

Abstract
Radiation-induced bone loss is a potential health concern for cancer patients undergoing radiotherapy. Enhanced bone resorption by osteoclasts and decreased bone formation by osteoblasts were thought to be the main reasons. In this study, we showed that both pre-differentiating and differentiating osteoclasts were relatively sensitive to X-rays compared with osteoblasts. X-rays decreased cell viability to a greater degree in RAW264.7 cells and in differentiating cells than than in osteoblastic MC3T3-E1 cells. X-rays at up to 8 Gy had little effects on osteoblast mineralization. In contrast, X-rays at 1 Gy induced enhanced osteoclastogenesis by enhanced cell fusion, but had no effects on bone resorption. A higher dose of X-rays at 8 Gy, however, had an inhibitory effect on bone resorption. In addition, actin ring formation was disrupted by 8 Gy of X-rays and reorganized into clusters. An increased activity of Caspase 3 was found after X-ray exposure. Actin disorganization and increased apoptosis may be the potential effects of X-rays at high doses, by inhibiting osteoclast differentiation. Taken together, our data indicate high radiosensitivity of osteoclasts. X-ray irradiation at relatively low doses can activate osteoclastogenesis, but not osteogenic differentiation. The radiosensitive osteoclasts are the potentially responsive cells for X-ray-induced bone loss.
Keywords: ionizing radiation, osteoblasts, osteoclasts, differentiation
INTRODUCTION
Radiation therapy is usually used as part of adjuvant therapy in cancer treatment to destroy malignant cells. Side effects during radiation treatment include damage to the nearby tissues, including the bone [1]. It has been demonstrated that X-rays can induce bone loss and consequently increase the risk of fracture, especially insufficiency fracture [2]. These fractures also exhibit difficulty in healing, with delayed union [3]. The development of fracture after radiotherapy in cancer patients has been reported in several cancer types, such as breast cancer, cervical cancer, lung cancer, prostate cancer, anal cancer, rectal cancer, and other soft-tissue sarcomas [4–7]. Animal studies have also shown that ionizing radiation (IR) can induce bone loss, osteoradionecrosis, fracture and delayed bone healing [8–10].
The bone is a metabolically dynamic tissue. Continuous bone remodeling throughout the lifetime is essential for the maintenance of bone health and for integrity of the bone structure [11]. Disrupted balance between bone resorption by osteoclasts and bone formation by osteoblasts was found after IR treatment [12–27]. Decreased mineralization was found at sites of direct irradiation, with slightly decreased or similar osteoblast numbers [12–15]. However, osteoblasts are relatively resistant, and even a higher dose of 10 Gy did not induce apoptosis in isolated osteoblasts in calvarias [15]. Osteoblasts were still viable following X-ray irradiation at 30 Gy [16]. Furthermore, certain doses of X-rays stimulated osteoblast differentiation in in vitro studies [17–19]. These conflicting results indicated that the mineralization ability of osteoblasts might not be directly affected by IR.
Osteoclasts are responsible for resorbing bone matrix, a necessary event in fracture healing and in regulating the level of blood calcium [20]. Animal studies have shown that exposure to IR causes a rapid decrease in bone mass due to immediate activation of osteoclasts. Total body irradiation with 2 Gy X-rays, γ-rays, or heavy ions led to prompt loss in bone mineral density at 1 week after irradiation [21–25]. The osteoclasts were activated with an elevated tartrate-resistant acid phosphatase (TRAP) concentration in the serum, the osteoclast number, and the area of osteoclast surface [22]. Furthermore, expressions of pro-osteoclastic cytokines in marrow cells were increased [21]. The IR doses used in radiotherapy are high enough to cause DNA damage and then induce cell death [26]. Oest et al. reported that following a transient increase in osteoclast number after focal irradiation to the hindlimb, there was a persistent depletion in osteoclasts [27]. Long-term loss of osteoclasts may be due to the death of osteoclast progenitors. These studies indicate that activation of existing osteoclasts and loss of osteoclast progenitors are two important mechanisms for X-ray–induced bone damage.
Osteoblasts and osteoclasts apparently have different radiosensitivities. In the present study, we first compared the direct effects of X-rays on osteoclastogenesis in RAW264.7 pre-osteoclast cells and on mineralization in MC3T3-E1 osteoblast cells, as well as cell viability in both cell lines. Then, the relatively radiosensitive RAW264.7 cells were chosen for further investigation. Osteoclast differentiation occurs in four stages: pre-osteoclast proliferation, osteoclast formation, fusion, and bone resorption [20]. The cellular response of osteoclasts to IR may largely depend on the differentiation status. In addition, both oxidative stress and apoptosis induced by X-rays are two main causes of decreased cell survival. Therefore, we also investigated the influence of X-rays on survival, differentiation and apoptosis in both osteoclast precursors and in differentiated osteoclasts. Study of the biological effects of X-rays contributes to a better understanding of the mechanisms by which radiation induces bone damage. Such studies are also useful for the identification of radioresponsive cells, and for development of radiation protection.
MATERIALS AND METHODS
Cell culture and irradiation
The murine monocyte/macrophage cell line RAW264.7 was used in this study. The cells were purchased from the Cell Bank of the Chinese Academy of Sciences (CAS; Shanghai, China), and maintained in α-Minimum Essential Medium (α-MEM; Gibco, Grand Island, NY, USA), supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco) and 2 mM L-glutamine (Amresco, Solon, OH, USA). Murine osteoblastic cell line MC3T3-E1 Subclone 4 was also used in this study, which was kindly provided by Prof. & Dr Hong Zhou of the University of Sydney. The osteoblastic MC3T3-E1 cells were maintained in α-MEM, supplemented with 2 mM L-glutamine, 10% (v/v) FBS. Both cell lines were cultured in a 37°C incubator with a humidified atmosphere containing 5% CO2.
The cells were seeded in 35-mm dishes at a density of 9000 cells/cm2 and incubated overnight for 12 h prior to irradiation. The cells were irradiated with different doses from 1 Gy to 8 Gy using an X-ray linear accelerator (160 kV, 25 mA; RadSource, Suwanee, GA, USA) at a fixed dose rate of 1.15 Gy/min. The focus skin distance was 40 cm, and the irradiation field was 280 mm × 180 mm with a dose uniformity of >95%. Sham-irradiated cells were defined as the 0 Gy group.
