

In this study, Mazzucco et al. describe a panel of genetic screens to identify genes required for replicative senescence and uncover a role in senescence for the potent tumor suppressor and ATM substrate USP28. Their findings suggest a role for ubiquitination in senescence and imply a common node downstream from ATM that links the TP53 and GATA4 branches of the senescence response.
Keywords: GATA4, senescence, USP28
Abstract
Senescence is a terminal differentiation program that halts the growth of damaged cells and must be circumvented for cancer to arise. Here we describe a panel of genetic screens to identify genes required for replicative senescence. We uncover a role in senescence for the potent tumor suppressor and ATM substrate USP28. USP28 controls activation of both the TP53 branch and the GATA4/NFkB branch that controls the senescence-associated secretory phenotype (SASP). These results suggest a role for ubiquitination in senescence and imply a common node downstream from ATM that links the TP53 and GATA4 branches of the senescence response.
Limits on the replicative life span of human cells have evolved to prevent the propagation of damaged cells. Senescence is one such barrier that is executed by tumor suppressor and cell cycle regulatory pathways and is implicated in organismal aging (Baker et al. 2011, 2016). Senescence bypass leads to cancer predisposition (Campisi 2005). DNA damage is a unifying feature of many stimuli that trigger senescence (Karlseder et al. 2002; Bartkova et al. 2006; Di Micco et al. 2006). Cell cycle arrest, DNA damage response (DDR) activation, life span, and senescence-associated β-galactosidase (SA-β-Gal) activity are frequently used to identify senescent cells (Campisi et al. 1996; Serrano et al. 1997). The p53 and p16-Rb tumor suppressor pathways are the best-understood senescence regulators (Wright and Shay 1992; Smogorzewska and de Lange 2002; Beausejour et al. 2003). p53-mediated expression of the CDK inhibitor p21 is a critical mediator of p53's role to promote senescence in human cells (Brown et al. 1997). In addition, the GATA4/SASP (senescence-associated secretory phenotype) pathway forms a third branch that controls senescence (Kang et al. 2015). In response to ATM/ATR activation, GATA4 is stabilized and activates NFκB. NFκB mediates expression of secreted inflammatory cytokines (Courtois-Cox et al. 2006; Acosta et al. 2008; Chicas et al. 2010). The mechanisms by which senescence is established and maintained remain to be fully elucidated. Here we present genome-wide senescence bypass screens that uncover a potent tumor suppressor gene (TSG), USP28, which controls multiple branches of the senescence pathway.
Results and Discussion
To identify genes regulating senescence in primary human cells, we sensitized human BJ fibroblasts by expression of either the HPV E6 or E7 oncoprotein that selectively inhibits the p53 or Rb pathway, respectively (Supplemental Fig. S1A), followed by infection with retroviral MSCV-driven miR30-based shRNA libraries, including a pilot scale library consisting of 8203 shRNA and a whole-genome library (Fig. 1A; Supplemental Fig. S1B,C; Materials and Methods). E6 and E7 were used to reduce the contribution of these partially redundant branches to allow greater sensitivity in detecting defects in the remaining branch. Mid-passage BJ fibroblasts were transduced in triplicate with a shRNA library and serially passaged until the cells expressing a control shRNA (luciferase [Luc]) ceased population doublings (PDs). p53 was the positive control (Supplemental Figs. S1C, S2A). shRNAs were recovered by PCR, and the fold change of shRNAs between the start and end was used to generate a list of candidate senescence genes (Fig. 1A; Supplemental Fig. S2B,C; Supplemental Tables S1–S4; see the Materials and Methods).
Figure 1.
