

ABSTRACT
Non-small cell lung cancer (NSCLC) is a progressive malignant disease, involving the activation of oncogenes and inactivation of tumor suppressors. In this study, we identified and characterized a novel oncogene hematopoietic- and neurologic-expressed sequence 1-like (HN1L) in human NSCLC. Overexpression of HN1L was frequently detected in primary NSCLC compared with their non-tumor counterparts (P < 0.001), which was significantly associated with tumor size (P = 0.022). In addition, Kaplan–Meier analysis showed that upregulation of HN1L correlated with worse overall survival (P = 0.029) and disease-free survival (P = 0.011) for NSCLC patients. Both in vitro and in vivo studies demonstrated that inhibition of HN1L expression with shRNA dramatically inhibited cell growth, adherent and non-adherent colony formation, and tumorigenicity in nude mice. The positive correlation of HN1L expression and Ki67 level in a large NSCLC samples further suggested the key role of HN1L in the regulation of cell growth. Further study showed that knockdown of HN1L resulted in dramatic cell cycle arrest by interfering with MAPK pathway via interacting with RASA4 protein. In conclusion, HN1L plays a crucial role in the progression of NSCLC by contributing to malignant proliferation, with possible use as a new intervention point for therapeutic strategies.
KEYWORDS: HN1L, NSCLC, prognosis, proliferation, MAPK pathway
Background
Lung cancer is the most frequent malignancy and the leading cause of cancer deaths worldwide.1 Despite substantial progress in the understanding and therapy of lung cancer, the overall 5-year survival rate remains at 10–15%.2 Non-small cell lung cancer (NSCLC), which includes adenocarcinoma (ADC), squamous cell carcinoma (SCC) and large cell carcinoma, accounts for approximately 80% of all lung cancer cases. Rapid growth is an important malignant feature of NSCLC, which can be caused by genetic and external factors that alter intracellular mechanisms responsible for controlling cell proliferation. Investigating the roles of the various oncogenes that are involved in the aberrant proliferation of NSCLC is a valuable goal toward uncovering the mechanisms of tumorigenesis and identifying new therapeutic targets for NSCLC treatment.
Hematopoietic- and neurologic-expressed sequence 1 (HN1) encodes a 154-aa protein in human that is highly conserved among species. It shares 30% identity with hematopoietic- and neurologic-expressed sequence 1-like (HN1L) which is another number of the HN1 family.3 In rodents, HN1 is widely expressed in numerous tissues during embryonic development. HN1 depletion in a murine melanoma cell line B16.F10 promotes a differentiated phenotype that includes increased melanogenesis and cell cycle arrest.4 HN1 is upregulated in nervous tissues, including axotomized adult rodent facial motor and vagal nerves.5 HN1 protein was highly expressed in an immature retina, and the subcellular localization changed during this retinogenesis as observed in newt retinal regeneration.6 Additionally, intracranial implantation of HN1-silenced murine GL261 glioma cells into mice results in tumor volumes that are significantly smaller than the xenografts from HN1-expressing cells.7 One previous study reported that HN1could distinguish human ovarian carcinoma from healthy ovarian epithelial tissue.8 Moreover, overexpression of HN1 is significantly associated with poorer overall survival of breast cancer patients.9 Conversely, some studies showed that HN1 overexpression suppressed colony formation in prostate cancer cells.10 Knockdown of HN1 results in deregulating G2/M transition in prostate cells via deregulating the Akt-GSK3β-mediated signaling.11 These ports of HN1 in models of development, regeneration and cancer suggest that this gene is critical for regulating cell development and growth. HN1L, a homologous gene of HN1, was originally identified from a mouse fertilized egg cDNA library in 2000.12 The human HN1L gene, also known as C16orf34 or L11, encodes a 190-aa protein. HN1L protein mainly localized in the nucleus and cytoplasm, and specifically expressed in certain tissues, such as the liver, kidney, prostate, testis and uterus at varying levels. However, the physiological functions of HN1L remain unknown.13 HN1L overexpression in NSCLC was firstly identified by suppression subtractive hybridization,14 but its precise roles in cancer has not been determined.
In the present study, we investigated the expression levels of HN1L in NSCLC and their paired adjacent non-tumorous tissues, and further evaluated the correlation of HN1L expression with clinical parameters and its prognostic value in NSCLC. Using a highly efficient shRNA to inhibit the expression of HN1L in human NSCLC cells, we determined this gene functions to regulate cell malignant proliferation.
Materials and methods
Clinical samples and cell lines
A total of 45 fresh NSCLC tumor and non-tumor samples were snap-froze in dry ice after resection and then stored at −80°C for RNA extraction. A total of 109 NSCLC tumor and non-tumor specimens that were surgically removed and embedded in paraffin blocks were obtained from Sun Yat-sen University Cancer Center between December 2002 and November 2004. The enrollment criteria were as follows: (a) definitive NSCLC diagnosis by pathology based on WHO criteria; (b) exclusive treatment with chemotherapy or radiotherapy before surgery; (c) surgical resection, defined as complete resection of all tumor nodules with the cut surface being free of cancer by histologic examination; (d) complete clinicopathologic and follow-up data. In this study, non-tumorous lung tissues were defined as 2.0 cm from the tumor margin. All clinical samples used in this study were approved by the Committees for Ethical Review of Research at Sun Yat-sen University Cancer Center.
Human bronchial epithelial cell line BEAS-2B, and NSCLC cell lines A549, H1299, HCC827 and SK-MES-1, were purchased from the American Type Culture Collection (ATCC). Human NSCLC cell line LSC1 (also known as SCC210011) was established in our laboratory.15 BEAS-2B cells were cultured in Defined Keratinocyte-SFM (Gibco, Thermo Fisher Scientific) mixing EpiLife® Medium with 60 µM Calcium (Gibco,, Thermo Fisher Scientific) (1:1 mixture, serum-free). A549, H1299, SK-MES-1 and LSC1 cells were maintained in dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and HCC827 is cultured in RPMI-1640 (containing 10% FBS). The cells were incubated at 37°C in a humidified chamber containing 5% CO2.
RNA extraction and quantitative real-time PCR (qPCR)
Total RNA was extracted from cells or tissues using TRIzol (Roche) and was reverse transcribed using reverse transcriptase (Takara) according to the manufacturer's instructions. Real-time qPCR was carried out to detect levels of the corresponding HN1L, β-actin using SYBR Green SuperMix (Roche) and ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). β-actin was used as an internal control. The gene-specific primers for human HN1L and ACTB were as follows: HN1L: 5′-CCCAGGAGGAGAATCGAGCA-3′ (forward) and 5′-CGGGGGTTGATTCGTCAAAG-3′ (reverse); ACTB: 5′-CATGTACGTTGCTATCCAGGC-3′ (forward) and 5′-CTCCTTAATGTCACGCACGAT-3′ (reverse). The relative expression level (defined as fold change) of the target gene (2−ΔΔCt) was normalized to the endogenous ACTB reference (ΔCt).
