

ABSTRACT
During pregnancy, maternal β cells undergo compensatory changes including hypertrophy, hyperplasia, and increased glucose-stimulated insulin secretion (GSIS). Failure of these adaptations to occur can result in gestational diabetes mellitus. The secreted protein, Connective tissue growth factor (Ctgf), is critical for normal β cell development and promotes regeneration after partial β cell ablation. During embryogenesis, Ctgf is expressed in pancreatic ducts, vasculature, and β cells. In the adult pancreas, Ctgf is expressed only in the vasculature. Here, we report that pregnant mice with global Ctgf haploinsufficiency (CtgfLacZ/+) have an impairment in maternal β cell proliferation, while β cell proliferation in virgin CtgfLacZ/+ females is unaffected. Additionally, α-cell proliferation, β cell size, and GSIS were unaffected in CtgfLacZ/+ mice, suggesting that vascular-derived Ctgf has a specific role in islet compensation during pregnancy.
KEYWORDS: β cell proliferation, islets, gestational diabetes, pancreas, pregnancy
Introduction
Connective tissue growth factor (Ctgf) is critical for β cell specification and proliferation during islet development.3,8 Ctgf is a member of the CCN family (named after the first three members of the family discovered, CYR61, CTGF, and NOV) of secreted extracellular matrix-associated proteins and is expressed by a variety of cell and tissue types, including chondrocytes, endothelial cells, and cardiac myocytes.2 Depending on the cellular microenvironment, Ctgf modulates a variety of processes including proliferation, adhesion, migration, and extracellular matrix production.17 In the embryonic pancreas, Ctgf is expressed in β cells, ducts, and vasculature.3 Inactivation of Ctgf specifically from vascular endothelial cells results in decreased β cell proliferation during development, demonstrating that Ctgf can act in a paracrine manner on neighboring β cells.2 Conversely, transgenic over-expression of Ctgf in insulin-producing cells increases embryonic β cell proliferation.2 Treatment of isolated adult mouse islets with recombinant Ctgf induces a two-fold increase in β cell proliferation, suggesting that extra-islet cell types are not required for the effects of Ctgf on β cells.20 In vivo, Ctgf over-expression in adult β cells induces proliferation only in the setting of reduced β cell mass (for example, after 50% β cell ablation), promoting β cell mass regeneration through increased proliferation.20
A variety of stimuli, including high fat diet, partial pancreatectomy, and pregnancy, increase β cell proliferation in adult animals. Increased β cell proliferation under these circumstances facilitates β cell mass expansion, thus ensuring sufficient quantities of insulin are produced to properly regulate blood glucose homeostasis during times of physiological stress. Failure of sufficient β cell proliferation during pregnancy risks the development of gestational diabetes mellitus (GDM), a condition characterized by glucose intolerance without previously diagnosed diabetes. In addition to the acute dangers caused by loss of glucose homeostasis, women diagnosed with GDM are more likely to develop complications during delivery, including pre-eclampsia and Caesarian section.6 Likewise, both the mother and offspring of GDM pregnancies are more likely to become obese or develop type 2 diabetes (T2D) later in life.16 Despite occurring in approximately 7–10% of human pregnancies, little is known about the molecular mechanisms or causes of GDM.1 Although ethnicity, obesity, and family history are all associated with GDM, no single risk factor adequately predicts the development of the disease.16 It is inherently difficult to study β cell compensation in human females, thus animal models are critical for elucidating mechanisms of β cell compensation that occur during pregnancy.15
While genes that regulate β cell replication are often intrinsic to the endocrine cells, the islet vasculature can also contribute to pregnancy-induced β cell proliferation. Hepatocyte growth factor (HGF) is an endothelially-derived β cell mitogen that increases in the circulation during pregnancy.4 Conditional knockout mouse models revealed that HGF, through interaction with the c-Met receptor on β cells, induces β cell mass expansion during pregnancy.4 Reduction in vascular endothelial growth factor (VEGF)-A signaling can also impair glucose intolerance during pregnancy due to islet hypovascularization, further demonstrating the impact that the islet vasculature has on maternal islet function during pregnancy.23
In this study, we examined whether reduction of Ctgf affects β cell compensation during pregnancy. Using a Ctgf LacZ reporter allele, we show that Ctgf is expressed in the endothelial cells of the islets in adult mice, and that global Ctgf haploinsufficiency impairs pregnancy-induced maternal β cell proliferation. In contrast, α-cell proliferation, β cell hypertrophy, and glucose-stimulated insulin secretion were unaffected by Ctgf haploinsufficiency.
