

Abstract
Background
Chlamydia suis is an important, globally distributed, highly prevalent and diverse obligate intracellular pathogen infecting pigs. To investigate the prevalence and genetic diversity of C. suis in China, 2,137 nasal, conjunctival, and rectal swabs as well as whole blood and lung samples of pigs were collected in 19 regions from ten provinces of China in this study.
Results
We report an overall positivity of 62.4% (1,334/2,137) of C. suis following screening by Chlamydia spp. 23S rRNA-based FRET-PCR and high-resolution melting curve analysis and confirmatory sequencing. For C. suis-positive samples, 33.3 % of whole blood and 62.5% of rectal swabs were found to be positive for the C. suis tetR(C) gene, while 13.3% of whole blood and 87.0% of rectal swabs were positive for the C. suis tet(C) gene. Phylogenetic comparison of partial C. suis ompA gene sequences revealed significant genetic diversity in the C. suis strains. This genetic diversity was confirmed by C. suis-specific multilocus sequence typing (MLST), which identified 26 novel sequence types among 27 examined strains. Tanglegrams based on MLST and ompA sequences provided evidence of C. suis recombination amongst the strains analyzed.
Conclusions
Genetically highly diverse C. suis strains are exceedingly prevalent in pigs. As it stands, the potential pathogenic effect of C. suis on pig health and production of C. suis remains unclear and will be the subject of further investigations. Further study is also required to address the transmission of C. suis between pigs and the risk of 'spill-over' and 'spill-back' of infections to wild animals and humans.
Electronic supplementary material
The online version of this article (10.1186/s12917-017-1295-x) contains supplementary material, which is available to authorized users.
Keywords: Chlamydia suis, Pig, FRET-PCR, ompA, MLST, Tanglegram
Background
Obligate intracellular bacteria of the genus Chlamydia are the etiological agents of chlamydiosis in wild and domestic birds, mammals and humans [1–4]. Eleven recognized Chlamydia spp. (C. trachomatis, C. suis, C. muridarum, C. pneumoniae, C. abortus, C. caviae, C. felis, C. pecorum, C. psittaci, C. avium and C. gallinacea) are included in the single genus Chlamydia under the family Chlamydiaceae currently contains [5].
In pigs, four chlamydial species (C. pecorum, C. abortus, C. psittaci and C. suis) are regularly detected [6]. Of these, C. abortus and especially C. suis are the main species and most common involved in porcine chlamydial infections, with mixed infections occurring regularly [7–10] and only a limited number of reports finding a single species (C. suis) infecting cohorts of pig [10, 11].
In pigs, C. suis is mainly associated with subclinical infections, however it has also been found in association with respiratory disease, diarrhea, conjunctivitis and reproductive failure [3, 6]. Beyond pigs, DNA from this organism has also been detected in sheep, cattle, horses, cats, kestrels, and frogs [12–14]. Whether this PCR-based evidence represents the detection of an active infection versus exposure is, however. unknown [12–14].
Zoonotic potential of C. suis was suggested in the previous study, showing a single or mixed infection with C. trachomatis [15]. In addition, conjunctival swabs of employees in a Belgian pig slaughterhouse was identified to carry C. suis DNA [3] which was also found in the and pharyngeal and rectal swabs of Belgian pig farmers [10].
Consequently, pig pathogens may not only affect the pig production, but also potentially play a role in public health.
For decades, tetracyclines, as broad-spectrum antibiotics, have been extensively used in the pig industry for both prophylactic and therapeutic treatment. Interestingly, C. suis is the only chlamydial species to have naturally acquired genes encoding for antibiotic resistance. Over the past years, there have been accumulating reports on the occurrence of tetracycline-resistant C. suis strains in America, Europe and Asia [16–20]. This resistance pattern is associated with tet(C) islands. No standard antibiotic treatment of pigs (amoxicillin, chlortetracycline, chlortetracycline plus sulfadimidine, tylosin, trimethoprime plus sulfadimidin plus sulfathiazole) appear to be able to clear chlamydial infections at the herd level, despite individual pigs becoming negative [21]. Moreover, clear evidence of selective pressure was obvious as tetracycline treatment resulted in a higher number of tetracycline-resistant C. suis isolates [22]. Highlighting the additional concerns over the presence of this antibiotic-resistance genes in C. suis, transfer of tetracycline resistance from C. suis to C. trachomatis and C. muridarum, and between C. suis and C. trachomatis strains has been demonstrated in vitro [23].
Currently, there is no standardized typing scheme to evaluate the genetic diversity of C. suis. However, molecular typing schemes for other Chlamydiaceae are primarily based on characterizing epitopes in the major chlamydial outer membrane protein. For the closely related C. trachomatis, variants of this protein are subjected to selection and isolates of the same serovar may not be closely related [24, 25]. A recent study has revealed that C. suis had the highest whole genome recombination rate of Chlamydia species studied to date by whole genome sequencing [26] with ompA, as one of 77 C. suis core genes, showing significant evidence for intragenic recombination. As an alternative to ompA-based typing schemes, multilocus sequence typing (MLST) schemes that typically target fragments of five to seven housekeeping (HK) genes under neutral selection are widely used for determining bacterial population structures and barcoding isolates [27].
We have recently shown that, in China, a range of important livestock species, including poultry [28] and cattle [29], are infected with different endemic and epidemic strains of Chlamydia. Pig production is a major agricultural industry in China, however, little is known about the diversity of chlamydial infections that might impact on the health of pigs in this country. Therefore, the present study was undertaken to investigate the prevalence of Chlamydia spp. in pigs from China, with a particular emphasis on C. suis infections. Given the potential importance of tetracycline resistance for the latter species, we also investigated the presence of tetracycline resistance genes in these strains. To analyze diversity, phylogenetic and cluster analyses of C. suis was performed using both ompA and a C. suis-specific MLST typing scheme designed for the purpose of this study.
Methods
Ethics statement
Protocols for the collection of samples in this study were reviewed and approved by the Institutional Animal Care and Use Committee of Yangzhou University College of Veterinary Medicine (YZU-CVM#2015-057). The written permission was obtained from the owners of the pigs used in this study.
Sample collection
Nasal, rectal swabs, whole blood and lung samples
In this study, a total of 2,137 clinical samples (lung, whole blood, rectal swabs, nasal swabs, conjunctival swabs) were collected from pigs in the farms of 19 regions of 10 provinces of China between 2015 and 2016 (Table 1). A part of samples were collected from pigs in 9 regions of Jiangsu province: whole blood samples (n=308) and rectal swabs (n=55) were collected in Yangzhou City, whole blood samples (n=33) were from Nantong city, nasal swabs were from Nanjing city (n=180), Huaian city (n=70), Suqian city (n=41), Taizhou city (n=49), Wuxi city (n=36), Yancheng city (n=37), Changzhou city (n=23). In addition, nasal swabs (n=68) and rectal swabs (n=68) were collected from swine farms in Kunming city in Yunnan province; nasal swabs were from Shanghai municipality (n=52), Xihua City in Henan province (n=63), Changchun City in Jilin province (n=63), Jiangmen City in Guangdong province (n=40), Haerbin City in Heilongjiang province (n=60) and Jining City in Shandong province (n=60), respectively. Nasal swabs were also collected from Specific Pathogen Free (SPF) pigs (n=108) in two experimental animal companies in Nanjing City in Jiangsu province. Lung tissue samples (n=23) from an abattoir in Nanping city in Fujian province were also collected.
Table 1.
