

Summary
Naturally stalled replication forks are considered to cause structurally abnormal chromosomes in tumor cells. However, underlying mechanisms remain speculative, as capturing naturally stalled forks has been a challenge. Here, we captured naturally stalled forks in tumor cells and delineated molecular processes underlying the structural evolution of circular mini-chromosomes (double minute chromosomes, DMs). Replication forks stalled on the DM by the co-directional collision with the transcription machinery for long non-coding RNA. RPA, BRCA2, and DNA polymerase eta (Polη), were recruited to the stalled forks. The recruitment of Polη was critical for replication to continue, as Polη knockdown resulted in DM loss. Rescued stalled forks were error-prone and switched replication templates repeatedly to create complex fusions of multiple short genomic segments. In mice, such complex fusions circularized the genomic region surrounding MYC to create a DM during tumorigenesis. Our results define a molecular path that guides stalled replication forks to complex chromosomal rearrangements.
Keywords: Stalled replication forks, Gross chromosomal rearrangements, Double minute chromosomes, DNA damage tolerance, Replication-transcription conflicts
eTOC Blurb
Watanabe et al. monitor naturally stalled forks in cancer cells and define molecular mechanisms underlying fork stalling and rescue. The authors find that DNA polymerase η plays a critical role in rescuing stalled forks. Rescued forks were unstable and switched replication templates several times, creating complex fusions of multiple genomic segments.
Introduction
Genome instability is an enabling characteristic by which tumor cells acquire unlimited proliferation and metastatic potential. Genome instability frequently occurs at the chromosomal level and produces aberrantly structured chromosomes, including small, circular chromosomes (double minute chromosomes, DMs) (Wahl, 1989). DMs can originate from oncogene loci and contribute to oncogene amplification and tumor heterogeneity (Turner et al., 2017). DMs, like other chromosomes, replicate in S-phase and segregate into daughter cells. Because DMs lack centromeres (Turner et al., 2017), segregation can be random, resulting in the accumulation of DMs in a subset of cells. Cells with multiple DMs overexpress oncogenes, gain growth advantage, and undergo clonal expansion. This vital role in tumor development underscores the importance of understanding how DMs arise and persist in tumor cells at the molecular level.
Several models have been proposed to explain how DMs arise. Results from cytogenetic studies in rodent cell models and tumor cells suggest that a chromosome break triggers the deletion of a chromosomal region that becomes a circular DNA (Storlazzi et al., 2006). This mechanism predicts that DMs have simple structures with continuous genomic regions. Recent genomic data for the DMs arising from EGFR-loci in glioblastomas may support this model (Brennan et al., 2013; Sanborn et al., 2013). Also, some of the DMs exhibit complex circular structures with multiple, discontinuous genomic segments from a single chromosome (Northcott et al., 2012; Rausch et al., 2012; Sanborn et al., 2013; Stephens et al., 2011). Chromothripsis, a process of fragmentation of a chromosome in micronuclei or in cells undergoing telomere crisis (Maciejowski et al., 2015; Zhang et al., 2015), could be involved in the formation of complex DMs (Figure S1). Alternatively, complex DMs could arise when a replication fork switches replication templates multiple times. Unstable fork movements can create fusions of multiple discontinuous genomic segments (Fork-Stalling and Template Switching) (Zhang et al., 2009). This model, however, also awaits experimental evidence but could be plausible given that premalignant and tumor cells continuously experience problems during replication and exhibit constitutively-active DNA damage responses (Bartkova et al., 2005; Gorgoulis et al., 2005). Furthermore, DMs frequently undergo secondary rearrangements and become structurally diverse within a tumor cell population (L’Abbate et al., 2014; Wahl, 1989). Whether replication-based mechanisms are responsible for the frequent secondary rearrangements remains to be determined.
When cells duplicate DNA, replication machinery travels for a long distance until they meet with forks coming from the opposite direction (converging forks). There are many natural obstacles on the road that can block fork movements: DNA secondary structures, tight DNA-protein binding complexes, and conflicts with transcription (Bermejo et al., 2012; Gaillard et al., 2015; Mirkin and Mirkin, 2007; Zeman and Cimprich, 2014). When converging forks are not available, replication remains incomplete, resulting in abortive replication and DNA loss. There are two ways to continue replication when replication forks stall (DNA damage tolerance, DDT) (Branzei and Foiani, 2010; Sale et al., 2012). Replication can proceed through the obstacles either by employing specialized DNA polymerases to directly synthesize DNA across the stalled lesions (translesion DNA synthesis) or by switching replication template and copy sister chromatids to bypass the lesion (legitimate template switching). Cooperation between checkpoint kinases and DNA repair proteins also help to maintain replication fork integrity at stalled forks (Kondratova et al., 2015; Schlacher et al., 2011). However, prolonged fork block can lead to the loss of fork integrity and collapsed forks with broken DNA ends (Petermann et al., 2010). In such a case, replication forks need to be re-established from broken DNA ends. In simple organisms S. pombe and S. cerevisiae, re-established replication can occur from single-strand breaks (SSBs) or double-strand breaks (DSBs) and depends on recombination proteins (RAD51) and a replicative DNA polymerase complex (Polδ) (Anand et al., 2013; Lambert et al., 2005; Lydeard et al., 2007). However, re-established replication can be error-prone and switch replication templates ectopically, resulting in rearrangements (Miyabe et al., 2015; Mizuno et al., 2013; Sakofsky et al., 2015). Whether re-established replication produces chromosomal rearrangements in tumor cells is not known.
Among natural obstacles, transcription machinery competes with replication machinery for the same DNA template in S phase and thus can be a common cause of stalled forks (Aguilera and Garcia-Muse, 2012; Gaillard et al., 2015). Replication-transcription conflicts constitute a new class of fragile sites in the mammalian genome (Barlow et al., 2013). Although both replication and transcription are tightly regulated temporally and spatially (Wei et al., 1998), collisions between two types of machinery do occur (Azvolinsky et al., 2009; Helmrich et al., 2011). Both head-on and co-directional collisions result in stalled replication forks. In addition, nascent RNA transcripts can block fork movements by forming stretches of RNA:DNA hybrids (R-loops) (Aguilera and Garcia-Muse, 2012; Bhatia et al., 2014; Hatchi et al., 2015; Huertas and Aguilera, 2003). R-loops can be processed by RNaseH1, a nuclease that degrades explicitly RNA of RNA: DNA hybrids.
Given the high loads of stalled/collapsed forks in tumor cells, re-established replication could be prevalent and be a common cause of structurally abnormal chromosomes, such as DMs. In this study, we sought for such evidence and molecular mechanisms by capturing naturally stalled forks.
Results
DNA rearrangements associated with the structural evolution of DMs
To obtain insight into the mechanisms underlying the structural evolution of DMs, we first characterized the very early rearrangement of DMs in the Colo320DM colon cancer cell line. In Colo320DM, the 1.6 Mb region at 8q24.1 was highly amplified, with MYC close to the telomeric end of the amplicon (Figure 1A, left). A small, 8.5 kb genomic segment that is located at 1.3 Mb telomeric side from the telomeric end of the amplicon was also amplified to an equal level (50-fold, Figure 1A, right). Fluorescence in situ hybridization (FISH) showed that the majority (63.4%) of DMs carried both MYC (red) and the small 8.5 kb segment (green) (Figure 1B and Figure S2A), indicating that the fusion between the two segments was a very early rearrangement. PCR-sequencing revealed microhomology (6 and 2 base pairs)-mediated fusions of the small segment (8.5 kb) into the 1.6 Mb circular chromosome; the centromeric side of the 8.5 kb segment (B) was fused to the centromeric side of the 1.6 Mb region (A), whereas the telomeric side of the 8.5 kb segment (C) was fused to the telomeric side of the 1.6 Mb region (D) (Figure 1C and Figure S2B). This organization could not arise by the initial circularization of the 2.9 Mb genomic segment followed by the deletion of the intervening 1.3 Mb segment, as such a process should have created the A–C fusion for the circularization and the B–D fusion for the following deletion (Figure S2B, right).