These studies emphasize that non-endocrine cells regulate β cell proliferation during pregnancy, emphasizing the importance that paracrine factors can have on islet compensation in response to increased insulin demand.
Materials and methods
Experimental animals
CtgfLacZ/+ mice have been described previously.3 Wild-type, age matched, sex matched siblings were used as controls for experiments using CtgfLacZ/+ mice. All mice were maintained on a C57BL/J6 background. Analyses were performed when mice were 10 weeks of age. All procedures were approved and performed in accordance with the Vanderbilt Institutional Animal Care and Use Committee under the supervision of the Division of Animal Care. Mice were housed in a controlled-temperature environment with a 12 hr night/day cycle and ad libitum access to high energy diet (11% kcal from fat; 5LJ5, Purina, St. Louis, MO) food and water except when otherwise noted.
Intraperitoneal glucose tolerance test (IP-GTT)
Animals were fasted for 16 hr prior to an IP-GTT. Fasting blood glucose was measured from 2 μl of tail vein blood with an Accu-check glucometer and glucose strips (Roche, Indianapolis, IN) as described previously.9
Intraperitoneal insulin tolerance test (IP-ITT)
Animals were fasted for 6 hr prior to an IP-ITT. Fasting blood glucose was measured as described above, and animals received an intraperitoneal injection of recombinant human insulin (Novo Nordisk, Princeton, NJ) at a dosage of 0.5 U/1000 g body weight. Insulin was diluted in filter-sterilized 1x PBS.
Islet isolation and perifusion
Mouse islets were isolated by intraductal infusion of collagenase P.13,21 Islet function was studied in a dynamic cell perifusion system as described previously.12
X-gal staining
Pancreata were dissected and fixed in 4% paraformaldehyde for 4 hr at 4°C. Tissues were washed in 1x PBS, cryoprotected with 30% sucrose in PBS for 24 hr and snap frozen in FSC 22 frozen section compound (Leica, Buffalo Grove, IL). 10 μm frozen sections were cut with a Leica CM3050S cryostat and attached to charged X-tra microscope slides (Leica, Buffalo Grove, IL). Slides were washed three times in LacZ wash buffer (2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, in 100 mM sodium phosphate buffer, pH 7.3), and then incubated in X-gal staining solution (2 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 1 mg/ml X-gal, in PBS) at 37°C for 30 mins. Sections were then washed in LacZ wash buffer and counterstained in Nuclear Fast Red (VECTOR, Burlingame, CA) for 5 mins and mounted with SHUR/Mount liquid mounting media (TBS, Durham, NC). Images were acquired using a ScanScope CS bright field microscope (Aperio Technologies, Vista, CA). Slides from three different mice per cohort were analyzed. Wild-type virgin and wild-type pregnant gestational day 14.5 (GD14.5) slides were run as a negative control for X-gal staining.
Immunolabeling
Pancreata were dissected and fixed for 4 hr in 4% paraformaldehyde, dehydrated in an ethanol series, and embedded in paraffin. Serial sections were cut at 5 μm and placed on glass slides. Indirect protein localization was obtained by incubation with the following primary antibodies: guinea pig anti-Insulin (Dako, Carpinteria, CA; 1:500), rabbit anti-Ki67 (Abcam, Cambridge, MA; 1:500), rat anti-CK19 (TROMA III) (Developmental Studies Hybridoma Bank, Iowa City, IA; 1:500), rabbit anti-CD31 (Abcam, Cambridge, MA; 1:100), guinea pig anti-glucagon (EMD Millipore, Bellerica, MA; 1:500) and chicken anti-β-galactosidase (Abcam, Cambridge, MA, 1:500). Primary antibodies were detected with the following species-specific donkey secondary antibodies: Peroxidase-conjugated anti-guinea pig (Jackson ImmunoResearch, West Grove, PA, 1:400), Cy2-conjugated anti-guinea pig (Jackson ImmunoResearch, West Grove, PA, 1:400), Cy3-conjugated anti-rabbit (Jackson ImmunoResearch, West Grove, PA, 1:400), Cy3-conjugated anti-chicken (Jackson ImmunoResearch, West Grove, PA, 1:400), Cy3-conjugated anti-rat (Jackson ImmunoResearch, West Grove, PA, 1:400). Nuclei were labeled with DAPI (0.5 μg/mL for 2 minutes; Thermo Scientific, Rockford, IL). Imaging was with a ScanScope FL scanner (Aperio Technologies, Vista, CA) and quantified using MetaMorph 6.1 (Molecular Devices, Sunnyvale, CA).