Prevalence and copy numbers of C. suis of pigs from provinces of China
| Province | City | Sample type | Age | Positivity | Copy /ml or swab, log10 |
|---|---|---|---|---|---|
| Fujian | Nanping | Lung | NA | 26.1% (6/23) | 3.72 (40mg) |
| Jiangsu | Yangzhou | Whole blood | Finisher pig | 33.4 % (103/308) | 2.79 |
| Rectal swab | Finisher pig | 98.2%(54/55) | 4.50 | ||
| Nantong | Whole blood | Suckling pig | 0.0% (0/11) | 0.00 | |
| Weaned pig | 20.0% (2/10) | 3.17 | |||
| Sow | 8.3% (1/12) | 2.85 | |||
| Nanjing | Nasal swab | Weaned pig* | 86.1% (93/108) | 3.65 | |
| 35d | 95.0%(57/60) | 2.61 | |||
| 49d | 76.7%(46/60) | 1.85 | |||
| 63d | 91.7%(55/60) | 4.19 | |||
| Huaian | Nasal swab | Suckling pig | 40.9% (9/22) | 2.16 | |
| Weaned pig | 18.8% (3/16) | 1.99 | |||
| Finisher pig | 58.3% (7/12) | 2.11 | |||
| Sow | 15.0% (3/20) | 1.99 | |||
| Suqian | Nasal swab | Suckling pig | 40.0% (6/15) | 2.68 | |
| Weaned pig | 38.9% (7/17) | 3.07 | |||
| Sow | 12.5% (1/9) | 2.20 | |||
| Taizhou | Nasal swab | Suckling pig | 69.2% (18/26) | 2.25 | |
| Weaned pig | 69.2 (9/13) | 2.91 | |||
| Finisher pig | 100.0% (5/5) | 3.67 | |||
| Sow | 0.0% (0/5) | 0.00 | |||
| Wuxi | Nasal swab | Suckling pig | 17.6% (3/17) | 1.99 | |
| Weaned pig | 100.0% (5/5) | 3.16 | |||
| Finisher pig | 100.0% (5/5) | 3.13 | |||
| Sow | 0.0% (0/9) | 0.00 | |||
| Yancheng | Nasal swab | Suckling pig | 43.8% (7/16) | 2.13 | |
| Weaned pig | 87.5% (7/8) | 2.95 | |||
| Finisher pig | 100.0% (3/3) | 2.89 | |||
| Sow | 30.0% (3/10) | 2.31 | |||
| Changzhou | Nasal swab | Suckling pig | 100% (7/7) | 3.48 | |
| Weaned pig | 100.0% (16/16) | 3.81 | |||
| Shanghai | Shanghai | Nasal swab | Suckling pig | 66.7% (12/18) | 2.60 |
| Weaned pig | 57.1% (8/14) | 2.12 | |||
| Finisher pig | 100.0% (10/10) | 3.89 | |||
| Sow | 20.0% (2/10) | 3.00 | |||
| Yunnan | Kunming | Nasal swab | Suckling pig | 92.3% (12/13) | 2.91 |
| Weaned pig | 100.0% (9/9) | 3.80 | |||
| Finisher pig | 86.7% (13/15) | 2.25 | |||
| NA | 90.3% (28/31) | 3.12 | |||
| Rectal swab | Suckling pig | 84.6% (11/13) | 3.03 | ||
| Weaned pig | 100.0% (9/9) | 4.14 | |||
| Finisher pig | 66.7% (10/15) | 2.58 | |||
| NA | 93.6% (29/31) | 4.54 | |||
| Zhejiang | Shaoxing | Nasal swab | Suckling pig | 89.0% (89/100) | 3.55 |
| Weaned pig | 95.0% (95/100) | 4.26 | |||
| Finisher pig | 82.0% (41/50) | 3.27 | |||
| Sow | 46.0% (23/50) | 2.97 | |||
| Shaoxing | Nasal swab | Sow | 32.0%(16/50) | 2.74 | |
| Conjunctival swab | Sow | 26.0% (13/50) | 2.56 | ||
| Rectal swab | Sow | 20.0% (10/50) | 2.89 | ||
| Whole blood | Sow | 0.0% (0/50) | 0.00 | ||
| Ningbo | Nasal swab | Weaned pig | 100.0% (50/50) | 4.86 | |
| Conjunctival swab | 100.0% (50/50) | 4.87 | |||
| Rectal swab | 100.0% (50/50) | 5.83 | |||
| Whole blood | 16.0% (8/50) | 3.50 | |||
| Henan | Xihua | Nasal swab | Sow | 28.1%(9/32) | 3.13 |
| Weaned pig | 96.8%(30/31) | 3.42 | |||
| Jilin | Changchun | Nasal swab | Weaned pig | 81.1%(53/63) | 3.94 |
| Guangdong | Jiangmen | Nasal swab | Weaned pig | 100.0%(20/20) | 4.86 |
| Sow | 90.0%(18/20) | 3.68 | |||
| Heilongjiang | Haerbin | Nasal swab | Suckling pig | 71.7%(43/60) | 2.07 |
| Shandong | Jining | Nasal swab | Suckling pig | 53.3%(16/30) | 3.68 |
| Sow | 6.7%(2/30) | 2.84 |
*represents SPF weaned pig from two experimental animal companies. NA: not applicable.
Nasal swabs, conjunctival and rectal swabs were collected into sterile Eppendorf tubes (Eppendorf, Shanghai, China) containing 400 μl DNA/RNA stabilization buffer (Roche Molecular Biochemicals, Indianapolis, IN, USA). Blood samples were collected into EDTA tubes (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and transported and stored at room temperature.
Nasal swabs from pigs of different ages
To compare and analyze differences in positivity and infectious load of C. suis among suckling pigs (age before 25 days old), weaned pigs (age after 25 d), finisher pigs (age after 100 d) and sows (age after 210 d), nasal swabs were collected from animals in each age group from Shaoxing city in Zhejiang Province: suckling pigs (n=100), weaned pigs (n=100), finisher pigs (n=50) and sows (n=50).
Nasal, conjunctival, rectal swabs and whole blood samples from pigs on two pig farms in Zhejiang province
To compare and analyze differences in the shedding of C. suis from different anatomical sites of animals, multiple samples (nasal, conjunctival, rectal swabs and whole blood) were collected randomly from 50 sows in a pig farm in Shaoxing of Zhejiang province and 50 weaned pigs in Ningbo of Zhejiang province. For the former, 50 sows in two blocks were randomly chosen out of 160 sows on pig farms in Shaoxing. For the latter, 50 weaned pigs in 10 blocks were chosen out of 200 weaned pigs on pig farms in Ningbo for this study. Whole blood samples (around 2 ml) were collected in EDTA tubes, nasal, conjunctival, rectal swabs were collected as above mentioned.
DNA extraction from swabs, whole blood and lung samples
The High-Pure PCR Template Preparation Kit (Roche Molecular Biochemicals, Indianapolis, IN, USA) was used to extract total nucleic acids from nasal, conjunctival, rectal swabs and whole blood and lungs from pigs, according to the manufacturer's instructions and described before [30]. The extracted DNA was eluted in 200 μl elution buffer. In this study, swabs obtained weekly from research laboratory were processed for Chlamydia qPCR to verify free of carry-over contamination occurred. Furthermore, diethylpyrocarbonate (DEPC)-treated ddH2O served as a negative control to confirm that contamination between samples did not occur during the DNA extraction.
Chlamydia FRET-PCR
The FRET-PCR was performed in a LightCycler 480-II real-time PCR platform., and the protocol of the PCR in this study followed what described [28, 31]. This 23S rRNA-based FRET-PCR was able to detect all 11 Chlamydia species and had a detection limit of single copies/reaction. The PCR products were further verified by electrophoresis followed by DNA sequencing (BGI, Shanghai, China).
Tet(C) and tetR(C) PCR
Whole blood samples of both C. suis-positive (n=45) and C. suis-negative (n=40) from Yangzhou in Jiangsu province and rectal swabs of both C. suis-positive (n=54, 45 from Kunming in Yunnan province and 9 from Shaoxing in Zhejiang province) and C. suis-negative (n=53, 38 from Kunming in Yunnan province and 15 from Shaoxing in Zhejiang province) were examined for presence of the tetracycline resistance gene, tet(C), and the tetracycline repressor gene, tetR(C), by PCR as previously described [17].
C. suis-specific ompA-PCRs
To investigate the polymorphisms in the C. suis ompA gene, a set of primers were designed using Vector NTI to amplify the ompA VD 1-2 (amplicon size: 491bp) to interrogate 108 C. suis -positive samples (44 whole blood, 3 lungs samples, 12 rectal swabs, 1 conjunctival swabs, 48 nasal swabs). Twenty μl PCRs were prepared containing 10.0 μl DNA template, 0.2μl forward primer (100 μM), 0.2μl reverse primer (100 μM,), 4.0 μl 5 x PCR buffer, 0.4 μl 10 μM dNTP, 0.3 μl 5 U/μl Taq DNA polymerase and 4.9 μl Ultrapure H2O. In addition, two sets of primers were designed to specifically amplify the ompA gene of C. abortus and C. pecorum (Table 2). Along with a set of specific primers for C. psittaci [29], these assays were used to confirm the detection of each species’ DNA from the Chlamydia FRET-qPCR. PCR amplification was performed in a LightCycler 480-II real-time PCR platform using a high-stringency 18-cycle step-down temperature protocol: 6 x 1 sec @ 95oC, 12 sec @64oC, 8 sec @ 72oC; 9 x 1 sec @ 95oC, 12 sec @ 62oC, 8 sec @ 72oC; 3 x 1 sec @ 95oC, 12 sec @ 60oC, 8 sec @ 72oC; followed by 30 low-stringency cycles: 30 x 1 sec @ 95oC, 12 sec @ 55oC, 30 sec @ 67oC, and 10 sec @ 72oC. The PCR products were further verified, purified and sequenced as mentioned above.