Cell viability assay
The viability of RAW264.7 cells was quantified by the Cell Counting Kit-8 (CCK8; Beyotime, Shanghai, China) assay. According to the manufacturer's instructions, cells were incubated with 10% CCK8 for 1 h. The absorbance was measured at 450 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
Caspase 3 activity and expression assay
The Caspase 3 activity was detected on the basis of the conversion of Ac-DEVD-pNA (acetyl-Asp-Glu-Val-Asp p-nitroanilide) to pNA by a Caspase 3 Activity Assay Kit (Beyotime). Briefly, the cells were harvested with lysis buffer. Ac-DEVD-pNA (acetyl-Asp-Glu-Val-Asp p-nitroanilide) was added and incubated at 37°C for 1 d. The activities of Caspase 3 were determined based on the absorbance at 405 nm by a microplate reader (Bio-Rad Laboratories). The total protein content was used to normalize the obtained values.
Expression of caspase 3 was measured by western blotting. Total protein was loaded on 10% SDS-PAGE gels. After transfer, polyvinylidene fluoride (PVDF) membranes were blocked with 5% non-fat dry milk and incubated with caspase 3 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, Danvers, MA) primary antibody overnight at 4°C, followed by dilution of HRP-conjugated second antibody (Beyotime). Expression of caspase 3 was normalized to GAPDH.
Osteoclast differentiation assay
Osteoclastogenesis in RAW264.6 cells was induced in the presence of 50 ng/ml receptor activator of NF-kappaB ligand (RANKL) (R&D Systems, Minneapolis, MN, USA). The formed multinucleated osteoclasts were determined by TRAP staining using a Leukocyte Acid Phosphatase kit (Sigma-Aldrich, St Louis, MO, USA). Osteoclasts, defined as TRAP-positive cells with more than three nuclei, were analyzed by ImageJ software (National Institutes of Health, USA; http://imagej.nih.gov/ij/). TRAP activity was evaluated by a TRAP Assay Kit (Beyotime). For bone resorption assay, osteoclasts were seeded on a bone-mimetic surface (Corning Osteo Assay plate; Corning, Tewksbury, MA, USA) in medium supplemented with RANKL. The plates were bleached with 10% sodium hypochlorite, washed with distilled water and dried at room temperature. The resorption pits were visualized on a light microscope (Leica Microsystems GmbH, Wetzlar, Germany) and analyzed by ImageJ software (National Institutes of Health).
Mineralization assay
The MC3T3-E1 cells (5 × 104 cells/cm2) were seeded into 35 mm petri-dishes. At confluence, osteogenesis by osteoblast MC3T3-E1 was induced by cell culture medium with ascorbic acid (50 μg/ml; Sigma-Aldrich) and β-glycerophosphate disodium salt hydrate (10 mM; Sigma-Aldrich). The cells were then exposed to different doses of radiation, and osteogenic media was changed every 48 h. For the mineralization assay, mineralized osteoblast cultures were fixed in 4% paraformaldehyde and then stained by 0.1% alizarin red S (Sigma-Aldrich). Positive alizarin red staining for calcium represented the calcium phosphate of osteoblast culture mineralization. Alizarin red–stained osteoblast cultures were photographed by a scanner, and the total area of red calcified nodules was measured by Image J software (National Institutes of Health).
Staining of F-actin and β-tubulin with immunofluorescence
Cell were fixed in 4% paraformaldehyde, permeabilized in PBS-0.5%Triton, and then blocked in 5% non-fat milk in PBS-0.5% Triton. After that, the cells were incubated with monoclonal anti–β-tubulin antibody (1:1000; mouse anti-mouse; RLM3135, Ruiying Biological, Suzhou, China) or rhodamine-labeled phalloidin (1:20; R415, Molecular Probes, Carlsbad, CA, USA). After being washed with PBS-0.1%Tween, the cells were incubated with FITC-labeled secondary antibody (1:1000; goat anti-mouse; A0568, Beyotime) to stain the tubulin. The nuclei were counterstained with DAPI (Molecular Probes), and images were taken using a confocal microscopy (Leica Microsystems GmbH).
Quantitative real-time PCR
Total RNA was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA), reversely transcribed, and processed for PCR reactions according to the manufacturer's protocol (TaKaRa, Dalian, China). qPCR was performed with the following primer pairs: GAPDH, RANK, MMP9, integrin β3, vacuolar-type H (+)-ATPase (V-ATPase), carbonic anhydrase II (Car2) and cathepsin K (CTSK). The sequences of the primers are listed in Table 1. The amplification program was as follows: pre-denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, annealing temperature for 20 s and 72°C for 20 s. The data were normallized to GAPDH and analyzed via the 2−ΔΔCt method.
Table 1.
Primers sequences used for quantitative real-time PCR
| Gene name (Genebank No.) | Primer sequences (5′–3′) | Annealing temperature (°C) |
|---|---|---|
| Car2 (NM_009801.4) | Forward: CATTACTGTCAGCAGCGAGCA | 54 |
| Reverse: GACGCCAGTTGTCCACCATC | ||
| CTSK (NM_007802.4) | Forward: CAGCAGAACGGAGGCATTGA | 54 |
| Reverse: CCTTTGCCGTGGCGTTATAC | ||
| Integrin β3 (NM_016780.2) | Forward: CCACCTTCACCAATATCAC | 55 |
| Reverse: CCAAATCCCACCCATACAC | ||
| MMP9 (NM_013599.3) | Forward: GCCCTGGAACTCACACGACA | 56 |
| Reverse: TTGGAAACTCACACGCCAGAA | ||
| GAPDH (NM_008084.2) | Forward: TGCACCACCAACTGCTTAG | 51 |
| Reverse: GGATGCAGGGATGATGTTC | ||
| RANK (NM_009399.3) | Forward: CTTGGACACCTGGAATGAAG | 52 |
| Reverse: CAGCACTCGCAGTCTGAGTT | ||
| V-ATPase (NM_175406.3) | Forward: CCACTGGAAGCCCAGTAAACAGA | 55 |
| Reverse: GAACGTATGAGGCCAGTGAGCA |
Reactive oxygen species assay
Cell total reactive oxygen species (ROS) was detected by a Reactive Oxygen Species Assay Kit (Beyotime). Briefly, the cells were incubated with DCFH-DA at 37°C for 20 min. After two washes with PBS, the cells’ DCF fluorescence was determined by a fluorescence microplate reader with temperature maintained at 37°C. The excitation filter was set at 485 nm and the emission filter was set at 528 nm. The total protein content was used to normalize the obtained values.
