

In this study, Morgan et al. identify an evolutionarily conserved factor, BRWD2/PHIP, that localizes with histone H3K4 methylation genome-wide in human cells, mouse embryonic stem cells, and Drosophila. Depletion of the Drosophila sole homolog dBRWD3 results in altered histone H3 Lys27 acetylation patterns at enhancers and promoters and changes in gene expression, suggesting a cross-talk between these epigenetic modifications and transcription through the BRWD family.
Keywords: BRWD, PHIP, COMPASS, histone methylation, Tudor domain, H3K4
Abstract
Histone H3 Lys4 (H3K4) methylation is a chromatin feature enriched at gene cis-regulatory sequences such as promoters and enhancers. Here we identify an evolutionarily conserved factor, BRWD2/PHIP, which colocalizes with histone H3K4 methylation genome-wide in human cells, mouse embryonic stem cells, and Drosophila. Biochemical analysis of BRWD2 demonstrated an association with the Cullin-4–RING ubiquitin E3 ligase-4 (CRL4) complex, nucleosomes, and chromatin remodelers. BRWD2/PHIP binds directly to H3K4 methylation through a previously unidentified chromatin-binding module related to Royal Family Tudor domains, which we named the CryptoTudor domain. Using CRISPR–Cas9 genetic knockouts, we demonstrate that COMPASS H3K4 methyltransferase family members differentially regulate BRWD2/PHIP chromatin occupancy. Finally, we demonstrate that depletion of the single Drosophila homolog dBRWD3 results in altered gene expression and aberrant patterns of histone H3 Lys27 acetylation at enhancers and promoters, suggesting a cross-talk between these chromatin modifications and transcription through the BRWD protein family.
Methylation of histone H3 on Lys4 (H3K4) is a chromatin modification associated with promoters and transcriptional cis-regulatory elements, and misimplementation of H3K4 methylation patterns is implicated in a range of human pathologies (Morgan and Shilatifard 2015; Piunti and Shilatifard 2016). Histone H3K4 trimethylation (H3K4me3) occurs predominantly at gene promoters near their transcription start sites (TSSs) (Shilatifard 2012). Recently, it was demonstrated that intergenic CpG island enhancers also carry H3K4me3 in mouse embryonic stem cells (ESCs) (Shen et al. 2016; Hu et al. 2017). In contrast to H3K4me3, H3K4me1 levels are low at promoters, and this modification is distributed primarily at intragenic and intergenic sites, some of which act as enhancers (Piunti and Shilatifard 2016). Enhancers containing H3K4me1 can be further categorized as active or poised based on their levels of H3K27 acetylation (H3K27ac) (Heintzman et al. 2007; Creyghton et al. 2010). Moreover, combinations of histone H3K4 methylation with H3 or H4 acetylation can contribute to multivalent binding of effector proteins (Ruthenburg et al. 2007, 2011; Li et al. 2016; Savitsky et al. 2016). Defining both the implementation and recognition mechanisms of H3K4 methylation is essential for understanding this modification's molecular function.
Histone H3K4 methylation is catalyzed by the COMPASS family of SET domain methyltransferase enzymes, which in mammals comprises six proteins: SET1A, SET1B, MLL1, MLL2, MLL3, and MLL4 (Shilatifard 2012; Piunti and Shilatifard 2016). These enzymes differ in their product specificity and genome-wide localization. SET1A is responsible for bulk promoter H3K4me3 in mammalian cells (Shilatifard 2012). In contrast, MLL2 is responsible for H3K4me3 at “bivalent” chromatin in ESCs and controls H3K4me3 at the aforementioned intergenic CpG island enhancers (Piunti and Shilatifard 2016; Hu et al. 2017). MLL3 and MLL4 uniquely catalyze H3K4me1 at intergenic sites (Hu et al. 2013a). Recent cancer genomic studies have identified frequent mutations in MLL3, MLL4, and their cofactor, UTX, a in broad spectrum of human malignancies (Morgan and Shilatifard 2015).
Methylation of histone H3K4 triggers the recruitment of specific effector proteins that mediate downstream processes (Kouzarides 2007). Known H3K4 methyl-binding proteins, such as ING2, BPTF, CHD1, TAF3, and JMJD2A, recognize various methylation states through their chromodomain, Tudor domain, and plant homeodomain (PHD) histone-binding modules (Sims and Reinberg 2006; Kouzarides 2007). A number of methyl histone-binding modules, including the Tudor domain, plant agenet, chromodomain, PWWP, and MBT domains, constitute a class known as the Royal Family (Maurer-Stroh et al. 2003). Royal Family domains encode dissimilar amino acid sequences but nevertheless adopt a common conserved three-dimensional fold (Maurer-Stroh et al. 2003). Defining the full repertoire of H3K4 methylation-interacting factors is particularly important, as recent studies have revealed methylation-dependent and methylation-independent functions for COMPASS proteins (Terranova et al. 2006; Hu et al. 2017).
Here, we report the identification of BRWD2/PHIP as a H3K4 methylation-binding protein that recognizes this modification through a previously uncharacterized Tudor-like module, which we named the CryptoTudor domain. CRISPR–Cas9 knockout of the COMPASS histone H3K4 methyltransferases demonstrates that these enzymes control BRWD2/PHIP chromatin occupancy in a context-dependent manner. Finally, we demonstrate that the depletion of the sole Drosophila homolog dBRWD3 alters the distribution of histone Lys27 acetylation (H3K27ac) at promoters and enhancers, suggesting the existence of a cross-talk between these epigenetic modifications of histones that is mediated by the CryptoTudor family of effectors.
Results
BRWD2/PHIP colocalizes with histone H3K4 methylation in mammalian cells
We and others have detected BRWD2/PHIP in association with active mammalian chromatin by mass spectrometry (MS) (Herz et al. 2014; Ji et al. 2015; Surface et al. 2016). To explore BRWD2/PHIP's genome-wide localization pattern in detail, we generated a Flag-tagged BRWD2/PHIP HEK293 cell line and performed ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) (Fig. 1A). This revealed a striking correlation between Flag-BRWD2/PHIP occupancy and histone H3K4 methylation (Fig. 1A,B) and prompted us to generate homemade antibodies against endogenous BRWD2/PHIP. As an antibody specificity control, we generated CRISPR–Cas9 knockout clones of BRWD2/PHIP in HCT116 cells (Supplemental Fig. 1A–C). Approximately 95% of BRWD2/PHIP ChIP-seq peaks detected in parental HCT116 cells are lost in BRWD2/PHIP knockout cells, indicating that the vast majority of signal is specific (Supplemental Fig. 1D–F). Endogenous BRWD2/PHIP colocalizes with H3K4 methylated chromatin (Fig. 1A,B). Genome-wide analysis revealed that BRWD2/PHIP binds to both intergenic and intragenic sites enriched for H3K4me1 as well as promoters that contain high levels of H3K4me3 and H3K27ac (Fig. 1B). The majority of BRWD2/PHIP peaks (95%) in HEK293 cells coincides with H3K4me1 peaks, whereas ∼52% overlap with H3K27ac (Fig. 1C). To examine the properties of BRWD2/PHIP in another cellular context, we performed ChIP-seq in mouse v6.5 ESCs. Similarly to HCT116 cells, we also observed a strong correlation between BRWD2/PHIP ChIP signal and H3K4 methylation in mouse ESCs (Fig. 1D,E). In ESCs, a subset of BRWD2/PHIP-bound promoters contains H3K4me3 and low levels of H3K27me3 and likely represents “bivalent” chromatin (Fig. 1E; Piunti and Shilatifard 2016). In contrast, intergenic BRWD2/PHIP peaks were enriched for H3K4me1 but lack H3K27me3 signal in ESCs (Fig. 1E). Similar to HEK293 cells, in ESCs, the majority of BRWD2/PHIP peaks coincides with H3K4me1 and/or H3K4me3 (Fig. 1F).
