact, in conjunction with the phenotypes of a number of Int mutants, suggests a dual role for the alternative stacking arrangements of β7. This also suggested an attractive explanation for the findings that a carboxy-terminal deletion of seven residues (commencing with Trp350), and mutations involving residues in or around β7 (all of which were expected to untether the Tyr342) abolished recombination but enhanced the topoisomerase activity of monomers (77, 78, 79). Because these same mutations decrease recombinase activity, the C-terminal tail could also be important in coordinating the catalytic activities of adjacent protomers, as seen in the X-ray crystal structure of the tetrameric higher order recombination complex (Fig. 4C) (44, 80).
FIGURE 4.
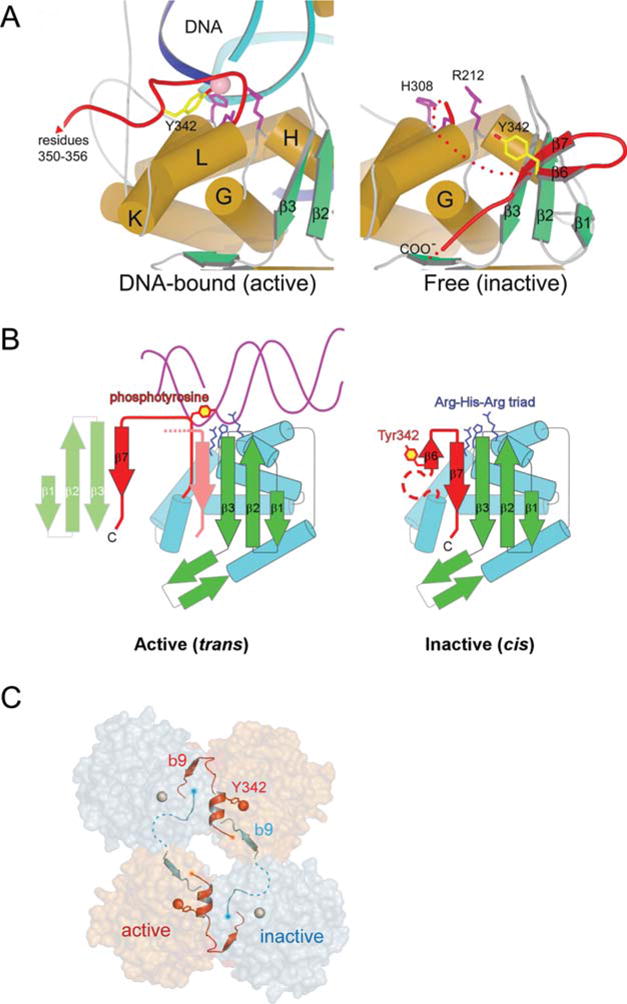
A Remodeling of Int’s active site switches DNA cleavage activity on and off. (A) A comparison of the DNA-bound (left), and unbound (right), structures of λ Int shows a dramatic reorganization of the C-terminal region spanning residues 331 to 356 (red). In the absence of DNA, Y342 (yellow) is far from the catalytic triad of R212, H308, and R311 (magenta side chains). In the DNA complex (left panel), Y342 has moved into the active site. Another consequence of the DNA-bound conformation is that the extreme C-terminal residues 349 to 356 extend away from the parent Int molecule and pack against another molecule in trans. (B) A cartoon illustrating how the DNA-bound conformation of Int positions the Y342 for cleavage of DNA. The isomerization from the inactive form, in which Y342 is held some distance from the catalytically important Arg212-His308-Arg311 triad (65), to the active conformation seen in complex with DNA, is accompanied by the release of strand β7 and its repacking in trans against a neighboring molecule. (C) The assembly of active (orange) and inactive (gray) catalytic sites results from a skewed packing arrangement of λ Int subunits (residues 75 to 356) in the tetramer. The scissile phosphates bound by active and inactive subunits are shown as red and gray spheres, respectively. Reprinted with permission from references 44 and 45. doi:10.1128/microbiolspec.MDNA3-0051-2014.f4
In contrast to the large movement of the Tyr342 nucleophile in transitioning from the unliganded to the liganded Int, the other four catalytically important residues, R212, K235, H308, and R311, show less than 1 Å movement on average between the two structures, as is also true for most of the other residues in the catalytic domain. The role of these residues in catalysis was established by mutational analyses of several tyrosine recombinase family members, biochemical analyses (especially of topoisomerase I), sequence comparisons of other family members, and shortly thereafter, comparisons with the X-ray crystal structures of other DNA-bound family members (41, 43, 81, 82, 83, 84, 85, 86, 87, 88).
ROLE OF THE Int NTD
The following experiments were carried out to prove it was possible to “de-tune” a monovalent recombinase, for example, Cre, and convert it to a regulated unidirectional recombinase by appending an NTD (89). Cre recombinase is bidirectional, unregulated, does not require accessory proteins, and has a minimal symmetric DNA target. Rather than de-tuning the Cre recombinase its DNA target was attenuated: a single base pair change, previously shown to weaken the interaction between Cre and its DNA binding site (90) was introduced into each of the inverted repeat Cre binding sites and the DNA sequence and spacing between the DNA cleavage sites (the “overlap” region) was changed to the canonical seven base pair sequence of the λ att sites. λ P and P′ arms were appended to the modified Cre target sites to generate analogs of the four λ att sites.
To complete the recombination pathway, a gene fusion encoding the first 74 residues of λ Int was fused to Cre. The resulting chimeric Cre protein product carried out recombinations between the analogs of the four λ att sites with all of the properties of canonical λ Int-dependent pathways: reactions were dependent upon IHF, Xis was required for the excision reaction but inhibited the integration reaction, integrative recombination required the P1 but not the P2 sites, and the excisive reaction required P2 but not P1 (cf. Fig. 1).
It appears from these experiments that the regulated directionality of the λ Int pathway has been conferred on Cre by the appended 74 N-terminal residues of λ Int coupled with the reduction in DNA binding efficiency between Cre and its DNA target sites. These experiments suggest that two simple steps, in no specified order, are all that is required for the evolution of the heterobivalent recombinases from their monovalent siblings. However, they do not rule out an alternative evolutionary trajectory in which the monovalent and heterobivalent site-specific recombinases evolved in parallel from a common, less efficient, precursor.
While the NTD of λ Int was able to confer regulated directionality on the Cre recombinase, it is possible, and even likely, that not all of the λ NTD functions were revealed in these experiments. For example, effects resulting from any interactions between the NTD and the CTD were not studied in those experiments and they would not likely even be manifest in the hybrid protein. One example of such interactions came from studies on the context-dependent effects of the NTD. These studies were prompted by the unexpected finding that the Int CTD (residues 65 to 356, called C65) is more active as a topoisomerase, in binding to core-type sites, cleaving DNA, and resolving synthetic Holliday junctions, than the full length Int. In other words, the NTD is an inhibitor of the primary Int functions (49). Equally surprising was the fact that when the cloned and purified NTD (residues 1 to 65) was added to the cloned and purified CTD, it stimulated all of the primary Int functions, well beyond the levels observed for either CTD or full length Int. In other words, when present in cis (i.e., in full length Int), the NTD is an inhibitor of Int functions, but when present in trans, it is a stimulator. Resolution of the apparent paradox came with the finding that addition of an oligonucleotide encoding the arm-type DNA sites (P′1–P′2) to full length Int abolished the cis NTD inhibition and resulted in the formation of a ternary complex between Int and core and arm-type DNAs.
These results led to the hypothesis of an enhanced dual role for the DNA bending accessory proteins. In addition to their structural function in facilitating the Int-mediated arm-core bridges that comprise the higher-order structure of recombinogenic complexes, they should also be viewed as a requirement to overcome the N-domain inhibition of recombinase functions (49). These data and the resultant hypothesis are consistent with the finding of mutants in one domain that effect the activity of the other (72, 77), and the important observation of Richet et al. that Int does not bind well to attB unless it part of a higher-order attP complex (91).
Residues Met1 to Leu64 comprise the minimal Int fragment that binds to arm-type sites and it does so with almost the same efficiency as full length Int (66, 92). However, an additional six residues are required (Met1 to Ser70) for cooperative binding to the adjacent arm-type sites P′1, P′2, and P′3. The greatest coopera-tivity in binding, which is between sites P′2 and P′3, depends upon the single bp between them and is resistant to an unopposed three base bulge in the top strand but not in the bottom strand. The asymmetric effect of the unopposed bulge is consistent with DNA bending upon Int binding to the P′ arm sites. Int’s affinity for the single sites P′1 or P1 exceeds its net affinity for P′2–P′3 (44, 93). It is interesting that the two lowest affinity arm-type sites, P2 and P′3 are each required for only one of the two recombination reactions, excision and integration, respectively, and are also the outermost sites in their respective pathways. Int binding at P2 is greatly enhanced by its cooperativity with Xis binding at X1, and Int binding at P′3 is enhanced (to a lesser extent) by its cooperativity with Int binding at P′2, thus, rendering the excisive reaction very sensitive to Xis concentration and the integrative reaction more sensitive to Int concentration (66). The latter fit nicely with a very early observation by Enquist et al. that integrative recombination is more sensitive than excisive recombination to decreased intracellular levels of Int (94). The Int 1–70 NTD is also equally as competent as full length Int for cooperative interactions with Xis when the two are bound at P2 and X1, respectively (66).
STRUCTURE OF THE NTD
The first view of the NTD structure came from a nuclear magnetic resonance (NMR) analysis of the Met1-Leu64 peptide, which revealed a fold structurally related to the three-stranded β-sheet family of DNA-binding domains. However, it was supplemented with a disordered 10 residue amino-terminal basic tail, that was shown to be important for arm binding by its loss of function upon removing a single positive charge (G2KΔ2R) (92). The importance and role of the amino-terminal basic tail was clearly shown in the subsequent NMR structure of the NTD in complex with its DNA target site (95). Only two other proteins containing this fold have been visualized in complex with their DNA targets: the N-terminal domains from the Tn916 Int protein (96) and from the ethylene responsive factor from Arabidopsis thaliana (AtERF1) (97). All three proteins recognize DNA via their unique three-stranded antiparallel β-sheet that is inserted into the major groove of their respective DNA targets. The smaller size of the β-sheet-DNA interface in the λ NTD, relative to the other two proteins, is presumably compensated by the additional contacts of the 11 residue amino-terminal tail that projects deep into the minor groove (95).
STRUCTURE OF A FULL Int TETRAMER COMPLEX
A structural view of the full λ Int did not come until it was cocrystallized with DNA bound at the NTD and CTD, recognition domains for the arm- and core-type DNA sites, respectively. These studies by the Ellenberger laboratory were particularly informative because they represented Int-DNA complexes at three different steps along the recombination pathway (44). One of the structures, a synaptic complex between two COC’ core-type sites bound by four CTDs (residues 75 to 356), represented an early step after the first DNA cleavage but before strand exchange. A second structure, with full length Ints in which the cleaved strands had exchanged but ligation was prevented by a modified DNA substrate, represented a post strand-exchange complex. And the third structure was a synthetic Holliday junction intermediate bound by four full length Ints, carrying the Tyr342Phe mutation, that were thus unable to cleave the DNA into products. In the last two structures, the NTDs of the full length Ints were bound to short oligonucleotides containing tandem P′1–P′2 arm-type DNA binding sites. It is likely that the presence of this arm-type DNA occupying the NTD domains was a critical factor in the successful crystallization of the full length Int, and additionally imposed a facilitating (albeit unnatural) 2-fold symmetry. The other factor critical for crystallization was the stable tetrameric arrangement of protomers within each complex.
The tetrameric complexes with full length Int assemble into three distinct layers. The NTD (residues 1 to 63) that binds to arm-type sites is joined to the core-binding domain (CB domain; residues 75 to 175) by a short α-helical segment (residues 64 to 74), and this, in turn, is connected to the C-terminal catalytic domain (residues 176 to 356) through another linker (residues 160 to 176). Together, the three domains of each Int form an ensemble that engages the core and arm DNA targets to form a tightly knit but flexible tetrameric complex (Fig. 5) (44).
FIGURE 5.
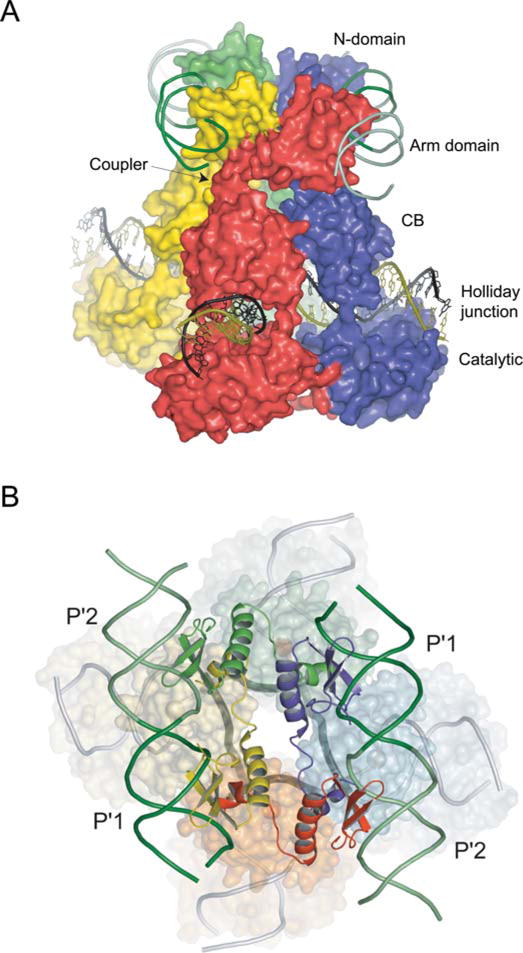
Structure of the λ Int tetramer bound to a Holliday junction and arm DNAs. (A) The domains of Int pack together as three stacked layers, with the NTDs cyclically swapped onto neighboring subunits. The NTD layer embraced by two antiparallel arm DNAs is linked through short α-helical couplers to the CTD, which encircles the branches of the Holliday junction. The active subunits are colored red/green and the inactive subunits are blue/yellow. (B) The 2-fold symmetry of the NTD layer is reflected in the skewed arrangement of the CTDs and the shape of the four-way junction (thick dark gray lines) in the bottom strands reactive isomer. Reprinted with permission from reference 44. doi:10.1128/microbiolspec.MDNA3-0051-2014.f5
The four NTDs are bound by two antiparallel arm DNAs that slightly bend towards each other, with each pair binding the adjacent P′1–P′2 binding sites. The basic N-terminal segment (residues 2 to 10), that was disordered in the NMR structure but shown to be required for recombination activity (92), tucks into the minor groove adjacent to the 3′ side of the arm-type consensus sequence (44).