Immunohistochemistry (IHC)
A total of 109 formalin-fixed, paraffin-embedded NSCLC tumor specimens were deparaffinized in xylene, rehydrated through a graded alcohol series (100%, 95%, 75% and 50%) and incubated with 3% hydrogen peroxide. For antigen retrieval, the slides were boiled in 10mM citrate buffer (pH = 6.0) for 40 minutes. Nonspecific binding was blocked with 5% bull serum albumin (BSA) for 30 minutes. After incubation with primary antibodies against HN1L (Sigma, HPA041908, 1:5000 dilution) or Ki67 (Abcam, ab16667, 1:400 dilution) at 4°C overnight in a humidified chamber, the slides were incubated with horseradish peroxidase-conjugated secondary antibody and stained with the DAB substrate. An immunoreactivity score system was applied as described previously.16 The percentage of HN1L-positive cells was scored as 0, <5%; 1, 5%–25%; 2, 25%–50%; 3, 50%–75%; 4, 75%–100%. The intensity of HN1L-positive staining was scored as 0, negative; 1, weak; 2, moderate; 3, strong. The total score was determined by the following formula: Staining index = positive percentage × intensity. To evaluate the Ki67 expression scores, we selected the 10 fields in the representative area of the tumors considering the heterogeneity, and calculated the ratios of Ki67-positive cells to the total cells (>1000 cells) in each field. The average ratio of Ki67-positive cells was scored as 0, <5%; 1, 5%–15%; 2, 15%–25%; 3, 25%–35%; 4, 35%–45%; 5, 45%–100%. The cutoff value for Ki67 level was 3, so score ≤3 was considered as low level, and score >3 was considered as high level.
Lentiviral mediated HN1L knockdown
In brief, a shRNA specifically targeting HN1L (5′-GGCGTAAGCAGAAACACTAAC-3′) cloned into the psi-LVRU6P vector were bought from GeneCopoeia. The recombinant construct was co-transfected together with three lentivirus packaging vectors, pLp1, pLp2 and pLp-VSVG (Invitrogen), into 293FT cell line (Invitrogen) using the HilyMax transfection reagent (Dojindo Corp. Japan). A psi-LVRU6P-scrambled shRNA (GeneCopoeia) was used as a negative control. Lentiviral particles were harvested to infect NSCLC cells. Stable clones were selected using puromycin (Sigma).
Cell growth assay
Cells were seeded at a density of 1000 per well in 96-well plate. The cell growth rate was monitored using a Cell Counting Kit-8 (CCK-8) assay kit (Dojindo Corp. Japan) according to the manufacturer's instruction. Triplicate independent experiments were done.
Colony formation assay
Anchorage-dependent growth was assessed by foci formation assay. Briefly, 1 × 103 cancer cells were seeded in a 6-well plate and cultured for one week. The surviving colonies (>50 cells per colony) were counted after crystal violet (Sigma) staining. Anchorage-independent growth was assessed by colony formation ability in soft agar. Briefly, 1 × 103 cells were suspended in soft agar mixture (DMEM, 10% FBS and 0.6% SeaPlaque agarose) and were subsequently overlaid on the solidified 0.4% agar base. After 2 weeks, colonies (≥10 cells) were counted under the microscope in 10 fields per well. Triplicate independent experiments were performed.
Tumorigenicity assay
All animal experiments were approved by Animal Ethics Committee at Sun Yat-sen University Cancer Center. The same number of parental cells, shRNA-silenced cells and sh-control cells (3 × 106 cells per mouse) were inoculated subcutaneously into the right dorsal flank of 4-week-old female nude mice (5 animals per group), respectively. After cells injection, the length (L) and width (W) of tumor were measured every week by calipers, and tumor volumes were calculated as volume (mm3) = L × W2 × 0.5. All mice were sacrificed after five weeks.
Bromodeoxyuridine (BrdU) incorporation assay
Cells were seeded on glass slides and grown until they reached about 50% confluence. The cell culture medium was then aspirated and labeling medium containing BrdU (Roche) was added, after which the cells were incubated at 37°C in a humidified chamber containing 5% CO2 for 30 minutes. The BrdU labeling medium was then aspirated and the slides were washed three times with phosphate buffered saline (PBS). The attached cells were fixed with 70% ethanol for 20 minutes at -20°C. After washing the cells with PBS, the slides were incubated with primary antibody against BrdU (Roche) at 37°C for 30 minutes. The cells were then incubated with an Alexa Fluor® 594 conjugated secondary antibody (Invitrogen) and mounted with anti-fade reagent with DAPI (Abcam). The results were evaluated under an OLYMPUS FV1000 fluorescence microscope.
Cell cycle assay
1 × 106 tumor cells were pre-fixed with cold alcohol (70%) at 4°C for 12 hours. The cells were washed twice with PBS and stained with propidium iodide (PI, Sigma, 50 μg/ml) containing RNase A (Sigma, 100 μg/ml) and 0.2% Triton X-100 (Sigma) at 37°C for 30 minutes. Cell cycle distribution was analyzed by FACS (Beckman) and ModFit LT software (Verity Software House).
Cell apoptosis test
Tumor cells were determined using an Annexin V-FITC/PI Apoptosis Detection Kit (Sigma) according to the manufacturer's instructions. Apoptotic cell proportion were tested by flow cytometry (Beckman) and data were analyzed by FlowJo v10.1 software.
Senescence detection
The cell senescence was tested using a Senescence β-Galactosidase (β-Gal) Staining Kit (C0602, Beyotime, China). Ten fields in the representative area of the each cells were selected, and calculated the average ratio of β-Gal positive cells to the total cells (>1000 cells).
Western blotting
Proteins were extracted from cells and tissues using cold RIPA buffer (Cell Signaling Technology) containing a protease inhibitor cocktail (Roche) and a phosphatase inhibitor PhosSTOP (Roche). Western blotting was performed according to the standard protocol using the specific antibodies listed in Table S1. β-Tubulin, β-Actin or GAPDH were used as loading controls.