Figure 1.

BRWD2/PHIP colocalizes with histone H3K4 methylation. (A) An example University of California at Santa Cruz (UCSC) genome browser track of ChIP-seq for BRWD2/PHIP and histone modifications in HEK293 cells. (B) Genome-wide occupancy plots for BRWD2/PHIP and histone modifications in HEK293 cells. The two left columns indicate whether peaks overlap with promoter TSSs or are intergenic/intragenic (non-TSS). Signal is centered on BRWD2/PHIP peaks and extends 5 kb in each direction. (C) Venn diagram displaying the overlap of H3K4me1 (light green), H3K27ac (orange), and BRWD2/PHIP (purple) peaks in HEK293 cells. (D) An example UCSC genome browser track of BRWD2/PHIP and histone modification ChIP-seq in mouse v6.5 ESCs. (E) Genome-wide occupancy plots of BRWD2/PHIP and histone modifications in v6.5 mouse ESCs. The two left columns indicate whether peaks overlap with a promoter TSS or are intergenic/intragenic (non-TSS). Signal is centered on BRWD2/PHIP peaks and extends 5 kb in each direction. (F) Venn diagram displaying the overlap of H3K4me1 (light green), H3K4me3 (dark green), and BRWD2/PHIP (purple) peaks in v6.5 mouse ESCs.
BRWD2/PHIP associates with Cullin-4 (CUL4)–RING ubiquitin E3 ligase-4 (CRL4), chromatin regulators, and active chromatin marks
To gain further insight into the function of BRWD2/PHIP, we immunoprecipitated Flag-tagged BRWD2/PHIP from non-cross-linked micrococcal nuclease (MNase)-digested chromatin and identified interacting proteins by tandem MS (Fig. 2A,B; Supplemental Fig. 2A). We observed a strong enrichment of the CRL4, which comprises CUL4A/B and DDB1 and is a previously reported BRWD2/PHIP-interacting partner (Fig. 2B; Supplemental Fig. 2A; Angers et al. 2006; Jin et al. 2006; Ozturk et al. 2013; Raman et al. 2015). In addition, BRWD2/PHIP pull-downs contain the COP9 signalosome (CSN), a CRL4 regulatory complex that removes the Nedd8 modification from active CUL4 (Duda et al. 2008; Lydeard et al. 2013; Cavadini et al. 2016). We probed the BRWD2/PHIP CRL4 interaction by purifying complexes from cells pretreated with combinations of the CUL inhibitor MLN4924 (Pevonedistat) (Soucy et al. 2009) and the proteasome inhibitor MG132 (Fig. 2B; Supplemental Fig. 2B,C). MLN4924 treatment blocks Nedd8 attachment to CUL4 and disrupts BRWD2/PHIP association with the CSN complex and the Nedd8-binding protein UBXD7 (Fig. 2B; Supplemental Fig. 2B; Soucy et al. 2009; den Besten et al. 2012; Cavadini et al. 2016). In contrast, BRWD2/PHIP association with CUL4A and DDB1 still occurs after MLN4924 treatment, although CUL4A levels appear slightly reduced (Supplemental Fig. 2C). Chromatin remodelers and histone-binding proteins such as CHD4, PHF6, and FACT were also detected in BRWD2/PHIP purifications; however, these interactions are not altered by MLN4924 treatment (Fig. 2B,C; Supplemental Fig. 2B). We interrogated the modification state of the histones associated with BRWD2/PHIP by Western blotting and found that they contain marks of active chromatin (H3K27ac, H3K4me1, H3K4me3, and H2A.Z), whereas the repressed chromatin mark H3K27me3 was barely detectable (Fig. 2D).
Figure 2.
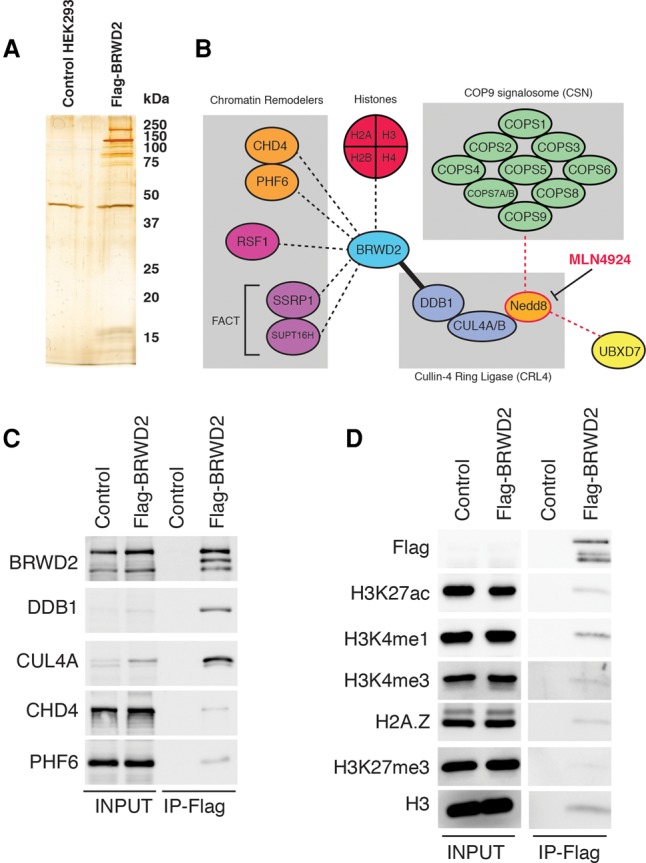
BRWD2/PHIP associates with CRL4, chromatin regulators, and active chromatin marks. (A) Silver-stained SDS-PAGE gel of Flag immunoprecipitations from control parental HEK293 TRex-expressing and Flag-BRWD2/PHIP-expressing cells. (B) Diagram summarizing BRWD2/PHIP protein–protein interactions. The solid black line indicates a strong interaction between BRWD2/PHIP and DDB1–CUL4A/B. Black dashed lines indicate interactions with chromatin remodelers and histones. Red dashed lines indicate interactions sensitive to treatment with MLN4924. (C) Western blot confirming BRWD2/PHIP interaction with CRL4 (DDB1 and CUL4A) and chromatin remodelers (CHD4 and PHF6). The middle band detected in the BRWD2/PHIP Western likely represents a BRWD2/PHIP protein cleavage product that occurs during immunoprecipitation incubation. (D) Western blot detecting histone modifications (H3K27ac, H3K4me1, H3K4me3, and H3K27me3) and histone variants (H2A.Z) associated with BRWD2/PHIP. Note that Flag-BRWD2/PHIP is expressed at near-endogenous levels and is not detectable with anti-Flag in the input but is detected after enrichment by immunoprecipitation.