β cell mass
Six to 12 slides per animal (1 to 2% of entire pancreas) were immunolabeled for insulin, visualized via the DAB Peroxidase Substrate Kit (Vector Laboratories), and counterstained with eosin. Quantification of β cell mass was performed as described previously.7 Briefly, one pancreatic section per slide was scanned using a ScanScope CS scanner (Aperio Technologies, Vista, CA). Images from each experiment were processed identically with the ImageScope Software (Aperio Technologies, Vista, CA). β cell mass was measured by obtaining the ratio of insulin-positive area to total pancreas area of all scanned sections per animal and multiplied by the pancreatic wet weight.
β cell and α-cell proliferation
Five slides (at least 250 μm apart) per animal were immunolabeled for insulin or glucagon and Ki67 using sodium citrate antigen retrieval (10 mM sodium citrate). A minimum of 4,000 β cells or 1,500 α-cells were counted, and the percentage of proliferating cells was determined by dividing the number of Ki67+, hormone+ cells by the total number of hormone+ cells.
Individual β cell size
Five slides (at least 250 μm apart) per animal were immunolabeled for insulin using sodium citrate antigen retrieval (10 mM sodium citrate). Individual β cell size was determined by dividing the total β cell insulin positive size by the number of β cell nuclei. A minimum of 125 islets were quantified per sample.
Islet vascularization
Sections were immunolabeled for insulin and CD31 using TEG (1 mM Tris, 0.5 mM EGTA; pH 9.0) antigen retrieval. CD31 positive area of at least 100 islets, measured across five slides (at least 250 μm apart) per animal was measured. Percent of islet vascularization was determined by dividing total CD31+ area by total insulin+ and CD31+ area.
Statistics
Statistical significance for IP-GTTs and IP-ITTs was calculated using two-way ANOVA and Bonferroni post hoc test. For β/α-cell proliferation assays, β cell mass, β cell size, and islet vascularization analysis, statistical significance was determined using one-way ANOVA and Tukey HSD test. Statistical significance for individual fractions of perifusion analysis was calculated using Student's t-test. Statistical analysis was conducted using GraphPad Prism 6 software (GraphPad, San Diego, CA); statistical tests utilized were determined using recommendations provided by GraphPad Prism 6 software. Statistical significance was set at P < 0.05.
Results
Ctgf is expressed in the pancreatic vasculature of virgin and pregnant female mice
Previous studies from our lab demonstrated that Ctgf is important during times of β cell expansion including embryonic development and regeneration after partial β cell ablation.2,8,16,19 However, it was unknown what role, if any, Ctgf has in β cell biology during pregnancy. We thus examined where Ctgf was expressed in adult female pancreata. As currently available commercial antibodies are not specific for Ctgf on tissue sections, we utilized mice heterozygous for a LacZ knockin Ctgf loss-of-function allele (3 hereafter referred to as CtgfLacZ/+) to determine the expression pattern of Ctgf in pancreata from virgin and pregnant female mice. X-gal staining revealed that Ctgf expression was detected in structures consistent with vasculature in both virgin and gestational day 14.5 (GD14.5) pregnant mice, but was not observed in any other structure (Fig. 1). To confirm these findings, we performed immunolabeling with a β-galactosidase antibody and CK19, insulin, or CD31 antibodies to determine if Ctgf was expressed in the ducts, β cells, or endothelial cells, respectively. No co-localization was observed between β-galactosidase and CK19 or insulin (Fig. 2A–A”, B–B”, D–D”, E–E”) in virgin or GD14.5 samples. In contrast, co-localization was observed between β-galactosidase- and CD31 (Figure C-C”, F-F”) in both cohorts. As the endothelial cells of the vasculature are the predominate cell type of the pancreas that expresses CD31, our findings suggest that the primary source of Ctgf expression in the adult pancreas is the vasculature. β-galactosidase and CD31 sub-cellular localization did not completely overlap, likely due to the transmembrane domain present in the CTGF/β-galactosidase fusion protein, which causes it to form aggregates in the plasma membrane; CD31 has a more uniform localization throughout endothelial cells. Vasculature immediately adjacent to the islets (arrows, Fig. 1) and microvasculature within the islets themselves (arrowheads, Fig. 1) also displayed β-galactosidase activity, indicating that β cells in adult female mice are exposed to Ctgf protein in a paracrine manner. Using a conditional Ctgf allele, we previously showed that specific inactivation of Ctgf from vascular endothelial cells results in a decrease in β cell proliferation during embryonic development, demonstrating that non-endocrine sources of Ctgf are necessary for normal levels of β cell proliferation.16 Thus, it was possible that endothelial-derived Ctgf could influence maternal β cells during pregnancy.
