

Xiao et al. report a new function for the budding yeast Cse4/CENP-A histone-fold domain interacting with inner kinetochore protein Mif2/CENP-C. They also discovered that AT-rich centromere DNA has an important role for Mif2 recruitment.
Keywords: AT-rich DNA, Cse4/CENP-A, Mif2/CENP-C, budding yeast, centromere, nucleosome
Abstract
Histone CENP-A-containing nucleosomes play an important role in nucleating kinetochores at centromeres for chromosome segregation. However, the molecular mechanisms by which CENP-A nucleosomes engage with kinetochore proteins are not well understood. Here, we report the finding of a new function for the budding yeast Cse4/CENP-A histone-fold domain interacting with inner kinetochore protein Mif2/CENP-C. Strikingly, we also discovered that AT-rich centromere DNA has an important role for Mif2 recruitment. Mif2 contacts one side of the nucleosome dyad, engaging with both Cse4 residues and AT-rich nucleosomal DNA. Both interactions are directed by a contiguous DNA- and histone-binding domain (DHBD) harboring the conserved CENP-C motif, an AT hook, and RK clusters (clusters enriched for arginine–lysine residues). Human CENP-C has two related DHBDs that bind preferentially to DNA sequences of higher AT content. Our findings suggest that a DNA composition-based mechanism together with residues characteristic for the CENP-A histone variant contribute to the specification of centromere identity.
Kinetochores are large subcellular assemblies of ∼75 polypeptides that assemble on chromosome centromeres to enable the faithful segregation of replicated daughter chromosomes during cell division (Fukagawa 2004; Biggins 2013). The discovery of three centromere-specific human autoantigens, designated CENP-A (the histone variant of CenH3), CENP-B (a sequence-specific helix–turn–helix DNA-binding factor), and CENP-C (a component of the inner kinetochore), paved the way toward a molecular characterization of centromeric chromatin and its role in kinetochore assembly (Fukagawa and Earnshaw 2014). At the fundamental level of the nucleosome, the replacement of canonical histone H3 with CENP-A in centromere-specific nucleosomes creates a platform for the recruitment of CENP-C (Carroll et al. 2010; Gascoigne et al. 2011). CENP-C is colocalized with CENP-A at metazoan centromeres and serves as a structural link between CENP-A nucleosomes and outer kinetochore proteins, thereby connecting centromeres to microtubules of the spindle apparatus for chromosome segregation (Moroi et al. 1980; Earnshaw and Rothfield 1985; Saitoh et al. 1992; Sullivan et al. 2001; Biggins 2013; Fukagawa and Earnshaw 2014). CENP-C is a key component of a multisubunit CCAN (constitutive centromere-associated network) complex establishing the inner kinetochore (Weir et al. 2016 and references 3–6 therein). Hence, the molecular basis of mutual recognition between CENP-C and the CENP-A nucleosome is central to understanding how the inner kinetochore engages with centromeric chromatin.
The core of the CENP-C polypeptide harbors three conserved domains important for centromere targeting: a highly conserved “CENP-C signature motif” that contacts the hydrophobic C terminus of CENP-A (Carroll et al. 2010; Kato et al. 2013), a central DNA-binding region that contains a CENP-C-like motif, and a homodimerization domain whose crystal structure displays a “cupin” or jelly roll structural motif (Fig. 1A; Supplemental Fig. S3A; Brown 1995; Lanini and McKeon 1995; Yang et al. 1996; Sugimoto et al. 1997; Politi et al. 2002; Cohen et al. 2008; Milks et al. 2009; Trazzi et al. 2009; Kato et al. 2013). The budding yeast CENP-C homolog Mif2 (Meeks-Wagner et al. 1986) lacks the vertebrate-specific DNA-binding region but contains one CENP-C motif and one “AT hook” (see Fig. 1A; Brown 1995; Huth et al. 1997; Reeves 2000; Cohen et al. 2008). Early studies have shown that the AT hook of Mif2 contributes to centromere targeting and chromosome segregation in vivo (Brown 1995; Lanini and McKeon 1995; Meluh and Koshland 1995; Cohen et al. 2008). Furthermore, an N-terminal domain in Mif2 associates with two kinetochore proteins (Ame1–Okp1) to facilitate outer kinetochore assembly (Hornung et al. 2014).
Figure 1.
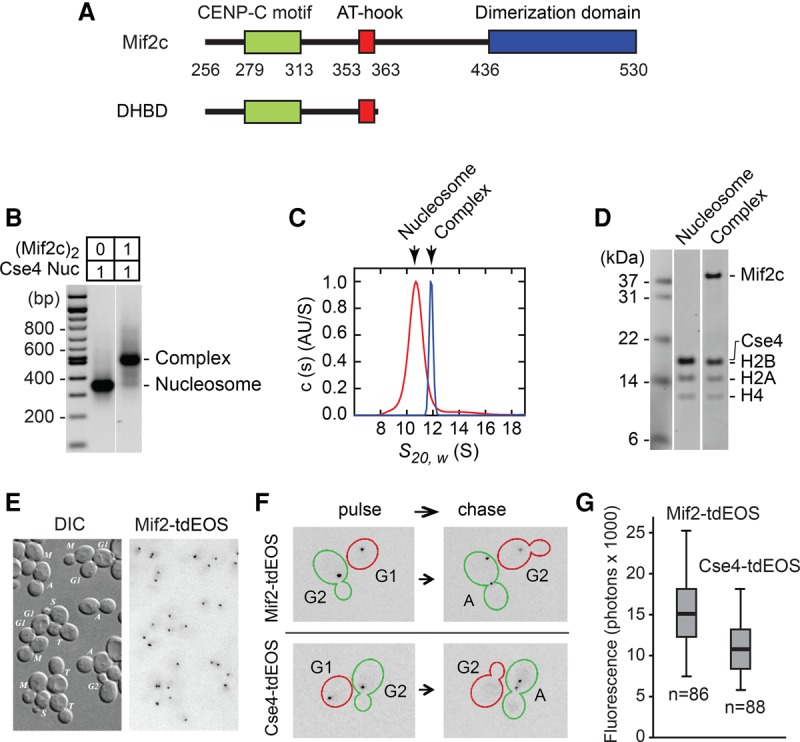
The Mif2 core (Mif2c) dimer binds stoichiometrically to a Cse4-containing nucleosome. (A) Diagram of Mif2c, the conserved core region of fungal Mif2 and the defined DNA- and histone-binding domain (DHBD) (see also Supplemental Fig. S7; Brown 1995; Cohen et al. 2008). (B) Native agarose gel electrophoresis of reconstituted Cse4/CEN3 (centromere 3) nucleosome and its complex with the Mif2c dimer. “Cse4” refers to the conserved histone-fold domain (residues 130–229) fused C-terminal to H3 tail residues 1–39. To assay for the formation of the Mif2c dimer–nucleosome complex, equimolar amounts (1:1, as indicated) of the purified Mif2c dimer and the reconstituted Cse4/CEN3 nucleosome were mixed and analyzed on a 1.3% native agarose gel. All reconstituted nucleosomes used in this figure and all other figures were analyzed on native agarose gels to assure nucleosome quality and reconstitution efficiency (see Supplemental Fig. S4A). (C) Analytical ultracentrifugation of the Cse4/CEN3 nucleosome and its complex with the Mif2c dimer as in B. Normalized absorbance c(s) distributions obtained for the Cse4/CEN3 nucleosome (red) and its complex with the Mif2c dimer (blue), each at 340 nM. The Cse4/CEN3 nucleosome yields a sedimentation coefficient of 10.69 S (estimated molar mass 185 kDa) (see also Supplemental Fig. S1A). The Mif2c dimer–Cse4/CEN3 complex has a sedimentation coefficient of 11.67 S (estimated molar mass 230 kDa). (D) SDS-PAGE of sucrose gradient peak fractions of the Cse4/CEN3 nucleosome and its complex with the Mif2c dimer. Quantification showed molar ratios of Cse4/H2B:H2A:H4 of 1:1:0.97 and of the DNA:histone octamer of 1:0.98 for free nucleosomes and of Cse4/H2B:H2A:H4:Mif2c dimer of 1:1.03:0.98:1 and of the DNA:histone octamer of 1:0.94 for Mif2c dimer-bound nucleosomes. (E) Differential interference contrast and fluorescence microscopy of Mif2-tdEos foci in G1, S, G2, and mitosis. (M) Metaphase; (A) anaphase; (T) telophase. Photoconverted (red-emitting) tdEos is shown in a negative format. (F) The persistence of Mif2-tdEos and turnover of Cse4-tdEos through S phase. Cells were imaged immediately after photoconversion (pulse) and after cell cycle progression (40- to 90-min chase). Representative cells are shown. (Red) G1-to-G2 progression through S phase; (green) G2 to anaphase of mitosis. (G) Fluorescence of Mif2-tdEos or Cse4-tdEos clusters in telophase after photoconversion (minimum, first quartile, median, third quartile, and maximum values are plotted). Parenthetically, an earlier report (Lawrimore et al. 2011) likely underestimated the Mif2:Cse4 ratio due to reliance on a malfunctioning version of Cse4 carrying a GFP tag immediately following critical QFI residues at the C terminus (Wisniewski et al. 2014).