Statistical analysis
All experiments were performed in triplicates, and there were more than three samples in each trial. Statistical analysis was performed using the GraphPad Prism software (GraphPad, La Jolla, CA, USA) with one-way ANOVA with Newman–Keuls. The data were expressed as mean ± standard deviation, and differences with P < 0.05 were considered statistically significant.
RESULTS
X-rays significantly reduced cell viability of both RAW264.7 and RAW-osteoclasts
RAW264.7 cells were exposed to X-rays at 1 Gy, 2 Gy, 4 Gy, 6 Gy and 8 Gy, followed by incubation under normal conditions for 3 days. The cell morphology was observed at 4 h and on Day 2. Cell viability was measured on Days 1, 2 and 3. Caspase 3 was examined on Days 2 and 4. At 4 h after X-ray treatment, the cells had not detached and still exhibited rounded shapes. On Day 2, some of the cells showed a flattened and vacuolated phenotype, similar to M1 macrophages, in the 4, 6 and 8 Gy groups (Fig. 1A). M1 macrophages may be activated by cellular debris derived from apoptotic cells [28]. The CCK8 method was adopted to measure cell viability. On Day 1, X-rays did not affect the cell viability in RAW264.7. However, in the presence of RANKL (50 ng/ml), IR showed a significantly negative influence on differentiating osteoclasts on Days 1–3. On Days 2 and 3, X-rays decreased the viability of RAW264.7 cells, both in the presence and absence of RANKL. Furthermore, the relative cell viability compared with non-irradiated controls of RAW-osteoclasts was much lower than that of RAW264.7 cells (Fig. 1B and C). These results showed that X-rays markedly reduced the cell viability of both RAW264.7 cells and differentiating RAW-osteoclasts compared with controls. X-rays posed more severe toxic effects on differentiating RAW-osteoclasts than on RAW264.7 cells.
Fig. 1.
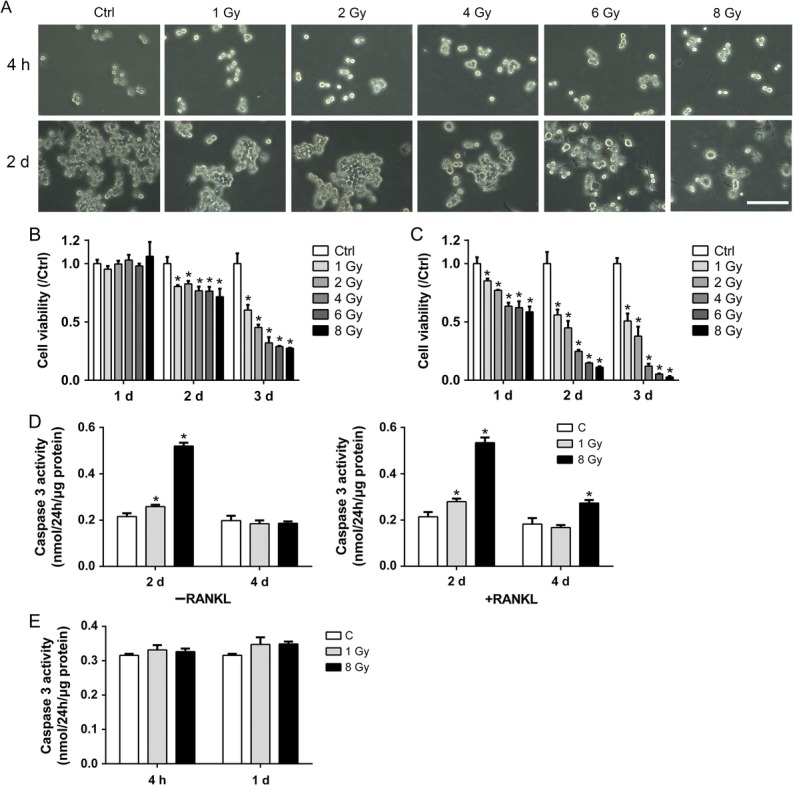
Effects of X-rays on cell viability of RAW264.7 cells. (A) Typical morphology of RAW264.7 cells 4 h and 2 days after exposure to various doses of X-rays. Scale bar, 100 μm. (B–C) The CCK 8 method was used to evaluate the cell viability relative to control in the presence or absence of RANKL (n = 3). (D) IR increased caspase 3 activity in both RAW264.7 and differentiating osteoclasts on Day 2 and 4 (n = 3). (E) IR did not altered caspase 3 activity in RAW264.7 cells at 4 h or on Day 1 (n = 3). All X-ray groups were compared with controls. Data shown are in the form of mean ± SD. *P < 0.05.
Caspase 3 is involved in the induction of cell apoptosis. We thus investigated whether the toxic effects of X-rays were related to caspase 3 activity. The activity of Caspase 3 was measured 2 or 4 days after exposure to 1 or 8 Gy of X-rays (Fig. 1D). The results showed that X-rays significantly increased the activity of Caspase 3 in pre-osteoclasts on Day 2, but not on Day 4. In the presence of RANKL, a high Caspase 3 activity was observed in the differentiating osteoclasts 2 days after exposure to X-rays. On Day 4, the differentiating osteoclasts still exhibited a higher activity of Caspase 3 in the 8 Gy group, but not in the 1 Gy group. However, Caspase 3 activity was not altered at 4 h or on Day 1 (Fig. 1E).
X-rays had limited effects on cell viability and differentiation of MC3T3-E1 cells
The MC3T3-E1 morphology was examined to determine the health status of the cells. At 4 h or 2 days after X-ray treatment, the cells were not detached but became thinner (Fig. 2A). Cell viability was measured with the CCK8 assay. Three days after X-ray exposure, the viability of both the differentiating and non-differentiating MC3T3-E1 cells was slightly decreased compared with that of the non-irradiated group. There were no alterations in cell viability in the 1 Gy group (Fig. 2B and C). The reduced folds of cell viability relative to control were considerably lower than those of RAW264.7 cells.
Fig. 2.