Table 2.
ompA and MLST primers used in this study
| Chlamydia | Target | Primer/probe | Sequence (5'-3') | Amplicon Size (bp) | MLST fragment (bp) |
|---|---|---|---|---|---|
| C. suis |
Cs_ompA
VD 1-2 |
UP | TTGAACATTTGGGATCGTTTTGA | 491 | _ |
| DN | CCAATGTAAGGAGTGAACATATTTAATCTG | ||||
| gatA | UP | TAAAAGTGCTTTAGAATTAAGAGATGCTGT | 539 | 425 | |
| DN | AGATGCTGGCTGACGAATCGA | ||||
| oppA_3 | UP | AGATATCAGTGGGAATCTGCTTGC | 674 | 468 | |
| DN | TAAGGATTTTTTTCCAATTTAAGCCAT | ||||
| hflX | UP | CTCTCCCTCTCAACAACGGAACTT | 616 | 435 | |
| DN | TTCAATAACATGCAGCAAAATATCCTC | ||||
| gidA | UP | TTTGGGAGTTTCTACGAAGGAAGG | 570 | 474 | |
| DN | ATAATTTCATATTGTACATCGAAAGGCAT | ||||
| enoA | UP | TCTCGGGGTCTCTTTAGCATTAGC | 590 | 381 | |
| DN | CTCCAACGAGTTGAATACGATCTCC | ||||
| hemN | UP | GAAGAGCTTGCTATTGAATTTGATCC | 608 | 432 | |
| DN | CGTTTTGTAGATAGATTCCTCGAATGA | ||||
| fumC | UP | CTCTTATGGGAAAGAATTGATGCCT | 641 | 465 | |
| DN | TACTTTCTCTACAAAACCTTCAGGAACATT | ||||
| C. abortus | Cab_ompA | UP | TACAAGCCTTGCCTGTAGGGAAC | 360 | _ |
| DN | CAGAAAATATCAAAGCGATCCCAG | ||||
| C. pecorum | Cpe_ompA | UP | ATGAAAAAACTCTTAAAATCGGCGT | 420 | _ |
| DN | CAGAAAATATCAAAGCGATCCCAG |
C. suis-specific MLST analysis
In this study, a C. suis-specific MLST typing scheme based on a previously published Chlamydiales MLST scheme [32, 33] was developed. The scheme was designed to target the partial fragments of seven C. suis HK genes. The selected genes are not adjacent to putative outer membrane, secreted, or hypothetical and are separated widely on the chromosome. In addition, each locus for the selected genes demonstrated a similar degree of nucleotide substitutions to provide consistency [27].
Amplification primers were designed based on the genome sequence of C. suis MD56 to amplify fragments of the genes encoding aspartyl/glutamyl-tRNA amidotransferase subunit A (gatA) (amplicon length: 539 bp), oligobinding protein (oppA) (674 bp), GTP-binding protein (hflX) (616bp), tRNA uracil-5-methyl transferase (gidA) (570 bp), enolase (enoA) (590 bp), Coproporphyrinogen III oxidase (hemN) (608bp), and fumarate hydratase class II (fumC) (641bp) (Table 2). PCR amplification of the seven HK genes was performed in a LightCycler 480-II real-time PCR platform following the above-mentioned protocol. Each sequence run was performed from a different PCR amplicon and sequence traces were obtained with automated DNA sequencing. Briefly, forward and reverse chromatograms for each sequenced HK gene fragment were aligned and trimmed, and the fragment sequence for that allele was obtained. Sequences for each gene were then aligned in MEGA 6.0 using the ClustalW multiple alignment algorithm and trimmed to appropriate lengths for Chlamydiales MLST fragments [32].
After the optimization and development, the C. suis-MLST was successfully applied to C. suis positive samples from nasal (n=12) and rectal (n=7) swabs, 6 whole blood (n=6) and 1 lung samples (n=1) from 26 pigs. Phylogenetic analyses were performed using DNASp 5.0 [34] and Geneious 9 [35]. DnaSP 5.0 was used to analyze the sequence polymorphisms: by determining the number of synonymous (dS) and non-synonymous (dN) substitutions per site, Jukes-Cantor corrected, the number of polymorphic sites, haplotypes and haplotypesper allele. Allele and sequence type (ST) assignation for 26 C. suis strains described in this study and one Italian strain-MD56 were determined and deposited at http://pubmlst.org/chlamydiales/ [36].
Phylogenetic analysis
Phylogenetic analyses were performed using the 489 bp variable region of the ompA gene and the concatenated C. suis MLST sequences. For ompA, a total of 127 sequences consisting of 50 publicly available sequences obtained from GenBank and 77 sequences from this study were aligned using the ClustalX 1.83. Based on these alignments, the NJ phylogenetic trees were constructed using the Kimura 2-parameter model. The MEGA 6.0. Bootstrap values were calculated by the use of 500 replicates.
A Bayesian phylogenetic tree using an alignment of a total of 40 concatenated MLST sequences from the 26 C. suis strains from this study, and 14 additional strains from Switzerland, USA, Italy and Austria. C. suis isolates was constructed with MRBAYES [37] with the GTR+G model, as implemented in Geneious 9. Run parameters included four Markov Chain Monte Carlo (MCMC) chains with a million generations, sampled every 1000 generations and with the first 10000 trees were discarded as burn-in. C. trachomatis MLST sequences were used as an outgroup to determine the mid-point root of the C. suis phylogeny. MLST gene fragments were extracted from publicly available genomes of the strains SWA-2 (GenBank acc. No. NZ_LT821323), 14-23b (NZ_FSSG01000023), 1-28b (NZ_FTQD01000002), 3-25a (NZ_FTQO01000003), 10-26b (NZ_FTQJ01000001), 5-22b (NZ_FTQB01000001), 8-29b (NZ_FTQU01000001), 3-29b (NZ_FTPY01000001- NZ_FTPY01000002), S45 (SRA accession: SRX1868493), R19 (SRX1868490), Rogers130 (SRX1868491), R1 (SRX1868495), R16 (SRX1868494), MD56 (NZ_AYKJ00000000).
Minimum spanning tree and identification of clonal complexes (CCs) using the goeBURST algorithm [38] on the C. suis MLST data from this study was performed using the https://online.phyloviz.net [39]. The MLST profiles were clustered into CCs under a user-defined threshold level of identity. In this study, we used relaxed triple locus variant (TLV) level where CCs were defined as groups of sequencing types (STs) which share four out of seven alleles with at least one other ST in the group, while the satellite STs were defined as STs that differ by at least three alleles from all other STs. A 'putative progenitor' in CCs is a ST that has the most single locus variant (SLV) links to other STs.
In order to compare phylogenetic inferences based on MLST and ompA alignments, we have constructed a tanglegram from cladograms of Bayesian C. suis MLST and ompA phylogenetic trees, and a strict consensus tree, computed with Dendroscope 3 [40]. For this analysis, we have also constructed a mid-point rooted Bayesian phylogenetic tree using the 489 bp ompA fragment alignment from the same 40 (26 Chinese and 14 global) C. suis strains used for MLST-derived phylogeny. The tree parameters included GTR+I model and four Markov Chain Monte Carlo (MCMC) chains with a million generations, sampled every 1000 generations and with the first 10000 trees were discarded as burn-in.
In order to infer recombination break points within each alignment, we used Dual-Brothers recombination detection with default setting (as implemented in Geneious 9). DualBrothers detects recombination based on the dual Multiple Change-Point (MCP) model which finds changes in topology and evolutionary rates across sites in a multiple sequence alignment [41].
Statistical analysis
The Chi-squared Test was used to compare the positivity of C. suis infection in different samples of pigs. The two-tailed Tukey honest significant difference (HSD) test in one-way ANOVA was used to analyze the differences of C. suis copy numbers in different samples. Differences at P≤0.05 were considered significant.