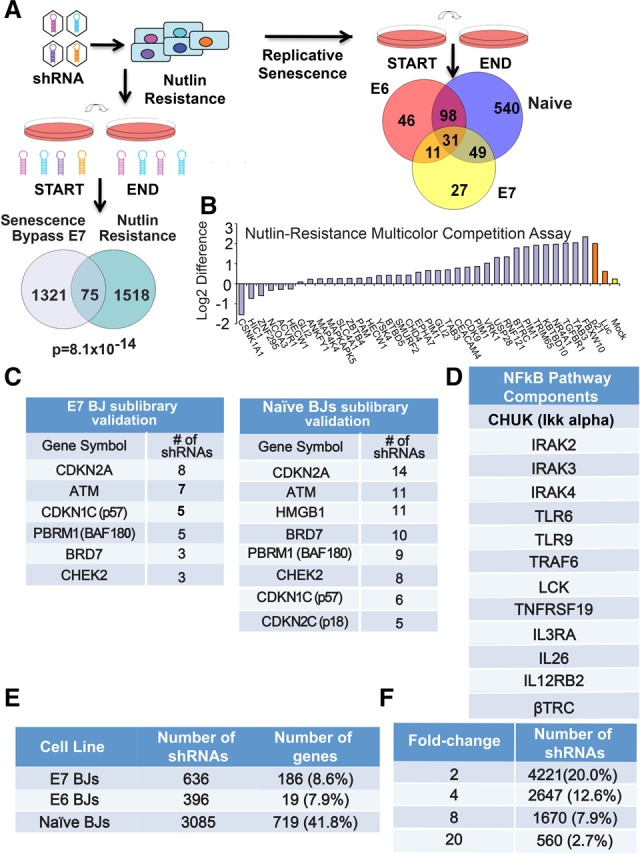
Loss-of-function screens identify high-confidence candidate genes. (A) Schematic showing screen design and library pool deconvolution for the senescence bypass and nutlin resistance screens. (Bottom left) Venn diagram showing the overlap between the senescence bypass screen candidate gene list in E7 BJ fibroblasts and the nutlin resistance screen performed in human mammary epithelial cells (HMECs) for the genome-scale library. The probability of overlap was calculated by hypergeometric probability distribution. (Right) Venn diagram showing the gene overlap between the senescence resistance validation screens in E6, E7, and naïve BJs. The numbers shown refer to the number of genes that were enriched twofold or greater and targeted by three or more shRNAs. (B) Representative validation data from nutlin resistance screens as shown by multicolor competition assays in HMECs in the presence or absence of nutlin-3a for three PDs. Cells expressing either Luc shRNA or mock-infected served as negative controls in HMECs expressing GFP (green fluorescent protein). Cells expressing shRNAs against p21 served as positive controls. Data are plotted as the difference between the log2 ratios of untreated and nutlin-treated cells. (C) Validation rescreen data indicating the number of selected shRNAs that scored to twofold or greater above the average of 100 negative control shRNAs for the indicated subset of genes for the sublibrary validation screen in E7 BJ fibroblasts (left) and naïve BJs (right). (D) Components of the NFκB pathway that scored in our screens in E6 BJ fibroblasts. (E) Table showing the number of selected shRNAs that scored twofold or greater with three or more shRNAs. (F) Table showing the number of shRNAs that scored above each threshold relative to the average of the negative control shRNAs in the validation screen performed in naïve BJ fibroblasts.
Both the pilot and genome-scale E6 BJ screens recovered known TSGs, including RB1, p57KIP2, p16INK4a, p18INK4c, and PTEN, suggesting targeting of the RB pathway and genes controlling p53-independent growth arrest (Supplemental Fig. 2D,E; Supplemental Tables S1, S2). This data set showed statistically significant overlap with targets of recurrent cancer-associated mutations (P = 0.035) (Forbes et al. 2011). As expected, p53 shRNAs were not recovered, but p21 shRNAs were, suggesting a p53-independent function in senescence.
Likewise, both E7 BJ screens recovered multiple shRNAs against the known p53 pathway, DNA damage, and cell cycle components such as p53, p21CIP1, p14ARF, βTRCP, PIAS1, ATR, TGFβ1, and TGFβR1 (Supplemental Fig. S2E,F; Supplemental Tables S3, S4). This data set was enriched for cell cycle-related (P = 0.047) and cancer-related genes (P = 0.041) using KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway annotation (Kanehisa 1997) and targets of recurrent cancer-associated mutations (P = 0.040) (Forbes et al. 2011), suggesting that some genes may be TSGs. Of the genes meeting our criteria, 10% were targeted by two or more shRNA in the E6 BJ screen, and 18% were targeted by two or more shRNA in the E7 BJ screen, highlighting higher-confidence candidates.