Figure 1. Stalled re[plication forks.
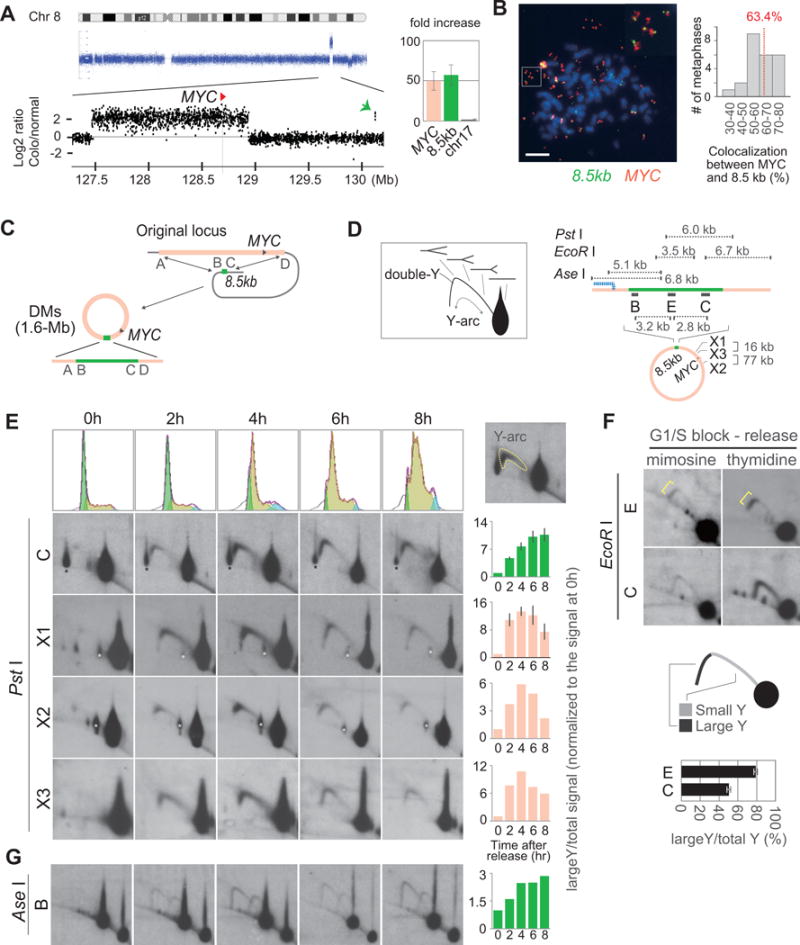
(A) Array comparative genomic hybridization showing the amplification of a small genomic segment (green arrow). Each dot represents log2 ratio (Colo320DM/normal fibroblast IMR90) of signal intensity for a single probe. (right) Relative copy numbers of the small segment and MYC in Colo320DM to a locus on chromosome 17. Error bars represent standard deviations.
(B) Metaphase FISH (left) and the histogram showing the co-localization between MYC (red) and the small segment (green) in 24 metaphases. The fraction of co-localized extrachromosomal elements to the total number of extrachromosomal elements in each metaphase is shown.
(C) A diagram showing the fusions between the MYC region and the 8.5 kb segment. The 8.5-kb segment is indicated in green.
(D) Schematic presentation of two-dimensional (2D) DNA agarose gel electrophoresis (left). The 1n spot represents either molecule before replication or molecule that have already completed replication across the restriction fragments. The Y-arc indicates replication intermediates (RIs). The double-Y indicates the converging forks coming from the opposite direction. The location of probes and a restriction map (8.5-kb region) for the analysis are also shown (right). The rearranged fragments on the A-side of the A–B fusion point is shown as a blue dotted line.
(E–G) Analysis of replication intermediates (RIs) digested with either PstI (D), EcoRI (E), or AseI (F). (E) FACS analysis indicates the S-phase progression of cells released from G1/S arrest with mimosine (top panels). Relative intensities of RI signals (Y-arc normalized to 1n linear molecule) at each time point to the relative intensities obtained at 0 hour time point (right) were plotted. Error bars represent standard deviations. Black stars (probe C) represent 1n spots from the normal allele. White stars (probe X1 and X2) represent residual signals from a prior probe due to incomplete stripping. (F) 2D images of EcoRI-digested fragments at 4 hours after either mimosine or double thymidine block (DTB) release. The ratios of large-/small-Y signals are shown on the bottom. Error bars represent SD. (G) RIs traveling on the two alternative structures of DMs. Because of a rearrangement in the flanking region of the B-side (Figure 1D and Figure S2C), two Y-arcs of equal intensities are seen throughout the time course.
We hypothesized that the 8.5 kb fragment could carry a functional feature that promotes the initial rearrangement of DMs. Such a feature could also promote the secondary rearrangements during the evolution of DM structure. Consistent with the idea, additional rearrangements were associated with the 8.5-kb segment and flanking region. At the A–B fusion, there was a rearrangement (Figure S2C, a broken blue line), in which an 11.9 kb EcoRV fragment and a 12.9 kb KpnI fragments were seen as distinct fragments from the EcoRV (6.5 kb) and KpnI (9.3 kb) fragments generated by A–B fusion. Furthermore, large SalI restriction fragments probed with the probe in the 8.5-kb segment (C) showed a 350 kb fragment (asterisk) which was smaller than the expected size (520 kb) (Figure S2D). Because SalI is a methylation-sensitive enzyme, restriction fragments larger than 520 kb can arise either by CpG methylation at the SalI restriction site GTCGAC or by rearrangements. However, a smaller fragment can only arise by the deletion involving the region close to the 8.5 kb segment. Thus, the 8.5 kb segment could play a functional role in the rearrangements of the DMs.
A natural fork stalling sites
In S. pombe, replication forks restarted from a site-specific protein-DNA fork barrier are prone to switch templates (Miyabe et al., 2015; Mizuno et al., 2013). For humans, the fork stalling and template switching (FoSTeS) mechanism has been proposed for the microhomology-mediated, complex genomic rearrangements (microhomology-mediated break-induced replication, MMBIR) in patients with genomic disorders (Lee et al., 2007; Liu et al., 2011; Zhang et al., 2009). We hypothesized that replication forks could stall in the 8.5-kb segment and switch replication templates, resulting in the rearrangements of DMs.
To test this possibility, we examined the kinetics of replication intermediates (RIs) by two-dimensional agarose gel electrophoresis (2D analysis) (Figure 1D and E, and Figure S3), as previously employed for the analysis of replication slow zones in S. cerevisiae (Cha and Kleckner, 2002). Although the 2D analysis has been very challenging in human cells due to the large genome size, we were able to visualize RIs clearly due to the 50-fold amplification of the MYC locus in Colo320DM. We arrested cells at the G1/S boundary by inhibiting replication initiation with mimosine (Kubota et al., 2014), released cells into S phase and then collected cells at several time points for 2D analyses with PstI-digested DNA. Strong Y-arc signals representing RIs, appeared very early in S phase, at 2 hours after release. In the control regions (X1, X2, and X3), the intensities of the Y-arcs reached their peaks at 4 hours and began to diminish at 6 hours (Figure 1E and Figure S3B). In contrast, in the 8.5-kb segment, the intensities continued to increase up to 8 hours, indicating the delayed clearance of RIs (fork stalling). We also noted the delayed clearance of RIs in cells released from the double thymidine block, in which Y-arcs disappeared 2–4 hours later in the 8.5-kb segment than in control regions (Figure S3C).
We dissected the fork movement in detail within the 8.5-kb segment. When we examined EcoRI-digested fragments with the probe in the middle of the 8.5 kb segment (probe E), the intensity between large- and small-Y was skewed in both mimosine and thymidine arrested/released cells (Figure 1F). The intensity of large Y was higher than those of small Y, with 80% of total Y-arc signals in large Y (large Y/total Y=0.8). In contrast, when we used the probe in the periphery of the 8.5 kb segment (probe C), the intensity was homogenous throughout the Y-arcs (large Y/total Y=0.5). Thus, large Y molecules accumulated more in the middle than in the periphery (C-side) of the 8.5 kb segment, indicating that replication forks stalled in the middle, not at the C-side of the 8.5 kb segment.