Statistical analysis
All data analyses were performed using SPSS 18.0 software. The independent Student's t test or ANOVA were used to assess the statistical significance between any two preselected groups. The Chi-squared or Fisher's exact test were applied for comparison of dichotomous variables. Univariate analysis was conducted by the log-rank test and the Cox proportional hazards model was used for the multivariate analysis. The significance of Kaplan-Meier survival analysis was determined using log-rank tests. P-values ≤ 0.05 (two-sided) were considered statistically significant.
Results
HN1L was frequently overexpressed in NSCLC
The expression levels of HN1L were studied by qRT-PCR in 45 pairs of primary NSCLC and their paired adjacent non-tumor tissues. The result revealed that overexpression of HN1L (defined as > 2-fold increase) was detected in 23/45 (51.1%) NSCLC tissues compared with the matched non-tumorous tissues (P < 0.001, Fig. 1a). The average level of HN1L expression in cancer samples was significantly higher than that in non-tumor tissues (P < 0.001, Fig. 1a). Western blotting also showed that HN1L was dramatically overexpressed in 5/8 (62.5%) of randomly selected NSCLCs (Fig. 1b). IHC demonstrated the cancer-specific high expression of HN1L in SCC and ADC (Fig. 1c). Moreover, the expression levels of HN1L in five NSCLC cell lines and one immortalized lung bronchial epithelial cell line (BEAS-2B) were tested by qRT-PCR and western blotting. Compared with BEAS-2B cells, upregulation of HN1Lwas detected in all NSCLC cells (Fig. 1d).
Figure 1.
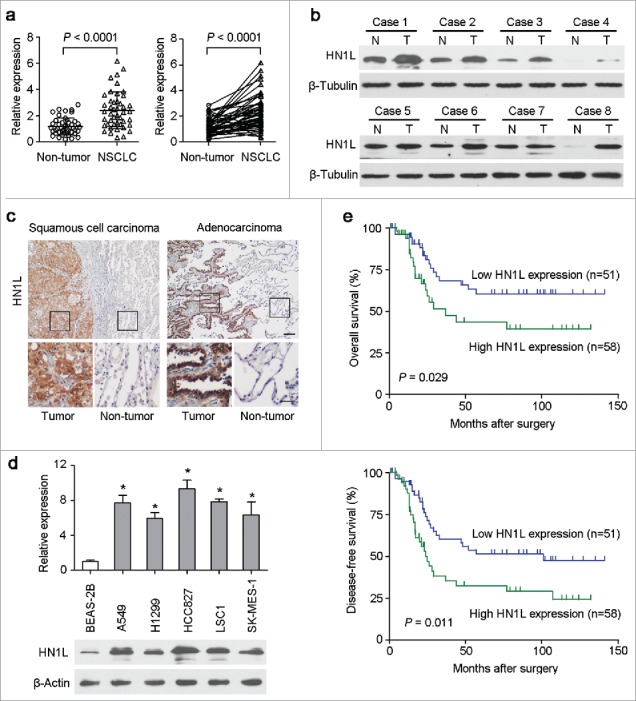
Overexpression of HN1L associated with poorer outcomes of NSCLCs. (a) Relative expression levels of HN1L detected by qPCR in 45 pairs of NSCLC tissues. (b) Western blot analysis of HN1L expression in matched primary tumor (T) samples and its corresponding non-tumor (N) tissues of 8 NSCLC cases. β-Tubulin was used as a loading control. (c) Representative IHC staining figures of HN1L expression in lung squamous cell carcinoma and adenocarcinoma. Scale bar, top figures, 100 μm; bottom figures, 50μm. (d) Relative expression levels of HN1L detected by qPCR and western blotting in an immortalized bronchial epithelial cell line (BEAS-2B) and other five NSCLC cell lines. Data represent mean ± SD. * P < 0.05. β-Actin was used as a loading control. (e) Kaplan-Meier analysis indicating the correlation of HN1L overexpression with poorer overall survival and disease-free survival rates of NSCLC patients.
Clinical significance of HN1L overexpression in NSCLC
HN1L expression was further studied in a large scale of NSCLCs by IHC. According to ROC curves analyses, the optimum cutoff value for HN1L was 6, so staining index ≤6 was considered as low expression, and staining index >6 was considered as high expression. Accordingly, the 109 patients were divided into two groups: the normal expression group (n = 51) and the overexpression group (n = 58). The correlations between HN1L expression in primary NSCLC and the clinicopathological features were summarized in Table 1. High expression of HN1L positively correlated with tumor size (P = 0.022). No correlation was observed between HN1L overexpression and other clinicopathological characteristics. Kaplan–Meier analysis revealed that overexpression of HN1L was associated with poorer overall survival (P = 0.029) and disease-free survival (P = 0.011) of NSCLC patients (Fig. 1e). Univariate analysis showed that lymph node metastasis, clinical stage and HN1L overexpression were prognostic factors for overall survival (Table 2). Further, multivariate Cox proportional hazard regression analysis showed that HN1L overexpression was an independent prognostic predictor for the overall survival of NSCLC patients (P = 0.046, Table 2). Altogether, we conclude that the tumoral overexpression of HN1L predicts a poor prognosis.
Table 1.
Correlation of HN1L expression with clinicopathological features in NSCLC patients.
| HN1L expression |
||||
|---|---|---|---|---|
| Clinical features | Cases | Normal (n = 51) | Upregulated (n = 58) | P value |
| Gender | 0.450 | |||
| Male | 90 | 44 (48.9%) | 46 (51.1%) | |
| Female | 19 | 7 (36.8%) | 12 (63.2%) | |
| Age (years old) | 0.833 | |||
| ≤65 | 77 | 37 (48.1%) | 40 (51.9%) | |
| >65 | 32 | 14 (43.7%) | 18 (56.3%) | |
| Smoking | 1.000 | |||
| Smokers | 29 | 14 (48.3%) | 15 (51.7%) | |
| Never smokers | 80 | 37 (46.2%) | 43 (53.8%) | |
| Histological subtype | 0.661 | |||
| SCC | 79 | 31 (39.2%) | 48 (60.8%) | |
| ADC | 30 | 10 (33.3%) | 20 (66.7%) | |
| Differentiated degree | 1.000 | |||
| I+II | 77 | 36 (46.8%) | 41 (53.2%) | |
| III | 32 | 15 (46.9%) | 17 (53.1%) | |
| Adjacent organs invasion | 0.830 | |||
| Negative | 30 | 15 (50.0%) | 15 (50.0%) | |
| Positive | 79 | 36 (45.6%) | 43 (54.4%) | |
| Tumor size (cm) | 0.022 | |||
| ≤4 | 47 | 28 (59.6%) | 19 (40.4%) | |
| >4 | 62 | 23 (37.1%) | 39 (62.9%) | |
| Lymph node metastasis | 0.567 | |||
| Negative | 55 | 24 (43.6%) | 31 (56.4%) | |
| Positive | 54 | 27 (50.0%) | 27 (50.0%) | |
| Clinical stage | 1.000 | |||
| I+II | 70 | 33 (47.1%) | 37 (52.9%) | |
| III+IV | 39 | 18 (46.2%) | 21 (53.8%) | □ |
SCC, squamous carcinoma; ADC, adenocarcinoma. Statistical significance (P < 0.05) is shown in bold.