While largely distinct, the E6 and E7 screens have more overlapping genes than expected randomly (P = 0.008), suggesting that the shared genes may act independently of both p53 and Rb. For example, we recovered multiple components of the interleukin-1 and NF-κB pathways known to function in the GATA4 branch, such as IL1RL1, IRAK2, IRAK4, IKBKB, JAK1, TNFRSF17, and MAPK8 (Fig. 1D; Orjalo et al. 2009). Furthermore, we recovered multiple shRNAs against CDKN2A encoding both p16 and ARF, which are in the Rb and p53 branches, respectively. Additionally, feedback signaling in the MAP kinase pathway is required for oncogene-induced senescence (Courtois-Cox et al. 2006), and we identified negative regulators of the MAP kinase pathway, including SMEK1, Sprouty2, RASGEF1B, DUSP1, DUSP3, DUSP11, DUSP13, and DUSP4, regardless of genetic background.
Identifying senescence genes connected to the p53 pathway
To ask which genes might function in the p53 branch, we screened HCT116 colon cells and normal human mammary epithelial cells (HMECs) for resistance to nutlin-3a, a molecule that activates p53 by inhibiting MDM2 (Vassilev 2004). Such genes would be potential effectors of p53 activation and might bypass senescence in BJ E7 cells. Nutlin3a-treated and control cells were passaged for 10 PDs and analyzed for shRNA abundance (Fig. 1A; see the Materials and Methods). We recovered shRNAs against p53 and pathway components such as p21CIP1, p14ARF, βTRCP, NR4A1, PIM1, PDCD4, and ZBTB4 (Supplemental Tables S5–S7). Many positive shRNAs were shared by both HCT116 cells and HMECs (P = 2.5 × 10−21) (Supplemental Table S8). We used a proliferation competition assay to validate individual shRNAs from our screen (Fig. 1B; Supplemental Fig. S2G; see the Materials and Methods). Testing 125 candidates, we had a validation rate of 37% (Fig. 1B; Supplemental Fig. S2H). Candidates showing a twofold or greater enrichment are listed in Supplemental Table S9. As expected, we found no significant overlap with the E6 BJ screen but a striking overlap with hits from E7 BJ cells (P = 8.10 × 10−14) (Fig. 1A, Venn diagram). A common data set from all of our library screens includes known p53 pathway components and many unstudied genes and represents a high-confidence data set for further studies of p53-mediated cell cycle arrest (Supplemental Table S10). Furthermore, our nutlin resistance data set had significant overlap with a data set of frequently occurring mutations associated with cancer (P = 0.032) (Forbes et al. 2011), suggesting putative TSGs in this data set.
Validation of senescence bypass candidate genes via sublibrary rescreening
To validate our data sets, we generated a library containing 12 shRNAs against the top 1800 scoring genes, including all genes that overlapped between the senescence and nutlin screens (Supplemental Table S11). Extremely potent genes such as CDKN1A or USP28 (discussed below) were excluded, as they overshadow less potent shRNAs. This library included 50 shRNAs targeting firefly Luc and 50 shRNAs targeting GFP (green fluorescent protein) as negative controls. Any shRNA enriching twofold or greater above the mean of these controls across all three replicates was considered a positive. We also screened naïve BJ fibroblasts to find genes that may extend life span in unperturbed cells.
The validation screens recovered multiple shRNAs against many known senescence, DDR, and cell cycle regulatory genes (Fig. 1C; Supplemental Tables S12–S14). We recovered several components of the interleukin-1 and NF-κB pathways known to control the SASP (Fig. 1D; Orjalo et al. 2009). Approximately 8% of genes identified in the primary screens scored with the stringent criteria of twofold or greater enrichment with three or more shRNAs and thus represent higher-confidence candidates (Fig. 1E). Increasing criteria stringency to fourfold or eightfold enrichment still maintains significant percentages of these shRNAs (Fig. 1F), suggesting that many exhibited robust performance.
We found genes that overlapped between two or more cell lines and also genes that scored in only one particular genetic background, supporting the value of our sensitized approach (Fig. 1A, Venn diagram). When comparing data sets between two lines or across all three lines, we recovered more shRNAs in common than predicted by chance (P = 4.3 × 10−7 for data sets recovered from E6 BJs and naïve BJs, P = 2.8 × 10−16 for data sets recovered from E7 BJs and naïve BJs, and P = 1.7 × 10−65 for E7 BJs and E6 BJs). The shRNAs identified genes that regulate either a third pathway, such as GATA4, or both the p53 and RB pathways. This analysis suggests that we successfully enriched for life span and proliferation regulators that may function in multiple genetic backgrounds.