We also determined the direction of replication within the 8.5 kb segment. Two replication forks, each of which represented an alternative structure by the rearrangement at the A–B fusion (Figure S2B, two EcoRV fragments with the probes A and B), appeared simultaneously (Figure 1G), suggesting that the two forks came from a single origin of the C-side. The modified 2D gel analysis procedure (Brewer and Fangman, 1987) supported the fork direction from C-side to B-side. In this procedure, the PstI fragments were digested with KpnI following the first-dimensional electrophoresis and were probed with C (Figure S3D). Because the KpnI site is located at the B-side of the 8.5 kb fragment, a fork that moves from C-side to B-side could produce two long linear molecules from large Y after KpnI digest (Figure S3E, left). If replication forks move from B-side to C-side, Y-arc signals would migrate faster in the electrophoresis of the second dimension but wouldn’t disappear by the KpnI digest (Figure S3E, right). We found that large Y molecules became linear DNA by the KpnI digest, which suggests the fork moving from C-side to B-side. These results collectively show that replication forks moved from the C-side into the 8.5-kb segment and stalled in the middle.
Impediment of replication forks by long non-coding RNA
A skewed intensity of the Y-arcs can occur by a co-directional conflict between transcription and replication in S. cerevisiae (Azvolinsky et al., 2009). MYC oncogene is surrounded by the so-called gene desert, where protein-coding genes are very sparse (Figure 2A, shaded in blue). Instead, long non-coding RNA genes (lncRNA) are very abundant in this locus and are often overexpressed in tumors (Huppi et al., 2012). We identified abundant lncRNA transcripts of various sizes within the 8.5 kb segment by Northern blot (with the probe C, Figure 2B, left). Non-coding transcripts were also detected in the 8.5 kb segment (B and C) by RT-PCR but were not detected in the control regions (MYC gene, X1, X2, and X3) (Figure 2B, right). lncRNA transcription was in the same direction with replication, as shown by the detection of the lncRNA only by a plus (+)-strand specific RT-PCR (Figure 2C). Thus, co-directionally transcribed lncRNA was associated with the stalled replication forks in the 8.5 kb segment.
Figure 2. Transcription-replication conflict.
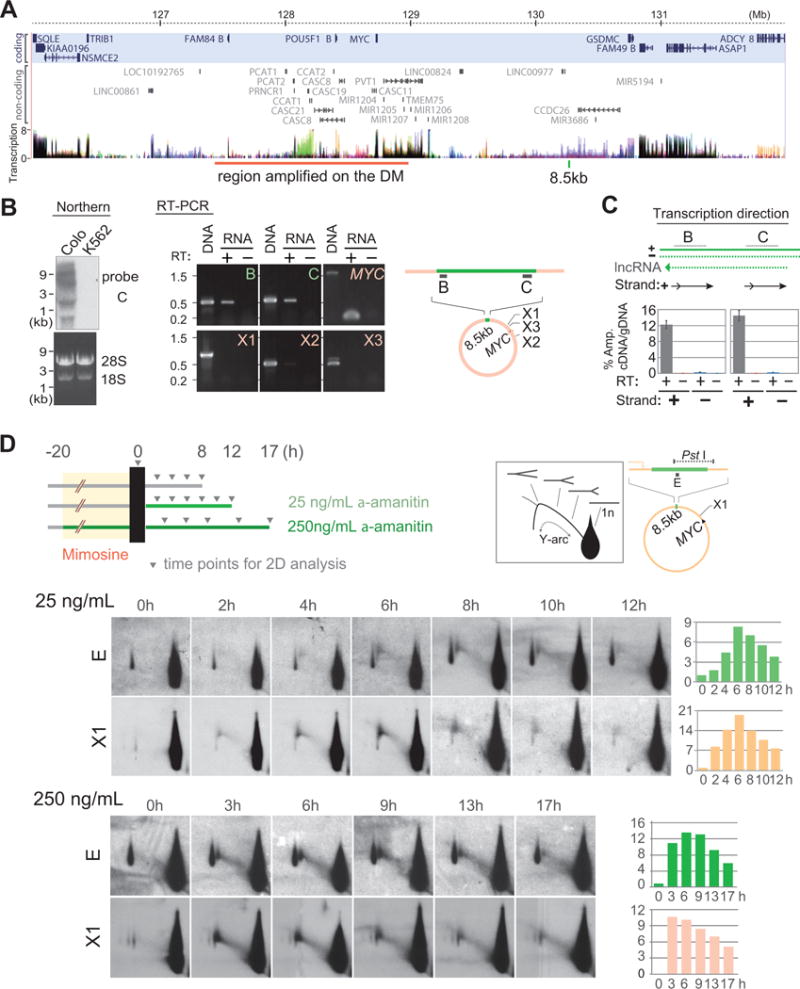
(A) A genomic view of the MYC locus (modified from UCSC genome browser). Protein coding genes (blue background), non-coding RNA genes and transcription activities are shown. Transcriptional activities show the levels of poly-adenylated RNA for nine cell types (the ENCODE Caltech RNA-seq project). Each color represents a transcription level of an individual cell line. Very dark colors derive from the overlapping transcription levels from multiple cell types. The regions amplified in the DM in Colo320DM cell line are shown on the bottom.
(B) Transcription of long non-coding RNA within the 8.5-kb segment in Colo320DM. Northern blotting using the probe C (left) and RT-PCR results for the transcripts of the 8.5-kb region (B, C) and four control regions (MYC, X1, X2, and X3) (right). K562, a CML cell line without MYC amplification.
(C) Strand-specific RT-qPCR defined the transcription of lncRNA from the leading strand. A schematic representation of the strategy (top). Transcript analysis for the regions B and C by strand-specific RT-qPCR (bottom). Amplification standard was first generated using genomic DNA as a template, and relative amount of amplification from cDNA was shown as % input. Error bars represent standard deviations.
(D) Time-course analysis of RIs after release from mimosine block in Colo320DM cells under either low (25 ng/ml, top) or high concentration (250 ng/ml, bottom) of α-amanitin treatment. Gray triangles indicate the time points when cells were collected. 2D gel analysis are shown for the 8.5 kb segment (probe E) and a control region (X1). Histograms show the relative intensities of RI signals, as described in Figure 1E.
To examine that transcription was the cause of stalled forks, we monitored RIs under the treatment of α-amanitin, an inhibitor of the largest subunit of RNA Polymerase II (Nguyen et al., 1996). We treated cells with either a low (25ng/ml media) or high (250ng/ml) concentration (Figure 2D). The low concentration (25ng/ml) of α-amanitin was added to the cell culture media after the release from mimosine arrest. The high concentration of α-amanitin (250ng/ml) was added with mimosine during both G1 arrest and S-phase. The kinetics of RIs (Y-arc signal/1n signal) were monitored for the PstI fragment of the 8.5 kb segment with probe E and the fragment in the control region (X1) for extended periods (12 hours for the low concentration α-amanitin and 17 hours for the high concentration α-amanitin), due to the slowed kinetics of RI appearance and disappearance. In both conditions, RIs appeared and disappeared at similar timings between the 8.5-kb segment and the control region (X1), suggesting that the inhibition of transcription could relieve the stalled forks at the 8.5 kb segment.
Molecular mechanisms underlying the stalled forks in the 8.5 kb segment
The stalled fork on the DMs provided us with a unique opportunity to define the temporal molecular processes occurring at naturally stalled forks. To do this, we employed chromatin immunoprecipitation (ChIP) for cells at 6 hours after release from the arrest (Figure 3). At 6 hours after release, Y-arcs persisted with high intensity in the 8.5 kb segment, but the intensities declined in the control regions (Figure 1E). We measured the markers representing replication-transcription conflicts as fractions of signals obtained from input DNA (% input). As a negative control, we measured the signals from DNA immunoprecipitated with non-specific IgG (gray bars). We monitor four locations within the 8.5 kb segment, with the #1 site close to the A–B fusion point, #2 and #3 in the middle, and #4 close to the C–D fusion point (Figure 3A). We also monitor a control locus (X1) and the promoter locus of MYC oncogene on the DM.