Genetic epistasis experiments indicated that the yeast CENP-A histone variant (named Cse4) is required for recruitment of Mif2 to the centromere (Collins et al. 2005), and biochemical studies revealed close association between the two proteins in native chromatin (Biggins 2013; Westermann and Schleiffer 2013; G Mizuguchi and C Wu, unpubl.). Moreover, high-resolution ChIP-qPCR (chromatin immunoprecipitation [ChIP] combined with quantitative PCR [qPCR]) located both Mif2 and Cse4 to the genetically defined 125-base-pair (bp) yeast centromere (Cohen et al. 2008). These findings imply that for each of the 16 budding yeast chromosomes, Mif2 is specifically targeted to a single Cse4 nucleosome over thousands of conventional nucleosomes (Biggins 2013). How this enormous selectivity of Mif2 for the Cse4 nucleosome is achieved is an important question in centromere biology.
Results
Reconstitution of a budding yeast centromeric nucleosome
Previous studies demonstrated that the conserved CENP-C motif makes direct contact with hydrophobic C-terminal amino acids of CENP-A (Carroll et al. 2010; Kato et al. 2013) and, furthermore, that CENP-C binding generally stabilizes and reshapes the CENP-A nucleosome (Falk et al. 2015, 2016). To further elucidate the selectivity of the budding yeast Mif2 for the Cse4 nucleosome, we examined biochemical interactions between a reconstituted centromeric nucleosome and a budding yeast Mif2 derivative harboring the major conserved motifs of CENP-C (Mif2 core [Mif2c]) (see Fig. 1A). To this end, we reconstituted a yeast centromeric nucleosome. The nucleosome reconstitution used the natural centromere 3 (CEN3) DNA sequence (Fig. 2A) and bacterially expressed yeast histones H2A, H2B, H4, and the entire Cse4 histone fold to the C terminus (but not the divergent N-terminal tail) (Fig. 1B; Supplemental Fig. S1). We first showed by analytical ultracentrifugation (AUC) that the reconstituted Cse4/CEN3 nucleosome has a molecular mass consistent with an “octasome,” comprising two each of the histones H2A, H2B, Cse4, and H4 (Fig. 1C; Supplemental Fig. S1A). Also, consistent with previous reports (Tachiwana et al. 2011; Xiao et al. 2011), the nucleosome protects ∼120–130 bp of DNA (Supplemental Fig. S1B,C). Furthermore, hydroxyl radical footprinting showed a cleavage pattern typical of a nucleosome for the reconstituted Cse4/CEN3 nucleosome, with the nucleosome dyad closely coinciding with the in vivo nucleosome center (Supplemental Fig. S2A–C). We refer to this nucleosome as the “Cse4” nucleosome, as it had the same affinity for the Mif2c dimer as one containing full-length Cse4 (data not shown).
Figure 2.

Both Cse4 histone and CEN DNA contribute to Mif2c dimer binding. (A) CEN3 and pericentric (periCEN) DNA coordinates from in vivo nucleosome positions (Cole et al. 2011). (B–E) Representative gel scans and accompanying data graphs of electrophoretic mobility shift assays (EMSAs). Reaction mixtures containing equal amounts of two differently colored nucleosomes (labeled with Alexa fluor 555 or Alexa fuor 647) bearing Cse4 or H3 on CEN3 or periCEN DNAs (see Supplemental Fig. S4B) were incubated for 40 min at room temperature and then resolved on 1.3% native agarose gels. Gels were scanned for two-color fluorescence (GE Typhoon) and analyzed by ImageQuant, values were imported to Prism, and the percentage of Mif2c dimer-bound nucleosome was graphed. (Top gel scans) Alexa fluor 555, with closed circles indicating Mif2c dimer-bound nucleosomes. (Bottom gel scans) Alexa fluor 647, with open circles indicating Mif2c dimer-bound nucleosomes. B and F represent Mif2c dimer-bound and free nucleosomes, respectively. The equation for calculating relative affinities/binding constant (relative Kd) is from Liu-Johnson et al. (1986) (see the Materials and Methods). The average relative Kd values and standard deviation/error bars were calculated using Microsoft Excel with four to 10 concentration points from two or more independent EMSAs. For E, higher Mif2c dimer concentrations generated a second bandshift of the H3/CEN3 nucleosome, suggesting binding of two Mif2c dimers.
The Mif2 homodimer forms a 1:1 complex with the Cse4 nucleosome
We first performed AUC to show that Mif2c is a dimer in solution and binds as a dimer to DNA (Supplemental Fig. S3C) or to the nucleosome (Fig. 1C). The Mif2c dimer binds with high affinity (Kd = 0.1 nM) to the Cse4/CEN3 nucleosome, as shown by an electrophoretic mobility shift assay (EMSA) (Supplemental Fig. S3D). Thus, at nanomolar concentrations, the Mif2c dimer and the Cse4/CEN3 nucleosome form a stable complex with 1:1 stoichiometry, as measured by EMSA and AUC (Fig. 1B,C) and further confirmed by sucrose gradient centrifugation and SDS-PAGE (Fig. 1D).
To examine the in vivo stoichiometry of Mif2 and Cse4 at centromeres, we tagged the endogenous proteins with photoconvertible fluorescent protein tdEos for expression under native promoter control. In live cells, Mif2 was localized to the cluster of 16 yeast centromeres linked to a spindle pole body, in a manner identical to Cse4 at every stage of the cell cycle (Fig. 1E; e.g., see Wisniewski et al. 2014). Strikingly, photoconverted Mif2 persists into the following cell cycle (Fig. 1F), a high level of stability distinct from the complete turnover and replacement of Cse4 with newly synthesized molecules at the start of S phase (Fig. 1F; Wisniewski et al. 2014). Because of slow fluorophore maturation (t1/2 ∼ 45 min) (Wisniewski et al. 2014) and the longevity of Mif2, Mif2-tdEos harbors more mature fluorophores than does Cse4-tdEos, accounting for ∼30% higher fluorescence intensity when measured at telophase (Fig. 1G). Hence, the observed intensities are consistent with a 1:1 stoichiometry of Mif2 and Cse4 at the centromeres as measured in vitro.
The Mif2c dimer binds with a more than 1000-fold selectivity for centromeric over pericentric nucleosomes
To investigate how Mif2 discriminates between centromeric and canonical nucleosomes, we first reconstituted centromeric (CEN) and pericentric (periCEN) nucleosomes on DNA fragments with different fluorescent labels (Fig. 2A). DNA sequences for reconstitution followed the in vivo map of the nucleosome positioned at CEN3 and the pericentric nucleosome to its left (Cole et al. 2011). We then performed competitive binding experiments comparing the association of the Mif2c dimer with the in vitro reconstituted Cse4/CEN3 and H3/periCEN nucleosomes by determining their relative affinities or relative binding constant (relative Kd) using gel mobility shift analysis of bound and free fractions (see the Materials and Methods; Liu-Johnson et al. 1986). Strikingly, the Mif2c dimer bound with a more than 1000-fold preference for the Cse4/CEN3 nucleosome over the H3/periCEN nucleosome (Fig. 2B). The affinity of the Mif2c dimer for the Cse4 nucleosome reconstituted on the CEN4 sequence was the same as for CEN3 (Supplemental Fig. S4C). The three orders of magnitude preference for centromeric over pericentric nucleosomes is consistent with Mif2's function in vivo, where it must target a single Cse4 nucleosome among thousands of canonical nucleosomes along a chromosome.
Both the Cse4 histone and centromere DNA contribute to Mif2 targeting
The basis for selective targeting of Mif2 could be due to unique features of the Cse4 histone and/or centromeric DNA. Accordingly, we performed competitive binding on reconstituted nucleosomes in which the Cse4 histone or CEN3 DNA component was individually replaced. Substitution of Cse4 for H3 resulted in a substantially lower preference for the Mif2c dimer, from >2000-fold to ∼100-fold (Fig. 2C), confirming that the Mif2c dimer specifically recognizes Cse4. Remarkably, substitution of CEN3 DNA for periCEN or the Widom 601 nucleosome positioning sequence also resulted in a substantially lower preference for the Mif2c dimer, from >2000-fold to ∼40-fold to 90-fold (Fig. 2D; Supplemental Fig. S4D). Corresponding replacement of periCEN with CEN3 DNA on the H3 nucleosome increased its affinity ∼60-fold for the Mif2c dimer (Fig. 2E). These results demonstrate that both the Cse4 histone and centromere DNA are important for, and act synergistically on, the selective binding of the Mif2c dimer.