USP28 is required for p53 regulation during replicative and IR-induced senescence
The deubiquitinating enzyme USP28 was among the most enriched genes in the senescence E7 BJ and nutlin screens, suggesting a role in p53 function. USP28 binds 53BP1 and has been implicated previously in the DDR and apoptosis following IR. It is required for proper induction of the proapoptotic p53 target PUMA (Zhang et al. 2006) and stabilization of MYC (Popov et al. 2007) but had not been implicated in senescence. USP28 depletion significantly increased the proliferation rate and strikingly delayed senescence (Fig. 2A–C; Supplemental Fig. S3A). The life span extension was comparable with p53 depletion. USP28 depletion also decreased SA-β-Gal 7 d after 12 Gy of IR in IMR90 fibroblasts (Fig. 2D), suggesting a role in IR-induced senescence; led to nutlin-3a resistance (Supplemental Fig. S3B); and resulted in life span extension in E7 BJ cells (Supplemental Fig. S3C,D).
Figure 2.
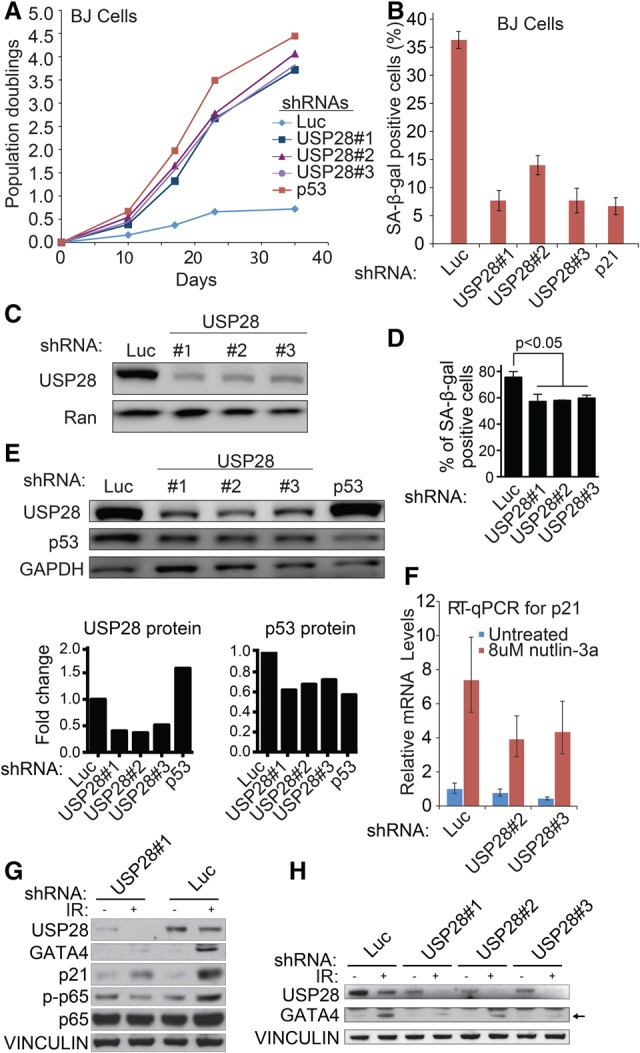
USP28 is required for p53, p21, and GATA4 induction during senescence. (A) Growth of late passage BJ cells expressing the indicated shRNAs. (B) Quantitation of SA-β-Gal staining in BJ cells expressing the USP28 shRNAs in A. (C) Western blot analysis of USP28 levels in BJ cells expressing the indicated shRNAs in A. (D) Quantitation of IR-induced SA-β-Gal staining in BJ cells expressing the indicated shRNAs. Data are mean ± SEM. Statistical significance was calculated by one-way analysis of variance (ANOVA). Data are representative of four independent experiments. (E) Western blot analysis of late passage BJ cells expressing the indicated shRNAs. (Below) Densitometric analysis to determine the USP28/GAPDH and p53/GAPDH ratios. (F) RT-qPCR for p21 expression in mid-passage BJ fibroblasts expressing the indicated shRNAs with and without 8 µM nutlin treatment at 6 h. (G) Western blot analysis of IMR90 cells expressing the indicated shRNAs 7 d after 12 Gy of IR treatment. (H) Western blot analysis of IMR90 cells expressing the indicated shRNAs 7 d after treatment with 12 Gy of IR. The arrow indicates GATA4 protein.