Figure 3.
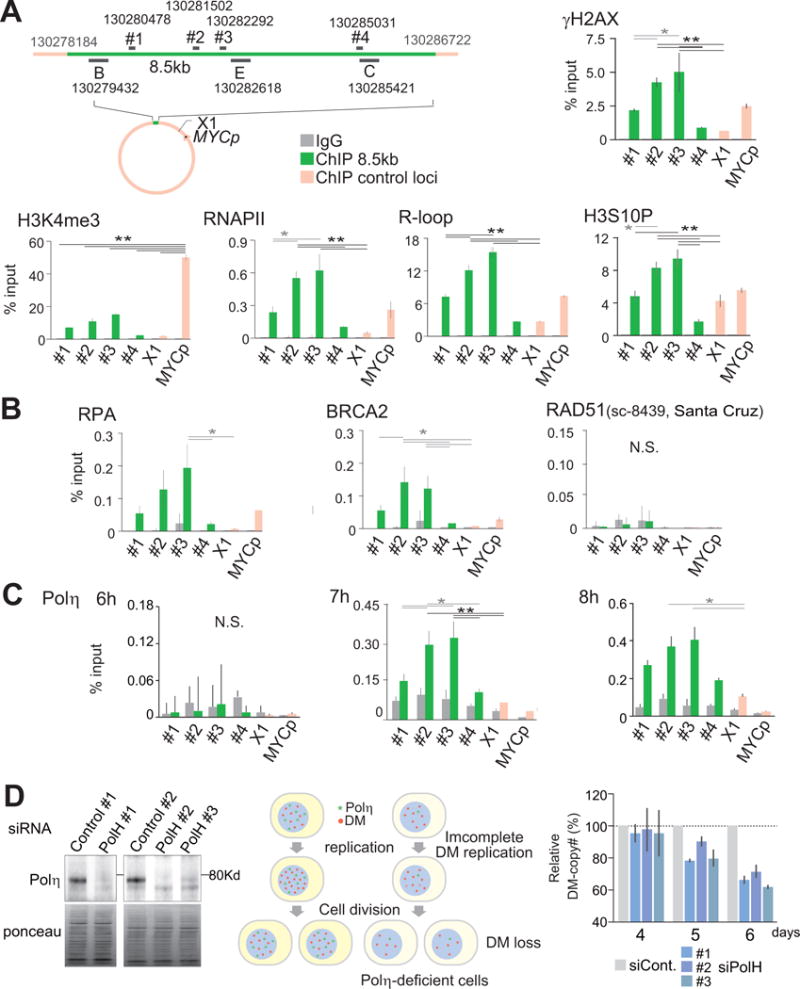
(A) Chromatin immunoprecipitation (ChIP)-qPCR analysis in S phase (6 hour after release from mimosine arrest) Colo320DM Loci analyzed by qPCR are shown, with their co-ordinates at the centromeric ends. Locations of the probes used for 2D gel electrophoresis are also shown. Results of the ChIP-qPCR for evaluating the mechanisms underlying stalled replication forks. Error bars represent standard deviations from four independent measurements. Significant differences of signals between the loci were examined by one-way ANOVA tests and Tukey multiple comparison tests, and are marked with either a single star (p<0.05) or two stars (p<0.01).
(B) ChIP-qPCR analysis of the recombination proteins RPA, BRCA2, and RAD51 (sc-8439, Santa Cruz).
(C) The recruitment of Polη to the stalled forks at 6, 7 and 8 hours after release from mimosine arrest. Error bars represent standard deviations from four independent measurements.
(D) Polη knockdown results in the copy number loss of DMs in Colo320DM cells. (left) Polη protein expression three days after the transfection of either control or POLH siRNA (three independent siRNAs #1, #2, and #3. (middle) Schematic representation of the processes leading to DM loss. Polη and DMs are shown green and red dots, respectively. (right) Real-time PCR-based copy number analysis in cells transfected with POLH siRNA at four, five and six days after transfection. The copy numbers for DMs were normalized to that of a locus in chromosome 17 within each sample. The fractions of the normalized copy number relative to that from the cells transfected with control siRNA (gray bars) are shown. Error bars represent standard deviations from four independent measurements.
We found significant enrichment of γ-H2AX, phosphorylated histone H2AX at the stalled forks (Figure 3A). γ-H2AX was more enriched in the middle of the 8.5 kb segment (#2 and #3) than in the peripheral regions (#1 and #4, p<0.05) (5-fold), which is consistent with the results from the 2D gel analysis (Figure 1F). RNA polymerase II (RNAPIIp) also accumulated in the middle of the 8.5 kb segment over peripheral regions (p<0.01). RNAPIIp was also more abundant at the stalled forks (#2 and #3) than at the active MYC promoter (MYCp) (p<0.05), whereas an epigenetic mark of active promoters (H3K4me3) was much less enriched at the stalled forks (p<0.01). The co-enrichment of RNAPIIp and γH2AX could indicate the collisions between transcription and replication machineries. R-loops, recognized by the S9.6 antibody (Bhatia et al., 2014), were also more abundant in the middle of the 8.5 kb segment (#2 and #3) than in both the peripheral and control regions (p<0.01). R-loops, RNA:DNA hybrids along with a displaced single-stranded DNA (ssDNA), can block replication forks and cause DNA damage (Aguilera and Garcia-Muse, 2012; Bermejo et al., 2012). The histone H3 phosphorylated at serine 10 (H3S10P), a histone mark for condensed chromatin, in the middle of the 8.5 kb segment (p<0.01) could indicate the formation of R-loops, as previously shown (Castellano-Pozo et al., 2013).
Transcription hampers fork movements in both head-on and co-directional collisions in S. cerevisiae (Azvolinsky et al., 2009). In head-on collisions, RNA:DNA helicase Senataxin can prevent stalled forks (Alzu et al., 2012), indicating that R-loops act as impediments. To determine whether R-loops blocked fork movements in the co-directional collision on the DMs, we overexpressed RNaseH1 in Colo320DM (Figure S4A). R-loops (S9.6 antibody ChIP) reduced to the level equivalent to the control regions in three RNaseH1-overexpressing cell clones. However, we did not observe the reduction of γ-H2AX in cells overexpressing RNaseH1. Thus, the removal of R-loops may not be sufficient to prevent stalled forks in this co-directional collision. In contrast, γ-H2AX became less enriched at the stalled forks by the α-amanitin treatment (Figure S4B). In this experiment, asynchronous Colo320DM cells were cultured with α-amanitin (250 ng/ml media) for either 2 or 6 hours. In both treatments, γ-H2AX became less enriched in the α-amanitin-treated cells than in the untreated cells.
Polη-dependent rescue of stalled forks
Stalled forks can be merged with converging forks to complete replication. However, this was unlikely for the stalled forks in the 8.5 kb segment because the double Y-pattern, which represents converging forks, was not clearly seen (Figure 1E). Therefore, to replicate DMs completely, re-establishing replication from stalled forks seems necessary. Re-establishment of replication from the experimentally induced stalled/collapsed forks primarily depends on the recombination proteins, in particular, RAD51 (Lambert et al., 2010; Petermann et al., 2010). However, we did not find the enrichment of RAD51 at the naturally stalled forks (Figure 3B and Figure S4C). We tested the RAD51 recruitment rigorously at several time points (Figure S4C) in cells released from mimosine arrest using antibodies that were validated for ChIP in the previous studies (Aymard et al., 2014). This lack of RAD51 recruitment was somewhat unexpected because the significant enrichments for recombination proteins RPA and BRCA2, which protect stalled forks and possibly recruit RAD51 for restart (Lambert et al., 2010; Schlacher et al., 2011), were readily detectable above the control IgG ChIP signals. Both RPA and BRCA2 were more enriched at the stalled forks (#3) than in the peripheral region (#4) and a control region (X1, p<0.05) (Figure 3B).