Figure 1.
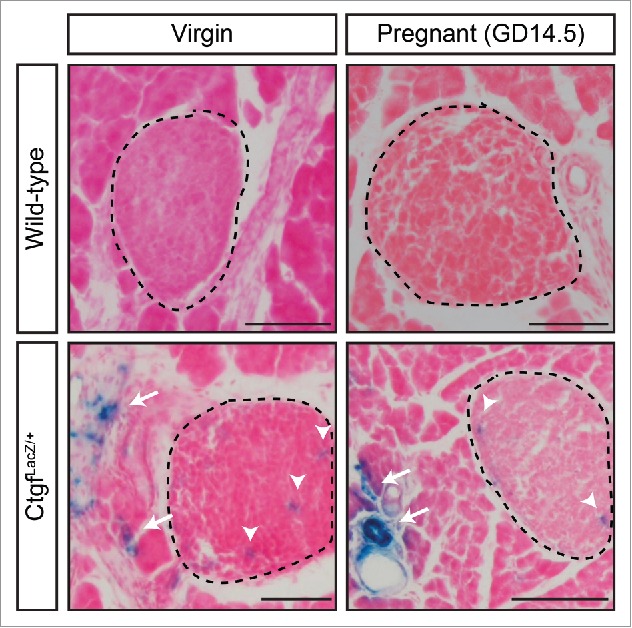
Ctgf is expressed in the pancreatic vasculature of adult virgin and pregnant female mice. X-gal staining of sections from virgin and pregnant (GD14.5) CtgfLacZ/+ female pancreata. Islets circled with dashed line. Arrows denote extra-islet X-gal stain; arrowheads denote intra-islet X-gal stain. Wild-type virgin and pregnant (GD14.5) samples were run as a negative control for X-gal stain. Tissue was counterstained with Nuclear Fast Red. n = 3/group. Scale bar represents 100 μm.
Figure 2.
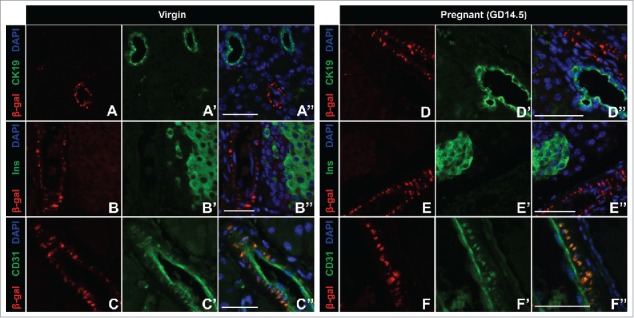
β-gal Ctgf reporter expression co-localizes with CD31-positive vascular endothelial cells in adult virgin and pregnant female mice. Dual labeling of Ctgf β-gal reporter and the ductal marker CK19 in (A-A”) virgin and (D-D”) pregnant gestational day 14.5 (GD14.5) mice. Dual labeling of Ctgf β-gal reporter with insulin in (B-B”) virgin and (E-E”) pregnant GD14.5 mice. Dual labeling with Ctgf β-gal reporter and CD31 in both (C-C”) virgin and pregnant (F-F”) GD14.5 mice. Nuclei are labeled with DAPI. n = 3/group. Scale bar represents 50 μm.
Ctgf haploinsufficiency reduces proliferation of β cells during pregnancy
Mice homozygous for the CtgfLacZ allele die shortly after birth due to respiratory failure.10 We therefore examined animals with global heterozygosity in Ctgf (CtgfLacZ/+ mice) to determine the effects on maternal β cell compensation at gestational day 14.5 (GD14.5) during pregnancy. GD14.5 was chosen as this is the time-point during pregnancy with the most profound increase in β cell proliferation compared to virgin animals.11,18 We observed no difference in β cell proliferation between virgin wild-type and virgin CtgfLacZ/+ females (Fig. 3A, B). In wild-type females, a more than two-fold increase in maternal β cell proliferation was observed at GD14.5 compared with virgin, consistent with previously published studies.11,25 Although pancreata from GD14.5 CtgfLacZ/+ females displayed an increase in β cell proliferation compared to CtgfLacZ/+ virgins, global Ctgf haploinsufficiency clearly impaired maternal β cell proliferation during pregnancy (Fig. 3A, B).