Table 2.
Univariate and multivariate analyses of overall survival in NSCLC patients.
| Univariate analysis |
Multivariate analysis |
||||||
|---|---|---|---|---|---|---|---|
| Variable | Hazard ratio | 95% CI | P value | Hazard ratio | 95% CI | P value | |
| Gender | 0.819 | 0.320–2.100 | 0.678 | ||||
| Age(years) | 1.030 | 0.998–1.064 | 0.069 | ||||
| Smoking history | 1.410 | 0.646–3.081 | 0.388 | ||||
| Histological subtype | 1.085 | 0.316–3.724 | 0.896 | ||||
| Differentiated degree | 0.911 | 0.549–1.509 | 0.717 | ||||
| Adjacent organs invasion | 0.989 | 0.385–2.539 | 0.982 | ||||
| Tumor size(cm) | 0.985 | 0.848–1.144 | 0.842 | ||||
| Lymph node metastasis | 1.847 | 1.290–2.646 | 0.001 | 1.243 | 0.611–2.528 | 0.548 | |
| Clinical stage | 1.948 | 1.325–2.866 | <0.001 | 1.536 | 0.742–3.176 | 0.247 | |
| HN1L expression | 0.496 | 0.260–0.946 | 0.033 | 0.542 | 0.283–1.040 | 0.046 | |
CI, confidence interval; Statistical significance (P < 0.05) is shown in bold.
Silencing HN1L inhibited tumorigenicity
To determine the oncogenic function of HN1L, RNA interference was used to knockdown HN1L expression. The introduction of shRNA into the NSCLC cell lines A549 and HCC827 dramatically decreased the expression level of HN1L relative to control cells that expressing a scramble shRNA or parental cells (Fig. 2a, Fig. S1). Functional assays revealed that knockdown of HN1L effectively suppressed the cell growth rate (Fig. 2b), foci formation efficiencies (Fig. 2c) and colony formation in soft agar (Fig. 2d) compared with control cells.
Figure 2.
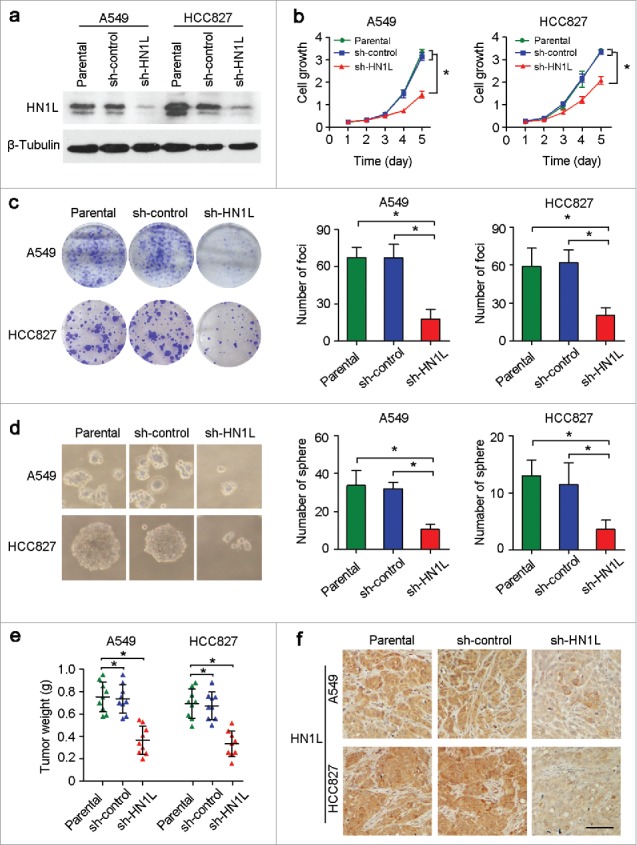
Knockdown of HN1L by shRNA supressed its malignancy in vitro and in vivo. (a) One shRNA against HN1L (sh-HN1L) effectively decreased HN1L expression as detected by western blotting. Scrambled shRNA (sh-control)-transfected cells and parental cells were used as negative controls. β-Tubulin was used as a loading control. Silencing HN1L expression could effectively inhibit cell growth (b), foci formation efficiencies (b), spheres formation in soft agar (d) in vitro. (e) The xenograft tumor weights were measured after surgery (n = 9). (f) Representative IHC images of HN1L expression in xenograft tumors. Scale bar, 100 μm. In all panels, data represent mean ± SD. *P < 0.05.
To further demonstrate whether HN1L silencing could inhibit tumorigenicity in vivo, the same number of parental cells and sh-HN1L/sh-control transfected cells (A549 and HCC827) were injected subcutaneously into the right dorsal flanks of nude mice (n = 9), respectively. The weights (Fig. 2e) of tumors that developed from sh-HN1L-transfected cells were significantly smaller than tumors originating from parental cells or sh-control cells. Results from the IHC staining confirmed the low expression of HN1L in xenograft tumors that developed from the sh-HN1L-transfected cells (Fig. 2f). These results suggest that HN1L has strong tumorigenic abilities in NSCLC.
HN1L promotes cell malignant proliferation
During DNA synthesis, BrdU can be incorporated into DNA in place of thymidine, which can then be used to measure the cell proliferation index.17 The BrdU incorporation study demonstrated that knockdown of HN1L in A549 and HCC827 cells dramatically inhibited cell proliferation compared with the sh-control transfected cells and parental cells (Fig. 3a). In addition, IHC staining confirmed that the proportion of Ki67-positive cells in the xenograft tumors originating from the sh-HN1L-transfected cells is lower than that in the sh-control and parental groups (Fig. 3b). To further validate the correlation of HN1L expression and cell proliferation, we evaluated the expression levels of HN1L and Ki67 in 109 NSCLC samples using IHC. The results indicated a significantly positive correlation between the level of HN1L and the percentage of proliferative cells in NSCLC (Table 3).
Figure 3.