The BRWD2/PHIP CryptoTudor domain mediates H3K4me interaction
To identify the regions of BRWD2/PHIP responsible for chromatin binding, we initially focused on its tandem bromodomains. Unexpectedly, we identified a sequence immediately upstream of the bromodomains that is highly conserved between humans and Drosophila (Supplemental Fig. 3A). Secondary structure prediction analysis using HHPRED and PHYRE2 (Kelley et al. 2015; Alva et al. 2016) suggested a similarity to the three-stranded β-barrel fold of Tudor Royal Family methyl-binding domains (Supplemental Fig. 3B,C; Maurer-Stroh et al. 2003). Because of its cryptic nature and predicted similarity to the Tudor domain, we named this region the CryptoTudor domain. We purified the BRWD2/PHIP CryptoTudor–bromodomain module (BRWD2–Crypt–bromo) from Escherichia coli, incubated it with mammalian nucleosomes, and subjected the captured histones to MS to quantify post-translational modifications (Fig. 3A–C). Consistent with our ChIP-seq studies, H3K4 methylated histones are enriched in BRWD2–Crypt–bromo pull-downs (Fig. 3C). H3K4me1 is the most abundant K4 methylation state, and its representation increases from 31% in the input chromatin to 48% of the BRWD2–Crypt–bromo pull-down material. H3K4me2 and H3K4me3 also exhibit enrichment from 0.8% (input) to 2.5% (pull-down) and from 0.2% (input) to 0.7% (pull-down), respectively. We also noted enrichment of specific acetylated histone peptides; namely, H3K9ac, H3K14ac, and H3K18ac. The most abundant of these is H3K14ac, which increased from 58% of total input histones to 84% of the BRWD2–Crypt–bromo-enriched material. Histone H3K9ac and H3K18ac are generally less abundant than H3K14ac but also showed enrichment. Histone H3K9ac increased from 3.2% (input) to 5.6% (pull-down), and H3K18ac increased from 1.9% (input) to 3.5% (pull-down). In contrast, H3K27ac was present at similar levels in both input (0.6%) and pull-down samples (0.5%). Modifications associated with transcriptional repression (H3K9me3 and H3K27me3) are depleted in BRWD2–Crypt–bromo pull-downs relative to input (Fig. 3D). Histone H3K9me3 decreased from 2.5% (input) to 1.7% (pull-down), and H3K27me3 decreased from 26% (input) to 19% (pull-down). To test direct interactions between the BRWD2–Crypt–bromo and histone modifications, we performed peptide pull-down assays. Both human and Drosophila Crypt–bromo modules interact with monomethylated, dimethylated, and trimethylated H3K4 but show no enrichment with H3K9me3 relative to unmodified H3 peptide (Fig. 3D). Isothermal titration calorimetry (ITC) experiments revealed that BRWD2–Crypt–bromo binds to H3K4me1 and H3K4me3 with Kd values in the low micromolar range, whereas minimal binding is detected with an unmodified peptide (Fig. 3E). Further characterization of the human BRWD2–Crypt–bromo module revealed that it binds weakly to a triple-acetylated (K9ac, K14ac, and K18ac) histone H3 peptide (Fig. 3F). This interaction is strongly enhanced when the H3 peptide contains H3K4me3 in addition to these three acetyl marks (Fig. 3F). In contrast, no appreciable binding is observed to peptides containing single acetyl modifications at H3K9, H3K14, H3K18, H3K27, or H4K20 (Fig. 3D,F,G), suggesting that H3K4 methylation, not H3 or H4 acetylation, is the primary determinant of BRWD2–Crypt–bromo binding to histone tails. Truncation experiments revealed that the CryptoTudor domain is responsible for binding to H3K4 methylation, whereas the isolated tandem bromodomains are unable to bind histone H3 tails (Fig. 3G). We then tested mutations at conserved aromatic residues within the CryptoTudor domain and found that a Trp1081-to-alanine mutation (W1081A) eliminates binding to H3K4 methylated peptides (Fig. 3H). Moreover, this position overlaps precisely with a homology model-predicted methyl-lysine-binding site (Fig. 3I). Collectively, our experiments suggest that BRWD2/PHIP associates with H3K4 methylated chromatin via a previously uncharacterized histone-binding domain related to the Tudor Royal Family, which we name the CryptoTudor domain.
Figure 3.

Characterization of the BRWD2/PHIP CryptoTudor domain. (A) Diagram depicting the recombinant BRWD2/PHIP CryptoTudor–bromodomain (BRWD2/PHIP–Crypt–bromo) construct used for binding studies. N-terminal 10X-Histidine (red box) and C-terminal 1XFlag (green box) tags were included for protein purification. (B) Coomassie stained SDS-PAGE gel of in vitro chromatin capture experiment using the BRWD2/PHIP–Crypt–bromo module. (Lane 1) Input MNase-digested chromatin. (Lane 2) Chromatin subjected to anti-Flag immunoprecipitation without BRWD2/PHIP–Crypt–bromo. (Lane 3) Chromatin incubated with recombinant BRWD2/PHIP–Crypt–bromo and subjected to anti-Flag immunoprecipitation. (C) Quantitative MS analysis of histones captured in B. Bars indicate the fraction of total histone represented by each modification state in the input (gray) or Flag immunoprecipitation material (red). (Un) Unmodified; (me1) monomethylated; (me2) dimethylated; (me3) trimethylated; (Ac) acetylated. (D) Coomassie-stained SDS-PAGE gel of histone peptide pull-downs performed with human (top) or Drosophila (bottom) Crypt–bromo constructs. Recombinant protein was incubated with streptavidin beads alone (SA-beads) or the biotinylated histone peptides indicated. Ten percent of input and 20% of each pull-down sample were loaded. (Bottom) Eluted proteins were subjected to dot blotting using streptavidin-HRP (SA-HRP). (E) ITC experiments with human BRWD2–Crypt–bromo titrated with H3 unmodified (left), H3K4me1 (middle), and H3K4me3 (right) peptides. (F) Coomassie-stained SDS-PAGE gel of pull-downs performed with human BRWD2/PHIP–Crypt–bromo and a panel of histone peptides. Ten percent of input and 20% of each pull-down sample were loaded. Streptavidin-HRP dot blot loading control is shown below the gel image. (G) Coomassie-stained SDS-PAGE gel of a panel of histone peptide pull-downs performed with human BRWD2–Crypt–bromo (top), the isolated CryptoTudor domain (middle), or the isolated tandem bromodomains (bottom). Ten percent of input and 20% of each pull-down sample were loaded. Dot blot loading control is shown below each gel image. (H) Coomassie-stained gels of histone peptide pull-downs performed with point mutations of the BRWD2 CryptoTudor domain. Dot blot loading controls are shown below the gels. (I) Homology model generated using Modeller and HHPRED alignments (Alva et al. 2016). Positions of residues predicted to be involved in methyl-lysine binding are highlighted in red.