USP28 depletion decreased p53 protein, but not p53 transcript, levels in BJ cells undergoing replicative senescence (Fig. 2E; Supplemental Fig. S3E,F). Furthermore, USP28 depletion reduced levels of basal and nutlin-3a-induced p21CIP1 expression (Fig. 2F), suggesting that USP28 may also affect p21CIP1 levels during replicative senescence, likely through p53. Importantly, p53 depletion leads to an increase in USP28, indicating the existence of a potential negative feedback loop (Fig. 2E; Supplemental Fig. S3H). Together, these results support the idea that USP28's role in senescence and growth arrest is due in part to a role in the p53 pathway.
The GATA4–SASP branch of the senescence pathway also requires USP28
To explore the p53-independent roles of USP28, we examined GATA4 levels after IR-induced senescence. USP28 depletion caused marked reduction of GATA4 accumulation (Fig. 2G,H). Thus, USP28 is required for both p53 and GATA4 activation during senescence. To test whether USP28 activity is sufficient for senescence induction, USP28 was overexpressed and found to induce senescence (as indicated by growth arrest and positive SA-β-Gal staining) (Fig. 3A,B; Supplemental Fig. S3I,J) comparable with p21CIP1 overexpression. USP28 overexpression increased the levels of multiple senescence markers, including p53, p21CIP1, and GATA4 proteins but not p16, again suggesting that USP28 may be inducing senescence in part through the p53 and GATA4 branches (Fig. 3C–E; Supplemental Fig. S3K,L). Although the magnitude of the SA-β-Gal effects vary based on infection efficiency, it was consistently observed using both constitutive and inducible USP28 expression systems (Fig. 3B; Supplemental S3L). USP28 overexpression does not increase GATA4 mRNA (Supplemental Fig. S3G), suggesting post-transcriptional regulation. Importantly, expression of a catalytically inactive mutant, USP28C171A, did not exert these effects, indicating that the deubiquitinating activity of USP28 is required for senescence (Fig. 3A–E; Supplemental Fig. S3I). Reduced growth and increased p53 and p21 levels were also observed in E7 BJs, again only upon overexpression of wild-type, but not catalytically inactive, USP28 (Supplemental Fig. S3J,K).
Figure 3.
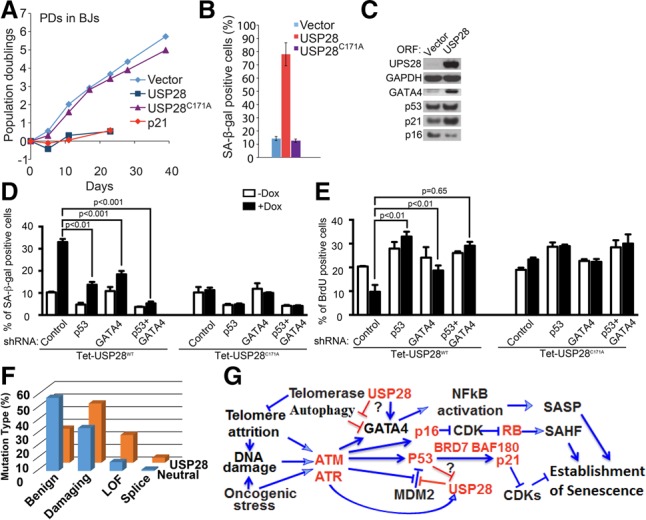
USP28 is a TSG that controls p53 and GATA4 in a catalytic activity-dependent manner. (A) Growth of mid-passage BJ fibroblasts expressing the indicated protein or empty vector. (B) Quantitation of SA-β-Gal staining in mid-passage BJ fibroblasts expressing the indicated protein or empty vector. (C) Western blotting of BJ cells expressing USP28 or an empty vector for 10 d. (D) SA-β-Gal staining on day 7 of doxycycline (Dox) treatment of BJ cells carrying a Dox-inducible vector (Tet-on) expressing either wild-type USP28 or the USP28C171A mutant and the indicated shRNAs. Data are mean ± SEM. Statistical significance was calculated by one-way ANOVA. Data are representative of three independent experiments. (E) Levels of BrdU-positive cells on day 7 of Dox treatment of the BJ cells described in F. Data are mean ± SEM. Statistical significance was calculated by ANOVA. Data are representative of three independent experiments. (F) The distribution of different classes of USP28 mutations in a collection of 8200 tumors compared with a typical neutral gene (Davoli et al. 2013). Polyphen2 was used to predict missense mutations as benign or damaging. A loss-of-function mutation is defined as either a stop codon or frameshift. (G) A schematic diagram indicating major senescence pathways driving cell cycle arrest and the formation of senescence-associated heterochromatic foci (SAHFs) and the SASP as well as points where USP28 acts within these pathways, with genes identified in this study indicated in red.