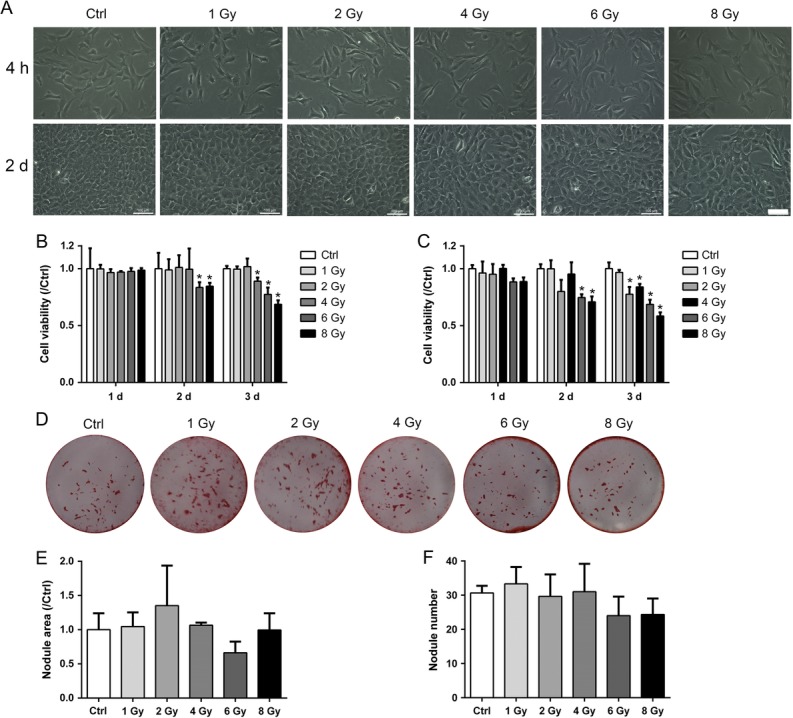
Effects of X-rays on cell viability and mineralization of MC3T3-E1 cells. (A) Typical morphology of MC3T3-E1 cells at 4 h and 2 days after exposure to various doses of X-rays. Scale bar: 100 μm. (B–C) The CCK 8 method was used to evaluate the cell viability relative to that of controls with or without the osteogenic medium (n = 3). (D) Osteogenic differentiation was confirmed by alizarin red S staining (D) and analyzed by nodule area and nodule number per dish (diameter: 35 mm), respectively (E–F) (n = 3). All X-ray groups were compared with controls. Data shown are in the form of mean ± SD. *P < 0.05.
Osteogenesis was determined by the mineralization assay. Matrix mineralization was characterized by analyzing the formation of calcified nodules. As shown in Fig. 2D, at 21 days in the presence of osteogenic medium after treatment, both the area and the number of the osteoblasts forming mineralized nodules remained unchanged (Fig. 2E and F). Compared with osteoclasts, X-rays had dual effects on osteoclastogenesis. X-rays at 1 Gy promoted osteoclast formation, and high X-ray doses had dramatically negative effects. However, there were no alterations in osteogenic differentiation after X-ray exposure. Taken together, X-rays had dual effects on osteoclastogenesis, but limited effects on osteogenesis.
X-rays affected osteoclast formation of RAW264.7 cells
After X-ray treatment, 50 ng/ml RANKL was added to induce osteoclastogenesis for 2 days. TRAP staining was performed to detect osteoclast formation (Fig. 3A). In accordance with the reduced cell viability, the number of TRAP-positive cells decreased in the 1, 6 and 8 Gy groups. The osteoclast fusion was evaluated by analyzing the formed osteoclast area. The fusion of pre-osteoclasts was enhanced with larger osteoclasts in the 1 Gy group, and suppressed in the 8 Gy group (Fig. 3B–D). The TRAP activity was not affected in any of the X-ray–treated groups on Day 2, but increased in the 1, 2 and 4 Gy groups on Day 4 (Fig. 3E).
Fig. 3.
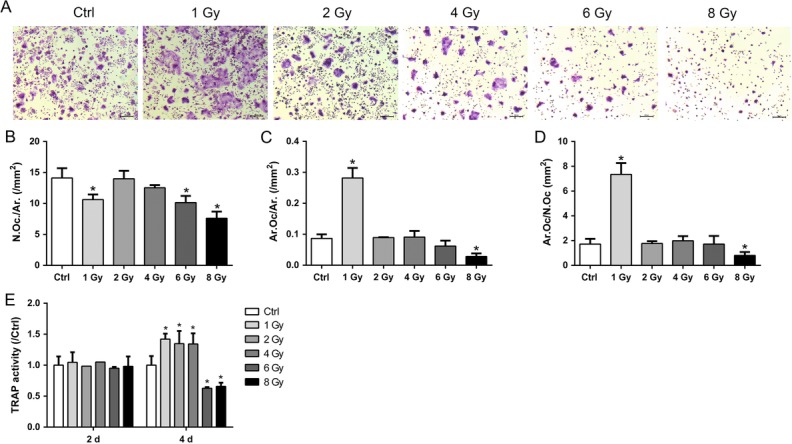
X-rays affected osteoclast formation. RAW264.7 cells were incubated with 50 ng/ml RANKL for 2 days after exposure to X-rays. (A) Osteoclast formation was identified by TRAP-positive cells with more than three nuclei and analyzed in osteoclast number per square millimeter (B), osteoclast area per square millimeter (C) and osteoclast number per single cell (D). (E) TRAP activity was measured on Days 2 and 4. Scale bar: 200 μm (n = 3). All X-ray groups were compared with controls. Data shown are in the form of mean ± SD. *P < 0.05.
In order to further examine the effects of X-rays on osteoclast fusion, the seeded RAW264.7 cells were first cultured in osteoclastogenic medium for 2 days, and then subjected to X-ray treatment. TRAP staining was performed on Day 4 to detect osteoclast fusion (Fig. 4A). The results showed that the number of osteoclasts decreased in all X-ray–treated groups (Fig. 4B). Compared with controls, the osteoclast area was increased in the 1 Gy group, but decreased in the 4, 6 and 8 Gy groups (Fig. 4C). The TRAP-stained area per osteoclast was elevated in the 1 and 2 Gy groups, and decreased in the 6 and 8 Gy groups (Fig. 4D). Taken together, these observations indicated that X-rays reduced the formation of TRAP-positive osteoclasts.The fusion of osteoclasts was accelerated by 1 Gy X-rays, but inhibited by higher doses.
Fig. 4.