The COMPASS family of histone H3K4 methylases differentially regulates BRWD2/PHIP chromatin occupancy
Members of the COMPASS enzyme family deposit H3K4 methylation at distinct sites in metazoan genomes (Shilatifard 2012; Piunti and Shilatifard 2016). Mammalian MLL1 and MLL2 catalyze H3K4me3 at promoters of developmentally regulated genes, whereas MLL3 and MLL4 mediate H3K4me1 at intergenic and intragenic enhancer regions (Herz et al. 2012; Hu et al. 2013a; Piunti and Shilatifard 2016). We tested the dependence of BRWD2/PHIP chromatin binding on the activity of these enzymes by performing ChIP-seq for BRWD2/PHIP and H3K4 methylation in different cell lines (HCT116 and v6.5 ESCs) carrying CRISPR deletions of various COMPASS family members. We compared parental HCT116 cells with cells null for MLL1 (MLL1 knockout) or cells lacking the MLL4 SET domain (MLL4-ΔSET) (Fig. 4A; Rickels et al. 2016). Deletion of MLL4 methyltransferase activity in HCT116 cells results in a dramatic loss of H3K4me1 from intergenic and intragenic regions accompanied by loss of BRWD2/PHIP binding (Fig. 4B,C). In contrast, MLL1 deletion causes only modest reductions in H3K4me1 and does not affect BRWD2/PHIP binding at most MLL4-dependent sites (Fig. 4B,C). Unexpectedly, a subset of sites (cluster 4) that lose H3K4me1 in MLL4-ΔSET mutant cells gain both H3K4me1 and BRWD2/PHIP binding when MLL1 is deleted in the same cell type, suggesting that the specific recruitment of BRWD2/PHIP to these new loci is caused by increased H3K4me1 levels (Fig. 4C; Supplemental Fig. 4A). Thus, it appears that the MLL1 and MLL4 complexes have opposite effects on H3K4me1 and BRWD2/PHIP occupancy at a discrete subset of loci. Similar to HCT116 cells, in v6.5 mouse ESCs, we observed loss of BRWD2/PHIP binding at sites where H3K4me1 is reduced in MLL4-ΔSET cells (Fig. 4 D,F; Supplemental Fig. 4B; Cao et al. 2017). In mouse ESCs, MLL2 controls H3K4me3 at both the promoters of bivalent genes and intergenic CpG island regulatory elements (Hu et al. 2013b; Hu et al. 2017). In MLL2 knockout ESCs (MLL2 knockout), we observed strong depletion of H3K4me3 and H3K4me1 from intergenic sites accompanied by loss of BRWD2/PHIP binding (Fig. 4E,F). Collectively, these experiments demonstrate that BRWD2/PHIP binding to chromatin is dependent on the COMPASS H3K4 methyltransferase family and that distinct family members regulate BRWD2/PHIP binding in a context-dependent manner.
Figure 4.

COMPASS complexes differentially regulate BRWD2/PHIP chromatin occupancy. (A) Diagram representing the experimental design for ChIP-seq in HCT116 parental, MLL1 knockout, and MLL4-ΔSET cells. (B) Representative UCSC genome browser track displaying a region between the FEM1C and CCDC112 genes at which H3K4me1 and BRWD2/PHIP chromatin occupancy is dependent on MLL4 enzymatic activity in HCT116 cells. Gray boxes indicate regions exhibiting diminished BRWD2/PHIP and H3K4me1 ChIP-seq signal in MLL4-ΔSET HCT116 cells. (C) Heat map displaying changes in ChIP-seq occupancy for BRWD2/PHIP, H3K4me1, H3K4me2, and H3K4me3 in the MLL1 knockout and MLL4-ΔSET cell lines relative to wild-type HCT116. Signal is centered on peaks of H3K4me1. Colors are scaled from a log2 fold change of −1.5 (green) to 1.5 (red). The two left columns indicate whether peaks overlap with a promoter TSS or are intergenic/intragenic (non-TSS). Peaks were separated into four clusters, indicated at the left. (D) Diagram representing the experimental design for ChIP-seq in v6.5 parental, MLL2 knockout, and MLL4-ΔSET ESCs. (E) Representative UCSC genome browser track displaying a region upstream of the Commd3 and Bmi1 genes at which H3K4me3, H3K4me1, and BRWD2/PHIP occupancy is dependent on MLL2 in v6.5 mouse ESCs. Gray boxes indicate regions that exhibit diminished BRWD2/PHIP, H3K4me1, and H3K4me3 ChIP-seq signal in MLL2 knockout v6.5 cells. (F) Heat map displaying changes is ChIP-seq occupancy for BRWD2/PHIP, H3K4me1, H3K4me2, and H3K4me3 in the MLL2 knockout and MLL4-ΔSET cells relative to wild-type v6.5 ESCs. Signal is centered on peaks of H3K4me1. Colors are scaled from a log2 fold change of −1.5 (green) to 1.5 (red). The two left columns indicate whether peaks overlap with a promoter TSS or are intergenic/intragenic (non-TSS). Occupancy was separated into three clusters, indicated at the left.
Drosophila dBRWD3 controls H3K27ac distribution at promoters and enhancers
The human genome contains three homologous BRWD genes (BRWD1/WDR9, BRWD2/PHIP, and BRWD3) with potential functional redundancy, whereas Drosophila harbors a single homolog, dBRWD3. We took advantage of this and performed RNAi against dBRWD3 in Drosophila S2 cells. dBRWD3 depletion appears to cause a slight increase in bulk levels of histone H3K4me1 and H3K27ac relative to control RNAi-treated cells (Fig. 5A). ChIP-seq studies revealed that the localization patterns of H3K4me3 and H3K4me1 were largely unaffected by dBRWD3 depletion, whereas H3K27ac displayed a notable alteration (Fig. 5B–D). Sharp peaks of H3K27ac typically located at promoters show a prominent reduction in signal, whereas many intergenic and intragenic sites displayed a spread in peak breadth (Fig. 5B–D; Supplemental Fig. 5A,B). RNA sequencing (RNA-seq) analysis revealed a correlation between these chromatin alterations and gene expression. Up-regulated genes (cluster 2) demonstrate increased intragenic H3K27ac (Fig. 5D,E). In contrast, down-regulated genes (cluster 1) exhibit loss of H3K27ac at their promoters (Fig. 5D,E).
Figure 5.