To further define Cse4 residues important for recognition by Mif2, we examined the Mif2c dimer binding to nucleosomes containing histone H3-Cse4 chimeras reconstituted on CEN3 DNA (Fig. 3A,B). Replacement of the H3 C-terminal segment from loop L2 to the C-terminal end with corresponding Cse4 sequences substantially enhanced Mif2c dimer binding to the hybrid nucleosome, from a difference of ∼100-fold to ∼14-fold (cf. Figs. 3C [chimera 1] and Fig. 2C). This is consistent with the importance of Cse4 C-terminal residues QFI (Kato et al. 2013). However, loop L1 and the α2 helix of the Cse4 histone fold were additionally required to restore full binding by the Mif2c dimer (Fig. 3D, chimera 2). A highly conserved feature of loop L1 among all CENP-A homologs is an extension of the loop by two or more amino acids (Cooper and Henikoff 2004; Baker and Rogers 2006). The three extra Cse4 L1 residues KDQ (Fig. 3A) form a distinct patch on the globular histone surface, lying orthogonal to the DNA superhelix on a surface model based on the crystal structures of the human CENP-A histone core and nucleosome (Figs. 3E, 4D, 6D [below]; Sekulic et al. 2010; Tachiwana et al. 2011). The extra loop L1 residues R80 and G81 of human CENP-A are solvent-accessible in the nucleosome structure and have been proposed for interaction with trans-acting factors (Tachiwana and Kurumizaka 2011). Thus, the extended L1 loop residues may present an additional cue for recognition by yeast Mif2 and human CENP-C. Interestingly, the L1–α2 region is required for the centromeric targeting of CENP-A (Black et al. 2004) by virtue of its association with the CENP-A chaperone HJURP/Scm3 (Zhou et al. 2011; Hong et al. 2013). Thus, loop L1 and the α2 helix of the histone fold serve two distinct functions for Cse4/CENP-A before and after histone deposition.
Figure 3.

Cse4 histone-fold loop 1–helix 2 are required for optimal binding by the Mif2c dimer. (A) Secondary structure elements and sequence comparison of the Cse4 and H3 histone-fold domains; divergent residues are highlighted in gray or colored. (B) Diagram of the H3 and Cse4 histone fold chimeras used for nucleosome reconstitution on CEN3 DNA. (C,D) EMSA showing the Mif2c dimer binding to mixtures of nucleosomes bearing Cse4 reconstituted on CEN3 DNA-Alexa fluor 555 (top gel scans; closed circles indicate Mif2c-bound) or H3-Cse4 chimeras reconstituted on CEN3 DNA-Alexa fluor 647 (bottom gel scans; open circles indicate Mif2c-bound). Binding reactions, gel electrophoresis, data collection, and quantification were performed as in Figure 2. (E) Surface model of the Cse4 core histone octamer (see the Materials and Methods; Sekulic et al. 2010; Tachiwana et al. 2011). Colors highlight the Cse4 surface residues diverged from H3 and in regions required for Mif2 binding. (Pink) Cse4 C-terminal 227F and 228I (Kato et al. 2013); (deep teal) L1 residue 171K; (red) L1 residue 172D; (orange) L1 residue 173E.
Figure 4.

Mif2c dimer footprints on one side of the Cse4 nucleosome on CEN3. (A) Nucleosomes reconstituted on 33P-labeled CEN3 DNA were incubated with purified Mif2c dimer and partially digested with DNase I, free and Mif2c dimer-bound nucleosomes were resolved on an agarose gel, bands were excised, and purified DNA was analyzed on a sequencing gel. (B) Autoradiograms and densitometry scans of top and bottom DNA strands of the Cse4/CEN3 nucleosome (see Fig. 6 for the Cse4/CEN10 nucleosome). Red lines represent free nucleosomes, and cyan lines represent Mif2c dimer-bound nucleosomes. (C) The CDEII sequence, with blue bars above and below indicating the span protected from DNase I cleavage. The triangle indicates the nucleosome dyad (see also Fig. 5; Shaytan et al. 2017). (D) Surface model of the Cse4 nucleosome (following Fig. 3F), with the DNase I footprint in blue.
Figure 6.

Mif2c dimer footprints on the other side of the Cse4 nucleosome on CEN10. (A) Nucleosomes reconstituted on 33P-labeled CEN10 DNA were incubated with purified Mif2c dimer and partially digested with DNase I, free and Mif2c-bound nucleosomes were resolved on an agarose gel, bands were excised, and purified DNA was analyzed on a sequencing gel. (B) Autoradiograms and densitometry scans of the top and bottom DNA strands of the Cse4/CEN10 nucleosome. Red lines represent free nucleosomes, and cyan lines represent Mif2c dimer-bound nucleosomes. (C) The CDEII sequence, with blue bars above and below indicating the span protected from DNase I cleavage. The triangle indicates the nucleosome center (Cole et al. 2011). (D) Surface model of the Cse4 nucleosome (following Fig. 3F), with the DNase I footprint in blue.
Site-specific binding of the Mif2c dimer on the centromeric nucleosome
The 125-bp yeast centromere DNA consists of three contiguous genetic elements: CDEI, CDEII, and CDEIII (Hegemann and Fleig 1993). We used DNase I footprinting to map the site for Mif2c dimer binding on the Cse4/CEN3 nucleosome to an ∼30-bp stretch within the ∼85-bp AT-rich CDEII element on one side of the nucleosome dyad toward CDEIII (Fig. 4). To further map precise Mif2–DNA contacts, we performed hydroxyl radical footprinting of the Mif2c dimer on the Cse4/CEN3 nucleosome. Since the hydroxyl radical is a small molecule, only sites of close contacts are protected from its attack. As shown in Figure 5, the Mif2c dimer protected ∼30 bp of DNA from hydroxyl radical cleavage, highly coincident with DNase I protection. Given the minimal binding site of 16–18 bp for the monomeric DNA- and histone-binding domain (DHBD) (Supplemental Fig. S7C), the 30-bp protection by the Mif2c dimer suggests that both of the DNA-binding domains from dimeric Mif2 engage on one side of the nucleosome. This raises the question of why a Mif2 dimer should bind to only one side of the nucleosome dyad when both sides consist of roughly equivalent AT percentage. We examined this issue and discovered a second, slower mobility shift complex upon a twofold to threefold increase of Mif2c dimer concentration in the binding reaction (Supplemental Fig. S5A). Hydroxyl radical footprinting shows that both sides of the nucleosome dyad of this complex are now protected from hydroxyl radical cleavage (Supplemental Fig. S5B–E). Thus, one Cse4/CEN3 nucleosome is capable of binding to two Mif2 dimers occupying each side of the nucleosome dyad. AT-rich DNA binding is still dominant, but the rules for the preferred binding to a specific AT-rich sequence remain unclear. We envision that the exact AT base pair arrangement could confer subtle differences in DNA bendability and/or conformation, making one AT-rich side of the dyad more attractive than the other.
Figure 5.

Hydroxyl radical footprinting of the Mif2c dimer on the Cse4/CEN3 nucleosome. (A) Nucleosomes reconstituted on 33P-labeled CEN3 DNA were incubated with purified Mif2c dimer and partially digested with iron(II)-EDTA, free and Mif2c-bound nucleosomes were resolved on an agarose gel, bands were excised, and purified DNA was analyzed on a sequencing gel. (B) Autoradiograms and densitometry scans of top and bottom DNA strands of the Cse4/CEN3 nucleosome. Red lines represent free nucleosomes, and cyan lines represent Mif2c dimer-bound nucleosomes. (C) Position of the dyad symmetry axis on the CEN3 DNA on Cse4/CEN3 nucleosome as identified by symmetry of hydroxyl radical footprinting patterns (see Shaytan et al. 2017). Top and bottom strands are shown in base-paired representation. The red arrowhead indicates the dyad at position 61.5, an ∼2-bp shift from the in vivo nucleosome center (see Fig. 4C; Supplemental Fig. S2A–C; Cole et al. 2011). (D,E) Hydroxyl radical cleavage intensity profiles of DNA strands at specific nucleotides in free Cse4/CEN3 nucleosomes and Mif2c dimer-bound nucleosomes. Intensity values of every profile were normalized from 0 to 1. The top strand plots are shown in a 5′-to-3′ direction, while the bottom strand plots are shown in a 3′-to-5′ direction. Blue bars indicate the span protected from hydroxyl radical cleavage conferred by binding of the Mif2c dimer to the nucleosome (see Supplemental Fig. S5 for footprinting of two Mif2c dimers on the Cse4/CEN3 nucleosome).