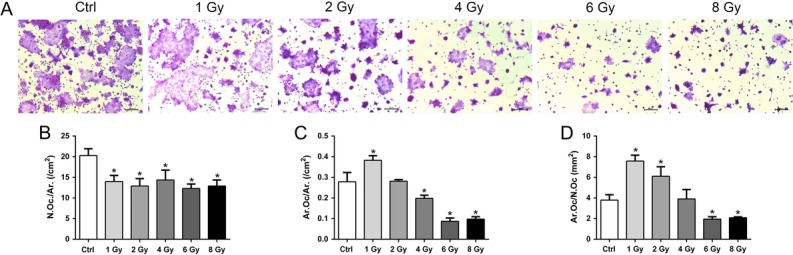
X-rays affected osteoclast fusion. RAW264.7 cells were first incubated with 50 ng/ml RANKL for 2 days and then exposed to X-rays. After exposure, the cells were cultured in osteoclastogenic medium for 2 more days. (A) Osteoclast formation was identified by TRAP-positive cells with more than three nuclei and analyzed in osteoclast number per square millimeter (B), osteoclast area per square millimeter (C) and osteoclast number per single cell (D). Scale bar: 200 μm. n = 3. All X-ray groups were compared with controls. Data shown are in the form of mean ± SD. *P < 0.05.
X-rays reduced the bone resorption potential of RAW-osteoclasts
Since X-rays had profound effects on osteoclast formation, we next examined their potential of bone resorption. RAW264.7 cells were seeded onto the Corning Osteo Assay plate. After X-ray treatment, the cells were cultured in the presence of RANKL for 10 days. X-rays at 4, 6 and 8 Gy significantly decreased the pit formation. Though 1 Gy X-rays markedly promoted the osteoclast fusion, there was no obvious alterations in the 1 and 2 Gy groups (Fig. 5A and B). X-rays at 4, 6 and 8 Gy had inhibitory effects. The surviving pre-osteoclasts after X-ray treatment still had the potential of bone resorption.
Fig. 5.
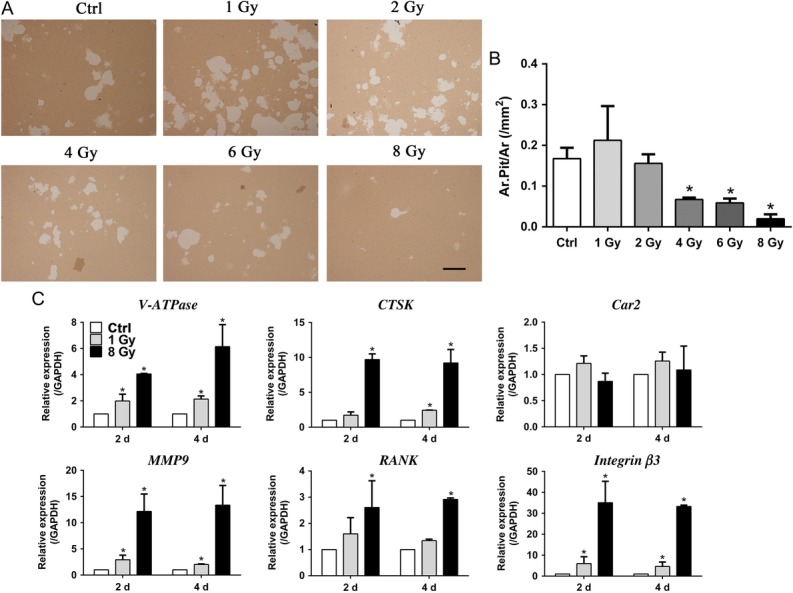
X-rays affected osteoclast resorption activity. (A–B) The level of bone resorption was determined by the pit formation assay and expressed as the area of absorbed pits per square millimeter. The white parts were pits resorbed by osteoclasts. Scale bar: 500 μm. (C) mRNA expression during osteoclast differentiation after IR treatment at Days 2 and 4 was examined, including expression of integrin β3, RANK, MMP9, V-ATPase, Car2 and CTSK (n = 3). All X-ray groups were compared with controls. Data shown are in the form of mean ± SD. *P < 0.05.
Our findings showed that 1 Gy of X-rays promoted spread and fusion of osteoclasts, while higher doses inhibited this process. Therefore, X-rays at 1 Gy and 8 Gy were used in further investigation. The influence of X-rays on the expression of osteoclast-related genes was examined on Days 2 and 4 after X-ray treatment (Fig. 5C). V-ATPase, CTSK, MMP9 and integrin β3, which play key roles in bone resorption, were induced significantly compared with in the non-irradiated group. Besides, compared with the 1 Gy group, the gene expression of bone resorption in the 8 Gy group was higher than that in the 1 Gy group. Nevertheless, there were no obvious X-ray effects on Car2 mRNA expression. X-rays stimulated high expression of RANK in the 8 Gy group, but not in the 1 Gy group.
X-rays influenced cytoskeleton distribution in RAW-osteoclasts
As mature osteoclasts generate actin-ring like structures at the periphery to ensure strong attachment to bone matrix, we next examined the distribution of the cytoskeleton on Days 2 and 4 after X-ray exposure (Fig. 6). Although no actin rings were observed in controls, osteoclasts began to form a large actin ring on Day 2 in the 1 Gy group. On Days 2 and 4, no intact actin rings were observed in the RAW-osteoclasts treated with 8 Gy. The actin filaments in the 8 Gy group were reorganized irregularly on Day 4.
Fig. 6.
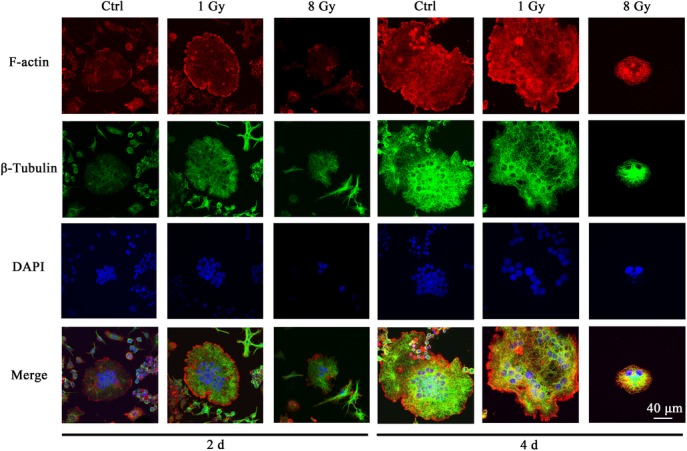
X-rays affected cytoskeleton distribution during osteoclast formation. RAW264.7 cells were incubated with 50 ng/ml RANKL for 2 days after exposure to X-rays. F-actin, tubulin and nuclei were stained with rhodamine-labeled phalloidin, anti–β-tubulin antibody and DAPI, respectively. Bar: 40 μm.