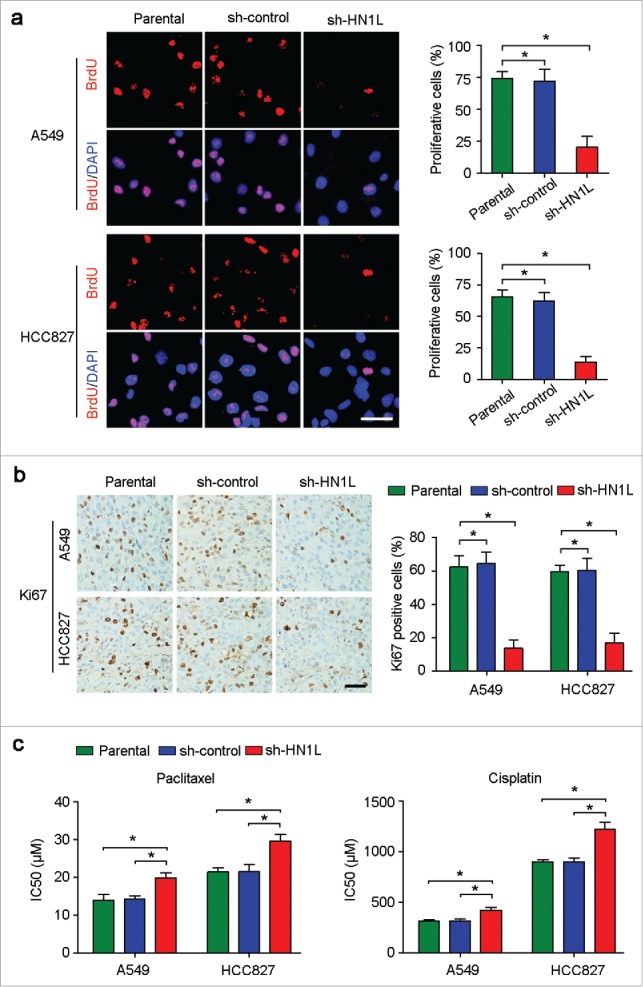
Aberrant HN1L expression indicated the malignant proliferation. (a) Knockdown of HN1L inhibited cell proliferation measured by BrdU incorporation assay. Scale bar, 50 μm. Data represent mean ± SD. *P < 0.05. (b) IHC staining showed the low-proportion of proliferative cells in tumor produced from HN1L silenced cells. Scale bar, 50 μm. Data represent mean ± SD. *P < 0.05. (c) The IC50 values of paclitaxel and cisplatin for A549 and HCC827 cells with or without HN1L silencing. Data represent mean ± SEM. *P < 0.05.
Table 3.
Correlation between HN1L and Ki67 expression in NSCLC patients.
| HN1L expression |
||||
|---|---|---|---|---|
| Ki67 expression | Cases | Low level (%) | High level (%) | P value |
| Low level | 55 | 37 (67.3%) | 18 (32.7%) | 0.004 |
| High level | 54 | 14 (25.9%) | 40 (74.1%) | |
Statistical significance (P < 0.05) is shown in bold.
Furthermore, to show the biological function of HN1L in cell proliferation, we also tested the drug sensitivity in A549 and HCC827 cells with or without HN1L silencing. Cell survival assay showed that the IC50 values of paclitaxel and cisplatin increased obviously after knockdown of HN1L in A549 and HCC827 cells (Fig. 3c), which indicated that silencing HN1L inhibited malignant proliferation and decreased the sensitivity to this cell cycle-associated chemotherapeutic agents. These evidences suggest the crucial role of HN1L in regulating cancer cell proliferation.
HN1L regulates cell growth via MAPK signaling pathway
To further understand the molecular mechanism of HN1L's contribution to cell growth, we analyzed the cell cycle distributions in sh-HN1L/sh-control transfected cells and parental cells. Knockdown of HN1L in A549 and HCC827 cells remarkably induced cell cycle arrest (Fig. 4a). Key regulators of the cell cycle were then analyzed by western blotting. Silencing HN1L notably decreased Cyclin D1, Cyclin E1, CDK4 and CDK6 protein levels, while the protein levels of p15, p21, p27 and p53 increased (Fig. 4b). IHC staining also showed the high expression of p21 and p53 in xenograft tumors that developed from the sh-HN1L-transfected cells (Fig. 4c), which indicated the crucial role of HN1L in regulating the cell cycle.
Figure 4.
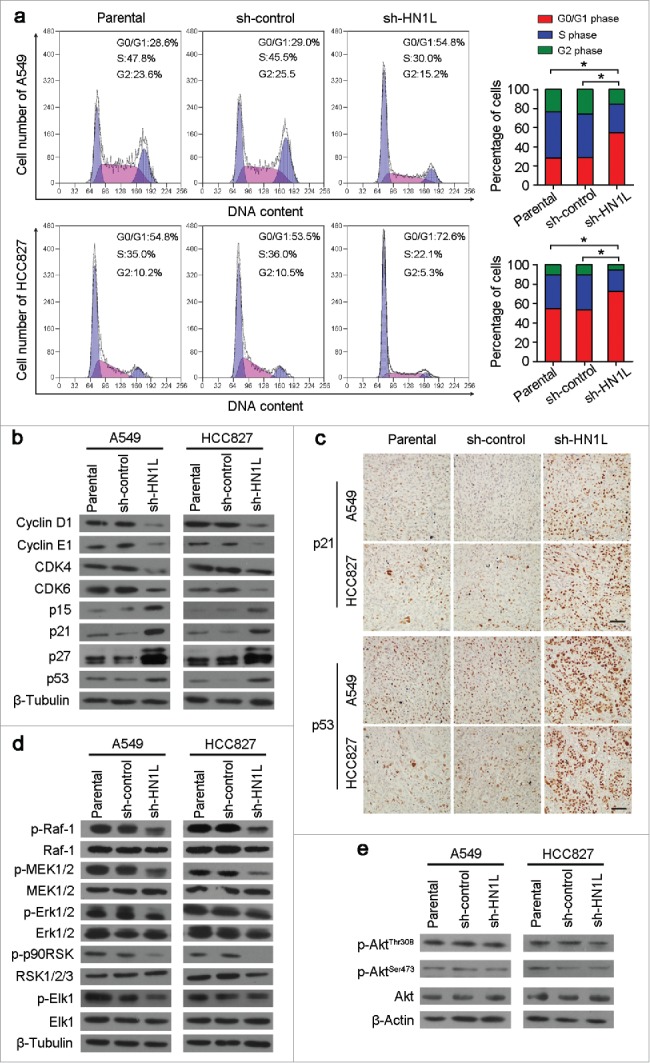
Silencing HN1L caused cell cycle arrest by interfering MAPK pathway. (a) Cell cycle distributions were analyzed by flow cytometry. *P < 0.05 (Chi-squared test). (b) Western blot analyzed the protein levels of several cell cycle regulators in sh-HN1L, sh-control and parental cells. β-Tubulin was used as a loading control. (c) Representative IHC images of p21 and p53 expressions in xenograft tumors. Scale bar, 150 μm. (d) Western blot analysis was applied to detect the phosphorylation levels of the key mediators in MAPK pathway (phospho-c-Raf(Ser259), phospho-MEK1/2(Ser217/221), phospho-Erk1/2(Thr202/204), phospho-p90RSK(Ser380) and phospho-Elk-1(Ser383)). β-Tubulin was used as a loading control. (e) The phosphorylation levels of Akt(Thr308/Ser473) was tested by western blotting. β-Actin was used as a loading control.