As noted above, the CB and catalytic domains (which are referred to together as the CTD in this review) are structurally analogous to the full-length monovalent tyrosine recombinases, Cre (42, 43), Flp (41), and XerC/D (98). Thus, it is not surprising that λ Int has a catalytic pocket that resembles the other family members with nearly identical conserved residues (Arg212, Lys235, His308, Arg311, and His333) that engage the scissile phosphate and Tyr342 nucleophile (44, 45, 65).
Among the factors likely to contribute to λ CTD’s lack of recombination function, is the linker (residues 160 to 176) between the CB and catalytic domains. In contrast to the analogous linker in Cre, it lacks the αE helix that contributes many intersubunit interactions that stabilize the Cre tetramer (42, 43). Consequently, the loosely packed CB domains of the λ Int tetramer are able to rotate against each other by as much as 30° in the different isomers that were crystallized (44).
It was particularly interesting that each of the three independent crystal structures determined by Biswas et al. (44) illustrates a different conformation of the core DNAs and different subunit packing interactions (Fig. 6). The skewed packing of protomers generates two very different subunit interfaces comprising active versus inactive catalytic sites. In the former, the Tyr342 helix is well ordered and stabilized by electrostatic interactions with two catalytically essential residues. In the latter, the β9 is incompatible with these stabilizing interactions and the region around Tyr342 is disordered (see also Fig. 4C). It should be noted that an α-helical conformation around Tyr342, that was not seen in the active conformation of the earlier crystal structure of the λ CTD (residues 75 to 356) (45), was confirmed by additional crystal structures (in the presence of orthovanadate) to likely be the true active conformation (44).
FIGURE 6.
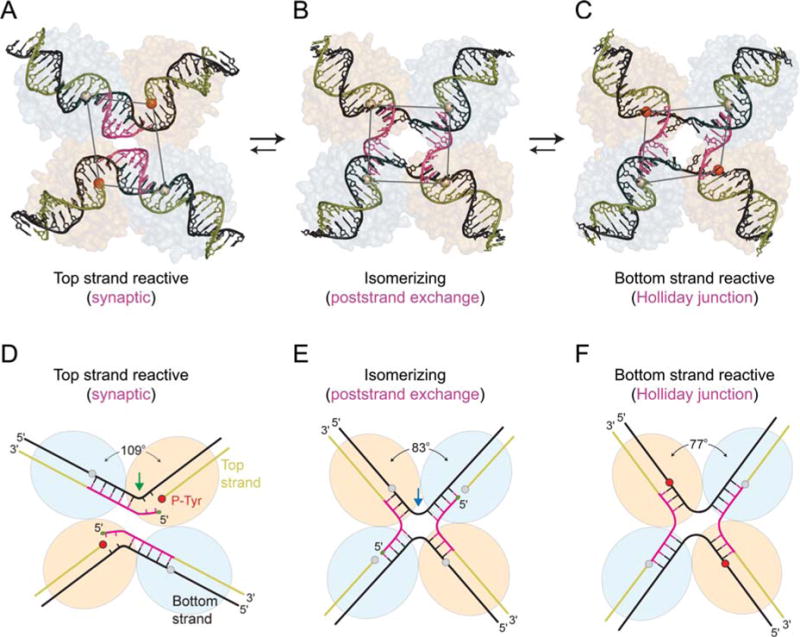
Three different conformations of λ Int tetramers representing distinct steps of the recombination reaction. The core DNAs within the λ-Int(75–356) synaptic complex (A, D), the λ-Int post- strand exchange complex (B, E), and the λ-Int Holliday junction complex (C, F) are shown along with schematic diagrams illustrating the interbranch angles and position of branch points. The pair of Int subunits in the active conformation (orange/red) is positioned closer to the center of each complex, whereas the inactive pair of subunits (gray) is further apart. Scissile phosphates (spheres) activated for cleavage are colored in red. Reprinted with permission from reference 44. doi:10.1128/microbiolspec.MDNA3-0051-2014.f6
In the crystal structure of the synaptic, prestrand-exchange, complex, the tetramer deviates strongly from 4-fold symmetry: the scissile phosphates (which can be visualized as the corners of a parallelogram) of the cleaved DNA strands are 39 Å apart while those of the uncleaved strands are 50 Å apart (Fig. 6A, D). This translational offset brings the cleaved 5′ ends closer to the phosphotyrosine of the synapsing partner, thus, facilitating strand exchange and ligation. In the post strand-exchange complex, the core DNAs resemble a HJ intermediate with approximate four-fold symmetry. Here the kink has moved to a more central position, 4 bp away from the cleaved site, bringing the cleavage sites of the bottom strands closer together, and possibly disfavoring reversal of the top strand cleavage (Fig. 6B, E). In the complex with a synthetic preformed HJ, the crossover point was fixed three nucleotides from one pair of cleavage sites, and consequently, these sites are used preferentially for resolution (67, 99). This complex is also highly skewed such that the scissile phosphates, bound by the active protomers, are brought close together (Fig. 6C, F). Although not apparent in the crystal structures, mutational analyses also reveal nonequivalent interactions between the NTDs of neighboring Int protomers during HJ resolution (100).
One of the features of the tetrameric Int crystal structures, which was also inferred from solution studies of these small complexes (101), is a cyclically permuted topology, in which each NTD packs on top of the neighboring CB domain. It is now thought that this 2-fold symmetric NTD arrangement does not reflect that of a bona fide (integrative or excisive) recombinogenic complex, but rather is a consequence of the symmetric arm-type sites that are not connected by DNA and bending proteins to the core region, as discussed further below.
INTEGRATION HOST FACTOR
Integration host factor (IHF) was discovered in the very early studies of λ site-specific recombination by virtue of its role as a host-encoded protein that was essential both in vitro and in vivo for integrative and excisive recombination (102, 103). Its specific architectural role was demonstrated by the observation that IHF bending at the H′ site could stimulate Int binding and cleavage at the low- affinity C′ core site (104). As has not infrequently been the case, an E. coli protein discovered for its role in a phage life cycle, turned out to be an important player in the physiology of the cell. IHF is involved in regulation of gene transcription, especially σ54 promoters (105), initiation of DNA replication (106), transposition (107), and phage packaging (108) (for reviews see references 109, 110, 111, 112, 113, 114, 115, 116).
To a large extent, the role and mechanisms of IHF in λ site-specific recombination, the crystal structure of IHF in complex with its DNA target, and the ways in which the physiology and biology of IHF in the host cell can impact recombination in vivo, have been reported and discussed prior to, and within, a previous review of λ site-specific recombination (27). More recent studies of IHF have centered on details of its interaction with DNA (117, 118, 119) and the mechanics and features of DNA bending by IHF (120, 121, 122, 123).
IHF is a hetero-dimeric protein consisting of two highly basic polypeptides, α and β, with molecular masses of 11,200 and 10,580 Da, respectively. These two subunits share approximately 30% homology to each other and also to the family of type II DNA-binding proteins that includes major histone-like proteins of E. coli such as HU.
The IHF structure, which is very similar to that of HU, is a compact, globular domain, consisting of symmetrically intertwined α and β subunits, from which two long β ribbon arms extend. The arms curl around the DNA and interact exclusively with the minor groove; most of the DNA bending (>160°) occurs at two large kinks, 9 bp apart (Fig. 7) (4). It has also been possible to construct a functional IHF in which the two chains have been fused into one (124).
FIGURE 7.
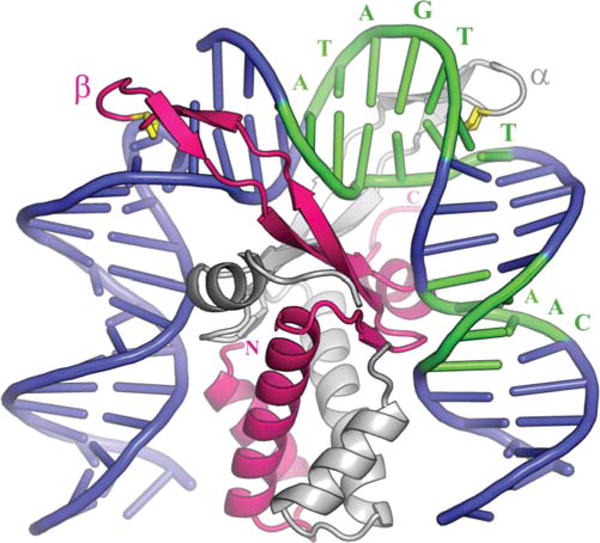
Complex of integration host factor with H′1N. The α and β subunits are shown in white and pink, respectively. The consensus sequence is highlighted in green and interacts mainly with the arm of α and the body of β. The yellow proline at the tip of each arm (P65 α/P64 β) is intercalated between bp 28 and 29 on the left side and 37 and 38 on the right. Reprinted with permission from reference 4. doi:10.1128/microbiolspec.MDNA3-0051-2014.f7
The sequence preference displayed by IHF does not come from specific side chain contacts: it makes no contacts at all within the major groove and only a few hydrogen bonds to positions in the minor groove. Rather its specificity comes from “indirect readout,” based on the sequence dependent structural parameters of its target DNA. Indeed, biochemical and structural studies of a relaxed-specificity mutant of IHF revealed how specificity is determined within the TTR portion of the consensus sequence (117). Within certain constraints, the structure of the DNA was driven by its own sequence and the protein side chains had readjusted to accommodate the different DNA structures.
Evidence that formation of a distinct DNA path was indeed the primary role of IHF came from “bend swap” experiments, where one or more IHF sites were replaced by unrelated DNA bending modules, either intrinsically bent DNA or different DNA bending proteins, such as HU (56, 125, 126). Although able to complement the lack of IHF-induced bending, none of the chimeric constructions performed as efficiently as the wild-type arrangement. The inability of the bend swap chimeras to achieve wild-type efficiency was evidently due to a requirement for considerable precision, as evidenced by the observation that an IHF bend wrongly positioned by just 1 bp, in a loop of constant length between C′ and P′1 of attL, could severely reduce excisive recombination efficiency (127).
It is attractive to propose that the evolution of Int’s dependence on host-encoded accessory proteins derives, at least in part, from the benefits of linking the regulation and direction of recombination to the physiology of the host cell (see also the discussion of Fis protein below). In this regard, the changing levels of intracellular IHF are potentially interesting. The relative abundance of IHF increases approximately 5- to 7-fold over a 6 h span after entry into stationary phase (128, 129), and decreases when stationary-phase cells are diluted into fresh medium and cell mass begins to double (130). It is interesting that in vitro high IHF concentrations tend to inhibit the excisive reaction (131). The in vivo downshift in IHF concentration is probably not due to increased protein degradation, as IHF is not unstable in its dimeric form (132, 133), but instead appears to be a consequence of arrested transcription upon entry into exponential phase and increased transcription of the individual subunits upon entry into stationary phase (134). Additionally, there is evidence that IHF may play an essential role in survival from cell starvation: not only is IHF critical for induction of 14 proteins from the glucose starvation stimulon but mutants lacking IHF appear to be severely compromised in their ability to survive glucose starvation (135).
Xis
As noted above, the small phage-encoded Xis protein is the key determinant of directionality in the λ pathway. Essential for the excisive reaction and stimulating more than 106-fold in vivo, it is also inhibitory for the integrative reaction (136, 137). The NMR structure of 1–55XisC28S revealed an unusual “winged”-helix structure formed by two α-helices that are packed against two extended strands. While this structure itself did not afford critical insights into how Xis plays such a critical role in the λ pathway, it did herald the start of a steady progression towards this goal by the Johnson and Clubb laboratories (138).
A 1.7 Å resolution cocrystal structure of 1–55XisC28S complexed with a 15 bp DNA fragment containing its cognate X2 binding site comprised the second step of the Johnson/Clubb progression and provided a detailed view of the complex, which was largely in accord with their proposals based on the NMR structure of the free protein (Fig. 8A) (139). Although, the Xis-X2 complex is bent only modestly (approximately 25°), and hardly enough to account for the strikingly large curvature observed for a larger Xis-attR complex (93), it did suggest a molecular model for the Xis stimulation of Int binding to the adjacent P2 arm-type site (Fig. 8B).
FIGURE 8.