Stalled forks can bypass damaged DNA by recruiting specialized polymerases (translesion DNA synthesis, TLS) (Branzei and Foiani, 2010; Sale et al., 2012). Among specialized polymerases, Polη is a good candidate for rescuing stalled forks in this setting because Polη has been shown to physically interact with BRCA2 and to co-localize at nuclear foci upon treatment with replication inhibitors (Buisson et al., 2014). Indeed, Polη was recruited to the middle of the 8.5 kb segment (#2 and #3) (Figure. 3C). We did not observe the significant enrichment of Polη over the control ChIP at 6 hours after release. However, both at 7 and 8 hours, Polη became significantly enriched over the IgG control. Polη signals were significantly more enriched at #2 and #3 than in the peripheral regions (#1 and #4, p<0.05 and p<0.01, respectively) and control regions (X1 and MYC, p<0.01).
Failure to rescue stalled/collapsed forks could result in abortive replication and DNA loss. If Polη plays a critical role in rescuing the stalled/collapsed forks, knocking down Polη would lead to the loss of the DMs. To test the possibility, we knocked down Polη in Colo320DM cells with siRNA and monitored DM copy numbers (Figure 3D). All of the three independent siRNAs (#1, #2 and #3) knocked down Polη very efficiently at 72 hours after transfection. We expected that DM loss would follow the Polη knockdown because the reduction of DM copy number/cell requires cell division after abortive replication of DMs. We evaluated the DM-specific copy number loss relative to the copy number of the chromosome 17. The DM copy number was almost equal between Polη-knockdown cells and control cells (transfected with non-targeted siRNA) until 96 hours (4th day) after transfection. We began to observe DM loss on the 5th day after transfection and, on the 6th day, nearly 40% of DMs were lost in all Polη–knockdown cell pools. These results support the role of Polη in rescuing the stalled forks on the DMs.
Ectopic template switching from a rescued replication fork
In both simple eukaryotes and mammalian cells, re-established replication can occur from stalled forks before being processed into DSBs (broken forks) (Anand et al., 2013; Lambert et al., 2005; Lydeard et al., 2007; Miyabe et al., 2015; Petermann et al., 2010). To better understand the process underlying Polη-dependent rescue, we examined de novo DSBs at or near the stalled forks in cells released from mimosine arrest (Figure S5). To avoid mechanical shearing of DNA, we mounted cells into agarose block and digested with EcoRI. In Southern blotting, replication-associated, de novo breaks should appear as smaller, fast-migrating fragments. Slowly-migrating fragments representing replication intermediates gradually increased during the time course (black bracket), as we observed in the 2D gel analysis (Figure 1E). However, fast-migrating fragments did not appear at the later time points. Thus, stalled forks were not processed into DSBs within 8 hours, suggesting that fork rescue may have occurred from stalled forks, but not from broken forks.
As was shown in S. pombe (Miyabe et al., 2015; Mizuno et al., 2013), rescued forks on the DMs could be error-prone. If so, stalled forks could restart into the B-side of the flanking region and switch replication templates ectopically. Indeed, we found multiple, discrete restriction fragments on the B-side of flanking region, but not on the C-side (Figure 4A). To further evaluate the rearrangement bias towards rescued forks, we conducted whole-genome sequencing and measured split reads (soft-clipped reads) across the 1.6 Mb region of DMs (Figure 4B). A split read consists of the fusion of two or more genomic segments (Wang et al., 2011) and thus, each split reads could represent an ectopic template switching event. We identified the number of reads that were mapped to the 1.6 Mb region and carried ectopic DNA sequence of 7 bp or longer on one end. We then plotted the fraction of split reads (the number of split reads/the number of total reads) for each 10 kb bin. On average, 3 % of the reads (gray dotted line) were split reads across the 1.6 Mb region. Split reads were more abundant in the region flanking the B-side of the 8.5 kb segment than average; 7.13 % of the reads were split reads within the 120 kb region (p<0.0003, Heteroscedastic t-test, Figure 5B, in red). This biased enrichment of split reads towards the region next to the B-side could be due to the increased rearrangements from the rescued forks.
Figure 4.
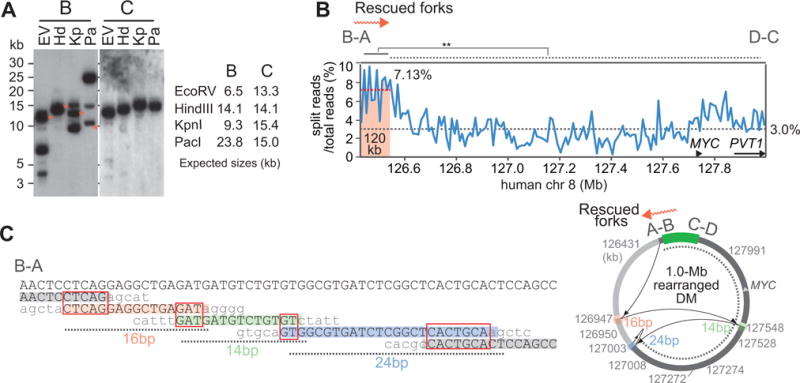
Error-prone rescued forks.
(A) DNA rearrangements occurred by the rescued replication forks. DNA was digested with a restriction enzyme (EV, EcoRV; Hd, HindIII; Kp, KpnI; Pa, PacI) and probed with B and C (left). Red stars represent fragments in the 1.0-Mb rearranged DM. White stars represent signals from the previous hybridization as a result of incomplete stripping. Expected fragment sizes in the 1.6-Mb original DM were also shown (right).
(B) We measured the number of reads that had DNA sequences (> 7 bp) from ectopic sites and present as the fractions of total reads.
(C) Sequence alignments of fusion points of the rearranged DM. The coordinate of each fusion point, as well as the distances of fork movement, are indicated. Red boxes mark microhomologies. A circular map of the DM and the rearranged DM is shown on the right.
Figure 5.
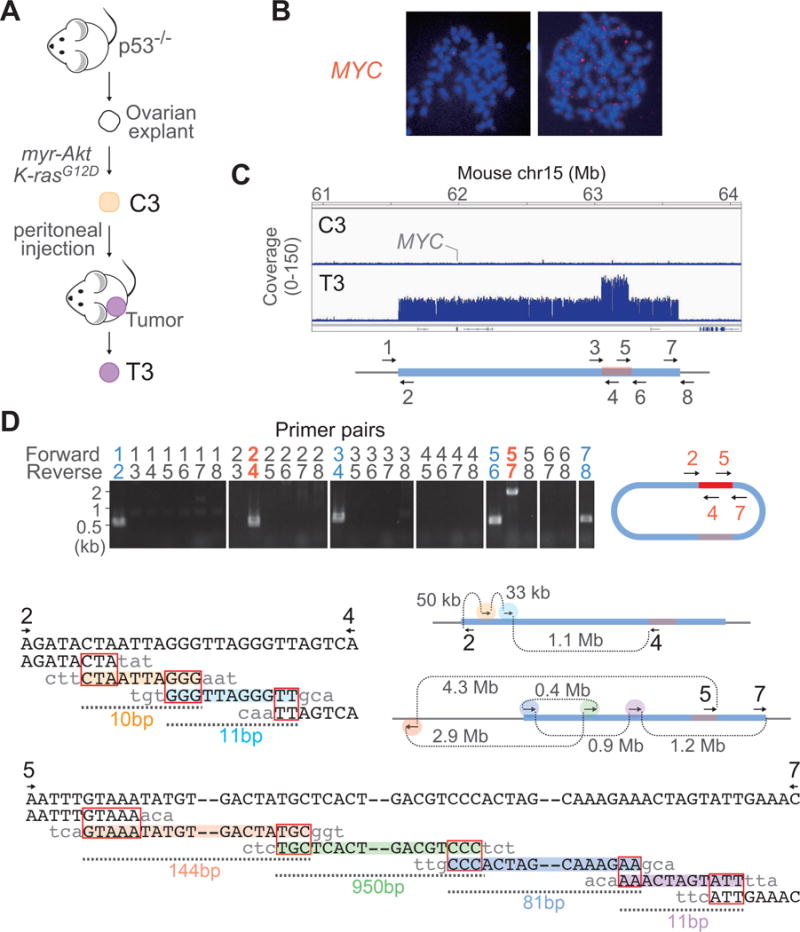
The de novo formation of a MYC-DM during tumorigenesis in the mouse by microhomology-mediated template switching.