To elucidate the molecular basis of proliferation promoted by HN1L, we analyzed the phosphorylation levels of several key mediators involved in MAPK and Akt signaling pathways18,19, which are two primary pathway in regulating proliferation. The results showed that phosphorylation levels of c-Raf(Ser259), MEK1/2(Ser217/221) and Erk1/2(Thr202/204), as well as two downstream targets, p90RSK(Ser380) and Elk1(Ser383), were remarkably decreased in HN1L-silenced A549 and HCC827 cells compared with control cells (Fig. 4d). However, knockdown of HN1L did not affect the phosphorylation level of Akt(Thr308/Ser473) in A549 and HCC827 cells (Fig. 4e). These results suggest that HN1L promotes cancer cell malignant proliferation by regulating the MAPK signaling pathway.
HN1L regulates Ras/MAPK pathway by the direct interaction of RASA4
To find the mediators that could regulate Raf/MEK/ERK pathway by HN1L suppression, we surveyed six different protein–protein interaction databases (IntAct, BioGRID, HPRD, StelzlLow, BioPlex and MINT) (Fig. 5a). And we found one significant HN1L interactive protein RAS P21 Protein Activator 4 (RASA4) that is a member of the GAP1 family of GTPase-activating proteins and suppresses the Ras/mitogen-activated protein kinase pathway.20 Co-immunoprecipitation (Fig. 5b) and immunofluorescent double-staining (Fig. 5c) also indicate the interaction of HN1L and RASA4 in NSCLC cells. We presume that this interaction may reduce the GTPase activity of RASA4, which further increase the Ras activity. Therefore, knockdown of HN1L dramatically suppress the activity of Ras (Fig. 5d) and the membrane localization of Raf-1 (Fig. 5e) in A549 and HCC827 cells. In conclusion, our mechanistic studies reveal that HN1L regulates Ras/MAPK pathway via interacting with RASA4.
Figure 5.
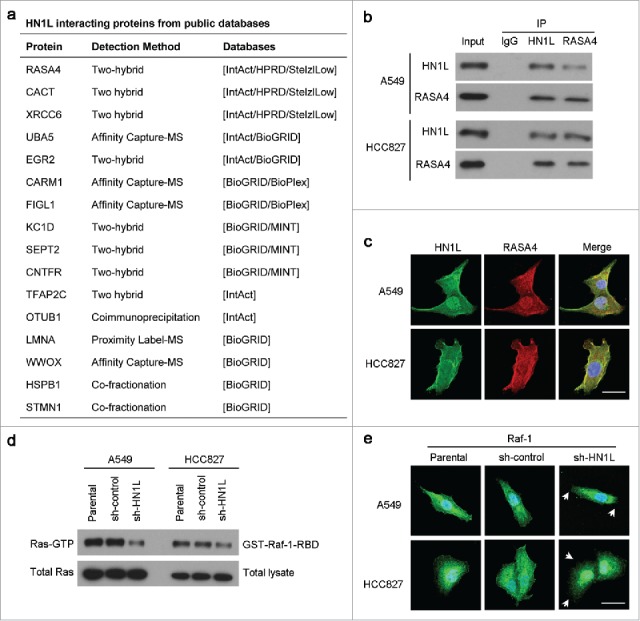
HN1L regulates Ras/MAPK pathway via interacting with RASA4. (a) List of the top HN1L-interacting proteins from the public databases. Co-immunoprecipitation (b) and immunofluorescent double-staining (c) indicate the direct interaction of HN1L and RASA4 in A549 and HCC827 cells. (d) GST-Raf1-RBD fusion protein is used to bind the activated form of GTP-bound Ras, which can then be immunoprecipitated with glutathione resin. Ras activation levels in A549 and HCC827 cells with or without HN1L silencing are then determined in western blot using a Ras monoclonal antibody. (e) Immunofluorescent staining showed the reduction of membrane localization of Raf-1 after knockdown of HN1L. Scale bar, 10 μm.
Apoptosis and senescence are not involved in HN1L function
Except for the regulation of proliferation, whether HN1L was related to apoptosis and senescence were also investigated. However, flow cytometry analysis showed the similar apoptosis cell proportions between HN1L silenced cells and control cells (Fig. 6a). Moreover, western blotting also showed that the expression of apoptosis-associated proteins, Caspase 3 (cleaved) and PARP (cleaved), were not changed obviously after HN1L silencing in A549 and HCC827 cells (Fig. 6b). Since cell senescence is closely related to cell cycle and HN1L plays a key role in proliferation, so we have also tested the activity of β-Galactosidase (β-Gal) that was specifically expressed in senescent cells in A549 and HCC827 cells before or after HN1L silencing.21 However, β-Gal staining showed no significant differences for the proportions of β-Gal positive cells between HN1L silenced cells and sh-control or parental cells (Fig. 6c). Furthermore, western blotting showed that the expression of p16Ink4a that was a key modulator in cellular senescence did not change after HN1L silencing in A549 and HCC827 cells (Fig. 6d).22 These results suggested that HN1L did not involve in cell apoptosis and senescence.
Figure 6.

Knockdown of HN1L could not induce cell apoptosis and senescence. (a) Apoptosis cells were analyzed by flow cytometry (Annexin V-FITC/PI staining). Data represent mean ± SD. (b) Western blot analyzed the levels of several apoptosis associated proteins in sh-HN1L, sh-control and parental cells. β-Actin was used as a loading control. (c) Cell senescence was tested by β-Galactosidase (β-Gal) staining. Representative β-Gal positve cells were indicaed by red arrows. Scale bar, 50 μm. Data represent mean ± SD. (d) Western blotting showed the protein level of p16Ink4a in A549 and HCC827 cells before or after HN1L silencing. β-Tubulin was used as a loading control.