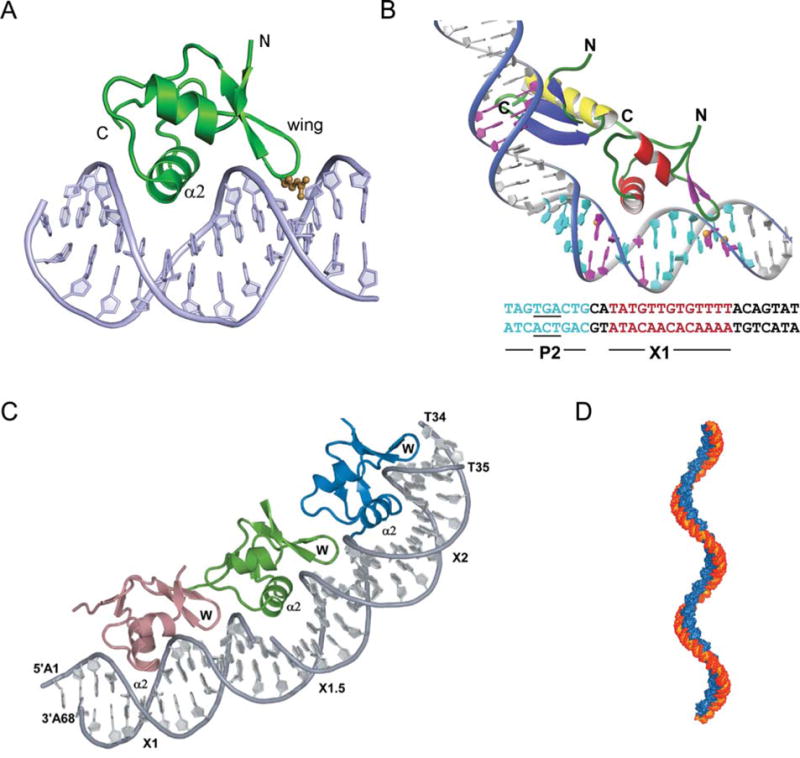
Complex of Xis with DNA. (A) The structure of 1–55XisC28S specifically bound to X2 DNA penetrates adjacent grooves of the duplex by fastening on the phosphodiester backbone. The major groove is filled primarily with helix α2 with the side chains of Glu19, Arg23, and Arg26 playing a major role in specific DNA recognition. The adjacent minor groove is contacted by the “wing” which does not contribute significantly to the specificity of complex formation but does contribute to binding affinity, although to a smaller extent than helix α2. The side-chain of Arg39 (brown) extends along the floor of the minor groove where it makes direct and water-mediated hydrogen bonds. (B) A model for the Int (NTD)-Xis-DNA ternary complex. The Int (NTD) is modeled to interact with the TGA trinucleotide (underlined) of the P2 site (blue) in the DNA major groove. Xis is modeled on the X1 site (magenta) in the same manner as observed in the complex with the X2 site. The C-terminal tail of Xis, which is disordered in solution (not shown), is located adjacent to the C-terminal helix of the NTD of Int to make a protein–protein interaction as shown by mutagenesis and NMR titration data (179). (C) X-ray crystal structure of Xis bound to the Xis binding region reveals the structural basis of cooperative binding. Xis monomers bound to the X1, X1.5, and X2 sites are colored dark salmon, green, and blue, respectively. (D) Structure-based model of an extended Xis-DNA filament. Units of the Xis-DNAX1–X2 crystal structure were stacked end-to-end by superimposing site X1 over X1.5 to assemble a pseudocontinuous helix with a pitch of ~22 nm. Proteins are blue; DNA is orange. Reprinted with permission from reference 139 (A and B) and reference 140 (C and D). doi:10.1128/microbiolspec.MDNA3-0051-2014.f8
A precursor to a larger and more informative co-crystal structure was the finding that, counter to the previous long-standing notion of two Xis binding sites (X1 and X2), a third Xis was bound at a site between them, called X1.5 (140, 141). The initial EMSA data were supplemented with protein–protein crosslinking experiments to further confirm the trimeric nature of the complex (140). More useful insights for understanding the role of Xis in directing recombination came from the 2.6 Å cocrystal structure of Xis bound to a larger DNA target comprising the entire 33 bp Xis binding region (Fig. 8C) (140). The three Xis proteins bind to this DNA in a head-to-tail orientation that generates a micronucleoprotein filament having approximately 72° of curvature and a slight positive writhe (Fig. 8D).
The differences in the specific interactions at X1 versus X2, combined with the observed nonspecific binding of Xis at the X1.5 site and the range of different interactions at ostensibly similar protein–protein interfaces, foreshadowed experiments showing that the flexible recognition surfaces of Xis result in a relatively promiscuous binder of DNA. The propensity for nonspecific DNA binding was further characterized in an Xis-DNA cocrystal with an 18 bp fragment of DNA (8). While this flexibility of DNA recognition is important for binding at the X1.5 site, where protein–protein interactions with the X1- and X2-bound Xis protomers provide additional stability, it also means that Xis is easily distracted from its attR target in vivo, where there is a huge excess of nonspecific DNA. Indeed, this latter point explains why excisive recombination in vivo is 50 to 200-fold lower in the absence of Fis (see also below) (8). Correspondingly, the Fis dependence of excisive recombination in vitro is only seen at limiting concentrations of Xis (142)
Fis
It is ironic, but understandable with hindsight, that although Fis protein was the first component of the λ recombination pathway to be crystallized (5, 6), it was the last component whose biological and molecular role was elucidated (8). Throughout this 16-year period (and beyond), the Johnson lab has played the leading role in studying the many roles and mechanisms of the Fis protein (112, 143, 144, 145). Fis was initially identified as a factor in promoting site-specific recombination by DNA invertases (146, 147) and was shortly thereafter shown to bind cooperatively with Xis at attR and to stimulate excisive recombination up to 20-fold when Xis is limiting (142). In vivo, the absence of Fis reduced attP formation from an induced lysogen by 100 to 1,000-fold (138, 148); it was also shown to be required along with Xis for binding to the attR region in the P22 challenge phage system (149).
Fis, like the other host-encoded accessory protein in the λ Int pathway, IHF, is a nucleoid associated protein of global, structural, and regulatory importance. Its role in determining overall chromosome structure is exerted by contributing to the looped-domain architecture of the nucleoid, and by influencing the regulation of genes encoding topoisomerases (150, 151, 152). Fis plays a role in the initiation of DNA replication, in several transposition reactions, and in the regulation of transcription at many different genes by several different mechanisms (for reviews, see references 112, 143, 151, and 153). The large number of critical sites of action for Fis becomes even more significant when considering how dramatically its intracellular levels vary as a function of cellular physiology.
Thompson et al. (142) showed that Fis levels drop dramatically when cells entered stationary phase and, more significantly, that occupation of the Fis binding site on att site DNA also drops in stationary phase cells. More detailed studies revealed that from these extremely low levels in stationary phase, Fis levels increase 500-fold during the initial lag phase when cells are diluted into fresh medium, and reach a peak of 50,000 to 100,000 copies per cell as the culture enters exponential phase. The control of Fis protein synthesis is at the level of mRNA where it is repressed by Fis protein (154, 155, 156) and stimulated by IHF (157). Transcription from the fis promoter, Pfis, is critically influenced by DksA, a component of the transcription initiation complex that is also required for negative regulation of rRNA promoters (158, 159). DksA, which acts in part by reducing the half-life of (unstable) RNA polymerase-Pfis promoter complexes, elevates the required concentration of the initiating NTP (CTP) and amplifies the inhibitory effect of ppGpp on Pfis (154, 155, 158, 160). In so doing, it constrains fis expression primarily to early log phase at high growth rates, and it inhibits expression at low growth rates or following amino acid starvation (154, 155). However, as normal growth phase-dependent regulation of fis is observed in a ΔrelA ΔspoT strain, other mechanisms can evidently compensate for the role of ppGpp in the pathway (154).
The crystal structures of Fis revealed a globular dimer composed of four tightly intertwined α-helices with two helix-turn-helix (HTH) motifs in each monomer (5, 6). One of the most striking features of the Fis structure was that the D helices, which were proposed to fit into adjacent major grooves of the DNA helix, are only 25 Å apart, approximately 10 Å shorter than the pitch of normal B DNA.
The long-standing hurdle to obtaining Fis-DNA co-crystals was the weak similarity among the many 15 bp sequences capable of forming stable complexes with Fis, thus, making it extremely difficult to derive an optimal consensus sequence for Fis binding. This obstacle was finally overcome by Stella et al. (7), who compiled the results of many analyses of Fis binding affinities into an informative hierarchy of DNA sequences, culminating in two 27 bp oligonucleotides whose Fis binding affinities were sufficiently optimized for crystallography (see Fig. 9A). Having established that compression of the central AT-rich minor groove is a critical feature of Fis binding, the authors went on to show that intrinsic DNA bends are unlikely to contribute significantly to Fis binding. Rather, they proposed that Fis initially searches for DNA with an intrinsically narrow minor groove, where AT composition, not sequence, is the critical determinant. Most recently, the Johnson lab has shown that the primary molecular determinant modulating minor groove widths is the 2-amino group on guanine (145).
FIGURE 9.
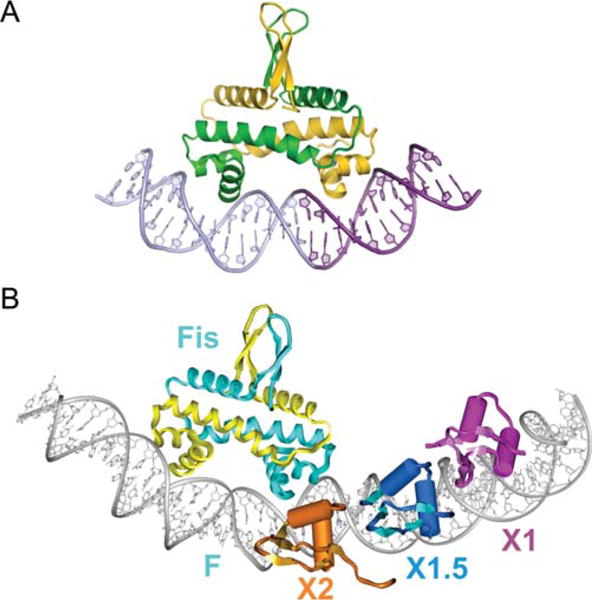
X-ray crystal structure of a Fis dimer complexed with DNA (A) and its relation to Xis binding (B). (A) The C-terminal helix representing the recognition helix of the HTH unit of each subunit is inserted into adjacent major grooves on the concave side of the 21 bp curved DNA. Only base contacts with a single residue, Arg85, are important for binding. The DNA undergoes substantial conformational adjustments, including adoption of ~65° overall curvature, to fit onto the Fis binding surface. The central 5 bp of the DNA interface are not contacted by Fis, but compression of the central minor groove to almost half the width of canonical DNA at the center enables the α-helices to insert into the adjacent major grooves, which do not show any appreciable change in width. (B) Model of the Fis-Xis cooperative complex. The X-ray crystal structure of three Δ55Xis monomers bound to the X1 (magenta), X1.5 (blue), and X2 (gold) binding sites was superimposed onto the model of the Fis K36E X-ray structure docked to DNA representing the F site. Fis subunits are cyan and yellow. The DNA recognition helices of Xis bound at X2 and the proximal Fis subunit nearly form a continuous protein surface within the major groove. Reprinted with permission from reference 7 (A) and reference 8 (B). doi:10.1128/microbiolspec.MDNA3-0051-2014.f9
While intrinsic DNA bends are not very important for targeting Fis binding, the bends induced by bound Fis are critical for its many functions, including DNA compaction, assembly of invertasomes, regulating transcription, and, most importantly for this article, directing the curvature of the attR complex. The Johnson lab proposed that cooperative DNA binding between Fis and its partners, which bind immediately adjacent to Fis but generate only a small number of interfacial amino acid residues, is likely facilitated by mutually compatible changes in DNA shape.
Early experiments indicating that the F and X2 sites on attR overlap one another had been interpreted to suggest that both sites could not be bound simultaneously by their cognate proteins. However, subsequent experiments using quantitative gel shifts, stoichiometry determinations, nuclease footprinting and protein–protein crosslinking clearly established that Fis and Xis bind to the F and X2 sites simultaneously and cooperatively (8, 141). Most interestingly, Papagiannis et al. showed that Fis binds to the attR site in vitro with approximately 100-fold greater affinity than Xis alone, and in vivo, the rate of excision is reduced approximately 100-fold when Fis is absent (8). They proposed that in vivo Fis targets the otherwise peripatetic Xis to the X2 site, which then recruits Xis to X1.5 and X1. Based on their Xis-DNA microfilament cocrystal structures and their Fis crystal structures they built a model of the Fis-Xis complex on attR DNA, in which their observed protein induced DNA distortions are proposed to favor the cooperative binding of Fis and Xis (Fig. 9B).
PATTERNS OF λ NTD BINDING AND BRIDGING
Prior to considering the patterns of λ NTD binding and bridging in recombination reactions between canonical pairs of att sites, it should be noted the λ Int is also capable of efficiently carrying out an IHF-dependent recombination between two identical attL sites lacking a P′1 arm site (55). The existence of such a bidirectional pathway lacking the usual complement of components raises interesting questions about the kinds of recombinogenic complexes Int is capable of forming (161, 162, 163) and also underscores the caveat of off-pathway reactions.
The caveat of off-pathway reactions was echoed by a caveat about the artificially imposed symmetry of the NTD domains of the complexes used for X-ray crystallography of Int tetramers bound to Holliday junctions (discussed above). Earlier genetic and nuclease protection experiments had suggested that the patterns of NTD binding to arm-type sites were asymmetric (see Fig. 1) (reviewed in reference 27) and these results were subsequently reinforced by nuclease protection studies on Holliday junction intermediates (trapped with a hexapeptide inhibitor [51]) and biotin interference assays (BIA) (164). The latter, which probe the requirements for protein binding at a particular DNA locus by obstructing the major groove with a biotin bound to the C5 position of designated thymines, was particularly compelling because it monitored a complete integrative or excisive recombination reaction. From these experiments, it became clear that any attempt to understand the architecture and function of canonical recombinogenic complexes would require an analysis of the full ensemble of proteins and DNAs.