Results
Prevalence of Chlamydia spp. in pigs
C. suis was found to be the only chlamydial species in all samples in this study with an overall positivity of 62.4% (1,334/2,137). In addition, 26.1% (6/23) of lung samples from Fujian province tested positive for C. suis (Table 1). Notably, a cohort of SPF pigs had 80.0% (52/65) of C. suis positivity in pigs less than two months old and 95.4% positivity (41/43) in pigs older than two months (Table 1).
Prevalence and copy numbers of C. suis affected by samples types
Nasal swabs, conjunctival swabs, rectal swabs and whole blood samples were collected from 100 pigs in Zhejiang province for C. suis detection. The prevalence and average bacterial genome numbers of whole blood samples was significantly lower than in other sample types (P<0.05). The C. suis positivity was 8.0% (8/100) in whole blood, 66.0% (66/100) in nasal swabs, 63.0% in conjunctival swabs (66/100), and 60.0% in rectal swabs (60/100). In addition, the average bacterial genome numbers of C. suis were 103.50±0.20 [SEM] per ml whole blood), 105.34±0.16 per rectal swab; 104.39±0.14 per conjunctival swab, and 104.37±0.14 per nasal swab (Fig. 1).
Fig. 1.
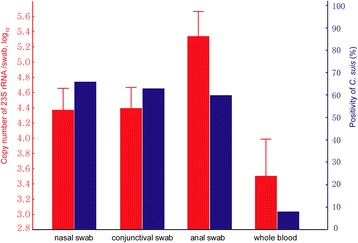
Prevalence and copy numbers of C. suis affected by samples type of pigs. Copy number (± SEM) is shown in the left ordinate (in red). Positivity is shown in the right ordinate (in blue). FRET-qPCR were performed to determine the positivity and copy number of C. suis from nasal, conjunctival, rectal swabs and whole blood samples of 50 sows and 50 weaned pigs in this study. The positivity of C. suis in nasal swabs (66%), conjunctival swabs (63%) and rectal swabs (60%) was significantly higher than whole blood (P<0.01, 8%). The average copy number of C. suis was highest in rectal swab (P<0.01, 105.34±0.16 [SEM] per swab), followed by conjunctival swab (104.39±0.14 per swab) and nasal swab (104.37±0.14 per swab), and whole blood (P<0.05, 103.50±0.20 per ml).
C. suis in pigs of different ages
C. suis positivity in nasal swabs was found to be similar in finisher pigs (84.0%; 84/100) and weaned pigs (84.0%; 262/312), which were significantly higher than in suckling pigs (69.4%, 225/324; P<0.01) and sows (31.3%, 61/195; P<0.01). Interestingly, the average copy number of C. suis was significantly higher in weaned pigs (P<0.01, 103.88±0.06 [SEM] per swab) than in sows (103.11±0.09 per swab), finisher pigs (103.09±0.07 per swab) and suckling pigs (102.94±0.05 per swab) (Fig. 2).
Fig. 2.

Prevalence and copy numbers of C. suis affected by ages of pigs. Copy number (± SEM) is shown in the left ordinate (in red). Positive rate is shown in the right ordinate (in blue). FRET-qPCR were performed to determine the positivity and copy number of C. suis from nasal swabs of suckling pigs (n=324), weaned pigs (n=312), finisher pigs (n=100) and sows (n=195) in this study. The positivity of C. suis was the lowest in sow (P<0.01, 31.3%), followed by finisher pigs (84.0%) and weaned pigs (84.0%), and suckling pigs (P<0.05, 69.4%). The average copy number of C. suis was significantly higher in weaned pigs (P<0.01, 103.88±0.06 [SEM] per swab) than sows (103.11±0.09 per swab), finisher pigs (103.09±0.07 per swab) and suckling pigs (102.94±0.05 per swab).
Tet(C) and tetR(C) genes
For C. suis-positive samples, 6/45 (13.3%) of whole blood samples from Yangzhou in Jiangsu, 39/45 (86.7%) of rectal swabs from Kunming in Yunnan and 8/9 (88.9%) of rectal swabs from Shaoxing in Zhejiang province contained the tet(C) gene while 15/45 (33.3%) of whole blood samples and 5/8 (62.5%) of rectal swabs samples were found to be positive for the tetR(C) gene while 6 of 45 (13.3%) whole blood samples and 47 of 54 (87.0%) rectal swabs samples contained the tet(C) gene. We also tested the presence of tet(C) genes in C. suis-negative samples in this study. For C. suis-negative samples, no (0/40; 0.0%) whole blood samples from Yangzhou in Jiangsu and 19/38 (50%) of 53 (54.7%) rectal swabs from Kunming in Yunnan and 10/15 (66.7%) of rectal swabs from Shaoxing in Zhejiang province contained the tet(C) gene. Both C. suis tet(C) and tetR(C) were detected in samples from pigs in 3 provinces sampled in this study. In a word, the tet(C) positivity in C. suis-positive samples was significantly higher than in C. suis-negative ones for whole blood from Jiangsu (13.3%, 6/45 vs. 0.0%, 0/40; P<0.01) and rectal swabs from Yunnan and Zhejiang (87.0%, 47/54 vs. 54.7%, 29/53; P<0.01).
C. suis ompA molecular typing
We obtained 77 distinct partial ompA sequences encompassing the variable domain 1 and 2 (VD1-2) from 108 pig C. suis strains (Fig. 3, Additional file 1). Phylogenetic analyses showed that the partial ompA VD1-2 sequences from C. suis strains from China identified in this study are highly polymorphic when compared with the existing ompA sequences deposited in GenBank. Percent sequence similarity observed within the Chinese cohort was 76.0%-100%, while between the Chinese cohort and the GenBank sequences, only 64.0%-100% sequence similarity could be observed. When compared with global ompA sequences, the Chinese C. suis ompA VD1-2 sequences could be resolved into four major clades of the global ompA VD1-2 phylogram, suggesting that the global diversity in C. suis strains is captured in the Chinese pig population (Figure 3). While considerable sequence variation was observed, some identical strains were also detected including the detection of at least one sequence (Ya/ChPP854/Nasal) in Zhejiang that was identical to the sequence from a C. suis strain (AB270743) deposited from Japan. Fine-detailed epidemiological analysis of these sequences further revealed that the same ompA sequences could be found in different cities and provinces, suggesting that some strains were more widely distributed than other strains.
Fig. 3.
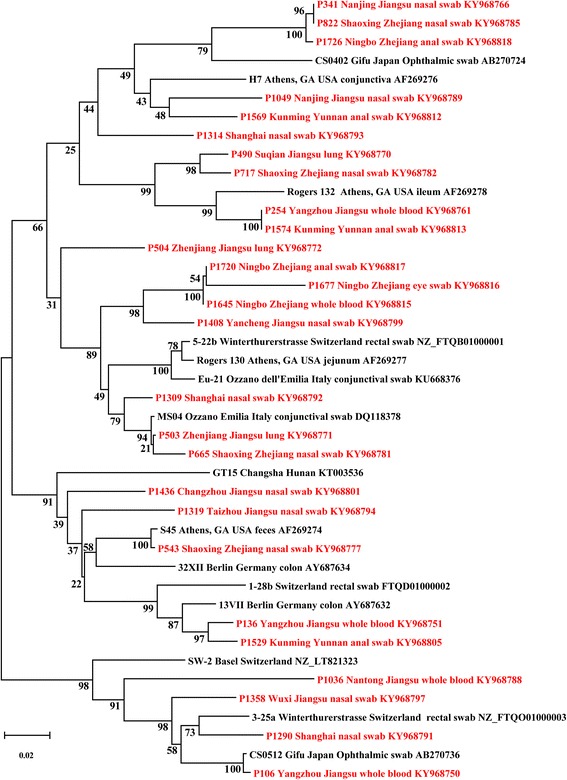
Mid-point rooted NJ phylogenetic tree of the ompA variable domains 1-2. A 489-bp fragment encompassing C. suis ompA VD1-2 of 27 porcine C. suis strains identified in this study (in red font; name of strain, countries, sample type and accession number) are compared with 15 other C. suis sequences deposited in GenBank from six countries (Germany, Switzerland, Italy, USA, Japan and China). Branch lengths are measured in nucleotide substitutions and numbers show branching percentages in bootstrap replicates. Scale bar represents the percent sequence diversity.