In order to further investigate how X-rays reorganized the actin filaments, the osteoclasts were first differentiated on Day 2 and then exposed to X-rays (Fig. 7). Actin dots at the cell periphery were observed in the 8 Gy group. Meanwhile, X-rays at 8 Gy induced smaller areas of osteoclasts with fewer nuclei than the non-irradiated and 1 Gy groups. These data suggest that X-ray irradiation at 1 Gy promotes osteoclast spreading with large actin rings, but, at a high dose, inhibits osteoclast fusion. No visible changes in tubulin distribution were observed in any of the X-ray–treated groups.
Fig. 7.
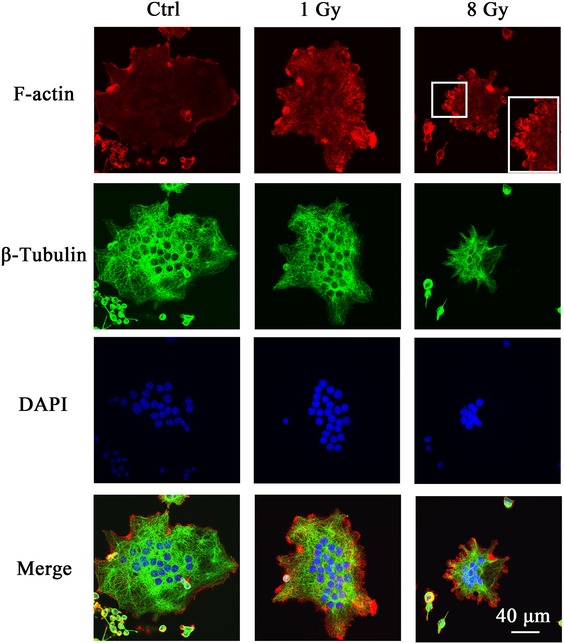
X-rays reorganized the actin filaments. RAW264.7 cells were first incubated with 50 ng/ml RANKL for 2 days and then exposed to X-rays. After irradiation, the cells were cultured in osteoclastogenic medium for 2 more days. F-actin, tubulin and nuclei were stained with rhodamine-labeled phalloidin, anti–β-tubulin antibody and DAPI, respectively. Bar: 40 μm.
ROS production was reduced in RAW-osteoclasts after X-ray treatment
ROS plays a critical role in osteoclast activation by RANKL [29]. However, the induction of ROS is an important mechanism for X-ray–induced cellular toxicity. ROS production can be stimulated by X-rays in bone marrow and causes cell apoptosis [30]. We then investigated whether X-rays affected osteoclast differentiation by altered generation of intracellular ROS (Fig. 8). The intracellular ROS assay was performed at different time-points (Days 2 and 4) with or without RANKL. The fluorescence intensity was quantified using a microplate reader because the differentiated RAW264.7 cells were huge multinucleated osteoclasts, and thus were not appropriate for flow cytometry. ROS production was not influenced by X-ray irradiation at 1 Gy or 8 Gy in the normal medium without RANKL. However, decreased ROS yields were found in the differentiated RAW-osteoclasts on Day 2 after X-ray treatment.
Fig. 8.

X-rays reduced total ROS production in the differentiating osteoclasts. Total cellular ROS was assessed by DCF fluorescence in the presence and absence of RANKL at Days 2 and 4 (n = 3). All IR groups were compared with controls. Data shown are in the form of mean ± SD. *P < 0.05.
DISCUSSION
In this study, we observed that exposure to X-rays caused a sharp change in both pre-osteoclasts and differentiating osteoclasts in a dose-dependent manner. X-rays can cause the transient induction of hematopoietic stem cell (HSC) apoptosis due to their high sensitivity [31]. In addition, almost all the blood cells, including the lymphoid and myeloid lineages, are extremely sensitive to X-rays [32]. In this study, we found that osteoclasts, which originated from radiosensitive HSCs, also showed high sensitivity to X-rays, in both progenitors and differentiating cells. The signaling involved in apoptosis (such as caspase 3) was upregulated by X-ray exposure. Compared with osteoclasts, we observed little decrease in cell viability of osteoblasts. Similarly, previous results demonstrated that osteoblasts, as the bone formation cells, were relatively resistant to X-ray induction of apoptosis [15]. Even following exposure to X-rays >10 Gy, osteoblasts remained viable [16, 33]. The rescued viability was caused by inhibition of cell proliferation, as indicated by arrests in the G2 phase [15, 16].
Previous in vivo and in vitro studies have reported that IR caused significant bone loss within 1 week, with elevated osteoclast numbers on the bone surface, expressions of pre-osteoclastic cytokines in bone marrow, and osteoclast marker genes in RAW264.7 cells [21, 22, 34]. The present study also demonstrated that osteoclast differentiation could be activated following X-ray exposure at relatively low doses, even though the cell viability was inhibited. Our results might partially explain why bone loss from radiation exposure occurred rapidly and then disappeared completely. In the case of osteoblasts, X-rays at relatively low doses increased osteogenic differentiation [17, 18] and even promoted bone fracture [19]. X-rays decreased viability to a greater degree in RAW264.7 cells an in differentiating cells than than in osteoblastic MC3T3-E1 cells. This indicated that osteoblasts and their progenitors were somewhat radioresistant. Taken together, radiosensitive osteoclasts and their progenitors, not osteoblasts, may be the predominant cells responsible for radiation-induced bone damage.
Interestingly, in the current study we found that X-rays had dual effects on osteoclast differentiation, in that higher doses of X-rays inhibited osteoclastogenesis, and low doses promoted it. X-rays promoted osteoclast formation at 1 Gy, had no effect at 2 and 4 Gy, and inhibited it at 8 Gy. The present study is consistent with existing reports that IR promotes osteoclastogenesis at doses of <2 Gy [21–23, 34]. Similar dose dependence was found in osteoblasts [19], though osteoblast mineralization was not noticeably altered for up to 8 Gy. The DNA repair machinery was activated in response to damage induced by IR [35, 36]. The reason why high-dose IR had a detrimental role on cells was thought to be due to a limit to the ability to repair DNA damage. As discussed above, osteoclasts and osteoblasts showed different responses to IR at the same X-ray dose when undergoing differentiation. Generally, osteoclastogenesis could be stimulated at a relatively low dose, but not osteogenic differentiation. Therefore, the mechanism for X-ray–induced bone damage may be complex in that activated osteoclasts may be the primary cause with relatively low dose X-ray irradiation, and inhibited osteoblasts may be the primary cause in the case of high-dose X-ray irradiation.