(A) Outline of the mouse ovarian tumorigenesis model.
(B) Images of fluorescence in situ hybridization for MYC (red) in C3 and T3 tumor cells, counterstained with DAPI (blue).
(C) Focal MYC-amplicon with an additional peak in T3 tumor cells detected by whole-genome sequencing.
(D) De novo formation of the DM. Genomic fusions in T3 tumor cells assessed by breakpoint PCR (top) and the sequence alignments of the fusion points at junctions, #2–4 and #5–7 (bottom). The distances of fork movements (DNA synthesis) and the microhomologies at the junctions (red rectangles) are indicated. The coordinates of each fusion point are described in Table S1. The shaded colors on the sequences correspond to the colors in the figures in which ectopic template switching events are described for each fusion points.
We next investigated rearrangements at nucleotide resolution and found a surprisingly unstable nature of a rescued fork. In a rearrangement representing an additional replication intermediate (Figure 1G), a fork moved from the B-side of the 8.5 kb segment into 980 bp in the flanking region (A-side). The fork then switched templates three times ectopically and replicated short genomic fragments (16, 14 and 24 bp) that were several 100 kb away from the 8.5 kb segment on the DM. These ectopic switching could result in the 600 kb deletion in the original DM (Figure 5C, left). All template switching events were microhomology-mediated (2–7 base pairs, Figure 5C, right, and Table S1).
Complex genomic fusions in the de novo formation of MYC-DM
We identified the microhomology-mediated fusion of multiple short genomic segments as a potential signature of rearrangements by rescued stalled forks. Rescued forks could also contribute to the formation of DMs. Identifying the signature in the initial rearrangement, from linear chromosomal DNA to circular DM, could support the possibility. To test the idea, we employed a mouse ovarian tumorigenesis system. In this system, we first transduced ovarian surface epithelial cells derived from p53 null mice in vitro with oncogenic myr-Akt and K-rasG12D (C3 cell line in Figure 5A) (Orsulic et al., 2002). This combination of genetic alterations was sufficient for continuous in vitro growth. We injected the C3 cells into the peritoneal cavity of a mouse. Tumors formed spontaneously after long latency (> 6 months), indicating that additional spontaneous events were required for in vivo tumorigenesis. We cultured one of a tumor in vitro and established a cell line (T3) (Figure 5A). Metaphase FISH using BAC clones covering the MYC genomic locus showed that MYC was amplified extrachromosomally in a number of small chromosomes (Figure 5B).
Whole-genome sequencing revealed that the 2.1 Mb genomic region harboring MYC was amplified approximately 30-fold in the T3 cell line relative to the parental C3 (Figure 5C). Among the 2.1 Mb amplicon, the 250 kb region within the amplicon (Figure 5C, in brown) was amplified 60-fold, approximately twice more than the other regions in the amplicon. We designed PCR primers for all the copy number breakpoints and tested every possible combination of primer pairs to identify de novo fusions (Figure 5D). We found that the centromeric end of the amplicon (PCR primer 2) was fused to the centromeric end of the 250 kb region (primer 4), whereas the telomeric end of the amplicon (primer 7) was fused to the telomeric end of the 250 kb region (primer 5) (Figure 5C). These fusions could interpose the 250 kb segment between the two ends of the amplified segment and create a circular chromosome of 2.3 Mb. By sequencing the PCR products, we identified the complex, microhomology-mediated fusions of short genomic segments in both fusions (Figure 5D and Table S1). The PCR fragment from 2–4 fusion point consisted of two short fragments from different genomic loci within the DM (Figure 5D). The PCR fragment from 5–7 fusion point was even more complex and harbored the segments from four different loci within the DM and a flanking region (11 – 950 bps) that were 400 kb – 4 Mb apart from each other. All of the fusions were microhomology-mediated (2–5 bp). The complexity by templated insertions of small fragments was a strong indication of replication-associated events (Lee et al., 2007; Liu et al., 2011; Yang et al., 2013; Zhang et al., 2009). Thus, rescued replication forks may be involved in the formation of DMs.
Discussion
In this study, we rigorously examined naturally-stalled replication forks in tumor cells. By applying 2D-gel electrophoresis, we observed clearance delay of replication intermediates (Y-arcs) within the 8.5 kb segment of the DMs (Figure 1E and Figure S3). The delay was associated with the enrichment of γ-H2AX (Figure 3A). These results strongly suggest that replication forks stalled naturally within the 8.5 kb segment. Several studies in the recent years addressed stalled forks in mammalian systems (Barlow et al., 2013; Helmrich et al., 2011). However, capturing naturally stalled forks with this resolution remains a challenge. Thus, we had a unique opportunity to address molecular processes occurring at stalled forks in a natural setting.
The 8.5 kb segment was highly transcribed (Figure 2B). These transcripts were long and heterogeneous in size and were transcribed from the leading strand. Because transcription inhibitors appeared to relieve stalled forks (Figure 2D and Figure S4C) and RNA pol2 accumulated in the middle of the 8.5 kb segment (Figure 3A), the co-directional collision with transcription machinery was likely a cause of the stalled replication forks. The 8.5 kb segment is also transcribed in a subset of ENCODE cell lines in which MYC locus is not amplified (GM12878 normal lymphoblastoid cell line and K562 myelogenous leukemia cell line, Figure S6). This result suggests that replication-transcription conflicts can also occur in the normal genomic context. The conflict in normal genomic context may have a significant implication in MYC-dependent tumorigenesis, given the fact that (1) MYC is one of the most commonly amplified genes in tumors (Beroukhim et al., 2010) and (2) the genomic region surrounding MYC harbors a number of lncRNA, including lncRNA genes, and highly transcribed super-enhancers (Hnisz et al., 2013; Huppi et al., 2012; Xiang et al., 2015). These lncRNAs could disturb replication fork movements and promote MYC amplification. Indeed, copy number breakpoints of the focal MYC amplification in lung tumors cluster within the lung cancer-specific super-enhancer and the nearby lncRNA gene PVT1 (Hnisz et al., 2013; L’Abbate et al., 2014; Northcott et al., 2012).
In our Chip-qPCR data, Polη was recruited to the stalled forks on the DMs. Polη recruitment appears to be required for rescuing stalled forks, as Polη knockdown resulted in DM loss (Figure 3C). How Polη rescues natural stalled forks on the DM needs further investigation (Figure 6). Polη is a specialized polymerase that can bypass damaged DNA (translesion DNA synthesis, TLS) (Haracska et al., 2000; Masutani et al., 1999) and catalyze strand displacement DNA synthesis in vitro (Buisson et al., 2014; McIlwraith et al., 2005; Sneeden et al., 2013). Strand displacement DNA synthesis can extend DNA synthesis from a strand-invasion intermediate, which is consistent with the role of Polη in homology-dependent DNA repair (Kawamoto et al., 2005). Such an activity of Polη may be critical when restarted forks switch replication templates and invade into ectopic locus. Without Polη, stalled forks on the DMs could collapse and become broken DNA ends. Nucleolytic attack to the broken DMs and/or cell division without completing DM replication would result in the loss of DMs. Alternative, recombination-based, (legitimate) template switching can also rescue stalled forks. However, template switching in yeast depends on RAD51 (Gonzalez-Prieto et al., 2013; Urulangodi et al., 2015) which was not recruited to the stalled forks on the DMs. We also observed the recruitment of BRCA2 to the stalled forks. BRCA2 is a crucial protein in the homology-dependent repair of DNA double-strand breaks as BRCA2 loads RAD51 onto ssDNA (Jensen et al., 2010; Liu et al., 2010). BRCA2 also protects nascent DNA strand at hydroxyurea-induced stalled forks by stabilizing RAD51-ssDNA nucleofilaments (Schlacher et al., 2011; Ying et al., 2012). The lack of RAD51 recruitment suggests that BRCA2 could execute other essential functions at the naturally stalled forks, including the recruitment of Polη (Buisson et al., 2014). BRCA2 could also regulate R-loops at the stalled forks so that R-loops alone did not cause fork stalling (Bhatia et al., 2014).