C. suis MLST analyses
In this study, we also assessed the genetic diversity of the C. suis strains detected using a C. suis-specific MLST scheme. The MLST of the 27 global C. suis strains confirmed high genetic diversity for this pathogen, identifying 26 novel STs among 27 strains (0.96 ST/strain). Sequence analysis of individual as well as concatenated C. suis MLST gene fragments from this study confirmed that the C. suis HK genes are conserved and under purifying selection with dN/dS ratios < 1, yet with high haplotype diversity. The number of mutations was comparable between the C. suis HK alleles, ranging from 29 SNPs in gidA to 12 SNPs in gatA, with most resulting mainly in synonymous substitutions (Table 3). Each MLST allele had 3 to 8 predicted putative recombination events.
Table 3.
Sequence analyses of the 27 C. suis MLST alleles and concatenated sequences
| Allele | Total number of polymorphisms (Δnt) | No. of non-synonymous substitutions | No. of synonymous substitutions | dN/dS ratio | Number of haplotypes |
|---|---|---|---|---|---|
|
gatA (425 bp) |
12 | 1 | 11 | 0.010 | 11 |
|
oppA_3 (468 bp) |
17 | 3 | 14 | 0.061 | 12 |
|
hflX (435 bp) |
24 | 2 | 22 | 0.022 | 18 |
|
gidA (474 bp) |
29 | 4 | 25 | 0.074 | 18 |
|
enoA (381 bp) |
23 | 1 | 22 | 0.007 | 23 |
|
hemN (432 bp) |
17 | 2 | 15 | 0.113 | 14 |
|
fumC (465 bp) |
14 | 2 | 12 | 0.024 | 14 |
| Concatenated (3080 bp) |
136 | 15 | 121 | - | 26 |
Due to its congruency with whole or core genome phylogeny, the concatenated MLST sequences have been previously used to infer phylogenetic relationships between the strains of other chlamydial species [42]. To assess the phylogenetic relationships between the Chinese and other global C. suis strains, a mid-point rooted Bayesian phylogenetic tree was constructed from an alignment of a total of 40 strains (Fig. 4a). Using C. trachomatis as an out-group, the Chinese isolates resolved into seven diverse larger clades, clustering on their own or with European and US isolates. In their own well-supported clades, Chinese C. suis strains formed many distinct lineages. When clustered with European and the USA isolates within a larger clade, Chinese isolates also grouped in separate but diverse sub-clades. Similarly, US and European strains also formed distinct well-supported clades (Fig. 4a).
Fig. 4.
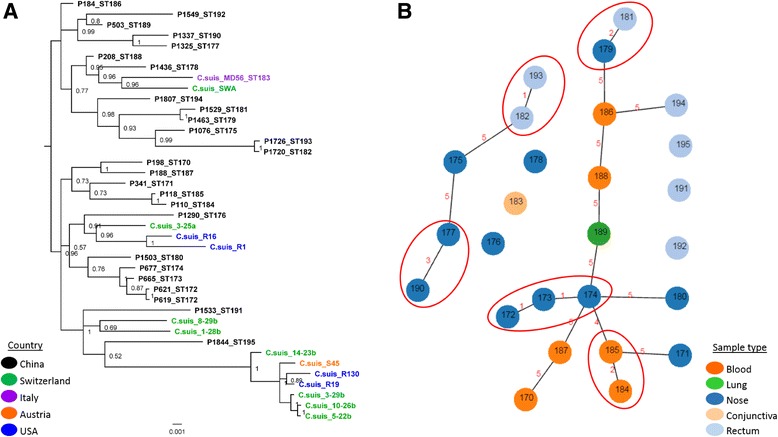
Phylogenetic and cluster analyses of C. suis STs. a: Bayesian phylogenetic analysis of the concatenated sequences of seven MLST fragments of 40 C. suis strains. Posterior probabilities are displayed on tree nodes, while the geographical origin of the strain is indicated by different colors as outlined in the legend; b: Cluster analyses of C. suis STs, N locus difference is indicated on the link between the nodes. Clonal complexes are circled in red. Sample types from which the C. suis STs were derived are indicated by the colors, as outlined in the legend.
The phylogenetic analyses further resolved the on-farm epidemiology of Chinese C. suis infections including evidence of sub-clades consisting of: a) related STs obtained from various anatomical sites of different hosts (as observed in the sub-clade consisting of nasal STs 190 and 177, rectal ST 192 and lung ST 189; and b) closely related STs obtained from the same anatomical site from different hosts (such as all nasal STs 172, 173, 174 and 180 sub-clade and blood STs 185 and 184, and 170 and 187 sub-clades).
The C. suis STs cluster analyses and minimum spanning tree also confirmed high ST diversity (Fig. 4b). Using a relaxed fit where STs must share at least 4/7 alleles, the 26 STs clustered into five clonal complexes (CCs, and 15 satellite STs (Figure 4b). Only two CCs resolved based on a stringent fit differing by a single locus. CC 1 consisted of nasal only isolates denoted STs 172, 173 and 174, with ST 172 as a predicted progenitor, while CC 2 consisted of rectal STs 182 and 193, with ST 182 predicted as a putative progenitor.
Comparison of C. suis MLST and ompA phylogenies
In order to further compare phylogenetic relationships of C. suis strains, we produced a tanglegram of both MLST and ompA trees to estimate phylogenetic positioning for each taxa (strain) (Fig. 5). As observed in Fig. 5, only six taxa, including the USA R1 and R16, Chinese P619 and P621; and Swiss 5-22b and 10-26b strains, maintained the same phylogenetic clustering in both trees, whereas all other taxa clustered differently in each tree.
Fig. 5.
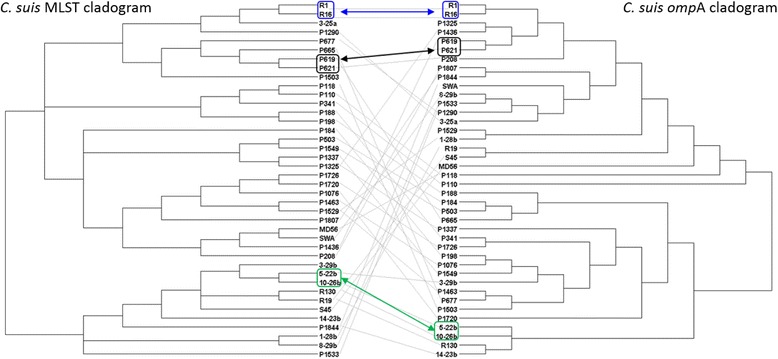
Tanglegram of C. suis MLST and ompA Bayesian trees, transformed as a cladograms, from this study. Same positioning of the taxa is indicated by colored squares and arrows, where blue indicates the USA C. suis isolates, black Chinese and green Swiss isolates.
We additionally tested for the recombination break points and changes in topology in the MLST and ompA alignments using DualBrothers. Based on observed break-points (Additional files 2, 3 and 4), we can infer at least one putative recombination event in the C. suis MLST alignment, and at least four in the ompA alignment.
Discussion
The present study investigated the prevalence of C. suis in five different sample types from 2,137 pigs from 19 regions of 10 provinces in China. The present PCR methodology represents a reliable diagnostic tool for rapid, highly sensitive and specific detection of C. suis, bypassing the need for C. suis cell culture [29, 31, 43]. We reported an overall prevalence of 62.4% (1,334/2,137) positive animals in the genus-specific FRET-PCR with our results confirming a high prevalence of C. suis in all pig herds. We failed to detect any evidence of C. abortus, C. pecorum and C. psittaci DNA. This study provides the first evidence for C. suis infection in commercially raised pigs in China. Furthermore, this is also the first report to find that 86.1% (93/108) SPF pigs were C. suis-positive.
Our study was the first to show the presence of C. suis in blood samples. For paired samples from the same host, nasal, conjunctival and rectal swabs were C. suis-positive when the whole blood samples were also C. suis-positive. Furthermore, 6/23 lung samples were C. suis-positive, suggesting that systemic infection is a common outcome of the haemtogenous spread of the bacteria.
In this study, generally, the infection rate of sows was significantly lower when compared with suckling, weaned and finisher pigs (Fig. 2). These observations might be attributed to antibiotic treatment and good housing conditions for sows or protective immunity in the adult animals. Housing conditions and management systems might contribute to controlling the transmission of chlamydial infections in pigs [10]. However, these results show that despite these practices, C. suis was prevalent in pigs at all stages of their production cycle.