Figure 3.
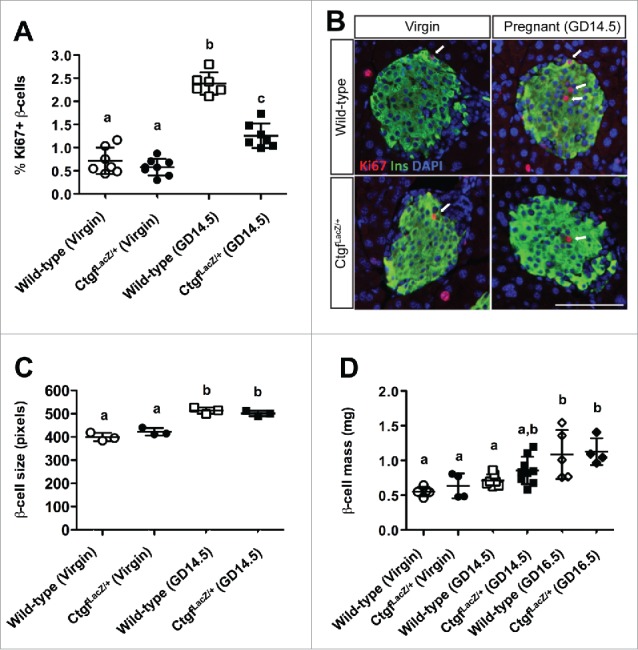
Global heterozygosity for Ctgf impairs pregnancy-induced β cell proliferation, but does not impact β cell hypertrophy or β cell mass. (A,B) Quantification and co-labeling of pancreas sections with insulin (green) and Ki67 (red) to quantify proliferating β cells in virgin and gestational day 14.5 (GD14.5) wild-type and CtgfLacZ/+ females. DAPI is in blue. Arrows indicate proliferating β cells. Scale bar represents 100 μm. (C) Individual β cell size in virgin and gestational day 14.5 (GD14.5) wild-type and CtgfLacZ/+ females. (D) β cell mass measured in virgin, GD14.5, and gestational day 16.5 (GD16.5) samples. Error bars represent standard deviation. Samples with the same letter are not significantly different from each other (P < 0.05).
Ctgf heterozygosity does not impair β cell mass, glucose-stimulated insulin secretion, whole body insulin tolerance, or α-cell proliferation
Since maternal β cell size and total β cell mass also normally increase during pregnancy, we investigated if these adaptations were impaired in pregnant CtgfLacZ/+ females.22 β cell hypertrophy was observed in CtgfLacZ/+ pancreata at GD14.5, suggesting that not all islet adaptations to pregnancy are impaired by Ctgf heterozygosity (Fig. 3C). The increase in β cell proliferation and size during pregnancy induces an overall increase in total β cell mass that reaches a maximum response by gestational day 16.5 (GD16.5). In our analysis, both wild-type and CtgfLacZ/+ females had a significant increase in β cell mass at GD16.5 compared to virgin controls; no significant difference in β cell mass was observed between wild-type and CtgfLacZ/+ females at this stage (Fig. 3D).
Furthermore, despite a relative deficiency in β cell proliferation in GD14.5 CtgfLacZ/+ females, there was no difference in glucose homeostasis between wild-type and CtgfLacZ/+ females as virgins or at GD14.5, likely due to the fact that β cell mass was still normal (Fig. 4A, B). Consistent with this finding, GSIS was not impaired in islets isolated from pregnant CtgfLacZ/+ females (Fig. 4C, Supplemental Table 1). Since CtgfLacZ/+ mice are globally heterozygous for the loss-of-function Ctgf allele, we conducted intraperitoneal insulin tolerance tests (IP-ITT) at GD14.5 to determine if whole-body insulin sensitivity is normal in the pregnant mice. No significant difference in blood glucose was detected in CtgfLacZ/+ at any time during the ITT (Fig. 4D).
Figure 4.