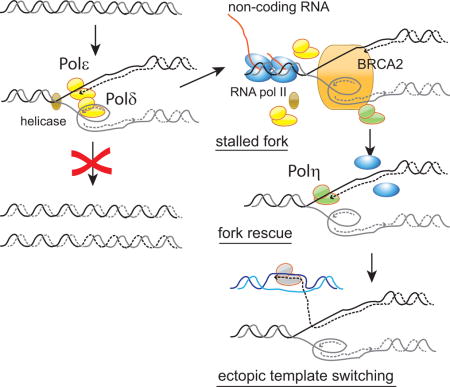
Figure 6.
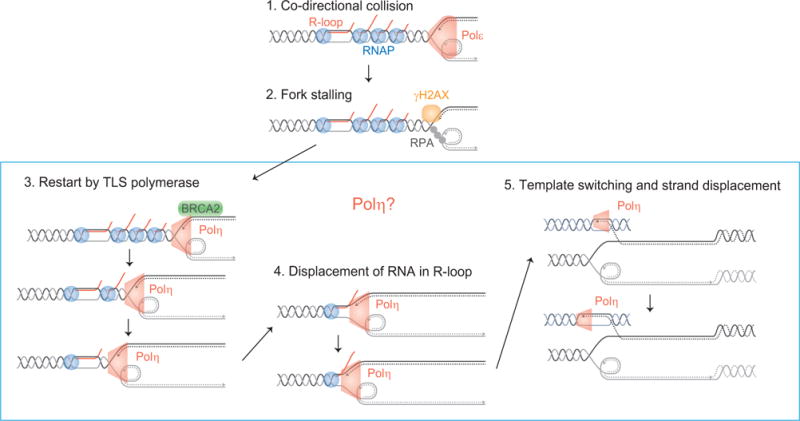
Molecular processes underlying the stalled forks on the DMs in Colo320DM cells and putative functions of Polη for error-prone fork restart (Model). Polη could rescue stalled forks by (1) facilitating fork restart and (2) displacing RNA from R-loops and synthesizing DNA. Polη could also execute its strand displacement DNA synthesis to replicate DNA following ectopic template switching.
Our data indicate that a single replication fork can be unstable and switch replication templates repeatedly to ectopic loci and created microhomology-mediated fusions of multiple genomic segments (Figure 4C). Re-established replication from stalled/collapsed forks could intrinsically be recombination-prone (Mizuno et al., 2013). Since the MYC locus lacks large inverted repeats, rescued forks can instead use microhomologies to switch templates and create microhomology-mediated fusions. Microhomology-mediated fusions are a common breakpoint feature of genome rearrangements in tumors (Stephens et al., 2011; Stephens et al., 2009). Considering the intense replication stress in tumor cells (Bartkova et al., 2005; Di Micco et al., 2006; Halazonetis et al., 2008), a significant proportion of microhomology-mediated fusions could derive from rescued forks.
Experimental Procedures
Detection of Replication Intermediates
Colo320DM cells (CVCL_0219) were obtained from ATCC. 2.4 × 106 cells were either arrested at the G1/S boundary using mimosine (0.5 mM, 20 hr) or at early S phase by a double thymidine block (2.5 mM, 17/14 hr) and collected at 0, 2, 4, 6, and 8 hours after release. DNA was isolated in gel blocks with 1% SDS and 1mg/mL proteinase K at 37°C for two days, washed with 50 ml of washing buffer (10mM Tris, 1mM EDTA, 120mM NaCl, pH 8.0) at 4°C for three days, and then digested overnight with restriction enzymes. Two-dimensional agarose gel electrophoresis was conducted as described previously (Brewer and Fangman, 1987), using 0.4% and 0.85% agarose gels for the first and second dimensions, respectively. RIs were detected by Southern hybridization with probes labeled with [α-32P]dATP (3000 Ci/mmol; Perkin Elmer) as described previously (Kondratova et al., 2015) and quantified using Image J software. Primers for preparing probes are listed in Table S2.
We inhibited transcription in Colo320DM cells using transcription inhibitors in two ways. In the first set of experiments, we treated cells after release from mimosine block with 25 ng/mL of α-amanitin after release. In the second set of experiments, we treated cells both before (for 20 hours) and after release using 250 ng/mL.
Chromatin immunoprecipitation (ChIP)
Colo320DM cells (1 × 107) were arrested at the G1/S boundary using mimosine (0.5 mM, 20 hr) and collected at 6 hr after release. Cells were cross-linked for 8 min with 1% formaldehyde, washed twice with cold PBS, and resuspended in 300 μl of lysis buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS, 1× Protease Inhibitor cocktail (Sigma, P8340)). The lysate was sonicated using a Bioruptor UCD-200 (Diagenode) at high intensity with 28 cycles of 30 s/90 s (on/off) and pre-cleared with anti-mouse or rabbit IgG conjugated Dynabeads (30 μL/sample, Life Technologies) overnight at 4°C. Dynabeads (50 μL/sample) were coupled at 4°C overnight with 4 μg of antibodies raised against RNA polymerase II CTD repeat phospho S2 (Abcam, ab5095), γH2AX (Abcam, ab2893), Histone H3 tri-methyl K4 (Abcam, ab8580), Histone H3 phospho S10 (Abcam, ab14955), RAD51 (Abcam, ab176458 and Santa Cruz, sc-8349, H-92), BRCA2 (Millipore, OP95), RNA/DNA hybrid (KeraFAST, ENH001, S9.6), RPA34 (Calbiochem, NA19L), and DNA Polymerase η (Abcam, ab17725). The pre-cleared lysate was diluted 10-fold with dilution buffer (50 mM Tris pH 8.0, 167 mM NaCl, 1.1% Triton X-100, 0.11% Na-deoxycholate, 1× Protease Inhibitor cocktail) and used for input (126 μL) and immunoprecipitation (IP, 1.82 mL). Each chromatin sample for IP was captured by antibody-coupled Dynabeads and washed twice with a series of buffers: low salt buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 0.1% Na-deoxycholate), high salt buffer (500 mM NaCl in Low salt buffer), LiCl buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% Na-deoxycholate) and TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Input and IP samples were treated with protease K at 55°C for two hr and uncrosslinked at 65°C for 12 hr. DNA was then isolated from the input and IP samples using the Illustra DNA Purification Kit (GE Healthcare). Quantitative-PCR was performed on a CFX384 Real-Time PCR Detection System (Bio-Rad), using either iQ SYBR Green Supermix (40 cycles of 95 °C for 15 sec and 60 °C for 1 min) or SsoFast EvaGreen Supermix (45 cycles of 98 °C for 10 sec and 60 °C for 10 sec) (Bio-Rad). All reactions were performed in quadruplicate. Data analysis was done using Bio-Rad CFX Manager Version 3.1. Primers are listed in Table S2. Statistical analyses were first carried out a one-way analysis of variance (ANOVA) to assess the overall statistical significance of differences and, if significant, performed multiple comparisons using Tukey test at the 95 or 99 % confidence level.
Transformation of Mouse Ovarian Surface Epithelial Cell Lines
All procedures in mice were approved by the Cedars-Sinai IACUC (protocol #5318) and performed in an AAALAC-accredited facility. Eight week-old female nude mice were used for intraperitoneal injections of mouse ovarian cancer cells. Ovarian explants from K5-TVA/p53−/− mice (Orsulic et al., 2002) were infected with RCAS viruses carrying mouse K-rasG12D and myr-Akt1 oncogenes. The resulting transformed mouse ovarian surface epithelial cell line (C3) was injected intraperitoneally into nude mice. The mouse ovarian tumor cells, T3, were derived from tumors generated by C3 injection.