A requisite step in moving towards a panoptic investigation of the recombinogenic complexes was the deciphering of which “core-type” and “arm-type” binding sites are joined to one another by Int-mediated bridges. Towards this end, a disulfide trapping technology (165, 166) was used, in conjunction with trapped Holliday junction complexes (see Fig. 2), to introduce disulfide crosslinks at the protein-DNA interfaces between an Int NTD and its cognate arm-type site, and between an Int CTD and its cognate core-type site (36). Trapped nucleo-protein HJ complexes doubly cross-linked to Int were only observed with those att sites in which cystamine-labeled arm site and the cystamine-labeled core site are “bridged” by the same Int molecule
From such analyses, it was concluded that the Int bridges between arm- and core-type sites in the integrative HJ recombination intermediate are: P′1–C′; P′2–C; P′3–B′; and P1–B. The Int bridges in the excisive HJ intermediate are: P′1–C′; P′2–B; and P2–B′. This leaves the C core site as the one that does not form an Int bridge with one of the three arm-type sites required for excisive recombination.
The Int bridges determined by site-directed cross-linking in HJ complexes were confirmed and complemented in full recombination reactions by a genetic approach using two chimeric recombinases. The first, called Crn1, consists of a Cre recombinase fused to the NTD of λ-integrase; it has all the properties of λ Int (described above) (89). This was complemented by construction of a second chimeric recombinase, Crn2, in which the NTD and CTD domains recognized different arm- and core-type DNA target sequences (36). A collection of hybrid att sites was constructed in which one of the bridged arm-core pairs (identified by the chemical crosslinking experiments) had the arm and core sequences recognized by Crn2, while the remaining arm-core bridges had the arm and core sequences recognized by Crn1. Using these substrates, it was shown that Crn1 could not carry out recombination unless Crn2 was also present (and vice versa). The results of the chimeric recombination reactions confirmed, and also provided information complementary to, the results from chemical crosslinking (as discussed below).
These results argue strongly against models in which regulated directionality of λ Int recombination depends upon some degree of Int bridge remodeling during the course of the reaction. Furthermore, the monogamous relationship of each arm-core bridged pair throughout the course of the recombination reaction makes it possible to extrapolate from the patterns observed in the HJ recombination intermediate to those predicted for the presynaptic recombination partners and the post HJ recombination products. Inspection of Fig. 10 reveals that for excisive recombination, the presynaptic partners have only intramolecular bridges, suggesting that Int bridging is not a driving force in synapsis of attL and attR. This is also likely to be the case for integrative recombination, even though the capture of a naked attB by a fully assembled (supercoiled) attP complex requires two intermolecular bridges (91). It was postulated that the reason attB cannot bind Int protomers unless they are part of a higher-order complex stems from the need to overcome the NTD inhibition of CTD function, described above (49), and not from any driving force by intermolecular bridges (see also discussion below).
FIGURE 10.
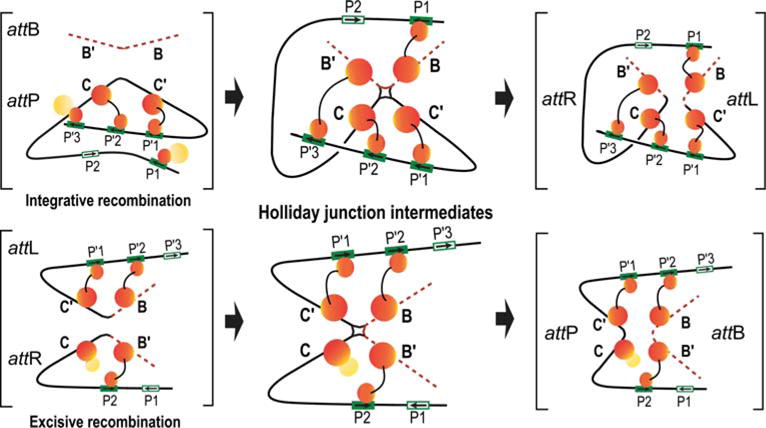
Schematic summary of the Int bridges in integrative and excisive recombination. The middle panel diagrams the Int bridges of the Holliday junction (HJ) recombination intermediates determined by Tong et al. (36). In the integrative complex, all four core sites and four of the five arm sites enjoy an Int bridge while the excisive complex engages three of the four core sites and three of the five arm sites. The flanking panels (brackets) depict extrapolations from the HJ complexes to the respective att site recombination partners (substrates) and recombinants (products) based on the deduction that Int bridges are not broken and reformed during recombination. doi:10.1128/microbiolspec.MDNA3-0051-2014.f10
ARCHITECTURES OF RECOMBINOGENIC COMPLEXES
In an attempt to derive architectural models for the HJ recombination intermediates, the Int bridging results were augmented with in-gel fluorescence resonance energy transfer (FRET) experiments (101, 167, 168, 169) to determine the apparent distances between selected positions within the excisive recombination HJ intermediate (37).
Using the Int bridging data, the apparent FRET distances for the HJ recombination complex, and the 3D structures for all of the protein components in their DNA-bound forms (4, 7, 8, 44, 45, 139, 140), it was possible to computationally build a model for the architecture of the λ excision complex (Fig. 11A, B, C). Insights gained from the excisive complex along with the integrative Int bridging data and 3D structures were used to generate a corresponding model of the integrative complex (Fig. 11D, E, F). Considered individually and together, the two architectures afford a number of interesting insights, as discussed below and in the figure legends (37).
FIGURE 11.
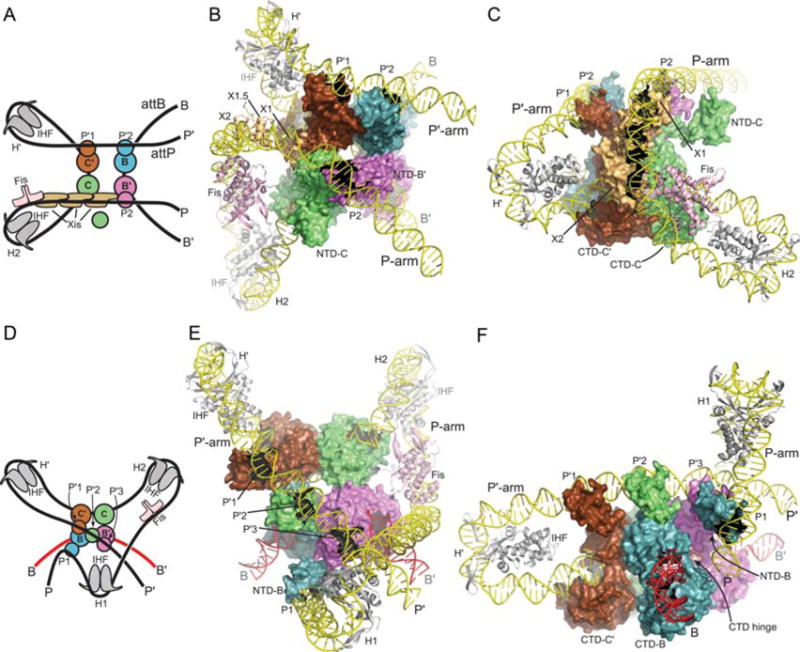
Models of the λ excisive and integrative recombination complexes. (A) Schematic representation of the excisive complex architecture. The excision reaction product resulting from Holliday junction (HJ) resolution is shown. Int subunits (blue, green, magenta, brown) are represented by a small circle (NTD) and a large circle (CTD). Integration host factor (IHF) heterodimers (gray) are shown bound to the H′ and H2 sites. Fis dimer (pink) and Xis (tan) subunits are indicated. (B) Model of the excisive complex in the same “top view” orientation as the schematic drawing in panel A. The NTD of the Int subunit bound at the C core site (NTD-C) is shown separated from the rest of the complex to improve clarity of the P-arm trajectory. (C) Side view of the excisive complex, highlighting the trajectory of the P-arm. IHF bending of the P′ arm at H′ directs the DNA over the CTD domains of the Int tetramer, facilitating engagement of the P′1 and P′2 arm sites by the Int subunits bound at the C′ and B core sites, respectively. In the P-arm of attR the phasing of the IHF-induced bend at H2 is different from that at H′; at H2, the P-arm is directed along the plane of the catalytic domain tetramer. An A-tract sequence that is stabilized by Fis binding (7, 8) directs the P-arm upwards, towards the Int CB domains. The cooperative Xis filament (8, 140) then redirects the P-arm across the top of the Int CTD domains, where the P2 site is bound by the Int subunit bound at the B′ core site. The Xis subunit bound at X1 resides close to the position where the NTD of the Int subunit bound at the C core site (Int-C) would be expected. The NTD of Int-C was not docked in a specific location of the excisive complex model, but it seems plausible, even attractive, that this domain could bind nonspecifically to the P-arm near the X1 site, perhaps interacting with Xis. (D) Schematic of the integrative complex architecture. The arm-type binding sites engaged by the four Int subunits are indicated. (E) Model of the integrative complex in the same “top view” as illustrated in panel B. In this orientation, the P-arm rises towards the viewer, crosses over the P′ arm, and is directed back towards the Int tetramer by the IHF bend at the H1 site. (F) Side view of the integrative model, looking approximately down the B core site. The NTD of the Int subunit bound at the B core site (NTD-B) is shown bound at the P1 site, on the flexible P-arm. The CB and catalytic domains of the Int subunit bound at the B site can be seen wrapped around the opposing face of attB, with the interdomain hinge indicated. The CTD-NTD linkers were not modeled and are not shown. IHF bending at H′ directs the P′ arm over the CTD domains of the Int tetramer, but in this case the P′1, P′2, and P′3 binding sites are engaged by the Int subunits bound to C′, C, and B′, respectively. As Xis is not present in the integrative complex, the P-arm is directed upwards, parallel to the Int tetramer, and as Fis stimulation of integration has been reported (180, 181), it was included in the model. IHF bound to the H1 site redirects the P-arm back towards the Int tetramer, crossing over the P′ arm in the process. The P1 arm-type site is thereby brought to a position where it can bind the NTD of the Int subunit poised for capture of the B core half-site (Int-B). Reprinted with permission from reference 37. doi:10.1128/microbiolspec.MDNA3-0051-2014.f11
In the excision complex, the P′ and B arms form a left-handed crossing, while the overall path of attR DNA indicates a left-handed, nucleosome-like, wrapping by IHF, Fis, Xis, and Int. The model thus predicts a negative DNA crossing node in attL and left-handed solenoidal wrapping in attR, both of which are consistent with negative supercoiling in the normal substrates. In the integrative complex the asymmetric mode of binding to C′, C, and B′ by the P′ arm requires considerable flexibility in the linker segments between the CTDs and NTDs, and this model explicitly predicts the formation of a negative DNA-crossing node in the recombination complex, where the P-arm crosses over P′. The model also features an unusual and flexible P-arm tether that positions Int-B for attB binding.
ASYMMETRY AND FLEXIBILITY
The architectures proposed for the recombinogenic complexes differ in several ways from the crystal structures of the HJ-bound Int tetramers bound to arm site DNA duplexes (44). While this would not necessarily have been predicted, it is not surprising, as the crystal structures did not include accessory DNA bending proteins or their cognate DNA sites, which join the core-and arm-type sites. An additional compromise required to form crystals was the substitution of a pair of P′1–P′2-containing oligonucleotides for the canonical asymmetric arrangement of arm-type binding sites. Indeed, subsequent experiments involving biotin-interference mapping of complete recombination reactions (described above) are more consistent with the asymmetric architectures than the symmetric arrangement in the smaller complexes designed for crystallization (164). In contrast to the symmetric and tightly packed NTD organization observed in crystal structures, the models for the architectures of the complexes feature highly asymmetric arrangements of the NTDs. In the former, the domains are swapped, with the NTD of one Int subunit located above the CB domain of an adjacent Int. The latter is incompatible with domain-swapped NTDs and implies considerable flexibility in the CB-NTD linkers.
TOPOLOGY
Excisive recombination between directly repeated attL and attR sites results in a large fraction of free circles when supercoiling levels are low, similar to that observed for the Cre and Flp recombinases (170). Integrative recombination between directly repeated attP and attB sites results in catenated circles for supercoiled substrates, implying that the recombination process itself imposes a strand crossing (170, 171, 172). Seah et al. (37) argue that the proposed architectures are consistent with these results and explain many of the other topological findings of Crisona et al. (170). Additionally, the tightly wrapped nature of the integrative complex model and the inclusion of a negative DNA crossing node are consistent with, and may explain, in part, the requirement for negative supercoiling for efficient integration (91).
CAPTURING THE HOST attB SITE
From the time, Richet and Nash (91) first showed that attB comes naked to a recombination with its fully decorated attP partner, and there has been considerable speculation about the details of this synaptic event. Because of the pseudodyad symmetry of the core-type sites the openings of the bound integrase C-clamps must face in opposite directions (45). While this is not a problem for the monovalent family members it implies that for the fully assembled attP complex one of the Int subunits (the one destined to bind the B core site of attB) must have the flexibility to wrap around the host chromosome from the opposite face.
Indeed, the architecture proposed for the integrative complex does contain an inherently flexible P-arm that tethers the Int-B subunit and allows for the dynamic binding required to engage the bacterial chromosome and ultimately lock onto the attB sequence. The model is also consistent with, and explains, a difference between the two kinds of Int bridging experiments reported by Tong et al. (36). Whereas chemical crosslinking of the P1–B Int bridge was the most robust of all the Int bridges, in the genetic analyses, the P1–B Int bridge was the weakest, precisely the difference expected for a flexible arm.