To determine the dependency of p53 and GATA4 in USP28-induced senescence, we combined doxycycline-inducible (Tet-on) USP28 overexpression with either p53 inhibition (via E6 expression) or GATA4 depletion in BJ fibroblasts. E6 or GATA4 depletion blunted USP28-induced senescence (Supplemental Fig. S4A). While either p53 or GATA4 depletion via shRNA reduced USP28-induced senescence as measured by SA-β-Gal activity, codepletion of p53 and GATA4 resulted in a synergistic effect (Fig. 3D). However, codepletion of p53 and RB (via E6 and E7 expression) did not exert a synergistic effect, consistent with our data showing that USP28 does not regulate p16 (Supplemental Fig. S4A). Last, while the percentage of BrdU-positive cells decreased significantly following USP28 overexpression, cells depleted of either p53 or GATA4 showed no significant reduction in BrdU staining (Fig. 3E). Together, these results indicate that p53 and GATA4 each independently contributes to USP28-induced senescence. Furthermore, as USP28, like p53 and GATA4, is regulated by ATM/ATR, our experiments pinpoint USP28 as a potential downstream effector of ATM and ATR that mediates GATA4 induction during senescence.
USP28 displays a strong mutational signature as a TSG
Given the strong connection between senescence and tumor suppression, we examined the possibility that USP28 is a TSG. Investigating a large-scale copy number analysis of tumor samples and cancer cell lines (Beroukhim et al. 2010), we found that USP28 was significantly deleted in cancers of multiple tissues types, falling within deletion peaks, including a significant number of focal deletions (Supplemental Fig. S4B). Frequent USP28 deletions were detected within a collection of multiple cancer types (21.7%) and epithelial cancers (25.6%) and also in specific tumor types (Supplemental Fig. S4C). Examination of The Cancer Genome Atlas data (Forbes et al. 2011; Davoli et al. 2013) revealed that the P-value for deletion significance is 1.1 × 10−88 with a q-value of 2.6 × 10−86. Analysis of mutational patterns in USP28 also supported a TSG role. In the somatic mutation database, the typical neutral gene shows a high percentage of benign (nondamaging missense and silent) mutations (54%) relative to high-functional-impact missense mutations (34%) or nonsense mutations (7%). In contrast, USP28 has only 27% benign mutations, 47% high-functional-impact missense mutations, and 22% nonsense mutations (Fig. 3F). According to our analysis using the TSG prediction algorithm TUSON Explorer, USP28 ranks as the 83rd strongest TSG candidate out of 18,680 genes, with a P-value of 9.6 × 10−06 and a q-value of 2.1 × 10−03. Unlike some TSGs (such as SMAD2, which is largely specific for colorectal and stomach carcinomas), USP28 mutations span all tumor types, much like p53 mutations (Supplemental Fig. S4D). Thus, USP28 appears to be a potent TSG that may operate by promoting replicative senescence.
Senescence control pathways
The overall structure of our screens allowed us to identify three classes of senescence factors: (1) those that work independently of p53, (2) those that work independently of RB, and (3) those that work independently of both p53 and Rb branches. The latter category could include genes that either form a third pathway, such as GATA4; induce telomerase or reduce telomere attrition; or act in both pathways, such as components of the DDR (i.e., ATM, ATR, and CHEK2, which trigger senescence). We found components of all three pathways, including a strong enrichment of inflammatory signaling components (Fig. 1C,D), particularly in the E6 BJ screen, supporting the notion that SASP reinforces the p53 branch of senescence. These findings could explain the observation that cancer cell lines have been shown to exhibit SASP, which could be of clinical relevance for both diagnostic and therapeutic purposes (Coppe et al. 2008; Ohanna et al. 2011). Through screens for regulators of p53, we identified a subset of these senescence genes that may function through the p53 pathway.