While C. suis was found to be the most common species in pigs [44], this organism was mostly often found in the intestine [45–47] and conjunctiva [48, 49], and in other sites, including the genital tract [50], nasal specimens [51], lung [52] and the aborted fetuses [53].
In terms of transmission, in this study, the prevalence of C. suis DNA in rectal swabs was significantly higher when compared to those taken from nose and eyes, with whole blood samples having the lowest C. suis DNA incidence. This result is most likely due to the gastrointestinal tract being the primary site infection and chlamydial replication. Alternatively, it is also possible that pigs can swallow C. suis eliminated from the respiratory tract by mucociliary clearance [51]. Furthermore, the incidence of C. suis DNA in rectal, nasal, conjunctival swab samples and whole blood samples was significantly higher for weaned pigs when compared with sows. Data of the present study may suggest the potential C. suis transmission via a fecal-oral route. Besides the fecal-oral route as a potential route of transmission, the transmission may presumably also occur via exchange of body fluids, particularly secretions from the conjunctiva and nose, which may also lead to aerosol formation [10]. Hamonic et al. [54] recommended that it is possible that viable C suis shed in boar semen could cross current biosecurity barriers, meaning vertical transmission may also need to be considered a route of transmission.
Since 1998, the number of reports of TcR C. suis infections in pigs are growing in the USA, Italy, Cyprus, Germany, Israel, Switzerland and Belgium [18–20, 22, 55]. In the present study, the tet(C) gene was detected in clinical samples from 3 provinces, which suggests that the presence of the tet(C) gene in C. suis is common in the Chinese domestic pig population. Selective pressure from tetracycline as drug may be responsible for recent bottlenecks in C. suis populations [26]. In this study, we were able to test only clinical samples instead of C. suis isolates. However, we cannot exclude that the tet(C) gene of other bacteria was detected, and 54.7% C. suis-negative rectal swab samples was found to be tet(C) positive in this study. For both whole blood and rectal swabs samples in this study, the tet(C) positivity of C. suis-positive samples were significantly higher than C. suis-negative samples, suggesting these C. suis strains contained tet(C) gene. This may be associated with the inclusion of 10-50 gram oxytetracycline in every 1,000 kilogram pig feed in the farms examined and in the Chinese pig industry. Given the apparently prevalence of this tet(C) positivity in C. suis, it is concerning to note that, Suchland et al. [23] previously demonstrated the in vitro horizontal transfer of tetracycline resistance from C. suis to clinical strains of C. trachomatis, an important human pathogen. As such, C. suis may transfer tetracycline resistance to other chlamydial species, through mixed infections of livestock or farmers. Therefore, for the Chinese pig industry and public health, effective preventive action, such as probiotics or vaccines, should be promoted to counteract it. Targeted surveillance of tetracycline-resistant C. suis strains may also be warranted to curb the spread of these unusual chlamydial strains.
Phylogenetic analyses showed that the ompA VD1-2 gene fragment of the C. suis strains is highly polymorphic (Fig. 3). C. suis MLST confirmed these observations, possibly suggesting a shared ancestry of some Chinese strains with those described in the USA and Europe. Combining phylogenetic and cluster analyses findings, and in absence of typing paired samples from a single host, C. suis epidemiology nevertheless appears complex.
The genome of C. suis is very plastic combining unprecedented diversity with significant levels of recombination predicted between strains as well as plasmid exchange [26, 56]. Although we only used MLST and a partial ompA sequence, phylogenetic and sequence analyses from the present study provided evidence of recombination in Chinese C. suis strains. We observed differing phylogenetic positioning of the strains when constructing phylogenies using different genes, and identified of putative change points in tree topology and sequence, all indicative of recombination in the Chinese strains as well. In combination with the extreme prevalence in swine herds, the well-known fusion of chlamydial inclusion in closely related C. trachomatis [23], but not other chlamydial lineages, may be the pre-requisite for the unprecedented genome-wide recombination frequency of C. suis. If so, these mechanisms would continuously create, even within single farms, new strains that explore the full evolutionary bandwidth and enable C. suis escape immune protection of the host. Systematic genome sequencing may reveal if C. suis strains of different geographic origin that are identical at, e.g., the ompA locus, such as Chinese strain P854 and Japanese strain AB270743, are truly identical or are the result of convergent evolution at this locus, but are otherwise divergent across the rest of the C. suis chromosome.
Conclusions
The epidemiological surveys in this study indicate that C. suis infection in Chinese pigs is common. Molecular typing of detected strains suggest that, like elsewhere, C. suis are genetically diverse and that the global diversity of this pathogen is reflected in the diversity of strains detected in Chinese pigs. Further research should be performed to study the route of transmission for C. suis, and the potential impacts of C. suis on pig production.
Additional files
Mid-point rooted NJ phylogeny of ompA variable domains 1-2. A 489 bp region encompassing ompA VD1-2 sequences of 77 porcine C. suis strains identified in this study (in red font; name of strain, countries, sample type and accession number) are compared with 50 other C. suis sequences deposited in GenBank from six countries: Germany, Switzerland, Italy, USA, Japan and China (in black font). Branch lengths are measured in nucleotide substitutions and numbers show branching percentages in bootstrap replicates. Scale bar represents the percent sequence diversity. (PDF 376 kb)
DualBrother recombination detection using the 489bp ompA fragment alignment of 26 Chinese C. suis strains. The top plot shows marginal posterior probabilities of the four most probable tree topologies, where break and change points of topologies are indicative of recombination. The last two plots show 95% Bayesian confidence interval (shaded in green) of the Kappa transition/transversion ratio, and average divergence Mu. (PDF 141 kb)
DualBrother recombination detection using the 489bp ompA fragment alignment of all 40 C. suis strains used in this study. The top plot shows marginal posterior probabilities of the four most probable tree topologies, where break and change points of topologies are indicative of recombination. The last two plots show 95% Bayesian confidence interval (shaded in green) of the Kappa transition/transversion ratio, and average divergence Mu. (PDF 157 kb)
DualBrother recombination detection using the concatenated 3,080 bp MLST alignment of all 40 C. suis strains used in this study. The top plot shows marginal posterior probabilities of the four most probable tree topologies, where break and change points of topologies are indicative of recombination. The last two plots show 95% Bayesian confidence interval (shaded in green) of the Kappa transition/transversion ratio, and average divergence Mu. (PDF 143 kb)
Acknowledgment
This publication made use of the PubMLST website (http://pubmlst.org/) developed by Keith Jolley (16) and hosted at the University of Oxford. The development of that website was funded by the Wellcome Trust.
Funding
This work was supported by a grant from the National Key R & D Program of China (2016YFD0500804) and a grant from the National Natural Science Foundation of China (NO: 31272575), and by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Yangzhou, Jiangsu, P. R. China.
Availability of data and materials
The ompA sequences generated in this study were deposited in GenBank with the accession numbers (Figure 3; Additional file-1). The datasets used and/or analysed during the current study available from the corresponding author on reasonable request
Abbreviations
- FRET
fluorescence resonance energy transfer
- MCMC
Markov Chain Monte Carlo
- ompA
out major protein A
Authors’ contributions
Experimental design by ML and CW. Project funding secured by CW. Sample collection by ML, FY, JG, ZF, CS, PJ, JL, JZ, YW, JW and XZ. Sample processing by ML, FY, JG, ZF, CS, PJ, JL, JZ, YW, JW and XZ. Data analysis by ML, MJ, BK, AP, YP, NB and CW. Manuscript by ML, MJ, BK, AP and CW. All authors have read and approved this manuscript.
Ethics approval and consent to participate
Protocols for the collection of samples in this study were reviewed and approved by the Institutional Animal Care and Use Committee of Yangzhou University College of Veterinary Medicine (YZU-CVM#2015-057). Written permission was obtained from the owners of the pigs used in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12917-017-1295-x) contains supplementary material, which is available to authorized users.
Contributor Information
Min Li, Email: limin6707@163.com.
Martina Jelocnik, Email: Martina.Jelocnik@research.usc.edu.au.
Feng Yang, Email: 674360059@qq.com.
Jianseng Gong, Email: jjsensen@163.com.
Bernhard Kaltenboeck, Email: kaltebe@auburn.edu.
Adam Polkinghorne, Email: apolking@usc.edu.au.
Zhixin Feng, Email: fzxjaas@163.com.
Yvonne Pannekoek, Email: y.pannekoek@amc.uva.nl.