To determine whether a site between the nucleosome dyad and CDEIII is generally preferred for Mif2 interactions on other centromeric nucleosomes, we performed footprinting experiments on nucleosomes reconstituted on CEN10. Interestingly, the Mif2c dimer footprint mapped to AT-rich sequences on the opposite side of the nucleosome dyad for CEN10, toward CDEI (Fig. 6). Thus, Mif2 interacts with the Cse4 nucleosome at a dyad-adjacent site within CDEII, but its binding orientation relative to the dyad axis appears to be centromere-specific. This implies that certain sequence arrangements of the underlying AT-rich DNA are preferable to Mif2. To examine whether the protected regions observed in nucleosomes reflect the sequence-specific binding of the Mif2c dimer to naked DNA, we performed DNase I footprinting reactions with naked CEN3 DNA. As shown in Supplemental Figure S6, we observed no specific footprints for the Mif2c dimer on naked CEN3 DNA despite assembly of a discrete Mif2c dimer–DNA complex as shown by EMSA. This indicates that site-specific localization of Mif2 on the nucleosome requires engagement by both DNA and the Cse4 histone cues described above.
Preference of the Mif2 DHBD for AT-rich centromere DNA
The CDEII elements are highly AT-rich (∼90%) and further distinguished by statistical overrepresentation of short An•Tn or Tn•An tracts (Baker and Rogers 2005) and ApT or TpA steps between tracts. Since AT-rich DNA is a known target for the AT-hook peptide motif (Reeves 2000), we tested binding of a Mif2cath mutant dimer in which the AT-hook residues GRPRGRPK were changed to AAADADAA (Fig. 7A). Despite complete mutation of the AT hook, the Mif2cath dimer still retained some selectivity for CEN3 over periCEN DNA as compared with the wild-type Mif2c dimer (∼11-fold vs. 110-fold) (see Fig. 7B,C), implicating the presence of additional motifs that bind selectively to AT-rich DNA. By analysis of deletion derivatives, we defined a Mif2 DNA-binding region to residues 256–356 that immediately precede the AT hook and contain the conserved CENP-A-binding CENP-C motif (Supplemental Figs. S3A,B, S7A,B; Kato et al. 2013). Accordingly, we refer to the Mif2 DNA- and CENP-A histone-binding domain plus the AT hook (residues 256–365) as the DHBD. AUC showed that the bacterially expressed DHBD is a monomer in solution and when bound to a 20-bp DNA (Supplemental Fig. S7C). EMSAs using dsDNA oligonucleotides showed that the minimal site for stable binding of the monomeric DHBD is 16–18 bp (Supplemental Fig. S7C). For longer DNA fragments used in EMSA, the ladder of shifted protein–DNA complexes represents multiple bindings of Mif2c dimers or DHBD monomers.
Figure 7.

Both the Mif2c dimer and the Mif2 DHBD monomer bind preferentially to CEN3 DNA. (A) The sequence of the Mif2 DHBD, with highlights for amino acid substitutions in the AT hook and RK clusters (clusters enriched for arginine–lysine residues). (B,C) EMSA of binding of the Mif2c dimer and its AT-hook mutant to CEN3 and periCEN DNA mixtures. The stepwise mobility shifts on the 150-bp DNA with increasing protein concentrations apparently reflect the association of multiple Mif2c dimers. (D–H) EMSA of binding of the Mif2 DHBD monomer and its AT-hook and RK cluster mutants to CEN3 and periCEN DNA mixtures. Data collection and quantification were performed as in Figure 2.
The DHBD binds preferentially to CEN3 over 601 DNA (98-fold) or periCEN DNA (32-fold) in competitive binding experiments (Fig. 7D; Supplemental Fig. S7E). Importantly, DHBDδATH with truncation of the AT hook (Supplemental Fig. S7E) and DHBDath with amino acid substitutions of the AT hook (Fig. 7E) still retained substantial levels of binding preference for CEN3 DNA. These results suggest the presence of additional DHBD elements that also contribute to selective binding to CEN3 DNA. Inspection of the Mif2 DHBD peptide sequence revealed three clusters enriched for arginine–lysine residues (referred to as RK clusters) that could be responsible for AT-rich DNA binding. RK1 and RK2 overlap the CENP-C motif, while RK3 is located near the AT hook (Fig. 7A). As shown in Figure 7, F–H, amino acid substitutions in these RK clusters resulted in significant reduction in both affinity and binding preference for CEN3 DNA. In particular, substitutions in RK3 drastically reduced DNA binding (Fig. 7H), while substitutions in RK1 and RK2 had lesser effects on DNA binding (Fig. 7F,G). Hence, among all DNA-binding elements in the DHBD, RK3 is the dominant CEN3 DNA-binding motif, with contributions from the AT hook and the other RK clusters to the overall affinity and binding preference for centromere DNA. The smearing of electrophoretic mobility shift patterns suggests lower stability for DHBD mutant–DNA complexes during native gel electrophoresis (Fig. 7E–H).
Preference of human CENP-C for α-satellite DNA
RK clusters overlapping and adjacent to the CENP-C motif are conserved in many phyla (Supplemental Fig. S8), suggesting that metazoan CENP-C proteins may use similar features for preferential binding to centromere DNA, such as α-satellite repeats. For human CENP-C, there are two DHBDs, HsDHBD-1 and HsDHBD-2, each harboring a CENP-A-binding CENP-C motif with overlapping and adjacent RK clusters but no AT hook (Supplemental Fig. S8; Supplemental Table S1). HsDHBD-2 overlaps a previously defined central DNA-binding domain of CENP-C (Supplemental Fig. S3A; Yang et al. 1996). To examine their DNA-binding preferences, we bacterially expressed and purified HsDHBD-1 and HsDHBD-2 (Supplemental Fig. S9A). As shown in Supplemental Figures S9, B–G, and S10, A and B, both human domains bind preferentially (threefold to fivefold) to sequences of higher AT content, such as human centromeric α-satellite DNA or yeast centromere DNA, compared with human DNA sequences of lower AT content. The levels of binding preference by the human DHBDs are similar to those by the budding yeast ScDHBD (Supplemental Fig. S10C–H). These results suggest that sequences of the yeast and human centromere may possess related compositional features important for CENP-C recognition despite gross differences in DNA sequence.
Discussion
Metazoan centromeres present a paradox in that their sequences are highly divergent even between related species despite functional conservation (Henikoff et al. 2001). Indeed, the elusive nature of a universal sequence identifier in the repetitive DNAs of active metazoan centromeres has led to broad acceptance that centromere identity is directed by epigenetic mechanisms; i.e., through the assembly and properties of the inherited CENP-A histone rather than the underlying centromere DNA sequence (Sullivan et al. 2001; Allshire and Karpen 2008; Black and Cleveland 2011). A conspicuous exception is the strictly genetic specification of budding yeast centromeres, where the nucleosomal Cse4/CENP-A histone is degraded at the G1–S transition of every cell cycle, precluding epigenetic transmission (Biggins 2013; Wisniewski et al. 2014). Instead, centromeric chromatin is re-established de novo by instructions from three specific sequence elements of the 120-bp yeast centromere and their multiple cognate factors (Westermann et al. 2007). Interestingly, pre-existing Mif2 at the budding yeast centromere is transmitted to daughter cells (Fig. 1F); the significance of this transmission remains to be explored.
Here we highlighted distinctive roles for the AT-rich yeast centromere DNA as well as Cse4-specific amino acid residues. Both DNA and histone variants contribute to the several thousand-fold preference of Mif2 for centromeric over pericentric nucleosomes. This vast preference likely underpins site specificity for kinetochore nucleation, although other mechanisms may contribute; e.g., interactions involving the N-terminal tail of Cse4 (Chen et al. 2000; Samel et al. 2012) and those between the Mif2 N terminus and kinetochore components CENP-UAme1–CENP-QOkp1 (Hornung et al. 2014). Further contributions to selectivity for CENP-A nucleosomes over H3 nucleosomes may also come from components of CCAN (Weir et al. 2016).
We confirmed the importance of the Cse4 hydrophobic C terminus for Mif2 interactions, concurring with previous findings (Carroll et al. 2010; Guse et al. 2011; Kato et al. 2013). However, while contributions from CENP-A histone-fold elements were previously excluded (Guse et al. 2011), we discovered a clear requirement for the L1 loop harboring three extra charged and polar residues (KDQ) and the α-2 helix of Cse4. The earlier use of a Xenopus egg extract and CENP-A nucleosomes reconstituted on noncentromeric DNA could have masked a subset of interactions. Consistent with our new findings, mutations of the extra L1 loop residues of Cse4 cause chromosome instability in budding yeast (Keith and Fitzgerald-Hayes 2000). Moreover, the CENP-A centromere targeting domain harboring the extended L1 loop is required for CENP-C recruitment in human cells (Logsdon et al. 2015). The distinct hydrophobic and electrostatic surfaces on the Cse4 nucleosome provide topographical coordinates for docking the Mif2 DHBD on either face of the Cse4 nucleosome. The proximity of the Cse4-binding CENP-C motif could align DNA-binding motifs (AT hook and RK clusters) toward DNA preferentially on one or the other side of the nucleosome dyad, as observed for CEN3 (Figs. 4C,D, 5B–E) and CEN10 (Fig. 6C,D). The function of AT-rich DNA on the unoccupied side of the dyad is unclear, but it could be a target for the DNA-binding domain of the Cse4-specific chaperone Scm3, which also associates with the Cse4 nucleosome in vivo and exhibits an AT-rich preference in vitro (Xiao et al. 2011).