ARCHITECTURAL BASIS FOR DIRECTIONALITY
The source of the strong bias towards the top strands being exchanged first in formation of the HJ (33, 35) is evident from the models in Fig. 11 and Fig. 12. During excisive recombination, both attL and attR are bent at their core sites in order to promote the bridging interactions that form between core and arm binding sites. The core site bend directions that lead to stable complexes are coupled to IHF-induced bends and commit both attL and attR to top strand cleavage upon synapsis of the sites. Similarly, only one bend direction of the attP core site will lead to stable bridging interactions between C/C′ core sites and P′1/P′2 arm sites. This direction commits attP to top strand cleavage in the synaptic complex with attB. Thus, the order of strand exchange in both pathways is determined prior to synapsis by formation of specific attL, attR, and attP complexes (34).
FIGURE 12.
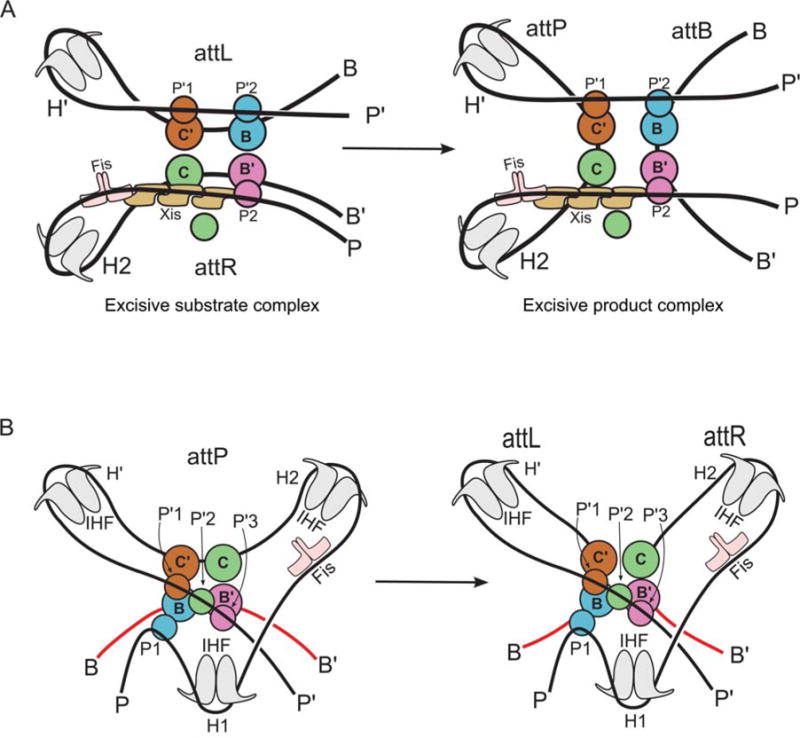
Schematic representation of the excisive and integrative reactions, based on the structural models shown in Fig. 13. Coloring of the protein subunits matches that shown in Fig. 11. Reprinted with permission from reference 37. doi:10.1128/microbiolspec.MDNA3-0051-2014.f12
The architecture of the excisive complex provides a bird’s eye view of how Xis mediates its critical role as the regulator of directionality (137, 173) (Fig. 13A). In the absence of Xis, the P-arm would not be directed across the top of the Int CTDs to make the required P2-B′ bridge and the P-arm would not be properly positioned to stabilize a functional attR. An additional critical role for Xis is to promote the cooperative binding of the Int NTD at P2 (66, 130, 174).
FIGURE 13.
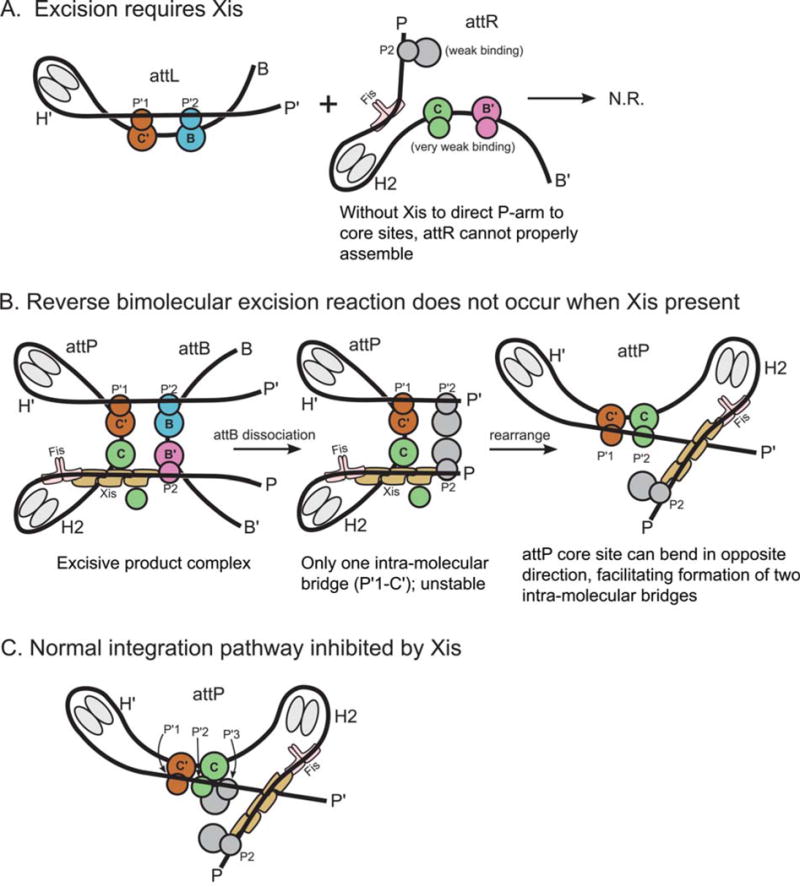
The basis for directionality in λ recombination. (A) An explanation for why Xis is required for excision. (B) Explanation for why the excision pathway is not run efficiently in reverse to perform integration. (C) Explanation for why Xis inhibits the normal integration reaction. The Xis, P2, and H1 sites cannot be occupied simultaneously (22). Schematics follow the same coloring scheme used in Fig. 11 and Fig. 12. Int subunits not bound to a core site are colored gray. doi:10.1128/microbiolspec.MDNA3-0051-2014.f13
The architecture of the excisive complex also explains the long-standing question of why the excision reaction does not run efficiently in reverse once attB is released (Fig. 13B). After dissociation of attB, the attP complex is expected to be less stable because it now only contains a single intramolecular bridge (P′1–C′). Furthermore, this complex has the potential to rearrange, such that the attP core bends in the opposite direction and facilitates the formation two intramolecular bridges (P′1–C′ and P′2–C). While this complex resembles a portion of the attP substrate complex, it is prevented from proceeding to a competent complex by the presence of Xis, which prevents the binding of IHF at H1 and Int at P1 (131), leaving the P-arm improperly positioned for the synaptic attP complex (cf. Fig. 12B). This same Xis-induced mispositioning of the P-arm is also responsible for the Xis inhibition of integrative recombination (Fig. 13C) (136). The architectures of the two complexes thus explain how the dramatically different roles of the P-arm in integrative versus excisive recombination provides the basis for an effective directionality switch that depends upon the presence or absence of Xis.
SUMMARY AND FUTURE DIRECTIONS
In the 12 years since publication of the last review that focused solely on the λ site-specific recombination pathway (in Mobile DNA II [27, 175]), the most dramatic advances in our understanding of the reaction have been in the area of X-ray crystallography. Protein-DNA structures have been determined for the CTD of Int (45), full Int in a tetrameric complex with HJ and arm site DNAs (44), Xis complexed with its DNA target (139), and Fis complexed with its DNA target (7). Including the earlier IHF-DNA structure (4), these results comprise a complete portfolio of all of the DNA-protein interfaces driving the Int pathway. Building on this foundation of structures, it has been possible to derive models for the assembly of components that determine the regulatory apparatus in the P-arm (8, 140), and for the overall architectures that define excisive and integrative recombinogenic complexes (36, 37). The most fundamental additional mechanistic insights derived from the application of hexapeptide inhibitors (53) and single molecule kinetics (34).
On the list of experiments needed to fill out and/or sharpen our understanding of the λ Int reactions, the answers to several structural questions are of high priority. Some of these are probably close at hand, such as structures for the interfaces between Xis and Fis on one side and Xis and the Int NTD on the other side. A more problematic, but very important structural question, concerns the conformations of the linker regions between the Int NTDs and CTDs within a functional recombinogenic complex. Architectures, or structures and stabilities, of the substrate and product complexes are important but will only be useful if accompanied by convincing demonstration they comprise part of the canonical pathway. If it would be too greedy to hope for X-ray crystal structures of the recombinogenic complexes, perhaps high-resolution cryo-electron microscopy might fill the bill. Additional single molecule analyses of the reactions, of HJ formation and resolution, and formation and stability of the products, should provide important mechanistic insights. Finally, without attempting to frame the specifics, it seems reasonable to predict the design of new applications of the λ Int pathway to study questions, and/or solve problems, unimagined at this time. In this vein one might ponder the question raised by J.K. King (176): “Is it possible or not, that in the random process of life, human creativity results in biological behavior related to Lambda genetics?”
Acknowledgments
This chapter is dedicated to the memory of Howard Nash and Bob Weisberg, who laid the foundations for studying the biochemistry and genetics, respectively, of the λ Int pathway. I am grateful to Greg Van Duyne, past and present members of my lab, and especially Reid Johnson, for many helpful discussions. I thank Reid Johnson, Tapan Biswas, Hideki Aihara, and Phoebe Rice for helpful comments and for making available figures depicting their work, Joan Boyles for administrative assistance, and especially Nicole Seah for help in preparing the manuscript. The work from my laboratory was supported by NIH grants GM062723 and GM033928.