dBRWD3 depletion alters chromatin modification patterns in Drosophila. (A) Western blotting of Drosophila S2 cells treated with RNAi targeting lacZ (control) or two independent RNAi sequences targeting dBRWD3. Western blotting confirmed efficient depletion of dBRWD3 and detected slightly elevated H3K4me1 and H3K27ac levels. Histone H3 and β-Tubulin served as loading controls. (B) UCSC genome browser track example for bBRWD3, H3K4me1, H3K4me3, and H3K27ac ChIP-seq in Drosophila S2 cells that displays a region where dBRWD3 depletion alters chromatin structure. The blue highlighted box indicates a region overlapping with a TSS where the H3K27ac signal is dramatically decreased upon dBRWD3 knockdown. The red highlighted box indicates an intragenic non-TSS where H3K27ac spreads in dBRWD3 knockdown cells. (C) Density bar plots displaying Z-scores centered at TSS or non-TSS peaks of H3K27ac. At TSSs, H3K27ac signal is strongly diminished, whereas at non-TSSs, this modification exhibits a spread in breadth. In contrast, H3K4me1 and H3K4me3 distributions are not dramatically affected by dBRWD3 depletion, although H3K4me1 appears to show a modest decrease at non-TSSs, and H3K4me3 shows a slight increase in breadth at TSSs. The Z-score color scale is at the right. (D) Metagene plots displaying the occupancy of H3K4me1, H3K4me3, and H3K27ac in control and dBRWD3 knockdown S2 cells. Green arrows above metagene plots indicate TSSs, and red filled circles indicate transcription termination sites. The color scale range for each pair of plots is indicated at the bottom. (E) Metagene plot displaying differential signal for H3K27ac between dBRWD3 and control knockdown S2 cells (gene order is identical to that in D). Green arrows above metagene plots indicate TSSs, and red filled circles indicate transcription termination sites. The color scale is ranged from a log fold change of −1.5 (green) to +1.5 (red). RNA-seq data for these genes are at the right. (Red) Up-regulated; (green) down-regulated. The genes were separated into three clusters (indicated at the far right) based on their ChIP-seq patterns.
Discussion
Here we identify BRWD2/PHIP and dBRWD3 as previously uncharacterized H3K4 methyl-interacting proteins in mammalian and Drosophila cells. We observed BRWD2/PHIP binding to chromatin at both enhancers marked by H3K4me1 and promoters, which carry predominantly H3K4me3 (Piunti and Shilatifard 2016). Our in vitro biochemical experiments further support the model that BRWD2/PHIP recognizes monomethylated, dimethylated, and trimethylated H3K4. This feature distinguishes BRWD2/PHIP from most other H3K4 methyl-binding proteins, which exhibit more restricted binding specificities. For instance, the TAF3–PHD (Vermeulen et al. 2007; van Ingen et al. 2008), ING2–PHD (Shi et al. 2006), BPTF–PHD (Wysocka et al. 2006; Ruthenburg et al. 2011), and JMJD2A double Tudor domains (Huang et al. 2006; Kim et al. 2006) show a preference for H3K4me3 over H3K4me2 and exhibit extremely weak or undetectable binding to H3K4me1. In contrast, the ZMYND8–PHD binds to unmethylated H3K4 and H3K4me1 but cannot interact with H3K4me2 and H3K4me3 (Li et al. 2016; Savitsky et al. 2016). Interestingly, the CHD1 double chromodomain possesses a binding profile similar to BRWD2/PHIP and binds to all three H3K4 methylation states, although CHD1 has a threefold higher affinity for H3K4me3 and H3K4me2 over H3K4me1 (Flanagan et al. 2005; Sims et al. 2005). Thus, BRWD2/PHIP represents a new member of a small category of chromatin-binding factors capable of interacting with all three forms of H3K4 methylation states.
We were initially surprised to discover that BRWD2/PHIP exhibits direct binding to methylated H3K4, considering that BRWD2/PHIP contains two bromodomains—a protein feature involved in binding to acetyl-lysine in histone and nonhistone proteins (Filippakopoulos et al. 2012; Fujisawa and Filippakopoulos 2017). Close analysis of the protein sequences adjacent to the BRWD2/PHIP bromodomains revealed a highly conserved upstream sequence with predicted similarity to the three-strand β-barrel fold of Royal Family Tudor domains (Maurer-Stroh et al. 2003). Many Royal Family proteins display minimal amino acid sequence similarity to each other but nevertheless adopt similar three-dimensional structures (Maurer-Stroh et al. 2003; Yap and Zhou 2010), which likely explains how the BRWD2/PHIP CryptoTudor domain escaped previous detection. Our experiments suggest that H3K4 methylation binding by the CryptoTudor domain acts as the primary determinant of BRWD2/PHIP binding to chromatin, as the isolated bromodomains exhibit no detectable interaction with a battery of histone peptides. The overwhelming majority of bromodomains contains an asparagine residue at the acetyl-lysine-binding site (Filippakopoulos et al. 2012); however, the second BRWD2/PHIP bromodomain contains a threonine substitution at this highly conserved position, raising the possibility that this atypical bromodomain is nonfunctional. When linked to the CryptoTudor domain, the presence of both bromodomains appears to enhance binding to histone H3 peptides containing H3K4 methylation in combination with acetylation at the K9, K14, and K18 positions. This is consistent with a multivalent mode of histone binding that has been observed for many chromatin proteins, including BPTF and ZMYND8 (Ruthenburg et al. 2007, 2011; Li et al. 2016; Savitsky et al. 2016). We speculate that the BRWD2/PHIP CryptoTudor domain is initially recruited to chromatin by interacting with methylated H3K4, and, subsequently, one or both bromodomains contact H3 acetylation and stabilize this interaction through multivalent effects. Determining the structural basis of BRWD2/PHIP interaction with the histone H3 tail is an important direction for future work.
The three mammalian BRWD proteins are implicated in a broad range of biological processes. BRWD1/WDR9 is required for gametogenesis and controls chromatin accessibility at the immunoglobulin κ locus during B-lymphocyte differentiation (Philipps et al. 2008; Mandal et al. 2015; Pattabiraman et al. 2015). BRWD2/PHIP was initially isolated as a regulator of the insulin–receptor signaling pathway and is required for postnatal survival in mice (Farhang-Fallah et al. 2000; Li et al. 2010). BRWD2/PHIP is highly expressed in human metastatic melanoma and promotes proliferation in this context (De Semir et al. 2012). A recent report also identified BRWD2/PHIP mutations in patients with severe intellectual disability (Webster et al. 2016). Similarly, mutations in BRWD3 cause an X-linked intellectual disability syndrome in humans, and mutant mice also display neural development defects (Field et al. 2007; Cox et al. 2010). Studies of the Drosophila homolog, dBRWD3/Ramshackle demonstrate that it also plays a complex role in diverse developmental contexts, such as organizing cell polarity and cytoskeleton, controlling circadian rhythm, positively regulating Jak/Stat and nuclear hormone receptor signaling, and counteracting Polycomb group function (Muller et al. 2005; D'Costa et al. 2006; Ozturk et al. 2013; Ihry and Bashirullah 2014; Shih et al. 2016). Our identification of the chromatin-binding ligand for BRWD2/PHIP and Drosophila dBRWD3 may shed light on the molecular mechanisms underlying these diverse developmental processes.