What special features of the 85-bp AT-rich CDEII favor Mif2 binding? The ∼30-bp footprint of the Mif2c dimer covers a large portion of one AT-rich side of the nucleosome dyad. CDEIIs for all 16 yeast centromeres contain multiple short An•Tn or Tn•An tracts plus ApT or TpA dinucleotide steps between tracts. The tract length and arrangement differ between centromeres (Baker and Rogers 2005). An•Tn and Tn•An tracts display a narrow minor groove and large propeller twist as well as cross-strand and bifurcated hydrogen bonds that improve base-stacking and helix rigidity (Coll et al. 1987; Nelson et al. 1987). On the other hand, TpA dinucleotide steps (but not ApT steps) show a widened minor groove at tract junctions (Stefl et al. 2004). Either one or both geometric features of AT-rich DNA may alter conventional helical parameters of nucleosomal DNA wound over the histone core (Bishop et al. 2011). For example, the crystal structure of one long A16•T16 tract on a canonical nucleosome has revealed local distortion of the nucleosomal DNA helix (Bao et al. 2006).
Given the correlation between Mif2 DHBD-binding preference and AT-rich DNA composition, we suggest that the conformations of AT-rich CDEII are recognized by the AT hook and RK clusters of the Mif2 DHBD. Mif2 carries one classic AT hook (GRPRGRPK) at the C-terminal end of its DHBD. Structural studies have revealed how individual AT-hook residues interact with narrow minor grooves of cognate AAAT and AATT elements (Reeves and Nissen 1990; Huth et al. 1997; Reeves 2000). Arginine and, to a lesser extent, lysine of RK clusters are widely used for protein–DNA recognition for not only the well-documented histone core of the nucleosome (Luger et al. 1997) but also metazoan sequence-specific transcription factors such as UBX, OCT1, the bacterial MogR repressor (Shen et al. 2009), and others (Rohs et al. 2009, 2010; Kong et al. 2015). We envision that the RK clusters, particularly RK3 of the Mif2 DHBD, exploit this mode of DNA interaction as part of its selective binding strategy. Accordingly, the physical structure of the Mif2 DHBD in complex with the Cse4 nucleosome should be an outstanding problem for future investigations.
We analyzed the phylogenetic conservation of RK clusters in the CENP-C family and showed that RK clusters are present in all CENP-C proteins in the tree of life. Moreover, by the number of RK clusters, CENP-C proteins rank within the top several percent across proteomes of eukaryotes (Supplemental Table S1). These RK cluster-rich proteins are prevalent among chromatin proteins, especially in heterochromatin and various chromatin remodeling complexes (see the gene ontology [GO] analysis in Supplemental Table S2). Strikingly, CENP-C proteins possess yet another type of DNA minor groove-binding motif—the SPKK/SPRR motifs (Churchill and Suzuki 1989)—found throughout metazoan and fungal lineages (but not in plants), which further supports the preference of CENP-C binding to narrow DNA minor grooves (Supplemental Fig. S8B). N-terminal histone tails in nucleosomes also harbor one or two RK clusters that were shown previously to interact dynamically with DNA minor grooves (Shaytan et al. 2016). The engagement of CENP-C RK clusters to bind nucleosomal DNA may displace histone tails and further promote their interactions with the negatively charged regions of CENP-C.
The human centromere α-satellite sequence (∼60% A+T) is not as AT-rich as yeast centromeres, but its AT content is still greater than the human promoter and intron sequences (Supplemental Fig. S11A). Early studies with bacterially expressed human CENP-C defined a central DNA-binding domain (residues 422–537) (Yang et al. 1996), which was subsequently also shown to exhibit CENP-A histone binding (Kato et al. 2013). ChIP-DNA blot analysis of CENP-C chromatin has demonstrated specific association of the domain with α-satellite DNA in vivo (Politi et al. 2002). However, it has been unclear whether CENP-C has a direct sequence preference for α-satellite DNA in addition to its preference for the variant CENP-A histone (Kato et al. 2013). We characterized DNA binding for the two CENP-C domains HsDHBD-1 and HsDHBD-2, the latter overlapping the previously defined central DNA-binding domain. By competitive EMSA studies of purified soluble HsDHBD-1 and HsDHBD-2 domains of CENP-C, we provide new evidence for direct preferential binding of both domains to the α satellite over sequences with lower AT content, implicating compositional features of human centromere DNA for CENP-C recognition. Supplemental Figure S11B shows the frequency distribution of A•T tract scores (fraction of bases occurring in an A•T tract 2–5 bases in length over a 150-bp window) for the entire human genome. This analysis shows that 63% of the genome has an A•T tract score less than that of the α satellite. These compositional differences may be exploited by human CENP-C, which does not possess either AT-hook or SPKK motifs but is enriched solely for RK residues (Fig. 7A; Supplemental Figs. S8, S9A; Supplemental Table S1). Despite a modest approximately threefold to fivefold preference shown by a single HsDHBD for α-satellite DNA, the compositional preference should be amplified by the presence of a total of four HsDHBDs on each CENP-C dimer, reinforced by the hundreds of CENP-A nucleosomes populating human centromeres. From this perspective, DNA composition could provide a complementary mechanism to the CENP-A histone-centric epigenetic process of centromere specification in humans.
Materials and methods
Plasmids for the expression of core histones and Mif2 proteins in Escherichia coli
Expression of core histones was described previously (Mizuguchi et al. 2007; Xiao et al. 2011). Plasmids expressing full-length Mif2 and truncated forms, residues 256–530 of the Mif2c dimer, residues 345–549, and residues 365–530, were a generous gift from S.C. Harrison (Cohen et al. 2008). Plasmids expressing Mif2 256–365 and 256–356 and the Mif2cath dimer with the AT-hook motif (residues 357–364) GRPRGRPK changed to AAADADAA were synthesized and cloned into the expression vector pET28a (Celtek Genes). Additional DHBD and human CENP-C motif constructs were also synthesized and cloned into pET28a (GenScript USA, Inc.). For more efficient removal of the His tag, the thrombin cleavage site of the original Mif2(256–530) plasmid was replaced with a precision proteinase cleavage site. Details are available on request.
Expression and purification of recombinant core histones and Mif2 proteins
Recombinant histone H3, histone H4, and histone variant Cse4 were expressed individually in E. coli and purified using established protocols (Dyer et al. 2004). Recombinant H2A–H2B dimers were expressed and purified as described previously (Mizuguchi et al. 2007; Xiao et al. 2011). His6-tagged Mif2c dimer and its derivatives were purified by Talon bead affinity chromatography followed by size fractionation on a Superdex 200 gel filtration column. To remove the His6 tag, affinity-purified proteins were incubated with Precision or thrombin proteinase followed by Talon bead affinity chromatography and size fractionation on a Superdex 200 gel filtration column.
Reconstitution of histone octamers
Core histone octamers were reconstituted using established protocols (Dyer et al. 2004). Equimolar amounts of purified recombinant histones (H2A, H2B, H3, and H4 or H2A, H2B, Cse4, and H4) were dissolved in unfolding buffer (7 M guanidine-HCl, 20 mM Tris-Cl at pH 7.5, 10 mM DTT) at 2 mg/mL. The mixtures were dialyzed against four changes of two liters each of refolding buffer (10 mM Tris-Cl at pH 7.5, 1 mM EDTA, 5 mM β-mercaptoethanol, 0.1 mM PMSF) containing 2 M NaCl for 2 d at 4°C. The mixture was then centrifuged at 15,000 rpm in a Tomy MX-300 microcentrifuge to remove any insoluble material. Soluble octamers were purified by size fractionation on a Superdex 200 gel filtration column.
Preparation of DNA fragments
The 201-bp 601 DNA fragment was prepared by PCR amplification followed by gel purification on a Bio-Rad model 491 Prep Cell and ethanol precipitation. The 150-bp 601 DNA and periCEN DNA were first PCR-amplified with an asymmetric AvaI site at both ends and cloned into a modified pUC19 vector that contained an asymmetric AvaI site. All other DNA fragments were synthesized with an asymmetric AvaI site and cloned into the pUC57 vector (GenScript USA, Inc.). The cloned DNA fragments were isolated and ligated into arrays of six to 10 tandem copies through the asymmetric AvaI site and then cloned in the modified pUC19 vector containing an asymmetric AvaI site. After restriction digestion, fragments were gel-purified as above. Thus, the purified DNA fragments had a 4-nucleotide overhang at both ends, and, for some applications, the sticky ends were filled in with Biotin-dNTPs (Roche), Alexa fluor dNTPs (Invitrogen), or 33P-dNTPs (Perkin Elmer) using Klenow polymerase. Alexa fluor-labeled DNA fragments were also prepared by PCR amplification with Alexa fluor-labeled primers and then gel-purified. Details of plasmid constructs and DNA fragment preparation are available on request.