References
- 1.Campbell AM. Episomes. In: Caspari EW, Thoday JM, editors. Advances in Genetics. Academic Press; New York, NY: 1962. pp. 101–145. [Google Scholar]
- 2.Nash HA. Purification of bactriophage λ Int protein. Nature. 1974;247:543–545. doi: 10.1038/247543a0. [DOI] [PubMed] [Google Scholar]
- 3.Nash HA, Robertson CA. Purification and properties of the Escherichia coli protein factor required for λ integrative recombination. J Biol Chem. 1981;256:9246–9253. [PubMed] [Google Scholar]
- 4.Rice PA, Yang SW, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 5.Kostrewa D, Granzin J, Koch C, Choe HW, Labahn J, Kahmann R, Saenger W. Three-dimensional structure of the E. coli DNA-binding protein FIS. Nature. 1991;349:178–180. doi: 10.1038/349178a0. [DOI] [PubMed] [Google Scholar]
- 6.Yuan HS, Finkel SE, Feng JA, Kaczor-Grzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant Fis protein: Relationship between mutational changes and recombinational enhancer function or DNA binding. Proc Natl Acad Sci USA. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stella S, Cascio D, Johnson RC. The shape of the DNA minor groove directs binding by the DNA-bending protein Fis. Genes Dev. 2010;24:814–826. doi: 10.1101/gad.1900610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papagiannis CV, Sam MD, Abbani MA, Yoo D, Cascio D, Clubb RT, Johnson RC. Fis Targets Assembly of the Xis Nucleoprotein Filament to Promote Excisive Recombination by Phage Lambda. J Mol Biol. 2007;367:328–343. doi: 10.1016/j.jmb.2006.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudy C, Taylor KL, Hinerfeld D, Scott JR, Churchward G. Excision of a conjugative transposon in vitro by the Int and Xis proteins of Tn 916. Nucleic Acids Res. 1997;25:4061–4066. doi: 10.1093/nar/25.20.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hickman AB, Waninger S, Scocca JJ, Dyda F. Molecular organization in site-specific recombination: The catalytic domain of bacteriophage HP1 integrase at 2.7 Å resolution. Cell. 1997;89:227–237. doi: 10.1016/s0092-8674(00)80202-0. [DOI] [PubMed] [Google Scholar]
- 11.Lewis JA, Hatfull GF. Control of directionality in L5 integrase-mediated site-specific recombination. J Mol Biol. 2003;326:805–821. doi: 10.1016/s0022-2836(02)01475-4. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Russell N, Orchard SS, Segall AM. Probing nucleoid structure in bacteria using phage lambda integrase-mediated chromosome rearrangements. In: Kelly TH, Stanley RM, editors. Methods Enzymol. Vol. 421. Academic Press; New York, NY: 2007. pp. 209–226. [DOI] [PubMed] [Google Scholar]
- 13.Van Houdt R, Leplae R, Lima-Mendez G, Mergeay M, Toussiant A. Towards a more accurate annotation of tyrosine-based site-specific recombinases in bacterial genomes. Mobile DNA. 2012;3:6. doi: 10.1186/1759-8753-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren D, Laxmikanthan G, Landy A. Integrase family of site-specific recombinases. Brenner’s Encyclopedia of Genetics. 2013;4:100–105. [Google Scholar]
- 15.Van Duyne GD. Protein-Nucleic Acid Interactions: Structural Biology. The Royal Society of Chemistry; Cambridge: 2008. Site-specific recombinases; pp. 303–332. [Google Scholar]
- 16.Rajeev L, Malanowska K, Gardner JF. Challenging a paradigm: the role of DNA homology in tyrosine recombinase reactions. Microbiol Mol Biol Rev. 2009;73:300–309. doi: 10.1128/MMBR.00038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segall AM, Craig NL. New wrinkles and folds in site-specific recombination. Mol Cell. 2006;19:433–435. doi: 10.1016/j.molcel.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Grindley ND, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 19.Rice PA. Resolving integral questions in site-specific recombination. Nat Struct Mol Biol. 2005;12:641–643. doi: 10.1038/nsmb0805-641. [DOI] [PubMed] [Google Scholar]
- 20.Lee L, Sadowski PD. Strand selection by the tyrosine recombinases. Prog Nucl Acid Res Mol Biol. 2005;80:1–42. doi: 10.1016/S0079-6603(05)80001-7. [DOI] [PubMed] [Google Scholar]
- 21.Williams KP. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 2002;30:866–875. doi: 10.1093/nar/30.4.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Duyne GD. A structural view of tyrosine recombinase site-specific recombination. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. ASM Press; Washington DC: 2002. pp. 93–117. [Google Scholar]
- 23.She Q, Brügger K, Chen L. Archaeal integrative genetic elements and their impact on genome evolution. Res Microbiol. 2002;153:325–332. doi: 10.1016/s0923-2508(02)01331-1. [DOI] [PubMed] [Google Scholar]
- 24.Dutton G. Site-specific recombinases. The Scientist. 2002;16:29–31. [Google Scholar]
- 25.Lewis JA, Hatfull GF. Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res. 2001;29:2205–2216. doi: 10.1093/nar/29.11.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobryn K, Chaconas G. The circle is broken: telomere resolution in linear replicons. Curr Opin Microbiol. 2001;4:558–564. doi: 10.1016/s1369-5274(00)00251-4. [DOI] [PubMed] [Google Scholar]
- 27.Azaro MA, Landy A. λ Int and the λ Int family. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. ASM Press; Washington DC: 2002. pp. 118–148. [Google Scholar]
- 28.Rutkai E, Gyorgy A, Dorgai L, Weisberg RA. Role of secondary attachment sites in changing the specificity of site-specific recombination. J Bact. 2006;188:3409–3411. doi: 10.1128/JB.188.9.3409-3411.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landy A. Dynamic, structural and regulatory aspects of lambda site-specific recombination. Annu Rev Biochem. 1989;58:913–949. doi: 10.1146/annurev.bi.58.070189.004405. [DOI] [PubMed] [Google Scholar]
- 30.Reese TA, Wakeman BS, Choi HS, Hufford MM, Huang SC, Zhang X, Buck MD, Jezewski A, Kambal A, Liu CY, Goel G, Murray PJ, Xavier RJ, Kaplan MH, Renne R, Speck SH, Artyomov MN, Pearce EJ, Virgin HW. Helminth infection reactivates latent γ-herpesvirus via cyto- competition at a viral promoter. Science. 2014;345:573–577. doi: 10.1126/science.1254517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu PL, Landy A. Resolution of synthetic att-site Holliday structures by the integrase protein of bacteriophage λ. Nature. 1984;311:721–726. doi: 10.1038/311721a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitts PA, Nash HA. An intermediate in the phage λ site-specific recombination reaction is revealed by phosphorothioate substitution in DNA. Nucleic Acids Res. 1988;16:6839–6856. doi: 10.1093/nar/16.14.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitts PA, Nash HA. Bacteriophage λ site-specific recombination proceeds with a defined order of strand-exchanges. J Mol Biol. 1988;204:95–107. doi: 10.1016/0022-2836(88)90602-x. [DOI] [PubMed] [Google Scholar]
- 34.Mumm JP, Landy A, Gelles J. Viewing single lambda site-specific recombination events from start to finish. EMBO J. 2006;25:4586–4595. doi: 10.1038/sj.emboj.7601325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nunes-Düby S, Matsumoto L, Landy A. Site-specific recombination intermediates trapped with suicide substrates. Cell. 1987;50:779–788. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- 36.Tong W, Warren D, Seah N, Laxmikanthan G, Van Duyne GD, Landy A. Mapping the Int bridges in the nucleoprotein Holliday junction intermediates of viral integrative and excisive recombination. Proc Natl Acad Sci USA. 2014;111:12366–12371. doi: 10.1073/pnas.1413007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seah N, Tong W, Warren D, Van Duyne GD, Landy A. Nucleoprotein architectures regulating the directionality of viral integration and excision. Proc Natl Acad Sci USA. 2014;111:12372–12377. doi: 10.1073/pnas.1413019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matovina M, Seah N, Hamilton T, Warren D, Landy A. Stoichiometric incorporation of base substitutions at specific sites in supercoiled DNA and supercoiled recombination intermediates. Nucleic Acids Res. 2010;38:e175. doi: 10.1093/nar/gkq674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun KQ. PhD Doctoral Thesis in Biochemistry. University of Illinois at Urbana-Champaign; Champaign, IL: 1990. A study of DNA-DNA interactions during bacteriophage lambda integrative recombination. [Google Scholar]
- 40.Conway AB, Chen Y, Rice PA. Structural plasticity of the Flp-Holliday junction complex. J Mol Biol. 2003;326:425–434. doi: 10.1016/s0022-2836(02)01370-0. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell. 2000;6:885–897. [PubMed] [Google Scholar]
- 42.Gopaul DN, Guo F, Van Duyne GD. Structure of the Holliday junction intermediate in Cre- loxP site-specific recombination. EMBO J. 1998;17:4175–4187. doi: 10.1093/emboj/17.14.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo F, Gopaul DN, Van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 44.Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by λ integrase. Nature. 2005;435:1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aihara H, Kwon HJ, Nunes-Düby SE, Landy A, Ellenberger T. A conformational switch controls the DNA cleavage activity of Lambda integrase. Mol Cell. 2003;12:187–198. doi: 10.1016/s1097-2765(03)00268-5. [DOI] [PubMed] [Google Scholar]
- 46.Tirumalai RS, Pargellis CA, Landy A. Identification and characterization of the NEM-sensitive site in lambda integrase. J Biol Chem. 1996;271:29599–29604. doi: 10.1074/jbc.271.47.29599. [DOI] [PubMed] [Google Scholar]
- 47.Pargellis CA, Nunes-Düby SE, Moitoso de Vargas L, Landy A. Suicide recombination substrates yield covalent λ integrase- DNA complexes and lead to identification of the active site tyrosine. J Biol Chem. 1988;263:7678–7685. [PubMed] [Google Scholar]
- 48.Tirumalai RS, Healey E, Landy A. The catalytic domain of λ site-specific recombinase. Proc Natl Acad Sci USA. 1997;94:6104–6109. doi: 10.1073/pnas.94.12.6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarkar D, Radman-Livaja M, Landy A. The small DNA binding domain of λ Int is a context-sensitive modulator of recombinase functions. EMBO J. 2001;20:1203–1212. doi: 10.1093/emboj/20.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SY, Aihara H, Ellenberger T, Landy A. Two structural features of integrase that are critical for DNA cleavage by multimers but not by monomers. Proc Natl Acad Sci USA. 2004;101:2770–2775. doi: 10.1073/pnas.0400135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cassell GD, Segall AM. Mechanism of inhibition of site-specific recombination by the Holliday junction-trapping peptide WKHYNY: Insights into phage λ integrase-mediated strand exchnage. J Mol Biol. 2003;327:413–429. doi: 10.1016/s0022-2836(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 52.Cassell G, Klemm M, Pinilla C, Segall A. Dissection of bacteriophage λ site-specific recombination using synthetic peptide combinatorial libraries. J Mol Biol. 2000;299:1193–1202. doi: 10.1006/jmbi.2000.3828. [DOI] [PubMed] [Google Scholar]
- 53.Boldt JL, Pinilla C, Segall AM. Reversible inhibitors of λ Int-mediated recombination efficiently trap Holliday junction intermediates and form the basis of a novel assay for junction resolution. J Biol Chem. 2004;279:3472–3483. doi: 10.1074/jbc.M309361200. [DOI] [PubMed] [Google Scholar]
- 54.Ghosh K, Lau CK, Guo F, Segall AM, Van Duyne GD. Peptide Trapping of the Holliday Junction Intermediate in Cre-loxP Site-specific Recombination. J Biol Chem. 2005;280:8290–8299. doi: 10.1074/jbc.M411668200. [DOI] [PubMed] [Google Scholar]
- 55.Segall AM, Nash HA. Architectural flexibility in lambda site-specific recombination: three alternate conformations channel the att L site into three distinct pathways. Genes Cells. 1996;1:453–463. doi: 10.1046/j.1365-2443.1996.d01-254.x. [DOI] [PubMed] [Google Scholar]
- 56.Segall AM, Goodman SD, Nash HA. Architectural elements in nucleoprotein complexes:interchangeability of specific and non-specific DNA binding proteins. EMBO J. 1994;13:4536–4548. doi: 10.1002/j.1460-2075.1994.tb06775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boldt JL, Kepple KV, Cassell GD, Segall AM. Spermidine biases the resolution of Holliday junctions by phage λ integrase. Nucl Acids Res. 2006;35:716–727. doi: 10.1093/nar/gkl1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajeev L, Segall A, Gardner J. The bacteroides NBU1 integrase performs a homology-independent strand exchange to form a Holliday junction intermediate. J Biol Chem. 2007;282:31228–31237. doi: 10.1074/jbc.M705370200. [DOI] [PubMed] [Google Scholar]
- 59.Klemm M, Cheng C, Cassell G, Shuman S, Segall AM. Peptide inhibitors of DNA cleavage by tyrosine recombinases and topoisomerases. J Mol Biol. 2000;299:1203–1216. doi: 10.1006/jmbi.2000.3829. [DOI] [PubMed] [Google Scholar]
- 60.Gunderson CW, Boldt JL, Authement RN, Segall AM. Peptide wrwycr Inhibits the Excision of Several Prophages and Traps Holliday Junctions inside Bacteria. J Bacteriol. 2009;191:2169–2176. doi: 10.1128/JB.01559-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ranjit DK, Rideout MC, Nefzi A, Ostresh JM, Pinilla C, Segall AM. Small molecule functional analogs of peptides that inhibit λ site-specific recombination and bind Holliday junctions. Bioorg Med Chem Lett. 2010;20:4531–4534. doi: 10.1016/j.bmcl.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bankhead TM, Etzel BJ, Wolven F, Bordenave S, Boldt JL, Larsen TA, Segall AM. Mutations at residues 282, 286, and 293 of phage λ integrase exert pathway-specific effects on synapsis and catalysis in recombination. J Bacteriol. 2003;185:2653–2666. doi: 10.1128/JB.185.8.2653-2666.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Y, Rice PA. New insight into site-specific recombination from Flp recombinase-DNA structures. Annu Rev Biophys Biomol Struct. 2003;32:135–139. doi: 10.1146/annurev.biophys.32.110601.141732. [DOI] [PubMed] [Google Scholar]