Here, we establish a direct link between COMPASS family-mediated H3K4 methylation and BRWD2/PHIP recruitment. This is of particular interest considering the comparison of the phenotypes associated with loss of COMPASS and BRWD2/PHIP complex components. Similar to BRWD2/PHIP and BRWD3, mutations of MLL4/KMT2D result in developmental abnormalities (Niikawa-Kuroki syndrome) that include intellectual disability (Ng et al. 2010). CUL4B, a strong BRWD2/PHIP-binding partner and component of CRL4, is mutated in an X-linked intellectual disability syndrome (Tarpey et al. 2007), and the BRWD2/PHIP-interacting protein PHF6 is mutated in another X-linked developmental disorder, Börjeson-Forssman-Lehmann syndrome (Lower et al. 2002). It is tempting to speculate that this phenotypic overlap may indicate a common pathway for these proteins in controlling neural development. We propose that H3K4me1 catalyzed through MLL3/4–COMPASS serves as a recruitment signal for BRWD2/PHIP, which in turn recruits the CRL4 ubiquitin ligase complex to chromatin. Intriguingly, CRL4 has established functions in cell cycle control and DNA damage responses (Jin et al. 2006; Lydeard et al. 2013); therefore, the molecular link that we established here may point to COMPASS functions beyond transcriptional regulation.
Materials and Methods
ChIP-seq
ChIP-seq was performed as described (Lee et al. 2006; Piunti et al. 2017; Vo et al. 2017).
Mammalian protein purification
Flag-tagged protein purifications from MNase-digested chromatin were performed as described (Herz et al. 2014; Piunti et al. 2017). MLN4924 (1 µM) and 10 µM MG132 were added 5 h prior to harvesting where indicated.
RNAi and ChIP in Drosophila S2 cells
Culture of S2 cells, RNAi, ChIP-seq, and RNA-seq were performed as described (Herz et al. 2012).
Antibodies
BRWD2/PHIP antibodies (Shilatifard, nos. 833 and 834) were raised against a protein fragment spanning BRWD2/PHIP amino acids 1681–1821 (sequence ID NP_060404.4). Drosophila dBRWD3 antibody (Shilatifard, no. 385) was raised against amino acids 976–1495 (sequence ID NP_732982.1). Proteins were expressed from the pET16 plasmid in Rosetta2 E. coli cells and purified using Ni-NTA agarose resin (Qiagen). Rabbit immunization was performed by Pocono Rabbit Farm and Laboratory. Other antibodies used in this study were H3K4me1 (Shilatifard Laboratory, no. 24), H3K4me2 (Shilatifard Laboratory, no. 27), H3K4me3 (Shilatifard Laboratory, no. 680), H3K27ac (Cell Signaling Technology, no. 8137), H2A.Z (Cell Signaling Technology, no. 2718), H3K27me3 (Shilatifard Laboratory, no. 67), DDB1 (Cell Signaling Technology, no. 6998), CUL4A (Cell Signaling Technology, no. 2699), CHD4 (Cell Signaling Technology #12011), PHF6 (Santa Cruz Biotechnology, no. sc-365237), β-Tubulin (E7, Developmental Studies Hybridoma Bank), Histone H3 (Shilatifard Laboratory, no. 42), and Flag-M2 (Sigma, no. F1804).
Recombinant protein purification from E. coli
Rosetta2 E. coli were transformed with pET-16 plasmids encoding N-terminal 10xHis-tagged and C-terminal 1xFlag-tagged constructs. Liquid cultures were grown to an OD600 of 0.6 and then induced with 1 mM IPTG for 24 h at 20°C. Proteins were extracted under native conditions in N-250 buffer (50 mM Tris at pH 7.5, 250 mM NaCl, 15 mM imidazole, 0.2% igepal, 0.2% Tween-20, 0.2% Triton X-100, 1× Sigma protease inhibitor no. P8849, 1 mM PMSF) supplemented with 1 mg/mL lysozyme and 25 U/mL benzonase nuclease (Sigma, no. E1014-25KU). Lysates were clarified by centrifugation at 40,000g for 20 min at 4°C (Beckman Coulter 45-Ti Rotor), and protein extracts were incubated with Ni-NTA agarose beads (Qiagen) for 3 h. Beads were washed three times with 50 bead volumes of N-250 buffer, and proteins were eluted with His elution buffer (50 mM Tris at pH 7.5, 250 mM NaCl, 300 mM imidazole, 0.2% igepal, 0.2% Tween-20, 0.2% Triton X-100, 25% glycerol, 1 mM PMSF, 1× Sigma protease inhibitor no. P8849). After elution, EDTA was added to 2 mM, and proteins were concentrated using Amicon Ultra centrifugal filters (Millipore)
Histone peptide pull-down assays
For each binding reaction, 5 µg of biotinylated histone peptide (Epicypher) was bound to 25 µL of streptavidin agarose beads (Thermo-Fisher, no. 20359) in N-250 buffer for 1 h. Beads were washed twice with 1 mL N-250 buffer to remove any unbound peptides and resuspended in N-250 buffer supplemented with 10 mM sodium butyrate and 1 mM EDTA. Approximately 150 µg of recombinant BRWD2/PHIP protein was added to the bead-bound peptides and incubated overnight at 4°C on a rotator. The following day, beads were washed five times with N-250 buffer. Bound proteins were eluted by heating for 10 min at 95°C in 4× SDS sample buffer (250 mM Tris at pH 6.8, 50% glycerol, 8% SDS). As a loading control, 0.25 µL of the eluted proteins was spotted onto a nitrocellulose membrane and detected using streptavidin-HRP (Thermo Scientific, no. 21130). To assay BRWD2/PHIP protein captured by the histone peptides, samples were supplemented with bromophenol blue and 2-mercaptoethanol and heated for 5 min at 95°C. Ten percent of input protein and 20% of the peptide pull-down were resolved by SDS-PAGE, and gels were stained with Coomassie blue.
ITC
BRWD2 protein isolated from E. coli was buffer-exchanged into ITC buffer (50 mM Tris at pH 7.4, 200 mM NaCl, 1 mM EDTA, 2% glycerol) using PD-10 columns (GE Healthcare). Histone peptides were purchased from Epicypher and dissolved in ITC buffer. ITC was performed on a MicroCal VP-ITC calorimeter (Malvern). BRWD2 protein concentration was 25 µM, and histone peptides were 1 mM. ITC was performed at 30°C using the following conditions: 5-µL injection volume, 10-sec injection duration, 300-sec injection spacing, and 10-sec filter period.