Discussion
Targeting oncogenic regulatory factors using monoclonal antibodies or small-molecule inhibitors is an emerging therapeutic strategy for cancers.23,24 At present, target therapy is part of the routine management of patients with advanced NSCLC, and has shown promising results.25–26 These therapies are developed and applied on the basis of a clear understanding of the relevant pathways that drive tumorigenesis and maintenance of the malignant phenotype. New therapeutic targets are needed to further improve the treatment of NSCLC.
A previous report revealed the upregulation of HN1L expression in NSCLC hinting its potential diagnostic and prognostic significance.14 Our data also confirmed the overexpression of HN1L in NSCLC, including SCC and ADC. Furthermore, elevated HN1L was significantly associated with tumor size and adverse outcomes in NSCLC patients. Notably, multivariate survival analyses identified HN1L as an independent prognostic factor, which suggests that assessment of HN1L abundance in NSCLC might provide valuable information regarding patient prognosis. Therefore, our data provided strong evidence that HN1L is a novel diagnostic and prognostic biomarker for NSCLC, which might be further translated and exploited in the clinic. This well-established clinical significance of HN1L overexpression in NSCLC strongly underscores its essential tumorigenic role. Moreover, high expression of HN1L correlated with a larger tumor volume, which suggested that HN1L might be a positive regulator of proliferation. Indeed, both in vitro and in vivo functional studies demonstrated that knockdown of HN1L with shRNA significantly impaired cell growth and decreased the tumorigenic potential of NSCLC cells, suggesting the essential oncogenic functions of HN1L in the development of NSCLCs. Additionally, BrdU incorporation assay in vitro showed the low percentage of proliferative cells in HN1L-silenced cells than that in control groups, which is consistent with the IHC staining of Ki67 in xenograft tumors in vivo. Furthermore, the positive correlation of HN1L expression and Ki67 expression is also identified in a large samples of human NSCLC. Thus, HN1L's involvement in the regulation of cancer cell proliferation is generally in line with HN1L's role as an oncogene.
The induction of cell cycle arrest at a specific checkpoint and thereby inducing apoptosis or senescence are common mechanisms for restraining tumor progression of tumor suppressors.27 In this study, we investigated cycle distribution using flow cytometry. The data showed that the proportion of G0/G1 cells increased after HN1L silence in A549 and HCC827 cell clines, indicating that knockdown of HN1L can induce G0/G1 arrest. Cell cycle progression is precisely regulated by a series of cell cycle checkpoint proteins, such as the cyclins and CDKs.28 Among these proteins, Cyclin D1 and Cyclin E1, together with CDK2, CDK4, or CDK6, play major roles in DNA replication and mitosis by regulating G0/G1 phase of the cell cycle.29 Therefore, we investigated the expression of these cell cycle regulation proteins, and found the decreased expression of Cyclin D1/E1 and CDK4/6 in HN1L-silenced cells, which contributed to G0/G1 arrest. In addition, we also showed that knockdown of HN1L arresting the cell cycle at G0/G1 phase partly through up-regulation of p15, p21, p27 and p53. However, HN1L silencing cannot lead to cell apoptosis and senescence. The underlying mechanism of HN1L's contribution to cell growth was also explored preliminarily. The MAPK and Akt pathway are two key signaling pathways involved in the progression of various tumors and is associated with cancer malignant proliferation.30,31 In this study, we demonstrated that silencing of HN1L significantly reduced the phosphorylation levels of c-Raf(Ser259), MEK1/2(Ser217/221) and Erk1/2(Thr202/204), as well as two downstream targets, p90RSK(Ser380) and Elk1(Ser383), that are involved in cell signal transduction in MAPK pathway. In addition, we also demonstrated that downregulation of HN1L did not affect the activation of the Akt pathway. Hence, we suggest that overexpression of HN1L, as novel upstream regulatory factor of MAPK pathway, results in aberrant activation of proliferation signals in NSCLC cells. However, the detailed mechanism of HN1L activating MAPK signaling pathway in NSCLC requires further study.
In summary, this study demonstrates that HN1L may be a useful diagnostic tool and prognostic marker for NSCLC. HN1L as a novel oncogene plays an important role in the development and progression of NSCLC by promoting cell malignant proliferation via activating MAPK signaling pathway. Further characterization of HN1L may lead to the identification of new therapeutic targets for better clinical management of NSCLC.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by funds from the Ph.D. Programs Foundation of Ministry of Education of China (20130171110084), and the National Natural Science Foundation of China (81272416, 81472250 and 81472255).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J. Clin. 2016;66(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Heist RS, Engelman JA. SnapShot: non-small cell lung cancer. Cancer Cell. 2012;21(3):448 e2. doi: 10.1016/j.ccr.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Varisli L, Ozturk BE, Akyuz GK, Korkmaz KS. HN1 negatively influences the beta-catenin/E-cadherin interaction, and contributes to migration in prostate cells. J. Cell. Biochem. 2015;116(1):170–8. doi: 10.1002/jcb.24956. [DOI] [PubMed] [Google Scholar]
- 4.Laughlin KM, Luo D, Liu C, Shaw G, Warrington KH Jr., Law BK, Harrison JK. Hematopoietic- and neurologic-expressed sequence 1 (Hn1) depletion in B16.F10 melanoma cells promotes a differentiated phenotype that includes increased melanogenesis and cell cycle arrest. Differentiation. 2009;78(1):35–44. doi: 10.1016/j.diff.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zujovic V, Luo D, Baker HV, Lopez MC, Miller KR, Streit WJ, Harrison JK. The facial motor nucleus transcriptional program in response to peripheral nerve injury identifies Hn1 as a regeneration-associated gene. J Neurosci. Res. 2005;82(5):581–91. doi: 10.1002/jnr.20676. [DOI] [PubMed] [Google Scholar]