Whole Genome Sequencing
The genomic DNA from Colo320DM cells was sequenced on Illumina HiSeq 2000 at the University of Chicago Genomics Facility. Sequencing reads were mapped to the human reference genome (hg38) using BWA to create a SAM file. Soft-clipped reads were identified based on the information from CIGAR string. For reads with longer than seven bp soft-clip on any of the ends were considered as real soft-clip reads. By using a 10kb bin to scan across the region from 126425748 to 127997618 in chromosome 8, all the reads in each bin were analyzed and counted to get the ratio of soft-clip reads against the total reads. To assess the statistical significance, the Tukey-Kramer multiple comparison tests were used at the 99 % confidence level.
The whole genomes of mouse C3 and T3 cell lines were sequenced at the Cedars-Sinai Genomics Core. Genomic DNA samples were used to prepare whole genome sequencing libraries using the Ion Xpress Plus Fragment Library Kit (Life Technologies) per manufacturer’s instructions. Final sequencing libraries were sequenced on the Ion Proton System (Life Technologies) using the Ion PI Template OT2 200 Kit v3 and Ion PI Sequencing 200 Kit v3 per manufacturer’s instructions. Raw sequencing reads were aligned to the UCSC mouse reference genome (mm10) and visualized with the integrative genomics viewer (IGV, Broad Institute).
Supplementary Material
Highlights.
Replication forks stall naturally under co-directional conflict with lncRNA.
Rescue of stalled forks depends on a specialized DNA polymerase Polη.
Rescued forks create complex microhomology-mediated fusions.
Complex fusions are instrumental to the formation of a double minute chromosome.
Acknowledgments
We thank Drs M-C Yao, T Horiuchi, M Hidaka, V Boener and A Rattray for comments on the manuscript. We also thank the Genomics Facility at the University of Chicago, Genomics Core and Flow Core at Cedars-Sinai Medical Center for technical support. This work is supported by the National Cancer Institute (Grant Number R01CA149385) and Cedars-Sinai Medical Center (to H.T.), Margie and Robert E. Petersen Foundation, The Fashion Footwear Charitable Foundation of New York, Inc., Avon Foundation and Associates for Breast and Prostate Cancer Studies (to A.E.G.), and Kanzawa Medical Research Foundation (to T.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
Colo320DM-NCBI BioProject (accession number PRJNA341527)
Mouse C3 and T3 cells - NCBI Sequence Read Archive under accession numbers SRR1980157 (C3) and SRR1982109 (T3)
Author contributions
Conceptualization, T.W., S.J.T. and H.T.; Methodology, T.W., M.M., R.S., X.C., L.M., and A.K. Investigation, T.W., S.O. and H.T.; Visualization, S.J.D. and A.N., Writing – Original Draft, T.W. and H.T.; Writing – Review & Editing, S.J.T. and S.O.; Resources, A.E.G. and H.T.; Supervision, H.T. and S.O.; Funding Acquisition, T.W. and A.E.G. and H.T.
References
- Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell. 2012;151:835–846. doi: 10.1016/j.cell.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand RP, Lovett ST, Haber JE. Break-induced DNA replication. Cold Spring Harb Perspect Biol. 2013;5:a010397. doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722–734. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710–718. doi: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–365. doi: 10.1038/nature13374. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Pauty J, Maity R, Zhao W, Coulombe Y, Sung P, Masson JY. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014;6:553–564. doi: 10.1016/j.celrep.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano-Pozo M, Santos-Pereira JM, Rondon AG, Barroso S, Andujar E, Perez-Alegre M, Garcia-Muse T, Aguilera A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol Cell. 2013;52:583–590. doi: 10.1016/j.molcel.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Prieto R, Munoz-Cabello AM, Cabello-Lobato MJ, Prado F. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J. 2013;32:1307–1321. doi: 10.1038/emboj.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Haracska L, Yu SL, Johnson RE, Prakash L, Prakash S. Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase eta. Nat Genet. 2000;25:458–461. doi: 10.1038/78169. [DOI] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell. 2015;57:636–647. doi: 10.1016/j.molcel.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–977. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell. 2003;12:711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ. The 8q24 gene desert: an oasis of non-coding transcriptional activity. Front Genet. 2012;3:69. doi: 10.3389/fgene.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N, et al. Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Mol Cell. 2005;20:793–799. doi: 10.1016/j.molcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Kondratova A, Watanabe T, Marotta M, Cannon M, Segall AM, Serre D, Tanaka H. Replication fork integrity and intra-S phase checkpoint suppress gene amplification. Nucleic Acids Res. 2015;43:2678–2690. doi: 10.1093/nar/gkv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Fukumoto Y, Ishibashi K, Soeda S, Kubota S, Yuki R, Nakayama Y, Aoyama K, Yamaguchi N, Yamaguchi N. Activation of the prereplication complex is blocked by mimosine through reactive oxygen species-activated ataxia telangiectasia mutated (ATM) protein without DNA damage. J Biol Chem. 2014;289:5730–5746. doi: 10.1074/jbc.M113.546655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Abbate A, Macchia G, D’Addabbo P, Lonoce A, Tolomeo D, Trombetta D, Kok K, Bartenhagen C, Whelan CW, Palumbo O, et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res. 2014;42:9131–9145. doi: 10.1093/nar/gku590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Freon K, Murray JM, Carr AM, Baldacci G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell. 2010;39:346–359. doi: 10.1016/j.molcel.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17:1260–1262. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell. 2005;20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabe I, Mizuno K, Keszthelyi A, Daigaku Y, Skouteri M, Mohebi S, Kunkel TA, Murray JM, Carr AM. Polymerase delta replicates both strands after homologous recombination-dependent fork restart. Nat Struct Mol Biol. 2015;22:932–938. doi: 10.1038/nsmb.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Miyabe I, Schalbetter SA, Carr AM, Murray JM. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013;493:246–249. doi: 10.1038/nature11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Giannoni F, Dubois MF, Seo SJ, Vigneron M, Kedinger C, Bensaude O. In vivo degradation of RNA polymerase II largest subunit triggered by alpha-amanitin. Nucleic Acids Res. 1996;24:2924–2929. doi: 10.1093/nar/24.15.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer cell. 2002;1:53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell. 2015;60:860–872. doi: 10.1016/j.molcel.2015.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn JZ, Salama SR, Grifford M, Brennan CW, Mikkelsen T, Jhanwar S, Katzman S, Chin L, Haussler D. Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res. 2013;73:6036–6045. doi: 10.1158/0008-5472.CAN-13-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeden JL, Grossi SM, Tappin I, Hurwitz J, Heyer WD. Reconstitution of recombination-associated DNA synthesis with human proteins. Nucleic Acids Res. 2013;41:4913–4925. doi: 10.1093/nar/gkt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi CT, Fioretos T, Surace C, Lonoce A, Mastrorilli A, Strombeck B, D’Addabbo P, Iacovelli F, Minervini C, Aventin A, et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet. 2006;15:933–942. doi: 10.1093/hmg/ddl010. [DOI] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017;543:122–125. doi: 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urulangodi M, Sebesta M, Menolfi D, Szakal B, Sollier J, Sisakova A, Krejci L, Branzei D. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev. 2015;29:2067–2080. doi: 10.1101/gad.265629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl GM. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989;49:1333–1340. [PubMed] [Google Scholar]
- Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R. Segregation of transcription and replication sites into higher order domains. Science. 1998;281:1502–1506. doi: 10.1126/science.281.5382.1502. [DOI] [PubMed] [Google Scholar]
- Xiang JF, Yang L, Chen LL. The long noncoding RNA regulation at the MYC locus. Curr Opin Genet Dev. 2015;33:41–48. doi: 10.1016/j.gde.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, Hsieh CH, Zhang C, Ren X, Protopopov A, Chin L, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153:919–929. doi: 10.1016/j.cell.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nature cell biology. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.