USP28 is a potent TSG controlling both p53 and GATA4
USP28 stood out in our screens as an extremely robust hit. USP28 had nearly as profound an effect on senescence as p53 and p21. Our analysis of somatic mutations in cancer suggest that USP28 is a frequent target of inactivation by either deletion or point mutation and signature, ranking within the top 0.5% of potential TSGs.
USP28 is required for p53 and p21 accumulation following p53 activation (Fig. 2E–G) and is phosphorylated on at least three sites by ATM in response to IR (Zhang et al. 2006). Since these DDR components are engaged in replicative senescence, USP28 function may be explained in part by its regulation of p53. However, USP28 also controls GATA4 accumulation. p53 negatively regulates USP28, which may explain p53's known feedback role in attenuating SASP (Coppe et al. 2008), much like miR146a's role in attenuating NF-κB signaling (Bhaumik et al. 2009). USP28 now stands as a major node in the senescence machinery, responsible for activation of two major independent branches controlling both the cell cycle arrest and secretory phenotypes, which are each critical to the overall senescence response (Fig. 3G). This dual role may explain both the robust behavior of USP28 in our screens and its frequent inactivation by deletion or mutation in human cancers that we reported here. These observations highlight a potential role for ATM/ATR in regulating both p53 and GATA4 via USP28 during senescence. The USP28–GATA4 axis described here may provide a mechanistic insight into GATA4 regulation. GATA4 is degraded by autophagy through association with the p62 adaptor (Kang et al. 2015). How GATA4 is recognized by p62 is not known, but p62 possesses a UBA domain through which it can bind ubiquitinated proteins (Long et al. 2008). As USP28 is a deubiquitinase and its activity is required for senescence through GATA4, this implies a role for ubiquitin in GATA4 regulation. This could be through direct ubiquitination of either GATA4 or an associated protein. A speculative model might be that in response to DNA damage, phosphorylation of USP28 might activate it to deubiquitinate either GATA4 or an associated factor, thereby preventing its recognition by p62 and autophagic degradation, leading to GATA4 stabilization and SASP activation.
Together, our results provide a foundation for the genetic dissection of the replicative senescence program. We identified novel senescence genes in several functional classes, including genes that are required for senescence and act to transmit the DNA damage signal, are required for proper p53 function, and activate the SASP via GATA4. Future understanding of how these proteins act and are regulated in response to senescence induction signals will provide a detailed picture of how replicative senescence is accomplished and how this tumor suppression program may be circumvented in cancer cells.
Materials and methods
Cell lines and virus production
HMECs were purchased from American Type Culture Collection (ATCC), immortalized with telomerase, and maintained in MEGM (Lonza). BJ and IMR90 cells were obtained from ATCC and maintained in 3% O2. Cells were grown in DMEM with 15% FBS, penicillin–streptomycin, 2 mM L-glutamine, and 0.1 mM nonessential amino acids (Invitrogen). Retroviruses were produced as described (Schlabach et al. 2008). HCT116 cells were grown in DMEM, 10% FBS, and penicillin–streptomycin. Viral supernatants were stored at −80°C. Nutlin-3a was obtained from Cayman Chemical. HCT116 cells were grown in McCoy's medium (Invitrogen) with 10% FBS.
Genome-wide senescence resistance and nutlin resistance screens
The genome-wide shRNA screens consisted of a total of 74,905 shRNAs targeting 19,011 genes in six independent pools of 12,800 shRNAs per pool. Mid-passage primary human BJ fibroblasts expressing either E6 or E7 were transduced with the MSCV-PM-shRNA retroviral library with a representation of ∼500 at a multiplicity of infection (MOI) of <1. The pilot screens used a previously described focused human shRNA library (Schlabach et al. 2008) of 8203 shRNAs. Initial reference samples of at least 1000 cells per shRNA were collected following selection in puromycin and propagated with a representation of ≥500 cells. End samples were collected after cells expressing control Luc shRNA ceased to grow, as indicated in Figure 1. Half-hairpin barcodes were PCR-amplified, labeled, and hybridized to microarrays as described in the Supplemental Material. In the case of nutlin resistance screens, HMECs were transduced with the library at a representation of 1000 and a MOI of <1. Initial reference samples of at least 1000 cells per shRNA were collected following selection in puromycin, and pools were cultured with or without 1.5 µM nutlin-3a at a representation of ≥500. End samples were collected after 10 PDs.