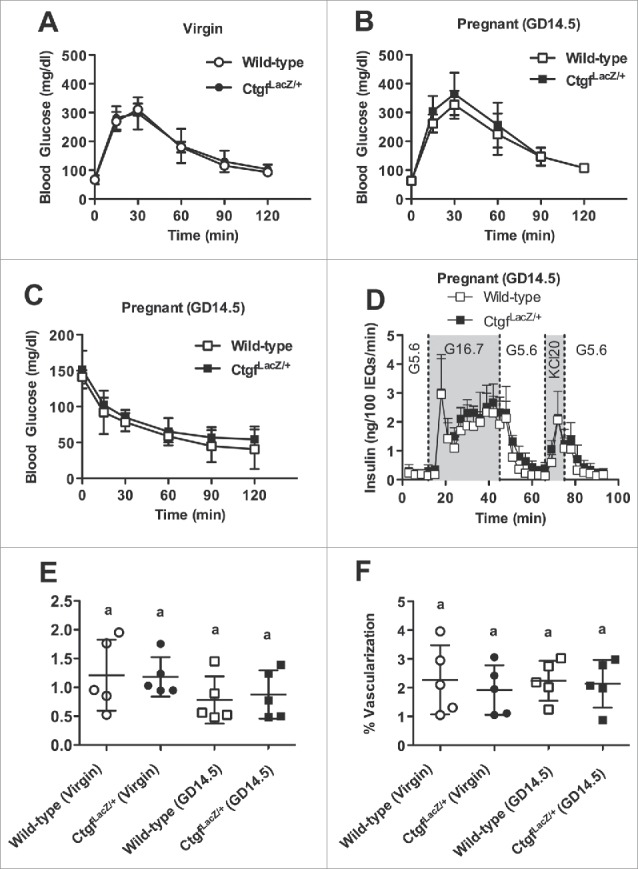
CtgfLacZ/+ females have normal glucose tolerance, insulin tolerance, α-cell proliferation, and islet vascularization. Whole-body glucose tolerance in (A) virgin and (B) gestational day 14.5 (GD14.5) wild-type and CtgfLacZ/+ females. Sample sizes were as follows: wild-type virgin n = 4; CtgfLacZ/+ virgin n = 11; wild-type pregnant (GD14.5) n = 8; CtgfLacZ/+ pregnant (GD14.5) n = 7. (C) Glucose-stimulated insulin secretion (GSIS) from whole intact islets isolated from pregnant (GD14.5) wild-type or CtgfLacZ/+ females (n = 3 mice/group) as assayed in a cell perifusion system. Islets were exposed to 5.6 mM glucose (G5.6), 16.7 mM glucose (G16.7), and 20 mM potassium chloride (KCL20). (D) Intraperitoneal insulin tolerance test in GD14.5 wild-type and CtgfLacZ/+ mice. Sample sizes were as follows: wild-type pregnant (GD14.5) n = 3; CtgfLacZ/+ pregnant (GD14.5) n = 5. Quantification of (E) α-cell proliferation and (F) islet vascularization in virgin and pregnant (GD14.5) wild-type and CtgfLacZ/+ mice. Error bars represent standard deviation. Samples with the same letter are not significantly different from each other (P < 0.05).
We previously showed that Ctgf induction during embryogenesis increased α-cell proliferation as well as β cell proliferation.8 Thus, α-cell proliferation was examined in virgin and GD14.5 wild-type and CtgfLacZ/+ pancreata. α-cell proliferation did not increase during pregnancy in wild-type mice and was not different in CtgfLacZ/+ mice (Fig. 4E).
Ctgf haploinsufficiency does not impair islet vascularization
The decrease in embryonic β cell proliferation observed with endothelial-specific Ctgf inactivation is accompanied by a reduction in overall islet vasculature.16 Since vascular endothelial cells produce factors that induce β cell proliferation (eg. HGF, Ctgf), we examined whether decreased islet vascularization could contribute to reduced maternal β cell proliferation in CtgfLacZ/+ pancreata during pregnancy.5 Pancreas sections from virgin and GD14.5 wild-type and CtgfLacZ/+ mice were co-labeled with insulin and CD31 to determine if there was a decrease in islet vascularization in CtgfLacZ/+ females. No differences in percentage of islet vascularization were observed between any of the four cohorts, indicating that maternal islet vascularization does not increase during pregnancy and arguing that impaired vascularization does not contribute to the decrease in maternal β cell proliferation in CtgfLacZ/+ pregnant females (Fig. 4F).