Nicole Borel, Email: n.borel@access.uzh.ch.
Chunlian Song, Email: 466569879@qq.com.
Ping Jiang, Email: jiangp@njau.edu.cn.
Jing Li, Email: lijing900401@163.com.
Jilei Zhang, Email: zhangjilei0103@163.com.
Yaoyao Wang, Email: 495930788@163.com.
Jiawei Wang, Email: 912911025@qq.com.
Xin Zhou, Email: zhouxin99@vip.126.com.
Chengming Wang, Email: wangche@auburn.edu.
References
- 1.Rohde G, Straube E, Essig A, Reinhold P, Sachse K. Chlamydial zoonoses. Dtsch Arztebl Int. 2010;107(10):174–180. doi: 10.3238/arztebl.2010.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reinhold P, Hartmann H, Constable PD. Characterisation of acid-base abnormalities in pigs experimentally infected with Chlamydia suis. Vet J. 2010;184(2):212–218. doi: 10.1016/j.tvjl.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 3.De Puysseleyr K, De Puysseleyr L, Geldhof J, Cox E, Vanrompay D. Development and validation of a real-time PCR for Chlamydia suis diagnosis in swine and humans. PLoS One. 2014;9(5):e96704. doi: 10.1371/journal.pone.0096704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hulin V, Oger S, Vorimore F, Aaziz R, de Barbeyrac B, Berruchon J, et al. Host preference and zoonotic potential of Chlamydia psittaci and C. suis in poultry. Pathog Dis. 2015;73(1):1–11. doi: 10.1093/femspd/ftv005. [DOI] [PubMed] [Google Scholar]
- 5.Sachse K, Bavoil PM, Kaltenboeck B, Stephens RS, Kuo CC, Rosselló-Móra R, et al. Emendation of the family Chlamydiaceae: proposal of a single genus, Chlamydia, to include all currently recognized species. Syst Appl Microbiol. 2015;38(2):99–103. doi: 10.1016/j.syapm.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Schautteet K, Vanrompay D. Chlamydiaceae infections in pig. Vet Res. 2011;42:29. doi: 10.1186/1297-9716-42-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanrompay D, Geens T, Desplanques A, Hoang TQ, De Vos L, Van Loock M, et al. Immunoblotting, ELISA and culture evidence for Chlamydiaceae in sows on 258 Belgian farms. Vet Microbiol. 2004;99(1):59–66. doi: 10.1016/j.vetmic.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Szeredi L, Schiller I, Sydler T, Guscetti F, Heinen E, Corboz L, et al. Intestinal Chlamydia in finishing pigs. Vet Pathol. 1996;33(4):369–374. doi: 10.1177/030098589603300401. [DOI] [PubMed] [Google Scholar]
- 9.Hoelzle LE, Steinhausen G, Wittenbrink MM. PCR-based detection of chlamydial infection in swine and subsequent PCR-coupled genotyping of chlamydial omp1-gene amplicons by DNA-hybridization, RFLP-analysis, and nucleotide sequence analysis. Epidemiol Infect. 2000;125(2):427–439. doi: 10.1017/S0950268899004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Puysseleyr L, De Puysseleyr K, Braeckman L, Morré SA, Cox E, Vanrompay D. Assessment of Chlamydia suis infection in pig farmers. Transbound Emerg Dis. 2017;64(3):826–833. doi: 10.1111/tbed.12446. [DOI] [PubMed] [Google Scholar]
- 11.Chahota R, Ogawa H, Ohya K, Yamaguchi T, Everett KD, Fukushi H. Involvement of multiple Chlamydia suis genotypes in porcine conjunctivitis. Transbound Emerg Dis. 2017; 10.1111/tbed.12645. [DOI] [PubMed]
- 12.Polkinghorne A, Borel N, Becker A, ZH L, Zimmermann DR, Brugnera E, et al. Molecular evidence for chlamydial infections in the eyes of sheep. Vet Microbiol. 2009;135(1-2):142–146. doi: 10.1016/j.vetmic.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 13.Pantchev A, Sting R, Bauerfeind R, Tyczka J, Sachse K. Detection of all Chlamydophila and Chlamydia spp. of veterinary interest using species-specific real-time PCR assays. Comp Immunol Microbiol. 2010;33(6):473–484. doi: 10.1016/j.cimid.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Lemus JA, Fargallo JA, Vergara P, Parejo D, Banda E. Natural cross chlamydial infection between livestock and free-living bird species. PLoS One. 2010;5(10):e13512. doi: 10.1371/journal.pone.0013512. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Dean D, Rothschild J, Ruettger A, Kandel RP, Sachse K. Zoonotic Chlamydiaceae Species Associated with Trachoma, Nepal. Emerg Infect Dis. 2013;19(12):1948–1955. doi: 10.3201/eid1912.130656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenart J, Andersen AA, Rockey DD. Growth and development of tetracycline-resistant Chlamydia suis. Antimicrob. Agents Chemother. 2001;45(8):2198–2203. doi: 10.1128/AAC.45.8.2198-2203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dugan J, Rockey DD, Jones L, Andersen AA. Tetracycline resistance in Chlamydia suis mediated by genomic islands inserted into the chlamydial inv-like gene. Antimicrob Agents Chemother. 2004;48(10):3989–3995. doi: 10.1128/AAC.48.10.3989-3995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Francesco A, Donati M, Rossi M, Pignanelli S, Shurdhi A, Baldelli R, et al. Tetracycline-resistant Chlamydia suis isolates in Italy. Vet Rec. 2008;163(8):251–252. doi: 10.1136/vr.163.8.251. [DOI] [PubMed] [Google Scholar]
- 19.Borel N, Regenscheit N, Di Francesco A, Donati M, Markov J, Masserey Y, et al. Selection for tetracycline-resistant Chlamydia suis in treated pigs. Vet Microbiol. 2012;156(1-2):143–146. doi: 10.1016/j.vetmic.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Schautteet K, De Clercq E, Miry C, Van Groenweghe F, Delava P, Kalmar I, et al. Tetracycline-resistant Chlamydia suis in cases of reproductive failure on Belgian, Cypriote and Israeli pig production farms. J Med Microbiol. 2013;62(Pt 2):331–334. doi: 10.1099/jmm.0.042861-0. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann K, Schott F, Donati M, Di Francesco A, Hässig M, Wanninger S, et al. Prevalence of chlamydial infections in fattening pigs and their influencing factors. PLoS One. 2015;10(11):e0143576. doi: 10.1371/journal.pone.0143576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wanninger S, Donati M, Di Francesco A, Hässig M, Hoffmann K, Seth-Smith HM, et al. Selective Pressure Promotes Tetracycline Resistance of Chlamydia suis in Fattening Pigs. PLoS One. 2016;11(11):e0166917. doi: 10.1371/journal.pone.0166917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suchland RJ, Sandoz KM, Jeffrey BM, Stamm WE, Rockey DD. Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob Agents Chemother. 2009;53(11):4604–11. doi: 10.1128/AAC.00477-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes JP, Bruno WJ, Nunes A, Santos N, Florindo C, Borrego MJ, et al. Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 2007;17(1):50–60. doi: 10.1101/gr.5674706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunelle BW, Sensabaugh GF. The ompA gene in Chlamydia trachomatis differs in phylogeny and rate of evolution from other regions of the genome. Infect Immun. 2006;74(1):578–585. doi: 10.1128/IAI.74.1.578-585.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph SJ, Marti H, Didelot X, Read TD, Dean D. Tetracycline Selective Pressure and Homologous Recombination Shape the Evolution of Chlamydia suis: A Recently Identified Zoonotic Pathogen. Genome Biol Evol. 2016;8(8):2613–2623. doi: 10.1093/gbe/evw182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- 28.Guo W, Li J, Kaltenboeck B, Gong J, Fan W, Wang C. Chlamydia suis, not C. psittaci, is the endemic chlamydial species in chicken (Gallus gallus) Sci Rep. 2016;6:19638. doi: 10.1038/srep19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Guo W, Kaltenboeck B, Sachse K, Yang Y, Lu G, et al. Chlamydia pecorum is the endemic intestinal species in cattle while C. gallinacea, C. psittaci and C. pneumoniae associate with sporadic systemic infection. Vet Microbiol. 2016;193:93–99. doi: 10.1016/j.vetmic.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Wei L, Kelly P, Zhang J, Yang Y, Zheng X, Tao J, et al. Use of a universal hydroxymethylbilane synthase (HMBS)-based PCR as an endogenous internal control and to enable typing of mammalian DNAs. Appl Microbiol Biotechnol. 2014;98(12):5579–5587. doi: 10.1007/s00253-014-5659-x. [DOI] [PubMed] [Google Scholar]