Given that osteoclasts were responsible for the relatively low-dose X-ray–induced rapid bone loss, bisphosphonate risedronate, an anti-resorptive agent, could reduce IR-induced osteoclastogenesis [37]. However, when locally irradiated with a high dose of X-rays at16 Gy, bisphosphonate alendronate failed to rescue IR-induced bone loss [12]. The apparently conflicting results obtained might be due to the high dose used in these studies. High-dose X-rays induced more apoptosis in pre-osteoclasts. This resulted in very few surviving cells, possibly not be enough for the formation of multinuclear osteoclasts. Our results also showed that the surviving cells still maintained the ability to differentiate into osteoclasts and the potential for bone resorption, as indicated by expressions of bone resorption–related genes.
The bone-resorption ability of osteoclasts is dependent on a special ring-like structure composed of actin filaments. During osteoclast differentiation, various actin structures based on podosomes are observed (cluster, ring and belt) [38]. In the current study, X-ray exposure was observed to lead to significant rearrangements of the actin filaments in mature osteoclasts. In particular, X-ray irradiation at 8 Gy destroyed actin ring formation, which inhibited bone resorption. When a differentiating osteoclast containing a formed actin ring was exposed to X-rays, actin dots, i.e. podosomes, were observed at the cell periphery. This indicated that X-rays could destabilize the actin ring; these podosomes were grouped in clusters. This observation may provide an explanation as to why X-ray exposure at a relatively high dose decreased the bone resorption ability of the osteoclasts, even though osteoclast fusion was promoted at 1 Gy. Dynamic reorganization of the actin cytoskeleton is a potential goal for IR, and needs to be further investigated.
Osteoclastogenesis was induced in the presence of RANKL, which stimulates the production of ROS [29, 39]. IR induced ROS immediately in the bone, causing reduced viability and increased apoptosis in bone marrow cells [25, 30]. However, in the present study, ROS production was decreased after IR treatment during osteoclast differentiation, and was unaltered in pre-osteoclasts. This indicated that high levels of ROS in bone marrow may not be generated in pre-osteoclasts. Other cells in bone marrow (such as HSCs, myeloid cells and lymphoid cells) may contribute to this oxidative stress. On the other hand, we also found that bone resorption was not altered after X-ray exposure at 1 Gy, though osteoclast formation was enhanced. Increased intracellular ROS are required for both formation and activation of osteoclasts [40, 41]. Since intracellular ROS can be greatly and rapidly produced by IR exposure [25, 30], the osteoclast formation could be promoted as an early response. However, the ROS level was decreased at Days 2 and 4 during osteoclast differentiation after IR exposure, which might negatively influence the capacity for bone resorption.
The present study (and other studies published in the literature) demonstrated that osteogenic differentiation can be stimulated in vitro [12–14], and decreased bone formation after IR with decreased or non-altered osteoblast number has been found in animal studies [22, 23]. This conundrum indicates that there is an intermediary cell transmitting signals for the bone-suppressive effects of IR to osteoblasts in the intact animal. The reduced osteoblastic bone formation in vivo following IR may be due to the regulatory role of osteoclasts. Accumulating evidence indicates that osteoclasts can regulate osteogenic differentiation via cell–cell contact [42, 43], cytokines [44] and exosomal microRNA [45]. On the other hand, irradiation-caused inflammation and production of ROS has deleterious effects at not only local, but also distant skeletal sites [15, 46]. For radiosensitive osteoclasts, whether IR stimulates the generation of the molecular mediators responsible for inhibited bone formation is yet to be clarified.
In summary, our research revealed that osteoclasts were relatively radiosensitive compared with osteoblasts. The osteoblast mineralization process was not influenced by X-ray irradiation for doses as high as 8 Gy. X-ray irradiation at 1 Gy promoted osteoclastogenesis, while higher doses exhibited inhibitory effects on osteoclastogenesis. Actin disorganization and apoptosis may be potential goals for X-ray irradiation, with high doses inhibiting osteoclast differentiation. These findings suggest that the radiosensitive osteoclasts can be transiently activated by X-ray irradiation, and provide new insights into mechanisms by which X-rays induce bone damage.
CONFLICT OF INTEREST
There are no conflicts to disclose.