Discussion
In this study, we investigated the importance of Ctgf in pregnancy-induced β cell proliferation and post-natal β cell function. Since the etiology behind GDM remains largely unknown, understanding factors that are necessary for islet compensation during pregnancy is critical to devising strategies to effectively prevent and treat this disease. Here we show that global reduction in Ctgf impairs pregnancy-induced β cell proliferation without impacting β cell hypertrophy, β cell mass, whole body glucose tolerance, insulin tolerance, α-cell proliferation, or islet vascularization.
The β cell phenotype observed in Ctgf mutant animals is highly dependent on pregnancy status. Virgin CtgfLacZ/+ females, which have a global reduction in Ctgf, display no decrease in β cell proliferation when compared to virgin controls; during pregnancy, an impairment in β cell proliferation is observed. Given that Ctgf expression was limited to the CD31+ cells of the pancreas in adult mice, we conclude that it is the loss of vasculature-derived Ctgf that causes the phenotype. As CtgfLacZ/+ mice have a global reduction in Ctgf, loss of the gene from non-pancreatic cell types could be influencing the decrease in pregnancy-induced β-cell proliferation. However, we favor the hypothesis that vascular-derived Ctgf is affecting maternal β-cell proliferation for two reasons. First of all, previous studies from our lab demonstrated that conditional loss of Ctgf from just the endothelial cells of the vasculature is enough to cause a significant decrease in β-cell proliferation during embryogenesis.3 Secondly, insulin tolerance tests were no different from controls in pregnant Ctgf heterozygous mice, suggesting that changes in peripheral insulin tolerance could not explain the observed decrease in β-cell proliferation.
We hypothesize that total Ctgf ablation from the vasculature would further diminish pregnancy-induced β cell proliferation. We attempted to address this possibility using a tamoxifen-inducible Cre recombinase expressed in the vasculature. However, we found that tamoxifen administration completely prevented successful pregnancies, even when administered 10 weeks prior to mating. Administering a lower dosage of tamoxifen may allow for pregnancy to occur, but would reduce the efficiency of recombination of the conditional allele, potentially masking any phenotypes. Additionally, a recent report showed that tamoxifen itself suppresses pregnancy-induced β cell proliferation.24 We are therefore seeking other solutions to assess the impact of endothelial-derived Ctgf on post-natal β cell proliferation without interfering with normal islet function.
It currently remains unclear why no β cell proliferation phenotype is evident in virgin CtgfLacZ/+ females. Potentially, Ctgf-induced β cell proliferation may be dependent on the presence of co-factors that are regulated by pregnancy. For example, the HGF/c-Met signaling axis is upregulated during pregnancy and is necessary for β cell mass expansion.4 One possible scenario therefore is that HGF/c-Met signaling facilitates Ctgf-induced β cell proliferation, and that haploinsufficieny for Ctgf does not impair β cell proliferation in virgin adult females due to the relative low HGF/c-Met signaling activity in the islets of virgin mice. Interestingly, we previously observed that Ctgf-mediated β cell regeneration is accompanied by induction of HGF in islets.20 While vasculature-derived HGF is known to drive pregnancy induced β cell proliferation, it is currently unclear if this is the mechanism by which Ctgf regulates β cell replication, or if it acts through another pathway altogether.
Despite the significant reduction in β cell proliferation in pregnant CtgfLacZ/+ mice compared to pregnant wild-type mice, we did not observe an impairment in pregnancy-induced β cell mass expansion. It is likely that the modest increase in β cell proliferation in pregnant CtgfLacZ/+ mice (compared to virgin CtgfLacZ/+), together with β cell hypertrophy, is sufficient to cause an expansion in β cell mass indistinguishable from wild-type mice. This hypothesis is consistent with the normal glucose tolerance observed in pregnant CtgfLacZ/+ mice.
Since Ctgf is a secreted protein, we considered the possibility that proliferation of other endocrine cell types would also be regulated by Ctgf. In our analysis, CtgfLacZ/+ mice did not experience a decrease in α-cell proliferation compared to wild-type controls regardless of pregnancy status. Unlike the β cells, pregnancy did not elicit any increase in α-cell proliferation at GD14.5 compared to virgin controls. This indicates that the mechanism by which Ctgf induces proliferation is not ubiquitous to all adult islet endocrine cells, but has some degree of specificity for the β cells.