Reconstitution of nucleosomes
Purified core histone octamers and DNA were mixed in 50 µL of high-salt buffer (2 M NaCl, 10 mM Tris-Cl at pH 7.5, 1 mM EDTA, 0.02% NP-40, 5 mM β-mercaptoethanol) supplemented with BSA at 400 g/mL. The mixture was transferred to a Slide-A-Lyzer minidialysis unit (Thermo Scientific). The dialysis unit was placed in a container with 600 mL of high-salt buffer and dialyzed for 30–60 min followed by salt gradient dialysis, during which a low-salt buffer (100 mM NaCl, 10 mM Tris-Cl at pH 7.5, 1 mM EDTA, 0.02% NP-40, 2 mM β-mercaptoethanol) was pumped into the container at 3.5 mL/min for 16 h. The dialysis unit was then transferred to low-salt buffer and dialyzed for 60 min. The dialysis was done at room temperature unless noted otherwise. Reconstituted nucleosomes were analyzed on native agarose gels to determine their quality and reconstitution efficiency (see Supplemental Fig. S4A,B).
EMSAs
Reconstituted nucleosomes and protein/DNA complexes were analyzed by electrophoresis at 120 V for 70–90 min on native agarose gels (Seakem ME and Lonza LE) in a buffer containing 25 mM Tris and 25 mM boric acid (Zimarino and Wu 1987; Huynh et al. 2005). After electrophoresis, gels were stained with SYBR Green I (Invitrogen) and visualized with a Fujifilm LAS-3000 camera. Images were exported into TIFF files for quantification using ImageQuant software (Amersham Biosciences). Gels of Alexa fluor-labeled samples were scanned on a Typhoon 9410 (Amersham Biosciences)/Typhoon FLA9500 (GE Biosciences) and quantified using ImageQuant software. Intensity data were imported into Prism and plotted. Relative affinities were calculated according to Liu-Johnson et al. (1986) (see below).
Determination of Kd and relative Kd
The gel shift assay (Fried and Crothers 1981) was used to determine the Kd of Mif2 bound to the Cse4/CEN3 nucleosome. The equation used to calculate the Kd of nucleosome A relative to nucleosome B [relative Kd = (BA/FA)/(BB/FB)] was adopted from Liu-Johnson et al. (1986). Nucleosomes were reconstituted on DNA fragments labeled with either Alexa fluor 555 or Alexa fluor 647. To calculate a relative Kd, two different color-labeled nucleosomes or free DNA fragments (A and B) were mixed in the same binding reaction with increasing concentrations of purified Mif2c dimer or its derivatives. The reaction mixtures were analyzed by EMSA, and bands representing Mif2-bound (B) and free (F) species were scanned and quantified for fluorescence intensity.
Sedimentation velocity
Sedimentation velocity experiments were carried out at 50,000 rpm at 20°C on a Beckman Coulter ProteomeLab XL-I analytical ultracentrifuge following standard protocols (Zhao et al. 2013a). Samples were loaded into 12-mm or 3-mm two-channel Epon centerpiece cells, depending on their concentration. Absorbance (230, 260, or 280 nm) and Rayleigh interference (655 nm) scans were collected at ∼7-min intervals, and data were analyzed in SEDFIT 14.1 (Schuck 2000) in terms of a continuous c(s) distribution with a resolution of 0.1 S and a confidence level 0.68. Scan file time stamps were corrected (Zhao et al. 2013b), and good fits were obtained with root-mean-square deviation corresponding to typical instrumental noise values. The solution density and viscosity were determined based on the solvent composition using Sednterp (http://sednterp.unh.edu) (Cole et al. 2008) or measured experimentally at 20°C on an Anton-Paar DMA 5000 density meter and an Anton Paar AMVn rolling ball viscometer. Protein partial specific volumes were calculated in Sednterp, a partial specific volume of 0.55 cm3g−1 was assumed for dsDNA, and the partial specific volumes for their complexes were calculated based on the expected composition. Mif2(256–365) at 14 and 60 µM and Mif2c(256–530) at 7 and 30 µM were analyzed in 1 M NaCl, 20 mM Tris-HCl (pH 7.5), and 2 mM β-mercaptoethanol. Twenty-base-pair dsDNA (AAAGTAAAAAATAAAAAGTA) and its complex with Mif2(256–365) were analyzed at nominal loading concentrations of 2.2 and 9.2 µM, respectively, in 0.2 M NaCl, 20 mM Tris-HCl (pH 7.5), and 2 mM β-mercaptoethanol. Forty-base-pair dsDNA (AAAGTAAAAAATAAAAAGTAGTTTATTTTTAAAAAATAAA) and its complex with the Mif2c dimer were analyzed at 1.9 and 2.0 µM, respectively, in 0.1 M NaCl, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM β-mercaptoethanol, and 0.02% (v/v) NP-40. Samples of the Cse4 octasome and its complex with the Mif2c dimer, each at a loading concentration of 340 nM, were studied in a buffer containing 54 mM NaCl, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 2 mM β-mercaptoethanol, and 0.02% (v/v) NP-40. Sedimentation experiments on the Cse4 octasome and its complex utsed water as a reference. In this case, to obtain better estimates of the sedimentation coefficient and molar mass, absorbance data were analyzed in Sedphat 10.55b (Schuck 2003) in terms of a hybrid model combining continuous c(s) distributions flanking the discrete species of interest. Furthermore, in cases where NP-40 was used, the analysis assumed that no detergent binding occurred to the samples of interest.
DNase I footprinting
To determine the site of Mif2 binding on Cse4 nucleosomal DNA, nucleosomes were reconstituted with DNA fragments end-labeled at 3′ with 33P-dTTP for the top strand and 33P-dATP for the bottom strand using Klenow polymerase. Purified Mif2c dimer (3 pM) was added to 6 pmol of end-labeled nucleosomes in the nucleosome reconstitution buffer (see above), and the volume was adjusted to 40–60 µL and incubated for 40 min at room temperature. Under these conditions, ∼50% of the nucleosomes are bound by the Mif2c dimer. The nucleosomes were then digested with 1 U of DNase I (Thermo Scientific, catalog no. 89835) for 80 sec at 23°C. The reaction products were resolved on a 1.3% native agarose gel, bands containing the free and Mif2c dimer-bound nucleosomes were visualized by SYBR Green staining and excised from the gel, and DNA was recovered from the gel slices and resolved on an 8% DNA sequencing gel (National Diagnostics, catalog no. EC-833). DNA mobility markers were G+A and C+T sequencing reactions of the same 33P-labeled DNA fragments performed as described (Molecular Cloning, Cold Spring Harbor Laboratory). Gels were transferred to a DEAE filter paper and dried under vacuum, and radioactive signals were captured using a PhosphorImager (Fuji Photo Film Co., Ltd.) and a Typhoon scanner (Typhoon 9410, Amersham Biosciences). Intensity data were imported into Prism, normalized, and plotted as percentage of relative cleavage. The data were also imported to Excel (Microsoft), and the relative mobility of the bands was transformed into base pair positions using the mobility standards.
Hydroxyl radical footprinting
Hydroxyl radical footprinting with iron(II)-EDTA was performed as described (Schwanbeck et al. 2004) under the same conditions as for DNase I footprinting (see above). PAGE images resulting from hydroxyl radical footprinting were analyzed using the Hydroid framework (available at https://github.com/ncbi/HYDROID) as described elsewhere (Shaytan et al. 2017).
Molecular modeling of Cse4/CEN nucleosomes
To generate a surface model of the Cse4/CEN3 nucleosome (Figs. 3–5), budding yeast Cse4 protein and CEN3 DNA sequences were substituted for the CENP-A and α-satellite DNA of the crystal structure of the CENP-A nucleosome (Tachiwana et al. 2011) using a protein model portal (http://www.proteinmodelportal.org/?pid=modelling_interactive).
Computational analysis of CENP-C motifs and RK clusters
The initial set of CENP-C sequences for selected species was handpicked from public databases and studies; these sequences were then used in BLAST searches against the NCBI nonredundant database to obtain a broader set of sequences that spanned 127 species from different taxonomic groups. For pan-proteome analysis, all protein records for a selected number of species were obtained from the NCBI RefSeq database (Pruitt et al. 2014). A motif search was done on the sequences with the in-house software using the following regular expression definitions extracted from reports: CENP-C motif, “R….P..[YFW]W” (Kato et al. 2013); AT hook, “..[RPKST]GRP[RPKS]” (Aravind and Landsman 1998); and SPKK motif, “SP[RK][RK]” (Churchill and Suzuki 1989), where “.” refers to any symbol, and square brackets denote that any of the amino acids listed within the brackets are allowed in this position. RK clusters were defined as nonoverlapping stretches of at least seven amino acid residues with a total formal charge of no less than +4, counted as the number of arginines and lysines minus the number of glutamates and aspirates in the stretch. Phyletic trees were visualized using ETE toolkit (Huerta-Cepas et al. 2016). Sequence alignments were visualized with TexShade (Beitz 2000). Enrichment of a given motif type in the CENP-C protein of a given species was calculated as the fraction of all known proteins for that species that had fewer occurrences of said motif than CENP-C. GO enrichment analysis was accomplished through PANTHER (Thomas et al. 2003).