FUNDING
This study was supported by the National Natural Science Foundation of China (31600674, 81602794, 11405235), the Natural Science Foundation of Jiangsu (BK20160334) , Project funded by China Postdoctoral Science Foundation (2017M610352), and by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
REFERENCES
- 1. Williams HJ, Davies AM. The effect of X-rays on bone: a pictorial review. Eur Radiol 2006;16:619–33. [DOI] [PubMed] [Google Scholar]
- 2. Oh D, Huh SJ. Insufficiency fracture after radiation therapy. Radiat Oncol J 2014;32:213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cannon CP, Lin PP, Lewis VO et al. . Management of radiation-associated fractures. J Am Acad Orthop Surg 2008;16:541–9. [PubMed] [Google Scholar]
- 4. Sivasubramanian H, Ee G, Srinivasaiah MG et al. . A subtrochanteric femoral fracture 15 years after radiotherapy: a case report. J Orthop Surg 2013;21:253–7. [DOI] [PubMed] [Google Scholar]
- 5. Higham CE, Faithfull S. Bone health and pelvic radiotherapy. Clin Oncol 2015;27:668–78. [DOI] [PubMed] [Google Scholar]
- 6. Wei RL, Jung BC, Manzano W et al. . Bone mineral density loss in thoracic and lumbar vertebrae following radiation for abdominal cancers. Radiother Oncol 2016;118:430–6. [DOI] [PubMed] [Google Scholar]
- 7. Chang CH, Chen SJ, Liu CY. Fracture risk and adjuvant therapies in young breast cancer patients: a population-based study. PLoS One 2015;10:e0130725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michel G, Blery P, Pilet P et al. . Micro-CT analysis of radiation-induced osteopenia and bone hypovascularization in rat. Calcif Tissue Int 2015;97:62–8. [DOI] [PubMed] [Google Scholar]
- 9. Wernle JD, Damron TA, Allen MJ et al. . Local irradiation alters bone morphology and increases bone fragility in a mouse model. J Biomech 2010;43:2738–46. [DOI] [PubMed] [Google Scholar]
- 10. Jegoux F, Malard O, Goyenvalle E et al. . Radiation effects on bone healing and reconstruction: interpretation of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;109:173–84. [DOI] [PubMed] [Google Scholar]
- 11. Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol 2011;6:121–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chandra A, Lin T, Tribble MB et al. . PTH1-34 alleviates radiotherapy-induced local bone loss by improving osteoblast and osteocyte survival. Bone 2014;67:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chandra A, Lin T, Zhu J et al. . PTH1-34 blocks radiation-induced osteoblast apoptosis by enhancing DNA repair through canonical Wnt pathway. J Biol Chem 2015;290:157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo C, Li C, Yang K et al. . Increased EZH2 and decreased osteoblastogenesis during local irradiation-induced bone loss in rats. Sci Rep 2016;6:31318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wright LE, Buijs JT, Kim HS et al. . Single-limb irradiation induces local and systemic bone loss in a murine model. J Bone Miner Res 2015;30:1268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szymczyk KH, Shapiro IM, Adams CS. Ionizing radiation sensitizes bone cells to apoptosis. Bone 2004;34:148–56. [DOI] [PubMed] [Google Scholar]
- 17. Park SS, Kim KA, Lee SY et al. . X-ray radiation at low doses stimulates differentiation and mineralization of mouse calvarial osteoblasts. BMB Rep 2012;45:571–6. [DOI] [PubMed] [Google Scholar]
- 18. Xu W, Xu L, Chen M et al. . The effects of low dose X-irradiation on osteoblastic MC3T3-E1 cells in vitro. BMC Musculoskelet Disord 2012;13:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen M Huang Q, Xu W et al. . Low-dose X-ray irradiation promotes osteoblast proliferation, differentiation and fracture healing. PLoS One 2014;9:e104016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 2003;423:337–42. [DOI] [PubMed] [Google Scholar]
- 21. Alwood JS, Shahnazari M, Chicana B et al. . Ionizing radiation stimulates expression of pro-osteoclastogenic genes in marrow and skeletal tissue. J Interferon Cytokine Res 2015;35:480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willey JS, Lloyd SA, Robbins ME et al. . Early increase in osteoclast number in mice after whole-body irradiation with 2 Gy X rays. Radiat Res 2008;170:388–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kondo H, Searby ND, Mojarrab R et al. . Total-body irradiation of postpubertal mice with 137Cs acutely compromises the microarchitecture of cancellous bone and increases osteoclasts. Radiat Res 2009;171:283–9. [DOI] [PubMed] [Google Scholar]
- 24. Schreurs AS, Shirazi-Fard Y, Shahnazari M et al. . Dried plum diet protects from bone loss caused by ionizing radiation. Sci Rep 2016;6:21343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kondo H, Yumoto K, Alwood JS et al. . Oxidative stress and gamma radiation–induced cancellous bone loss with musculoskeletal disuse. J Appl Physiol 2010;108:152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roos WP, Kaina B. DNA damage–induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 2013;332:237–48. [DOI] [PubMed] [Google Scholar]
- 27. Oest ME, Franken V, Kuchera T et al. . Long-term loss of osteoclasts and unopposed cortical mineral apposition following limited field irradiation. J Orthop Res 2015;33:334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee NK, Choi YG, Baik JY et al. . A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005;106:852–9. [DOI] [PubMed] [Google Scholar]
- 30. Cao X, Wu X, Frassica D et al. . Irradiation induces bone injury by damaging bone marrow microenvironment for stem cells. Proc Natl Acad Sci U S A 2011;108:1609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y, Schulte BA, LaRue AC et al. . Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood 2005;107:358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muralidharan S, Sasi SP, Zuriaga MA et al. . Ionizing particle radiation as a modulator of endogenous bone marrow cell reprogramming: implications for hematological cancers. Front Oncol 2015;5:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Despars G, Carbonneau CL, Bardeau P et al. . Loss of the osteogenic differentiation potential during senescence is limited to bone progenitor cells and is dependent on p53. PLoS One 2013;8:e73206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang B, Zhou H, Zhang XD et al. . Effect of radiation on the expression of osteoclast marker genes in RAW264.7 cells. Mol Med Rep 2012;5:955–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lau P, Baumstark-Khan C, Hellweg CE et al. . X-irradiation–induced cell cycle delay and DNA double-strand breaks in the murine osteoblastic cell line OCT-1. Radiat Environ Biophys 2010;49:271–80. [DOI] [PubMed] [Google Scholar]
- 36. McMahon SJ, Schuemann J, Paganetti H et al. . Mechanistic modelling of DNA repair and cellular survival following radiation-induced DNA damage. Sci Rep 2016;6:33290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willey JS, Livingston EW, Robbins ME et al. . Risedronate prevents early radiation-induced osteoporosis in mice at multiple skeletal locations. Bone 2010;46:101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jurdic P, Saltel F, Chabadel A et al. . Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol 2006;85:195–202. [DOI] [PubMed] [Google Scholar]
- 39. Callaway DA, Jiang JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Miner Metab 2015;33:359–70. [DOI] [PubMed] [Google Scholar]
- 40. Kim MS, Yang YM, Son A et al. . RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J Biol Chem 2010;285:6913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moon HJ, Kim SE, Yun YP et al. . Simvastatin inhibits osteoclast differentiation by scavenging reactive oxygen species. Exp Mol Med 2011;43:605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Irie N, Takada Y, Watanabe Y et al. . Bidirectional signaling through ephrina2-epha2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem 2009;284:14637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Negishi-Koga T, Shinohara M, Komatsu N et al. . Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med 2011;17:1473–80. [DOI] [PubMed] [Google Scholar]
- 44. Tang Y, Wu X, Lei W et al. . TGF-beta1–induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 2009;15:757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li D, Liu J, Guo B et al. . Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun 2016;7:10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jia D, Gaddy D, Suva LJ et al. . Rapid loss of bone mass and strength in mice after abdominal irradiation. Radiat Res 2011;176:624–35. [DOI] [PMC free article] [PubMed] [Google Scholar]