- 64.Tal A, Arbel-Goren R, Costantino N, Court DL, Stavans J. Location of the unique integration site on an Escherichia coli chromosome by bacteriophage lambda DNA in vivo. Proc Natl Acad Sci USA. 2014;111:7308–7312. doi: 10.1073/pnas.1324066111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kwon HJ, Tirumalai RS, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the λ integrase catalytic core. Science. 1997;276:126–131. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarkar D, Azaro MA, Aihara H, Papagiannis C, Tirumalai RS, Nunes-Düby SE, Johnson RC, Ellenberger T, Landy A. Differential affinity and cooperativity functions of the amino-terminal 70 residues of λ integrase. J Mol Biol. 2002;324:775–789. doi: 10.1016/s0022-2836(02)01199-3. [DOI] [PubMed] [Google Scholar]
- 67.Azaro MA, Landy A. The isomeric preference of Holliday junctions influences resolution bias by λ integrase. EMBO J. 1997;16:3744–3755. doi: 10.1093/emboj/16.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamadurai HB, Jain R, Foster MP. Crystallization and structure determination of the core-binding domain of bacteriophage lambda integrase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:470–473. doi: 10.1107/S174430910801381X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kovach MJ, Tirumalai RS, Landy A. Site-specific photo-crosslinking of lambda Int. J Biol Chem. 2002;277:14530–14538. doi: 10.1074/jbc.M108197200. [DOI] [PubMed] [Google Scholar]
- 70.Tirumalai RS, Kwon H, Cardente E, Ellenberger T, Landy A. The recognition of core-type DNA sites by λ Integrase. J Mol Biol. 1998;279:513–527. doi: 10.1006/jmbi.1998.1786. [DOI] [PubMed] [Google Scholar]
- 71.Ross W, Landy A. Patterns of λ Int recognition in the regions of strand exchange. Cell. 1983;33:261–272. doi: 10.1016/0092-8674(83)90355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheng Q, Swalla BM, Beck M, Alcaraz R, Jr, Gumport RI, Gardner JF. Specificity determinants for bacteriophage Hong Kong 022 integrase: analysis of mutants with relaxed core-binding specificities. Mol Microbiol. 2000;36:424–436. doi: 10.1046/j.1365-2958.2000.01860.x. [DOI] [PubMed] [Google Scholar]
- 73.Dorgai L, Yagil E, Weisberg RA. Identifying determinants of recombination specificity: Construction and characterization of mutant bacteriophage integrases. J Mol Biol. 1995;252:178–188. doi: 10.1006/jmbi.1995.0486. [DOI] [PubMed] [Google Scholar]
- 74.Yagil E, Dorgai L, Weisberg R. Identifying determinants of recombination specificity: Construction and characterization of chimeric bacteriophage integrases. J Mol Biol. 1995;252:163–177. doi: 10.1006/jmbi.1995.0485. [DOI] [PubMed] [Google Scholar]
- 75.Patsey RL, Bruist MF. Characterization of the interaction between the lambda intasome and att B. J Mol Biol. 1995;252:47–58. doi: 10.1006/jmbi.1995.0474. [DOI] [PubMed] [Google Scholar]
- 76.Lange-Gustafson BJ, Nash HA. Purification and properties of Int-H, a variant protein involved in site-specific recombination of bacteriophage λ. J Biol Chem. 1984;259:12724–12732. [PubMed] [Google Scholar]
- 77.Han YW, Gumport RI, Gardner JF. Mapping the functional domains of bacteriophage lambda integrase protein. J Mol Biol. 1994;235:908–925. doi: 10.1006/jmbi.1994.1048. [DOI] [PubMed] [Google Scholar]
- 78.Tekle M, Warren DJ, Biswas T, Ellenberger T, Landy A, Nunes-Düby SE. Attenuating functions of the C-terminus of λ Integrase. J Mol Biol. 2002;324:649–665. doi: 10.1016/s0022-2836(02)01108-7. [DOI] [PubMed] [Google Scholar]
- 79.Kazmierczak RA, Swalla BM, Burgin AB, Gumport RI, Gardner JF. Regulation of site-specific recombination by the carboxyl terminus of λ integrase. Nucleic Acids Res. 2002;30:5193–5204. doi: 10.1093/nar/gkf652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hazelbaker D, Radman-Livaja M, Landy A. Receipt of the C-terminal tail from a neighboring Int protomer allosterically stimulates Holliday junction resolution. J Mol Biol. 2005;351:948–955. doi: 10.1016/j.jmb.2005.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rowley PA, Kachroo AH, Ma C-H, Maciaszek AD, Guga P, Jayaram M. Electrostatic suppression allows tyrosine site-specific recombination in the absence of a conserved catalytic arginine. J Biol Chem. 2010;285:22976–22985. doi: 10.1074/jbc.M110.112292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grainge I, Jayaram M. The integrase family of recombinases: organization and function of the active site. Mol Microbiol. 1999;33:449–456. doi: 10.1046/j.1365-2958.1999.01493.x. [DOI] [PubMed] [Google Scholar]
- 83.Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol Cell. 2000;5:1035–1041. doi: 10.1016/s1097-2765(00)80268-3. [DOI] [PubMed] [Google Scholar]
- 84.Krogh BO, Shuman S. Proton relay mechanism of general acid catalysis by DNA topoisomerase IB. J Biol Chem. 2002;277:5711–5714. doi: 10.1074/jbc.C100681200. [DOI] [PubMed] [Google Scholar]
- 85.Yakovleva L, Lai J, Kool ET, Shuman S. Nonpolar nucleobase analogs illuminate requirements for site-specific DNA cleavage by vaccinia topoisomerase. J Biol Chem. 2006;281:35914–35921. doi: 10.1074/jbc.M608349200. [DOI] [PubMed] [Google Scholar]
- 86.Van Duyne GD. A structural view of Cre-loxP site-specific recombination. Annu Rev Biophys Biomol Struct. 2001;30:87–104. doi: 10.1146/annurev.biophys.30.1.87. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y, Rice PA. The role of the conserved Trp 330 in Flp-mediated recombination. Functional and structural analysis. J Biol Chem. 2003;278:24800–24807. doi: 10.1074/jbc.M300853200. [DOI] [PubMed] [Google Scholar]
- 88.Whiteson KL, Rice PA. Binding and catalytic contributions to site recognition by flp recombinase. J Biol Chem. 2008;283:11414–11423. doi: 10.1074/jbc.M800106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Warren D, Laxmikanthan G, Landy A. A chimeric Cre recombinase with regulated directionality. Proc Natl Acad Sci USA. 2008;47:18278–18283. doi: 10.1073/pnas.0809949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hartung M, Kisters-Woike B. Cre mutants with altered DNA binding properties. J Biol Chem. 1998;273:22884–22891. doi: 10.1074/jbc.273.36.22884. [DOI] [PubMed] [Google Scholar]
- 91.Richet E, Abcarian P, Nash HA. Synapsis of attachment sites during lambda integrative recombination involves capture of a naked DNA by a protein-DNA complex. Cell. 1988;52:9–17. doi: 10.1016/0092-8674(88)90526-0. [DOI] [PubMed] [Google Scholar]
- 92.Wojciak JM, Sarkar D, Landy A, Clubb RT. Arm-site binding by the lambda integrase protein: solution structure and functional characterization of its amino-terminal domain. Proc Natl Acad Sci USA. 2002;99:3434–3439. doi: 10.1073/pnas.052017999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson JF, Landy A. Empirical estimation of protein-induced DNA bending angles: Applications to λ site-specific recombination complexes. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Enquist LW, Kikuchi A, Weisberg RA. The role of λ integrase in integration and excision. Cold Spring Harb Symp Quant Biol. 1979;43:1115–1120. doi: 10.1101/sqb.1979.043.01.124. [DOI] [PubMed] [Google Scholar]
- 95.Fadeev EA, Sam MD, Clubb RT. NMR structure of the amino-terminal domain of the lambda integrase protein in complex with DNA: immobilization of a flexible tail facilitates beta-sheet recognition of the major groove. J Mol Biol. 2009;388:682–690. doi: 10.1016/j.jmb.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wojciak JM, Connolly KM, Clubb RT. NMR structure of the Tn 916 integrase-DNA complex. Nat Struct Biol. 1999;6:366–373. doi: 10.1038/7603. [DOI] [PubMed] [Google Scholar]
- 97.Allen MD, Yamasaki K, Ohme-Takagi M, Tateno M, Suzuki M. A novel mode of DNA recognition by a beta-sheet revealed by the solution structure of the GCC-box binding domain in complex with DNA. EMBO J. 1998;17:5484–5496. doi: 10.1093/emboj/17.18.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Subramanya HS, Arciszewska LK, Baker RA, Bird LE, Sherratt DJ, Wigley DB. Crystal structure of the site-specific recombinase, XerD. EMBO J. 1997;16:5178–5187. doi: 10.1093/emboj/16.17.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nunes-Düby SE, Yu D, Landy A. Sensing homology at the strand swapping step in λ excisive recombination. J Mol Biol. 1997;272:493–508. doi: 10.1006/jmbi.1997.1260. [DOI] [PubMed] [Google Scholar]
- 100.Lee SY, Radman-Livaja M, Warren D, Aihara H, Ellenberger T, Landy A. Non-equivalent interactions between amino-terminal domains of neighboring λ integrase protomers direct Holliday junction resolution. J Mol Biol. 2005;345:475–485. doi: 10.1016/j.jmb.2004.10.068. [DOI] [PubMed] [Google Scholar]
- 101.Radman-Livaja M, Biswas T, Mierke D, Landy A. Architecture of recombination intermediates visualized by In-gel FRET of λ integrase-Holliday junction-arm-DNA complexes. Proc Natl Acad Sci USA. 2005;102:3913–3920. doi: 10.1073/pnas.0500844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miller HI, Kikuchi A, Nash HA, Weisberg RA, Friedman DI. Site-specific recombination of bacteriophage λ: the role of host gene products. Cold Spring Harb Symp Quant Biol. 1979;43:1121–1126. doi: 10.1101/sqb.1979.043.01.125. [DOI] [PubMed] [Google Scholar]
- 103.Miller HI, Kirk M, Echols H. SOS induction and auto-regulation of the him A gene for site-specific recombination in Escherichia coli. Proc Natl Acad Sci USA. 1981;78:6754–6758. doi: 10.1073/pnas.78.11.6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moitoso de Vargas L, Kim S, Landy A. DNA looping generated by the DNA-bending protein IHF and the two domains of lambda integrase. Science. 1989;244:1457–1461. doi: 10.1126/science.2544029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sze CC, Laurie AD, Shingler V. In vivo and in vitro effects of integration host factor at the DmpR-regulated σ 54 -dependent Po promoter. J Bacteriol. 2001;183:2842–2851. doi: 10.1128/JB.183.9.2842-2851.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ryan VT, Grimwade JE, Camara JE, Crooke E, Leonard AC. Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol Microbiol. 2004;51:1347–1359. doi: 10.1046/j.1365-2958.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- 107.Crellin P, Sewitz S, Chalmers R. DNA looping and catalysis: The IHF-folded Arm of Tn 10 promotes conformational changes and hairpin resolution. Mol Cell. 2004;13:537–547. doi: 10.1016/s1097-2765(04)00052-8. [DOI] [PubMed] [Google Scholar]
- 108.Morse BK, Michalczyk R, Kosturko LD. Multiple molecules of integration host factor (IHF) at a single DNA binding site, the bacteriophage lambda cos 11 site. Biochimie. 1994;76:1005–1017. doi: 10.1016/0300-9084(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 109.Friedman DI. Integration host factor: a protein for all reasons. Cell. 1988;55:545–554. doi: 10.1016/0092-8674(88)90213-9. [DOI] [PubMed] [Google Scholar]
- 110.Goosen N, van de Putte P. The regulation of transcription initiation by integration host factor. Mol Microbiol. 1995;16:1–7. doi: 10.1111/j.1365-2958.1995.tb02386.x. [DOI] [PubMed] [Google Scholar]
- 111.Nash HA, Lin ECC, Lynch AS. The HU and IHF proteins: accessory factors for complex protein-DNA assemblies. In: Lin ECC, Lynch AS, editors. Regulation of Gene Expression in Escherichia coli. R.G. Landes Company; Austin, TX: 1996. pp. 150–179. [Google Scholar]
- 112.Johnson RC, Johnson LM, Schmidt JW, Gardner JF, Higgins NP. The Bacterial Chromosome. ASM Press; Washington DC: 2005. Major nucleoid proteins in the structure and function of the Escherichia coli chromosome; pp. 65–132. [Google Scholar]
- 113.Dame RT. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol Microbiol. 2005;56:858–870. doi: 10.1111/j.1365-2958.2005.04598.x. [DOI] [PubMed] [Google Scholar]
- 114.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 115.Travers A. DNA-protein interactions: IHF-the master blender. Curr Biol. 1997;7:R252–R254. doi: 10.1016/s0960-9822(06)00114-x. [DOI] [PubMed] [Google Scholar]
- 116.Rice PA. Making DNA do a U-turn: IHF and related proteins. Curr Opin Struct Biol. 1997;7:86–93. doi: 10.1016/s0959-440x(97)80011-5. [DOI] [PubMed] [Google Scholar]
- 117.Lynch TW, Read EK, Mattis AN, Gardner JF, Rice PA. Integration host factor: putting a twist on protein-DNA recognition. J Mol Biol. 2003;330:493–502. doi: 10.1016/s0022-2836(03)00529-1. [DOI] [PubMed] [Google Scholar]
- 118.Swinger KK, Lemberg KM, Zhang Y, Rice PA. Flexible DNA bending in HU-DNA cocrystal structures. EMBO J. 2003;22:3749–3760. doi: 10.1093/emboj/cdg351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vander Meulen KA, Saecker RM, Record MT., Jr Formation of a wrapped DNA-protein interface: experimental characterization and analysis of the large contributions of ions and water to the thermodynamics of binding IHF to H′ DNA. J Mol Biol. 2008;377:9–27. doi: 10.1016/j.jmb.2007.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dixit S, Singh-Zocchi M, Hanne J, Zocchi G. Mechanics of binding of a single integration-host-factor protein to DNA. Phy Rev Ltrs. 2005;94:118101–118104. doi: 10.1103/PhysRevLett.94.118101. [DOI] [PubMed] [Google Scholar]
- 121.Sugimura S, Crothers DM. Stepwise binding and bending of DNA by Escherichia coli integration host factor. Proc Natl Acad Sci USA. 2006;103:18510–18514. doi: 10.1073/pnas.0608337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kuznetsov SV, Sugimura S, Vivas P, Crothers DM, Ansarai A. Direct observation of DNA bending/unbending kinetics in complex with DNA-bending protein IHF. Proc Natl Acad Sci USA. 2006;103:18515–18520. doi: 10.1073/pnas.0608394103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sugimura S. Kinetic and steady-state studies of binding and bending of Lambda phage DNA by integration host factor. Ph.D. Yale University; New Haven, CT: 2005. [Google Scholar]