In vitro chromatin capture
In vitro chromatin capture was performed using an approach similar to the matrix-assisted reader chromatin capture (MARCC) method (Su et al. 2015). Native mononucleosomes were prepared by MNase digestion of 3 × 108 HEK293 nuclei as described (Herz et al. 2014; Piunti et al. 2017). After stopping the MNase reaction, 500 µg of recombinant His-Flag-tagged BRWD2/PHIP protein was added, and samples were incubated overnight at 4°C. The following day, samples were centrifuged to remove protein precipitate, and cleared supernatant was incubated with 500 µL of Flag-M2 agarose beads for 2 h at 4°C. Samples were washed (once with 5 mL and then five times with 1mL) with wash buffer (50 mM HEPES at pH7.9, 100 mM KCl, 100 mM NaCl, 10% glycerol, 1 mM EDTA, 2 mM EGTA, 0.1% Triton X-100, 10 mM sodium butyrate), and bound complexes were eluted with elution buffer (20 mM HEPES at pH 7.9, 100 mM NaCl, 100 µg/mL 3xFlag peptide [Sigma, F4799]). Input and Flag pull-down samples were extracted with 5 vol of 0.2 M H2SO4 to enrich histones (Shechter et al. 2007), precipitated with 20% trichloroacetic acid, washed with 100% acetone, and air-dried.
CRISPR–Cas9 gene editing
Plasmids expressing Cas9 and single-guide RNAs (sgRNAs) were constructed in pX330 (Cong et al. 2013). pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang (Addgene plasmid no. 42230). The sgRNA sequences used to delete the mouse Mll4 SET domain were GCAGCTTAAATTCCGGCCTTG and GAGGCGAGGGGCCCCGATTGA. Human MLL4 SET deletion sgRNAs were GTGGTGTCGGCGGGTTACTC and GCATCCATTTCCGACAATTCC. Human BRWD2/PHIP promoter deletion sgRNAs were GGATGTGTGTGTACTTGCGA and GAAGATGTTGATAACCGCTCA. HCT116 cells (1 × 107 cells) were electroporated with 30 µg of each sgRNA and 15 µg of pCAGGS-EGFP-IRES-Puro (a kind gift from Dr. Hitoshi Niwa). Beginning 1 d after electroporation, cells were selected with 1ug/mL puromycin for 24 h, allowed to recover for 4–5 d, and then plated at low density (1000 cells per 10-cm dish) to isolate single-cell-derived colonies.
MS of protein complexes
Sample preparation and MS of immunoprecipitated complexes were performed as described (Hickox et al. 2017).
MS of histone modifications
Acid-extracted histones were propionylated, trypsin-digested, and subjected to MS to quantify histone modifications as described previously (Zheng et al. 2013; Piunti et al. 2017). Raw MS files were imported and analyzed in Skyline with Savitzky-Golay smoothing (MacLean et al. 2010). All Skyline peak area assignments were confirmed manually. Total peak areas from SRM were used to plot bar graphs representing relative abundances of distinct histone modifications. The relative abundances were determined from the mean of three technical replicates, with error bars showing standard deviation.
Supplementary Material
Acknowledgments
We thank Dr. Dan Foltz and Dr. Edwin Smith for insightful discussions. We thank Jaydeep Singh and Annika Krueger for technical assistance. We thank the Proteomics Core Facility at Northwestern University, which is supported by National Cancer Institute (NCI) Cancer Center Support Grant P30CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center, and the National Resource for Translational and Developmental Proteomics supported by P41GM108569. The Savas laboratory is supported by an R00 DC-013805 award from the National Institutes of Health (NIH) National Institute on Deafness and Other Communication Disorders. Studies related to the COMPASS family in the Shilatifard laboratory are supported by a generous Outstanding Investigator Award from the NIH NCI (R35CA197569).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.305201.117.
References
- Alva V, Nam SZ, Soding J, Lupas AN. 2016. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res 44: W410–W415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Li T, Yi XH, MacCoss MJ, Moon RT, Zheng N. 2006. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443: 590–593. [DOI] [PubMed] [Google Scholar]
- Cao KX, Collings CK, Marshall SA, Morgan MA, Rendleman EJ, Wang L, Sze CC, Sun TJ, Bartom ET, Shilatifard A. 2017. SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev 31: 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith REJ, et al. 2016. Cullin–RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531: 598–603. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox BJ, Vollmer M, Tamplin O, Lu M, Biechele S, Gertsenstein M, van Campenhout C, Floss T, Kuhn R, Wurst W, et al. 2010. Phenotypic annotation of the mouse X chromosome. Genome Res 20: 1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Costa A, Reifegerste R, Sierra S, Moses K. 2006. The Drosophila ramshackle gene encodes a chromatin-associated protein required for cell morphology in the developing eye. Mech Dev 123: 591–604. [DOI] [PubMed] [Google Scholar]
- den Besten W, Verma R, Kleiger G, Oania RS, Deshaies RJ. 2012. NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat Struct Mol Biol 19: 511–U570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Semir D, Nosrati M, Bezrookove V, Dar AA, Federman S, Bienvenu G, Venna S, Rangel J, Climent J, Meyer Tamguney TM, et al. 2012. Pleckstrin homology domain-interacting protein (PHIP) as a marker and mediator of melanoma metastasis. Proc Natl Acad Sci 109: 7067–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. 2008. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134: 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhang-Fallah J, Yin XH, Trentin G, Cheng AM, Rozakis-Adcock M. 2000. Cloning and characterization of PHIP, a novel insulin receptor substrate-1 pleckstrin homology domain interacting protein. J Biol Chem 275: 40492–40497. [DOI] [PubMed] [Google Scholar]
- Field M, Tarpey PS, Smith R, Edkins S, O'Meara S, Stevens C, Tofts C, Teague J, Butler A, Dicks E, et al. 2007. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am J Hum Genet 81: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. 2012. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149: 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim YC, Minor W, Rastinejad F, Khorasanizadeh S. 2005. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438: 1181–1185. [DOI] [PubMed] [Google Scholar]
- Fujisawa T, Filippakopoulos P. 2017. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18: 246–262. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu YT, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu CX, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang KW, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A. 2012. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. 2014. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345: 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickox AE, Wong ACY, Pak K, Strojny C, Ramirez M, Yates JR, Ryan AF, Savas JN. 2017. Global analysis of protein expression of inner ear hair cells. J Neurosci 37: 1320–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DQ, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. 2013a. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 33: 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DQ, Garruss AS, Gao X, Morgan MA, Cook M, Smith ER, Shilatifard A. 2013b. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol 20: 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DQ, Gao X, Cao KX, Morgan MA, Mas G, Smith ER, Volk AG, Bartom ET, Crispino JD, Di Croce L, et al. 2017. Not all H3K4 methylations are created equal: Mll2/COMPASS dependency in primordial germ cell specification. Mol Cell 65: 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. 2006. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 312: 748–751. [DOI] [PubMed] [Google Scholar]