Yeast strains
All strains were derived by homologous recombination from Saccharomyces cerevisiae W1588-4C and had the following genotypes: JBY 119 (MATa ADE2 dynLC::hphMX4 Cse4::natMX4 can1-100 hiS2-11,15 leu2-3,112::LEU2-Cse4-tdEOS-Cse4,trp1-1,ura3-1 RAD5) (Wisniewski et al. 2014) and JBY 123 (MATa ADE2 dynLC::hphMX4 MIF2-tdEOS-kanMX4 can1-100 hiS2-11,15 leu2-3,112 trp1-1 ura3-1 RAD5).
Microscopy
A Hamamatsu C9100-13 camera (−94°C, 0.63 MHz, 16-bit ADC) was used typically with EM gain of 50 (conversion factor 0.044 e−/ADU, readout noise 0.470 e− RMS, thermal current 0.014 e−/sec, established experimentally) (see Berry and Burnell 2005). IR was blocked with a FF01-750/SP filter (Semrock). A Zeiss AxioObserver Z1 microscope (Carl Zeiss Microscopy) was equipped with a Zeiss plan-apochromat 150× NA1.35 glycerin immersion objective, P-737 piezoelectric stage (Physik Instrumente), Zeiss Colibri LED illuminator, and custom fluorescence cubes (see Supplemental Table S3 for light sources and filters used for wide-field fluorescence imaging). Yeast cells were grown in complete darkness in the CSM + adenine medium (250 rpm at 25°C, final OD600 ≤ 0.3), manipulated only under dim red light (660 nm), and imaged in CellAsic Y04C microfluidic chambers (CellAsic). Narrowband illumination (671 nm; Edmund Optics, no. 65-233) was used for differential interference contrast. To minimize phototoxicity, low-level excitation (∼7 W/cm2, 1- to 5-sec exposure) was used for fluorescence imaging, and 405-nm light (∼0.7 W/cm2, 7–10 sec) was used for tdEos photoconversion. Typically, Z-stacks consisted of 13 steps, 333-nm apart.
Image calibration and display
Raw 16-bit images were converted into FITS format and calibrated in 32-bit floating-point space using bias, thermal, and flat-field frames (AIP4WIN) (Berry and Burnell 2005). Z-stacks were reduced to the composite image only for presentation purposes by projecting individual layers (with centromeres in focus) onto a common plane.
Photometry
All intensity measurements were carried on calibrated unreduced Z-stacks with aperture photometry in AIP4WIN software using the typical FWHM of the centromere cluster (4 pixels = 428 nm) as a radius of measurement aperture and an outer background annulus (five pixels = 535 nm wide, area four times larger than measurement aperture) (see Berry and Burnell 2005 for discussion of photometry techniques). The background-corrected signal was converted into photoelectrons (equivalent of detected photons) using experimentally established camera parameters (see Berry and Burnell 2005 for details). Light sources and filters used for wide-field fluorescence imaging are listed in Supplemental Table S3.
Supplementary Material
Acknowledgments
We thank S.C. Harrison for providing Mif2 expression plasmids, R. Schwanbeck for consultations on data analysis of hydroxyl radical footprints, Z. Zhang for consultations on PAGE of footprinting reactions, and V. Nguyen, A. Ranjan, Y. Bai, and our colleagues for critical reading of the manuscript. This work was supported by the Center for Cancer Research, the National Cancer Institute (C.W., H.X., F.W., P.F., S.L., Y.H., and D.W.), the National Library of Medicine (A.K.S., D.L., and A.R.P.), the National Institute of Diabetes and Digestive and Kidney Diseases (R.G.), the Howard Hughes Medical Institute Janelia Research Campus (C.W. and J.W), and the Bloomberg Distinguished Professorship, Johns Hopkins University (C.W). H.X. and C.W. designed experiments and wrote the manuscript with contributions from other coauthors. H.X. designed expression plasmids; purified histones and recombinant Mif2 proteins; and performed nucleosome reconstitution, EMSA, and Mif2–DNA and Mif2–nuclesome interactions; hydroxyl radical and DNase I footprinting; and data analysis. F.W. constructed plasmids, expressed and purified recombinant Mif2, analyzed Mif2–DNA interactions, and performed molecular modeling. J.W. designed and performed yeast living-cell imaging experiments. A.K.S, A.R.P., and D.L. performed computational analysis of CENP-C motifs/RK clusters and analysis of hydroxyl radical footprints of Mif2 on nucleosomes. R.G. performed analytical ultracentrifugation experiments and data analysis. Y.H., D.W., and S.L. assisted in plasmid construction and purification of proteins and DNA fragments. P.F. performed bioinformatics on the human genome. All authors read the manuscript and approved the content.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.304782.117.
References
- Allshire RC, Karpen GH. 2008. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9: 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Landsman D. 1998. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res 26: 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RE, Rogers K. 2005. Genetic and genomic analysis of the AT-rich centromere DNA element II of Saccharomyces cerevisiae. Genetics 171: 1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RE, Rogers K. 2006. Phylogenetic analysis of fungal centromere H3 proteins. Genetics 174: 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, White CL, Luger K. 2006. Nucleosome core particles containing a poly(dA.dT) sequence element exhibit a locally distorted DNA structure. J Mol Biol 361: 617–624. [DOI] [PubMed] [Google Scholar]
- Beitz E. 2000. TEXshade: shading and labeling of multiple sequence alignments using LATEX2ε. Bioinformatics 16: 135–139. [DOI] [PubMed] [Google Scholar]
- Berry R, Burnell J. 2005. The handbook of astronomical image processing. Willmann-Bell, Richmond, VA. [Google Scholar]
- Biggins S. 2013. The composition, functions, and regulation of the budding yeast kinetochore. Genetics 194: 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop EP, Rohs R, Parker SC, West SM, Liu P, Mann RS, Honig B, Tullius TD. 2011. A map of minor groove shape and electrostatic potential from hydroxyl radical cleavage patterns of DNA. ACS Chem Biol 6: 1314–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Cleveland DW. 2011. Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 144: 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Foltz DR, Chakravarthy S, Luger K, Woods VL Jr, Cleveland DW. 2004. Structural determinants for generating centromeric chromatin. Nature 430: 578–582. [DOI] [PubMed] [Google Scholar]
- Brown MT. 1995. Sequence similarities between the yeast chromosome segregation protein Mif2 and the mammalian centromere protein CENP-C. Gene 160: 111–116. [DOI] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. 2010. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol 189: 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Baker RE, Keith KC, Harris K, Stoler S, Fitzgerald-Hayes M. 2000. The N terminus of the centromere H3-like protein Cse4p performs an essential function distinct from that of the histone fold domain. Mol Cell Biol 20: 7037–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill ME, Suzuki M. 1989. ‘SPKK’ motifs prefer to bind to DNA at A/T-rich sites. EMBO J 8: 4189–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RL, Espelin CW, De Wulf P, Sorger PK, Harrison SC, Simons KT. 2008. Structural and functional dissection of Mif2p, a conserved DNA-binding kinetochore protein. Mol Biol Cell 19: 4480–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL, Lary JW, P Moody T, Laue TM. 2008. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol 84: 143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole HA, Howard BH, Clark DJ. 2011. The centromeric nucleosome of budding yeast is perfectly positioned and covers the entire centromere. Proc Natl Acad Sci 108: 12687–12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll M, Frederick CA, Wang AH, Rich A. 1987. A bifurcated hydrogen-bonded conformation in the d(A.T) base pairs of the DNA dodecamer d(CGCAAATTTGCG) and its complex with distamycin. Proc Natl Acad Sci 84: 8385–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KA, Castillo AR, Tatsutani SY, Biggins S. 2005. De novo kinetochore assembly requires the centromeric histone H3 variant. Mol Biol Cell 16: 5649–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JL, Henikoff S. 2004. Adaptive evolution of the histone fold domain in centromeric histones. Mol Biol Evol 21: 1712–1718. [DOI] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. 2004. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375: 23–44. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Rothfield N. 1985. Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma 91: 313–321. [DOI] [PubMed] [Google Scholar]
- Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LE, Van Duyne GD, Vinogradov SA, et al. 2015. Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science 348: 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Lee J, Sekulic N, Sennett MA, Lee TH, Black BE. 2016. CENP-C directs a structural transition of CENP-A nucleosomes mainly through sliding of DNA gyres. Nat Struct Mol Biol 23: 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried M, Crothers DM. 1981. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res 9: 6505–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa T. 2004. Centromere DNA, proteins and kinetochore assembly in vertebrate cells. Chromosome Res 12: 557–567. [DOI] [PubMed] [Google Scholar]
- Fukagawa T, Earnshaw WC. 2014. The centromere: chromatin foundation for the kinetochore machinery. Dev Cell 30: 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, Cheeseman IM. 2011. Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145: 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. 2011. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477: 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann JH, Fleig UN. 1993. The centromere of budding yeast. Bioessays 15: 451–460. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293: 1098–1102. [DOI] [PubMed] [Google Scholar]