- 124.Corona T, Bao Q, Christ N, Schwartz T, Li J, Droge P. Activation of site-specific DNA integration in human cells by a single chain integration host factor. Nucleic Acids Res. 2003;31:5140–5148. doi: 10.1093/nar/gkg711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goodman SD, Nicholson SC, Nash HA. Deformation of DNA during site-specific recombination of bacteriophage λ: replacement of IHF protein by HU protein or sequence-directed bends. Proc Natl Acad Sci USA. 1992;89:11910–11914. doi: 10.1073/pnas.89.24.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goodman SD, Nash HA. Functional replacement of a protein-induced bend in a DNA recombination site. Nature. 1989;341:251–254. doi: 10.1038/341251a0. [DOI] [PubMed] [Google Scholar]
- 127.Nunes-Düby SE, Smith-Mungo LI, Landy A. Single base-pair precision and structural rigidity in a small IHF-induced DNA loop. J Mol Biol. 1995;253:228–242. doi: 10.1006/jmbi.1995.0548. [DOI] [PubMed] [Google Scholar]
- 128.Ditto MD, Roberts D, Weisberg RA. Growth phase variation of integration host factor level in Escherichia coli. J Bacteriol. 1994;176:3738–3748. doi: 10.1128/jb.176.12.3738-3748.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murtin C, Engelhorn M, Geiselmann J, Boccard F. A quantitative UV laser footprinting analysis of the interaction of IHF with specific binding sites: Re-evaluation of the effective concentration of IHF in the Cell. J Mol Biol. 1998;284:949–961. doi: 10.1006/jmbi.1998.2256. [DOI] [PubMed] [Google Scholar]
- 130.Bushman W, Thompson JF, Vargas L, Landy A. Control of directionality in lambda site-specific recombination. Science. 1985;230:906–911. doi: 10.1126/science.2932798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thompson JF, Moitoso de Vargas L, Skinner SE, Landy A. Protein–protein interactions in a higher-order structure direct lambda site-specific recombination. J Mol Biol. 1987;195:481–493. doi: 10.1016/0022-2836(87)90177-x. [DOI] [PubMed] [Google Scholar]
- 132.Granston AE, Nash HA. Characterization of a set of integration host factor mutants deficient for DNA binding. J Mol Biol. 1993;234:45–59. doi: 10.1006/jmbi.1993.1562. [DOI] [PubMed] [Google Scholar]
- 133.Nash HA, Robertson CA, Flamm E, Weisberg RA, Miller HI. Overproduction of Escherichia coli integration host factor, a protein with nonidentical subunits. J Bacteriol. 1987;169:4124–4127. doi: 10.1128/jb.169.9.4124-4127.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aviv M, Giladi H, Schreiber G, Oppenheim AB, Glaser G. Expression of the genes coding for the Escherichia coli integration host factor are controlled by growth phase, rpoS, ppGpp and by auto-regulation. Mol Microbiol. 1994;14:1021–1031. doi: 10.1111/j.1365-2958.1994.tb01336.x. [DOI] [PubMed] [Google Scholar]
- 135.Nystrom T. Glucose starvation stimulon of Escherichia coli: Role of integration host factor in starvation survival and growth phase-dependent protein synthesis. J Bacteriol. 1995;177:5707–5710. doi: 10.1128/jb.177.19.5707-5710.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abremski K, Gottesman S. Purification of the bacteriophage λ xis gene product required for λ excisive recombination. J Biol Chem. 1982;257:9658–9662. [PubMed] [Google Scholar]
- 137.Nash HA. Integrative recombination of bacteriophage lambda DNA in vitro. Proc Natl Acad Sci USA. 1975;72:1072–1076. doi: 10.1073/pnas.72.3.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sam M, Papagiannis C, Connolly KM, Corselli L, Iwahara J, Lee J, Phillips M, Wojciak JM, Johnson RC, Clubb RT. Regulation of directionality in bacteriophage lambda site-specific recombination: structure of the Xis protein. J Mol Biol. 2002;324:791–805. doi: 10.1016/s0022-2836(02)01150-6. [DOI] [PubMed] [Google Scholar]
- 139.Sam MD, Cascio D, Johnson RC, Clubb RT. Crystal structure of the excisionase-DNA complex from bacteriophage lambda. J Mol Biol. 2004;338:229–240. doi: 10.1016/j.jmb.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 140.Abbani MA, Papagiannis CV, Sam MD, Cascio D, Johnson RC, Clubb RT. Structure of the cooperative excisionase (Xis)-DNA complex reveals a micronucleoprotein filament that regulates phage lambda intasome assembly. Proc Natl Acad Sci USA. 2007;104:2109–2114. doi: 10.1073/pnas.0607820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sun X, Mierke DF, Biswas T, Lee SY, Landy A, Radman-Livaja M. Architecture of the 99 bp DNA-Six-Protein regulatory complex of the λ att Site. Molecular Cell. 2006;24:569–580. doi: 10.1016/j.molcel.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thompson JF, Moitoso de Vargas L, Koch C, Kahmann R, Landy A. Cellular factors couple recombination with growth phase: characterization of a new component in the λ site-specific recombination pathway. Cell. 1987;50:901–908. doi: 10.1016/0092-8674(87)90516-2. [DOI] [PubMed] [Google Scholar]
- 143.Finkel SE, Johnson RC. The FIS protein: it’s not just for DNA inversion anymore. Mol Microbiol. 1992;6:3257–3265. doi: 10.1111/j.1365-2958.1992.tb02193.x. [DOI] [PubMed] [Google Scholar]
- 144.Aiyar SE, McLeod SM, Ross W, Hirvonen CA, Thomas MS, Johnson RC, Gourse RL. Architecture of Fis-activated transcription complexes at the Escherichia coli rrnB P1 and rrnE P1 promoters. J Mol Biol. 2002;316:501–516. doi: 10.1006/jmbi.2001.5390. [DOI] [PubMed] [Google Scholar]
- 145.Hancock SP, Ghane T, Cascio D, Rohs R, Di Felice R, Johnson RC. Control of DNA minor groove width and Fis protein binding by the purine 2-amino group. Nucleic Acids Res. 2013;41:6750–6760. doi: 10.1093/nar/gkt357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Johnson RC, Bruist MF, Simon MI. Host protein requirements for in vitro site-specific DNA inversion. Cell. 1986;46:531–539. doi: 10.1016/0092-8674(86)90878-0. [DOI] [PubMed] [Google Scholar]
- 147.Koch C, Kahmann R. Purification and properties of the Escherichia coli host factor required for inversion of the G segment in bacteriophage Mu. J Biol Chem. 1986;261:15673–15678. [PubMed] [Google Scholar]
- 148.Ball CA, Johnson RC. Efficient excision of phage λ from the Escherichia coli chromosome requires the Fis protein. J Bacteriol. 1991;173:4027–4031. doi: 10.1128/jb.173.13.4027-4031.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Numrych TE, Gumport RI, Gardner JF. A genetic analysis of Xis and FIS interactions with their binding sites in bacteriophage lambda. J Bacteriol. 1991;173:5954–5963. doi: 10.1128/jb.173.19.5954-5963.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Skoko D, Yoo D, Bai H, Schnurr B, Yan J, McLeod SM, Marko JF, Johnson RC. Mechanism of chromosome compaction and looping by the Escherichia coli nucleoid protein Fis. J Mol Biol. 2006;364:777–798. doi: 10.1016/j.jmb.2006.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schneider R, Travers A, Kutateladze T, Muskhelishvili G. A DNA architectural protein couples cellular physiology and DNA topology in Escherichia coli. Mol Microbiol. 1999;34:953–964. doi: 10.1046/j.1365-2958.1999.01656.x. [DOI] [PubMed] [Google Scholar]
- 152.Dorman CJ. Nucleoid-associated proteins and bacterial physiology. In: Laskin A, Sariaslani S, Geoffrey G, editors. Advances in Applied Microbiology. Vol. 67. Academic Press; New York, NY: 2009. pp. 47–64. [DOI] [PubMed] [Google Scholar]
- 153.Browning DF, Grainger DC, Busby SJW. Effects of nucleoid-associated proteins on bacterial chromosome structure and gene expression. Curr Opin Microbiol. 2010;13:773–780. doi: 10.1016/j.mib.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 154.Ball CA, Osuna R, Ferguson KC, Johnson RC. Dramatic changes in Fis levels upon nutrient upshift in Escherichia coli. J Bacteriol. 1992;174:8043–8056. doi: 10.1128/jb.174.24.8043-8056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ninnemann O, Koch C, Kahmann R. The E. coli fis promoter is subject to stringent control and autoregulation. EMBO J. 1992;11:1075–1083. doi: 10.1002/j.1460-2075.1992.tb05146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nilsson L, Verbeek H, Vijgenboom E, van Drunen C, Vanet A, Bosch L. FIS-dependent trans activation of stable RNA operons of Escherichia coli under various growth conditions. J Bacteriol. 1992;174:921–929. doi: 10.1128/jb.174.3.921-929.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pratt TS, Steiner T, Feldman LS, Walker KA, Osuna R. Deletion analysis of the fis promoter region in Escherichia coli: Antagonistic effects of integration host factor and Fis. J Bacteriol. 1997;179:6367–6377. doi: 10.1128/jb.179.20.6367-6377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mallik P, Paul BJ, Rutherford ST, Gourse RL, Osuna R. DksA is required for growth phase-dependent regulation, growth rate-dependent control, and stringent control of fis expression in Escherichia coli. J Bacteriol. 2006;188:5775–5782. doi: 10.1128/JB.00276-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Paul BJ, Ross W, Gaal T, Gourse RL. rRNA transcription in Escherichia coli. Annu Rev Genet. 2004;38:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- 160.Walker KA, Mallik P, Pratt TS, Osuna R. The Escherichia coli fis promoter is regulated by changes in the levels of its transcription initiation nucleotide CTP. J Biol Chem. 2004;279:50818–50828. doi: 10.1074/jbc.M406285200. [DOI] [PubMed] [Google Scholar]
- 161.Cassell G, Moision R, Rabani E, Segall A. The geometry of a synaptic intermediate in a pathway of bacteriophage λ site-specific recombination. Nucleic Acids Res. 1999;27:1145–1151. doi: 10.1093/nar/27.4.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Segall AM. Analysis of higher order intermediates and synapsis in the bent-L pathway of bacteriophage λ site-specific recombination. J Biol Chem. 1998;273:24258–24265. doi: 10.1074/jbc.273.37.24258. [DOI] [PubMed] [Google Scholar]
- 163.Segall AM, Nash HA. Synaptic intermediates in bacteriophage lambda site-specific recombination: Integrase can align pairs of attachment sites. EMBO J. 1993;12:4567–4576. doi: 10.1002/j.1460-2075.1993.tb06145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hazelbaker D, Azaro MA, Landy A. A biotin interference assay highlights two different asymmetric interaction profiles for lambda integrase arm-type binding sites in integrative versus excisive recombination. J Biol Chem. 2008;283:12402–12414. doi: 10.1074/jbc.M800544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huang H, Harrison SC, Verdine GL. Trapping of a catalytic HIV reverse transcriptase template:primer complex through a disulfide bond. Chem Biol. 2000;7:355–364. doi: 10.1016/s1074-5521(00)00113-7. [DOI] [PubMed] [Google Scholar]
- 166.Verdine GL, Norman DP. Covalent trapping of protein-DNA complexes. Annu Rev Biochem. 2003;72:337–366. doi: 10.1146/annurev.biochem.72.121801.161447. [DOI] [PubMed] [Google Scholar]
- 167.Lorenz M, Diekmann S. Quantitative distance information on protein-DNA complexes determined in polyacrylamide gels by fluorescence resonance energy transfer. Electrophoresis. 2001;22:990–998. doi: 10.1002/1522-2683()22:6<990::AID-ELPS990>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 168.Ramirez-Carrozzi VR, Kerppola TK. Long-range electrostatic interactions influence the orientation of Fos-Jun Binding at AP-1 sites. J Mol Biol. 2001;305:411–427. doi: 10.1006/jmbi.2000.4286. [DOI] [PubMed] [Google Scholar]
- 169.Lilley DMJ, Wilson TJ. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr Opin Chem Biol. 2000;4:507–517. doi: 10.1016/s1367-5931(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 170.Crisona NJ, Weinberg RL, Peter BJ, Sumners DW, Cozzarelli NR. The topological mechanism of phage λ integrase. J Mol Biol. 1999;289:747–775. doi: 10.1006/jmbi.1999.2771. [DOI] [PubMed] [Google Scholar]
- 171.Pollock TJ, Nash HA. Knotting of DNA caused by a genetic rearrangement: evidence for a nucleosome-like structure in site-specific recombination for bacteriophage lambda. J Mol Biol. 1983;170:1–18. doi: 10.1016/s0022-2836(83)80224-1. [DOI] [PubMed] [Google Scholar]
- 172.Nash HA, Pollock TJ. Site-specific recombination of bacteriophage lambda: The change in topological linking number associated with exchange of DNA strands. J Mol Biol. 1983;170:19–38. doi: 10.1016/s0022-2836(83)80225-3. [DOI] [PubMed] [Google Scholar]
- 173.Gottesman S, Gottesman M. Excision of prophage λ in a cell-free system. Proc Natl Acad Sci USA. 1975;72:2188–2192. doi: 10.1073/pnas.72.6.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Numrych TE, Gumport RI, Gardner JF. Characterization of the bacteriophage lambda excisionase (Xis) protein: the C-terminus is required for Xis-integrase cooperativity but not for DNA binding. EMBO J. 1992;11:3797–3806. doi: 10.1002/j.1460-2075.1992.tb05465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Craig NL, Craigie R, Gellert M, Lambowitz AM. Mobile DNA II. ASM Press; Washington DC: 2002. [Google Scholar]
- 176.King JK. Man the misinterpretant: will he discover the universal secret of sexuality encoded within him? Int J Humanit. 2007;4:1–19. [Google Scholar]
- 177.Craig NL, Nash HA. E. coli integration host factor binds to specific sites in DNA. Cell. 1984;39:707–716. doi: 10.1016/0092-8674(84)90478-1. [DOI] [PubMed] [Google Scholar]
- 178.Nunes-Düby S, Azaro M, Landy A. Swapping DNA strands and sensing homology without branch migration in λ site-specific recombination. Curr Biol. 1995;5:139–148. doi: 10.1016/s0960-9822(95)00035-2. [DOI] [PubMed] [Google Scholar]
- 179.Warren D, Sam M, Manley K, Sarkar D, Lee SY, Abbani M, Clubb RT, Landy A. Identification of the λ integrase surface that interacts with the Xis accessory protein reveals a residue that is also critical for homomeric dimer formation. Proc Natl Acad Sci USA. 2003;100:8176–8181. doi: 10.1073/pnas.1033041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Esposito D, Gerard GF. The Escherichia coli Fis protein stimulates bacteriophage λ integrative recombination in vitro. J Bacteriol. 2003;185:3076–3080. doi: 10.1128/JB.185.10.3076-3080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ball CA, Johnson RC. Multiple effects of Fis on integration and the control of lysogeny in phage λ. J Bacteriol. 1991;173:4032–4038. doi: 10.1128/jb.173.13.4032-4038.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]