- 6.Goto T, Tokunaga F, Hisatomi O. Hematological- and Neurological-Expressed Sequence 1 Gene Products in Progenitor Cells during Newt Retinal Development. Stem Cells Int. 2012;2012:436042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laughlin KM, Luo D, Liu C, Shaw G, Warrington KH Jr., Qiu J, Yachnis AT, Harrison JK. Hematopoietic- and neurologic-expressed sequence 1 expression in the murine GL261 and high-grade human gliomas. Pathol. Oncol. Res. 2009;15(3):437–44. doi: 10.1007/s12253-008-9147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu KH, Patterson AP, Wang L, Marquez RT, Atkinson EN, Baggerly KA, Ramoth LR, Rosen DG, Liu J, Hellstrom I, et al.. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin. Cancer Res. 2004;10(10):3291–300. doi: 10.1158/1078-0432.CCR-03-0409. [DOI] [PubMed] [Google Scholar]
- 9.Zhang ZG, Chen WX, Wu YH, Liang HF, Zhang BX. MiR-132 prohibits proliferation, invasion, migration, and metastasis in breast cancer by targeting HN1. Biochem. Biophys. Res. Commun. 2014;454(1):109–14. [DOI] [PubMed] [Google Scholar]
- 10.Varisli L, Gonen-Korkmaz C, Syed HM, Bogurcu N, Debelec-Butuner B, Erbaykent-Tepedelen B, Korkmaz KS. Androgen regulated HN1 leads proteosomal degradation of androgen receptor (AR) and negatively influences AR mediated transactivation in prostate cells. Mol. Cell. Endocrinol. 2012;350(1):107–17. doi: 10.1016/j.mce.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 11.Varisli L, Gonen-Korkmaz C, Debelec-Butuner B, Erbaykent-Tepedelen B, Muhammed HS, Bogurcu N, Saatcioglu F, Korkmaz KS. Ubiquitously expressed hematological and neurological expressed 1 downregulates Akt-mediated GSK3beta signaling, and its knockdown results in deregulated G2/M transition in prostate cells. DNA Cell Biol. 2011;30(6):419–29. doi: 10.1089/dna.2010.1128. [DOI] [PubMed] [Google Scholar]
- 12.Ko MS, Kitchen JR, Wang X, Threat TA, Wang X, Hasegawa A, Sun T, Grahovac MJ, Kargul GJ, Lim MK, et al.. Large-scale cDNA analysis reveals phased gene expression patterns during preimplantation mouse development. Development. 2000;127(8):1737–49. [DOI] [PubMed] [Google Scholar]
- 13.Zhou G, Wang J, Zhang Y, Zhong C, Ni J, Wang L, Guo J, Zhang K, Yu L, Zhao S. Cloning, expression and subcellular localization of HN1 and HN1L genes, as well as characterization of their orthologs, defining an evolutionarily conserved gene family. Gene. 2004;331:115–23. doi: 10.1016/j.gene.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 14.Petroziello J, Yamane A, Westendorf L, Thompson M, McDonagh C, Cerveny C, Law CL, Wahl A, Carter P. Suppression subtractive hybridization and expression profiling identifies a unique set of genes overexpressed in non-small-cell lung cancer. Oncogene. 2004;23(46):7734–45. doi: 10.1038/sj.onc.1207921. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Yang H, Chen L, Li Y, Zhu Y, Dai Y, Chen K, Ai J, Zeng T, Mao X, Liu L, Li X, Guan XY. Establishment and characterization of human non-small cell lung cancer cell lines. Mol. Med. Rep. 2012;5(1):114–7. [DOI] [PubMed] [Google Scholar]
- 16.Zhu YH, Yang F, Zhang SS, Zeng TT, Xie X, Guan XY. High expression of biglycan is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2013;6(11):2497–505. [PMC free article] [PubMed] [Google Scholar]
- 17.Gratzner HG. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: A new reagent for detection of DNA replication. Science. 1982;218(4571):474–5. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 18.Kirilov M, Clarkson J, Liu X, Roa J, Campos P, Porteous R, Schutz G, Herbison AE. Dependence of fertility on kisspeptin-Gpr54 signaling at the GnRH neuron. Nat. Commun. 2013;4:2492. doi: 10.1038/ncomms3492. [DOI] [PubMed] [Google Scholar]
- 19.Nakahata S, Ichikawa T, Maneesaay P, Saito Y, Nagai K, Tamura T, Manachai N, Yamakawa N, Hamasaki M, Kitabayashi I, et al.. Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat. Commun. 2016;5:3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lockyer PJ, Kupzig S, Cullen PJ. CAPRI regulates Ca(2+)-dependent inactivation of the Ras-MAPK pathway. Curr Biol. 2001;11(12): p. 981–6. doi: 10.1016/S0960-9822(01)00261-5. [DOI] [PubMed] [Google Scholar]
- 21.Fan DN, Schmitt CA. Detecting Markers of Therapy-Induced Senescence in Cancer Cells. Methods Mol Biol. 2017;1534: p. 41–52. doi: 10.1007/978-1-4939-6670-7_4. [DOI] [PubMed] [Google Scholar]
- 22.Cafarotti S, Lococo F, Froesh P, Zappa F, Andre D. Target Therapy in Lung Cancer. Adv. Exp. Med. Biol. 2016;893:127–36. [DOI] [PubMed] [Google Scholar]
- 23.Sculier JP, Berghmans T, Meert AP. Advances in target therapy in lung cancer. Eur Respir Rev. 2015;24(135):23–9. doi: 10.1183/09059180.00011014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desai A, Adjei AA. FGFR Signaling as a Target for Lung Cancer Therapy. J Thorac Oncol. 2016;11(1):9–20. [DOI] [PubMed] [Google Scholar]
- 25.Sharma SP. New therapeutic target for non-small-cell lung cancer. The Lancet Oncology. 2014;15(12):e533. doi: 10.1016/S1470-2045(14)70493-0. [DOI] [PubMed] [Google Scholar]
- 26.Smit EF, van den Heuvel MM. PD-L1 in non-small-cell lung cancer: the third target for immunotherapy. Lancet. 2016;387(10030):1795–6. doi: 10.1016/S0140-6736(16)00700-5. [DOI] [PubMed] [Google Scholar]
- 27.Li CW, Chen BS. Investigating core genetic-and-epigenetic cell cycle networks for stemness and carcinogenic mechanisms, and cancer drug design using big database mining and genome-wide next-generation sequencing data. Cell Cycle. 2016;15(19):2593–607. doi: 10.1080/15384101.2016.1198862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dominguez D, Tsai YH, Gomez N, Jha DK, Davis I, Wang Z. 2016. A high-resolution transcriptome map of cell cycle reveals novel connections between periodic genes and cancer. Cell Res. 26(8):946–62. doi: 10.1038/cr.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell prolif. 2003;36(3):165–75. doi: 10.1046/j.1365-2184.2003.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang KA, Piao MJ, Madduma Hewage SR, Ryu YS, Oh MC, Kwon TK, Chae S, Hyun JW. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the MAPK signaling pathway. Tumour Biol. 2016;37(7):9615–24. doi: 10.1007/s13277-016-4864-x. [DOI] [PubMed] [Google Scholar]
- 31.Zhen Y, Li D, Li W, Yao W, Wu A, Huang J, Gu H, Huang Y, Wang Y, Wu J, et al.. Reduced PDCD4 Expression Promotes Cell Growth Through PI3K/Akt Signaling in Non-Small Cell Lung Cancer. Oncol Res. 2016;23(1–2):61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.