- 31.De Graves FJ, Gao D, Hehnen HR, Schlapp T, Kaltenboeck B. Quantitative detection of Chlamydia psittaci and C. pecorum by high-sensitivity real-time PCR reveals high prevalence of vaginal infection in cattle. J Clin Microbiol. 2003;41(4):1726–1729. doi: 10.1128/JCM.41.4.1726-1729.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pannekoek Y, Morelli G, Kusecek B, Morré SA, Ossewaarde JM, Langerak AA, et al. Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 2008;8:42. doi: 10.1186/1471-2180-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pannekoek Y, Dickx V, Beeckman DS, Jolley KA, Keijzers WC, Vretou E, et al. Multi Locus Sequence Typing of Chlamydia Reveals an Association between Chlamydia psittaci Genotypes and Host Species. PLoS One. 2010;5(12):e14179. doi: 10.1371/journal.pone.0014179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 35.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huelsenbeck JP, Ronquist FMRBAYES. Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 38.Francisco AP, Bugalho M, Ramirez M, Carriço JA. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics. 2009;10:152. doi: 10.1186/1471-2105-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribeiro-Gonçalves B, Francisco AP, Vaz C, Ramirez M, Carriço JA. PHYLOViZ Online: web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 2016;44(W1):W246–W251. doi: 10.1093/nar/gkw359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61(6):1061–1067. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 41.Minin VN, Dorman KS, Fang F, Suchard MA. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics. 2005;21(13):3034–3042. doi: 10.1093/bioinformatics/bti459. [DOI] [PubMed] [Google Scholar]
- 42.Jelocnik M, Walker E, Pannekoek Y, Ellem J, Timms P, Polkinghorne A. Evaluation of the relationship between Chlamydia pecorum sequence types and disease using a species-specific multi-locus sequence typing scheme (MLST) Vet Microbiol. 2014;174(1-2):214–222. doi: 10.1016/j.vetmic.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 43.Sachse K, Grossmann E, Jäger C, Diller R, Hotzel H. Detection of Chlamydia suis from clinical specimens: comparison of PCR, antigen ELISA, and culture. J Microbiol Methods. 2003;54(2):233–238. doi: 10.1016/S0167-7012(03)00040-X. [DOI] [PubMed] [Google Scholar]
- 44.Longbottom D. Chlamydial infections of domestic ruminants and swine: new nomenclature and new knowledge. Vet J. 2004;168(1):9–11. doi: 10.1016/S1090-0233(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 45.Zahn I, Szeredi L, Schiller I, Straumann Kunz U, Bürgi E, Guscetti F, et al. Immunohistochemical determination of Chlamydia psittaci/pecorum and C. trachomatis in the piglet gut. Zentralbl Veterinarmed B. 1995;42(5):266–276. [PubMed] [Google Scholar]
- 46.Schiller I, Koesters R, Weilenmann R, Kaltenboeck B, Pospischil A. PCR detection of porcine Chlamydia trachomatis and ruminant Chlamydia psittaci serovar 1 DNA in formalin-fixed intestinal specimens from swine. Zentralbl Veterinarmed B. 1997;44(3):185–191. doi: 10.1111/j.1439-0450.1997.tb00963.x. [DOI] [PubMed] [Google Scholar]
- 47.Pollmann M, Nordhoff M, Pospischil A, Tedin K, Wieler LH. Effects of a probiotic strain of Enterococcus faecium on the rate of natural Chlamydia infection in swine. Infect Immun. 2005;73(7):4346–4353. doi: 10.1128/IAI.73.7.4346-4353.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker A, Lutz-Wohlgroth L, Brugnera E, ZH L, Zimmermann DR, Grimm F, et al. Intensively kept pigs predisposed to chlamydial associated conjunctivitis. J Vet Med A Physiol Pathol Clin Med. 2007;54(6):307–313. doi: 10.1111/j.1439-0442.2007.00963.x. [DOI] [PubMed] [Google Scholar]
- 49.Davidson HJ, Rogers DP, Yeary TJ, Stone GG, Schoneweis DA, Chengappa MM. Conjunctival microbial flora of clinically normal pigs. Am J Vet Res. 1994;55(7):949–951. [PubMed] [Google Scholar]
- 50.Kauffold J, Melzer F, Berndt A, Hoffmann G, Hotzel H, Sachse K. Chlamydiae in oviducts and uteri of repeat breeder pigs. Theriogenology. 2006;66(8):1816–1823. doi: 10.1016/j.theriogenology.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 51.Reinhold P, Liebler-Tenorio E, Sattler S, Sachse K. Recurrence of Chlamydia suis infection in pigs after short-term antimicrobial treatment. Vet J. 2011;187(3):405–407. doi: 10.1016/j.tvjl.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 52.Sachse K, Hotzel H, Slickers P, Ellinger T, Ehricht RDNA. microarray-based detection and identification of Chlamydia and Chlamydophila spp. Mol Cell Probes. 2005;19(1):41–50. doi: 10.1016/j.mcp.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 53.Schiller I, Koesters R, Weilenmann R, Thoma R, Kaltenboeck B, Heitz P, et al. Mixed infections with porcine Chlamydia trachomatis/pecorum and infections with ruminant Chlamydia psittaci serovar 1 associated with abortion in swine. Vet Microbiol. 1997;58(2-4):251–260. doi: 10.1016/S0378-1135(97)00154-5. [DOI] [PubMed] [Google Scholar]
- 54.Hamonic G, Pasternak JA, Käser T, Meurens F, Wilson HL. Extended semen for artificial insemination in swine as a potential transmission mechanism for infectious Chlamydia suis. Theriogenology. 2016;86(4):949–956. doi: 10.1016/j.theriogenology.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 55.Andersen A, Rogers D. Resistance to tetracycline and sulfadiazine in swine C. trachomatis isolates. Editors: Stephens, R. S. In: Ninth International Symposium on Human Chlamydial Infection. San Fransico: Conference Proceedings. 1998; pp. 313-316
- 56.Seth-Smith HM, Wanninger S, Bachmann N, Marti H, Qi W, Donati M, et al. The Chlamydia suis genome exhibits high levels of diversity, plasticity and mobile antibiotic resistance: comparative genomics of a recent livestock cohort shows influence of treatment regimes. Genome Biol Evol. 2017;9(3):750–760. doi: 10.1093/gbe/evx043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mid-point rooted NJ phylogeny of ompA variable domains 1-2. A 489 bp region encompassing ompA VD1-2 sequences of 77 porcine C. suis strains identified in this study (in red font; name of strain, countries, sample type and accession number) are compared with 50 other C. suis sequences deposited in GenBank from six countries: Germany, Switzerland, Italy, USA, Japan and China (in black font). Branch lengths are measured in nucleotide substitutions and numbers show branching percentages in bootstrap replicates. Scale bar represents the percent sequence diversity. (PDF 376 kb)
DualBrother recombination detection using the 489bp ompA fragment alignment of 26 Chinese C. suis strains. The top plot shows marginal posterior probabilities of the four most probable tree topologies, where break and change points of topologies are indicative of recombination. The last two plots show 95% Bayesian confidence interval (shaded in green) of the Kappa transition/transversion ratio, and average divergence Mu. (PDF 141 kb)
DualBrother recombination detection using the 489bp ompA fragment alignment of all 40 C. suis strains used in this study. The top plot shows marginal posterior probabilities of the four most probable tree topologies, where break and change points of topologies are indicative of recombination. The last two plots show 95% Bayesian confidence interval (shaded in green) of the Kappa transition/transversion ratio, and average divergence Mu. (PDF 157 kb)
DualBrother recombination detection using the concatenated 3,080 bp MLST alignment of all 40 C. suis strains used in this study. The top plot shows marginal posterior probabilities of the four most probable tree topologies, where break and change points of topologies are indicative of recombination. The last two plots show 95% Bayesian confidence interval (shaded in green) of the Kappa transition/transversion ratio, and average divergence Mu. (PDF 143 kb)
Data Availability Statement
The ompA sequences generated in this study were deposited in GenBank with the accession numbers (Figure 3; Additional file-1). The datasets used and/or analysed during the current study available from the corresponding author on reasonable request