- Ihry RJ, Bashirullah A. 2014. Genetic control of specificity to steroid-triggered responses in Drosophila. Genetics 196: 767–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Abraham BJ, Lee TI, Jaenisch R, Bradner JE, Young RA. 2015. Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc Natl Acad Sci 112: 3841–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JP, Arias EE, Chen J, Harper JW, Walter JC. 2006. A family of diverse Cul4–Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 23: 709–721. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 Web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. 2006. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep 7: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA. 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc 1: 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SA, Francisco AB, Han CC, Pattabiraman S, Foote MR, Giesy SL, Wang C, Schimenti JC, Boisclair YR, Long QM. 2010. The full-length isoform of the mouse pleckstrin homology domain-interacting protein (PHIP) is required for postnatal growth. FEBS Lett 584: 4121–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Li YY, Lv J, Zheng XD, Wen H, Shen HJ, Zhu GJ, Chen TY, Dhar SS, Kan PY, et al. 2016. ZMYND8 reads the dual histone mark H3K4me1–H3K14ac to antagonize the expression of metastasis-linked genes. Mol Cell 63: 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, Schelley S, Hoyme HE, White SM, Delatycki MB, et al. 2002. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet 32: 661–665. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Schulman BA, Harper JW. 2013. Building and remodelling Cullin–RING E3 ubiquitin ligases—‘ubiquitylation: mechanism and functions’ review series. EMBO Rep 14: 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Hamel KM, Maienschein-Cline M, Tanaka A, Teng G, Tuteja JH, Bunker JJ, Bahroos N, Eppig JJ, Schatz DG, et al. 2015. Histone reader BRWD1 targets and restricts recombination to the Igk locus. Nat Immunol 16: 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. 2003. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 28: 69–74. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Shilatifard A. 2015. Chromatin signatures of cancer. Genes Dev 29: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Kuttenkeuler D, Gesellchen V, Zeidler MP, Boutros M. 2005. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 436: 871–875. [DOI] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, et al. 2010. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42: 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozturk N, VanVickle-Chavez SJ, Akileswaran L, Van Gelderb RN, Sancar A. 2013. Ramshackle (Brwd3) promotes light-induced ubiquitylation of Drosophila cryptochrome by DDB1–CUL4–ROC1 E3 ligase complex. Proc Natl Acad Sci 110: 4980–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman S, Baumann C, Guisado D, Eppig JJ, Schimenti JC, De la Fuente R. 2015. Mouse BRWD1 is critical for spermatid postmeiotic transcription and female meiotic chromosome stability. J Cell Biol 208: 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipps DL, Wigglesworth K, Hartford SA, Sun FY, Pattabiraman S, Schimenti K, Handel M, Eppig JJ, Schimenti JC. 2008. The dual bromodomain and WD repeat-containing mouse protein BRWD1 is required for normal spermiogenesis and the oocyte-embryo transition. Dev Biol 317: 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Shilatifard A. 2016. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352: aad9780. [DOI] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma QH, Takahashi YH, Woodfin AR, et al. 2017. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23: 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Sergeev M, Garnaas M, Lydeard JR, Huttlin EL, Goessling W, Shah JV, Harper JW. 2015. Systematic proteomics of the VCP–UBXD adaptor network identifies a role for UBXN10 in regulating ciliogenesis. Nat Cell Biol 17: 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickels R, Hu DQ, Collings CK, Woodfin AR, Piunti A, Mohan M, Herz HM, Kvon E, Shilatifard A. 2016. An evolutionary conserved epigenetic mark of Polycomb response elements implemented by Trx/MLL/COMPASS. Mol Cell 63: 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. 2007. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8: 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li HT, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou YL, Muir TW, Patel DJ, et al. 2011. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145: 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky P, Krojer T, Fujisawa T, Lambert JP, Picaud S, Wang CY, Shanle EK, Krajewski K, Friedrichsen H, Kanapin A, et al. 2016. Multivalent histone and DNA engagement by a PHD/BRD/PWWP triple reader cassette recruits ZMYND8 to K14ac-rich chromatin. Cell Rep 17: 2724–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Dormann HL, Allis CD, Hake SB. 2007. Extraction, purification and analysis of histones. Nat Protoc 2: 1445–1457. [DOI] [PubMed] [Google Scholar]
- Shen HJ, Xu WQ, Guo R, Rong BW, Gu L, Wang ZT, He CX, Zheng LJ, Hu X, Hu Z, et al. 2016. Suppression of enhancer overactivation by a RACK7–histone demethylase complex. Cell 165: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XB, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. 2006. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442: 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HT, Chen WY, Liu KY, Shih ZS, Chen YJ, Hsieh PC, Kuo KL, Huang KH, Hsu PH, Liu YW, et al. 2016. dBRWD3 regulates tissue overgrowth and ectopic gene expression caused by polycomb group mutations. PLoS Genet 12: e1006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, Reinberg D. 2006. Histone H3 Lys 4 methylation: caught in a bind? Genes Dev 20: 2779–2786. [DOI] [PubMed] [Google Scholar]
- Sims RJ, Chen CF, Santos-Rosa H, Kouzarides T, Patel SS, Reinberg D. 2005. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J Biol Chem 280: 41789–41792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458: 732–736. [DOI] [PubMed] [Google Scholar]
- Su ZL, Boersma M, Lee JH, Liu SC, Garcia B, Denu J. 2015. MARCC (matrix-assisted reader chromatin capture): ChIP-less analysis of chromatin states. FASEB J 29: 877.7. [Google Scholar]
- Surface LE, Fields PA, Subramanian V, Behmer R, Udeshi N, Peach SE, Carr SA, Jaffe JD, Boyer LA. 2016. H2AZ1 Monoubiquitylation antagonizes BRD2 to maintain poised chromatin in ESCs. Cell Rep 14: 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, O'Meara S, Edkins S, Teague J, Butler A, Dicks E, Stevens C, Tofts C, Avis T, et al. 2007. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet 80: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova R, Agherbi H, Boned A, Meresse S, Djabali M. 2006. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc Natl Acad Sci 103: 6629–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ingen H, van Schaik FMA, Wienk H, Ballering J, Rehmann H, Dechesne AC, Kruijzer JAW, Liskamp RMJ, Timmers HTM, Boelens R. 2008. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure 16: 1245–1256. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WWMP, van Schaik FMA, Varier RA, Baltissen MPA, Stunnenberg HG, Mann M, Timmers HTM. 2007. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131: 58–69. [DOI] [PubMed] [Google Scholar]
- Vo BT, Li CL, Morgan MA, Theurillat I, Finkelstein D, Wright S, Hyle J, Smith SMC, Fan YP, Wang YD, et al. 2017. Inactivation of Ezh2 upregulates Gfi1 and drives aggressive Myc-driven group 3 medulloblastoma. Cell Rep 18: 2907–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster E, Cho MT, Alexander N, Desai S, Naidu S, Bekheirnia MR, Lewis A, Retterer K, Juusola J, Chung WK. 2016. De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb Mol Case Stud 2: a001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. 2006. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442: 86–90. [DOI] [PubMed] [Google Scholar]
- Yap KL, Zhou MM. 2010. Keeping it in the family: diverse histone recognition by conserved structural folds. Crit Rev Biochem Mol 45: 488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YP, Thomas PM, Kelleher NL. 2013. Measurement of acetylation turnover at distinct lysines in human histones identifies long-lived acetylation sites. Nat Commun 4: 2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.