Sublibrary validation screening for senescence resistance genes
A validation sublibrary was designed against ∼1800 high-confidence genes from the genome-wide and pilot screens. The sublibrary contained 12 new shRNAs per gene. Included were all shRNAs scoring in the primary screen targeting ∼1800 genes and 100 negative control shRNAs targeting Luc and EGFP. Screens were performed as described above.
Candidate gene criteria
For analysis of the E7 BJ fibroblast pilot, we used a SAM criteria of a 5% or less false discovery rate (FDR) and an average log2 cutoff of three or more across all three replicates (i.e., eightfold or greater enrichment), generating a list of 160 shRNAs (a hit rate of 1.2% of the library). For our genome-scale screen deconvoluted by sequencing, we used a sequencing read cutoff of 33 reads and ≥10-fold enrichment in any replicate, producing a list of 1396 shRNAs (a hit rate of 1.8%). Lists were combined to generate a single data set for the focused and genome screens in E7 BJ fibroblasts. We analyzed the focused set data from the E6 BJ fibroblasts using similar candidate criteria (≤5% FDR and an average log2 cutoff of three or more across three replicates) and obtained a list of 201 shRNAs (hit rate 1.5%). In the genome-scale screen performed in E6 BJ fibroblasts, we used criteria of ≥10-fold enrichment in two of three replicates to generate a list of 601 candidate shRNAs (hit rate of 0.8%). Lists were combined to generate a single data set for both screens in E6 BJ fibroblasts. Our validation sublibrary was generated using these compiled data sets. Statistical significance analysis of the overlap between data sets was calculated using hypergeometric distribution.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant GM44644 and Department of Defense grant W81XWH-15-0711 to S.J.E. S.J.E. is an Investigator with the Howard Hughes Medical Institute. A.S. is Howard Hughes Medical Institute Faculty Scholar.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.304857.117.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. 2016. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530: 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637. [DOI] [PubMed] [Google Scholar]
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. 2003. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22: 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J. 2009. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 4: 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Wei W, Sedivy JM. 1997. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277: 831–834. [DOI] [PubMed] [Google Scholar]
- Campisi J. 2005. Suppressing cancer: the importance of being senescent. Science 309: 886–887. [DOI] [PubMed] [Google Scholar]
- Campisi J, Dimri GP, Nehlin JO, Testori A, Yoshimoto K. 1996. Coming of age in culture. Exp Gerontol 31: 7–12. [DOI] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. 2010. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17: 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. 2006. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 10: 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. 2013. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155: 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638–642. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. 1997. A database for post-genome analysis. Trends Genet 13: 375–376. [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. 2015. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlseder J, Smogorzewska A, de Lange T. 2002. Senescence induced by altered telomere state, not telomere loss. Science 295: 2446–2449. [DOI] [PubMed] [Google Scholar]
- Long J, Gallagher TR, Cavey JR, Sheppard PW, Ralston SH, Layfield R, Searle MS. 2008. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J Biol Chem 283: 5427–5440. [DOI] [PubMed] [Google Scholar]
- Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, et al. 2011. Senescent cells develop a PARP-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev 25: 1245–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. 2009. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci 106: 17031–17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M. 2007. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol 9: 765–774. [DOI] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al. 2008. Cancer proliferation gene discovery through functional genomics. Science 319: 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. 2002. Different telomere damage signaling pathways in human and mouse cells. EMBO J 21: 4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT. 2004. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle 3: 419–421. [PubMed] [Google Scholar]
- Wright WE, Shay JW. 1992. The two-stage mechanism controlling cellular senescence and immortalization. Exp Gerontol 27: 383–389. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zaugg K, Mak TW, Elledge SJ. 2006. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 126: 529–542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.