- Hong J, Feng H, Zhou Z, Ghirlando R, Bai Y. 2013. Identification of functionally conserved regions in the structure of the chaperone/CenH3/H4 complex. J Mol Biol 425: 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung P, Troc P, Malvezzi F, Maier M, Demianova Z, Zimniak T, Litos G, Lampert F, Schleiffer A, Brunner M, et al. 2014. A cooperative mechanism drives budding yeast kinetochore assembly downstream of CENP-A. J Cell Biol 206: 509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Serra F, Bork P. 2016. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 33: 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth JR, Bewley CA, Nissen MS, Evans JN, Reeves R, Gronenborn AM, Clore GM. 1997. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat Struct Biol 4: 657–665. [DOI] [PubMed] [Google Scholar]
- Huynh VA, Robinson PJ, Rhodes D. 2005. A method for the in vitro reconstitution of a defined ‘30 nm’ chromatin fibre containing stoichiometric amounts of the linker histone. J Mol Biol 345: 957–968. [DOI] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y. 2013. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science 340: 1110–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith KC, Fitzgerald-Hayes M. 2000. CSE4 genetically interacts with the Saccharomyces cerevisiae centromere DNA elements CDE I and CDE II but not CDE III. Implications for the path of the centromere DNA around a cse4p variant nucleosome. Genetics 156: 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Liu J, Li L, Yue L, Zhang L, Jiang H, Xie X, Luo C. 2015. Functional interplay between the RK motif and linker segment dictates Oct4-DNA recognition. Nucleic Acids Res 43: 4381–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanini L, McKeon F. 1995. Domains required for CENP-C assembly at the kinetochore. Mol Biol Cell 6: 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrimore J, Bloom KS, Salmon ED. 2011. Point centromeres contain more than a single centromere-specific Cse4 (CENP-A) nucleosome. J Cel Biol 195: 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Johnson HN, Gartenberg MR, Crothers DM. 1986. The DNA binding domain and bending angle of E. coli CAP protein. Cell 47: 995–1005. [DOI] [PubMed] [Google Scholar]
- Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, Dawicki-McKenna JM, Heun P, Black BE. 2015. Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J Cell Biol 208: 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Meeks-Wagner D, Wood JS, Garvik B, Hartwell LH. 1986. Isolation of two genes that affect mitotic chromosome transmission in S. cerevisiae. Cell 44: 53–63. [DOI] [PubMed] [Google Scholar]
- Meluh PB, Koshland D. 1995. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol Biol Cell 6: 793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milks KJ, Moree B, Straight AF. 2009. Dissection of CENP-C-directed centromere and kinetochore assembly. Mol Biol Cell 20: 4246–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Xiao H, Wisniewski J, Smith MM, Wu C. 2007. Nonhistone Scm3 and histones CenH3-H4 assemble the core of centromere-specific nucleosomes. Cell 129: 1153–1164. [DOI] [PubMed] [Google Scholar]
- Moroi Y, Peebles C, Fritzler MJ, Steigerwald J, Tan EM. 1980. Autoantibody to centromere (kinetochore) in scleroderma sera. Proc Natl Acad Sci 77: 1627–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson HC, Finch JT, Luisi BF, Klug A. 1987. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature 330: 221–226. [DOI] [PubMed] [Google Scholar]
- Politi V, Perini G, Trazzi S, Pliss A, Raska I, Earnshaw WC, Della Valle G. 2002. CENP-C binds the α-satellite DNA in vivo at specific centromere domains. J Cell Sci 115: 2317–2327. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, et al. 2014. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42: D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves R. 2000. Structure and function of the HMGI(Y) family of architectural transcription factors. Environ Health Perspect 108: 803–809. [DOI] [PubMed] [Google Scholar]
- Reeves R, Nissen MS. 1990. The A·T–DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J Biol Chem 265: 8573–8582. [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. 2009. The role of DNA shape in protein-DNA recognition. Nature 461: 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. 2010. Origins of specificity in protein–DNA recognition. Annu Rev Biochem 79: 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Tomkiel J, Cooke CA, Ratrie H III, Maurer M, Rothfield NF, Earnshaw WC. 1992. CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell 70: 115–125. [DOI] [PubMed] [Google Scholar]
- Samel A, Cuomo A, Bonaldi T, Ehrenhofer-Murray AE. 2012. Methylation of CenH3 arginine 37 regulates kinetochore integrity and chromosome segregation. Proc Natl Acad Sci 109: 9029–9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. 2003. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem 320: 104–124. [DOI] [PubMed] [Google Scholar]
- Schwanbeck R, Xiao H, Wu C. 2004. Spatial contacts and nucleosome step movements induced by the NURF chromatin remodeling complex. J Biol Chem 279: 39933–39941. [DOI] [PubMed] [Google Scholar]
- Sekulic N, Bassett EA, Rogers DJ, Black BE. 2010. The structure of (CENP-A-H4)(2) reveals physical features that mark centromeres. Nature 467: 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaytan AK, Armeev GA, Goncearenco A, Zhurkin VB, Landsman D, Panchenko AR. 2016. Coupling between histone conformations and DNA geometry in nucleosomes on a microsecond timescale: atomistic insights into nucleosome functions. J Mol Biol 428: 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaytan AK, Xiao H, Armeev GA, Wu C, Landsman D, Panchenko AR. 2017. Hydroxyl-radical footprinting combined with molecular modeling identifies unique features of DNA conformation and nucleosome positioning. Nucleic Acids Res 45: 9229–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Higgins DE, Panne D. 2009. Recognition of AT-rich DNA binding sites by the MogR repressor. Structure 17: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl R, Wu H, Ravindranathan S, Sklenar V, Feigon J. 2004. DNA A-tract bending in three dimensions: solving the dA4T4 vs. dT4A4 conundrum. Proc Natl Acad Sci 101: 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Kuriyama K, Shibata A, Himeno M. 1997. Characterization of internal DNA-binding and C-terminal dimerization domains of human centromere/kinetochore autoantigen CENP-C in vitro: role of DNA-binding and self-associating activities in kinetochore organization. Chromosome Res 5: 132–141. [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Blower MD, Karpen GH. 2001. Determining centromere identity: cyclical stories and forking paths. Nat Rev Genet 2: 584–596. [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Kurumizaka H. 2011. Structure of the CENP-A nucleosome and its implications for centromeric chromatin architecture. Genes Genet Syst 86: 357–364. [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi-Takanaka Y, Oda T, Sato M, Park SY, et al. 2011. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature 476: 232–235. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trazzi S, Perini G, Bernardoni R, Zoli M, Reese JC, Musacchio A, Della Valle G. 2009. The C-terminal domain of CENP-C displays multiple and critical functions for mammalian centromere formation. PLoS One 4: e5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JR, Faesen AC, Klare K, Petrovic A, Basilico F, Fischbock J, Pentakota S, Keller J, Pesenti ME, Pan D, et al. 2016. Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 537: 249–253. [DOI] [PubMed] [Google Scholar]
- Westermann S, Schleiffer A. 2013. Family matters: structural and functional conservation of centromere-associated proteins from yeast to humans. Trends Cell Biol 23: 260–269. [DOI] [PubMed] [Google Scholar]
- Westermann S, Drubin DG, Barnes G. 2007. Structures and functions of yeast kinetochore complexes. Annu Rev Biochem 76: 563–591. [DOI] [PubMed] [Google Scholar]
- Wisniewski J, Hajj B, Chen J, Mizuguchi G, Xiao H, Wei D, Dahan M, Wu C. 2014. Imaging the fate of histone Cse4 reveals de novo replacement in S phase and subsequent stable residence at centromeres. Elife 3: e02203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Mizuguchi G, Wisniewski J, Huang Y, Wei D, Wu C. 2011. Nonhistone Scm3 binds to AT-rich DNA to organize atypical centromeric nucleosome of budding yeast. Mol Cell 43: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CH, Tomkiel J, Saitoh H, Johnson DH, Earnshaw WC. 1996. Identification of overlapping DNA-binding and centromere-targeting domains in the human kinetochore protein CENP-C. Mol Cell Biol 16: 3576–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, Schuck P. 2013a. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci 71: 20.12.1–20.12.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ghirlando R, Piszczek G, Curth U, Brautigam CA, Schuck P. 2013b. Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Anal Biochem 437: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Feng H, Zhou BR, Ghirlando R, Hu K, Zwolak A, Miller Jenkins LM, Xiao H, Tjandra N, Wu C, Bai Y. 2011. Structural basis for recognition of centromere histone variant CenH3 by the chaperone Scm3. Nature 472: 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimarino V, Wu C. 1987. Induction of sequence-specific binding of Drosophila heat shock activator protein without protein synthesis. Nature 327: 727–730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.