The impact of Ctgf on human β cell proliferation and function currently remains unexplored, and its expression pattern in the pancreas throughout human development, adulthood, and pregnancy is also unknown. Mutations in CTGF are not currently associated with any form of diabetes in human patients. T2D. The mechanism of β cell mass expansion during human pregnancy currently remains controversial, so it is unclear if loss of a β cell mitogen in a human patient could contribute to GDM. While GDM affects 7–10% of human pregnancies, to date only mutations in the human prolactin receptor gene (PRLR) have been linked specifically to GDM and not T2D, highlighting how little is known about the genetic causes of this disease.14,16 Our data highlight the fact that the vasculature likely plays a significant role in beta cell compensation during pregnancy.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Anastasia Coldren, Heather Durai, and Dr. Marcela Brissova in the Vanderbilt University Medical Center Islet Procurement and Analysis Core for assistance with islet isolation, perifusion, and data analysis. We thank the members of the Gannon laboratory for helpful discussions about the project.
Funding
This research involved use of the Islet Procurement and Analysis Core of the Vanderbilt Diabetes Research and Training Center for all islet isolations (Director: Dr. Marcela Brissova). This core is supported by National Institutes of Health grant DK020593. This work was supported by the U.S. Department of Veterans Affairs (grant 1BX000990–01A1 to M.G.), the Juvenile Diabetes Research Foundation International (1–2011-592 and 1-INO-2014–177-A-V to M.G.), the American Diabetes Association (1–16-IBS-100 to M.G.), and an American Heart Association Postdoctoral Fellowship (14POST20380262 to R.C.P.).
References
- [1].American Diabetes Association Gestational diabetes mellitus. Diabetes Care. 2004;27:s88–s90. doi: 10.2337/diacare.27.2007.S88. PMID:14693936 [DOI] [PubMed] [Google Scholar]
- [2].Brigstock DR. The CCN family: A new stimulus package. J Endocrinol. 2003;178:169–175. doi: 10.1677/joe.0.1780169. PMID:12904165 [DOI] [PubMed] [Google Scholar]
- [3].Crawford LA, Guney MA, Oh YA, Deyoung RA, Valenzuela DM, Murphy AJ, Yancopoulos GD, Lyons KM, Brigstock DR, Economides A, et al.. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and beta-cell proliferation during embryogenesis. Mol Endocrinol. 2009;23:324–336. doi: 10.1210/me.2008-0045. PMID:19131512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Demirci C, Ernst S, Alvarez-Perez JC, Rosa T, Valle S, Shridhar V, Casinelli GP, Alonso LC, Vasavada RC, Garcia-Ocana A. Loss of HGF/c-Met signaling in pancreatic beta-cells leads to incomplete maternal beta-cell adaptation and gestational diabetes mellitus. Diabetes. 2012;61:1143–1152. doi: 10.2337/db11-1154. PMID:22427375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Garcia-Ocana A, Takane KK, Syed MA, Philbrick WM, Vasavada RC, Stewart AF. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem. 2000;275:1226–1232. doi: 10.1074/jbc.275.2.1226. PMID:10625667 [DOI] [PubMed] [Google Scholar]
- [6].Gilmartin AB, Ural SH, Repke JT. Gestational diabetes mellitus. Reviews in obstetrics & gynecology. 2008;1:129–134. [PMC free article] [PubMed] [Google Scholar]
- [7].Golson ML, Bush WS, Brissova M. Automated quantification of pancreatic beta-cell mass. Am J Physiol Endocrinol Metab. 2014;306:E1460–1467. doi: 10.1152/ajpendo.00591.2013. PMID:24760991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guney MA, Petersen CP, Boustani A, Duncan MR, Gunasekaran U, Menon R, Warfield C, Grotendorst GR, Means AL, Economides AN, et al.. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proc Natl Acad Sci U S A. 2011;108:15242–15247. doi: 10.1073/pnas.1100072108. PMID:21876171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Henley KD, Gooding KA, Economides AN, Gannon M. Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. Am J Physiol Endocrinol Metab. 2012;303:E752–761. doi: 10.1152/ajpendo.00531.2011. PMID:22829579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. PMID:12736220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, Fontaine M, Yen MH, Kim SK. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–809. doi: 10.1126/science.1146812. PMID:17975067 [DOI] [PubMed] [Google Scholar]
- [12].Kayton NS, Poffenberger G, Henske J, Dai C, Thompson C, Aramandla R, Shostak A, Nicholson W, Brissova M, Bush WS, et al.. Human islet preparations distributed for research exhibit a variety of insulin-secretory profiles. Am J Physiol Endocrinol Metab. 2015;308:E592–602. doi: 10.1152/ajpendo.00437.2014. PMID:25648831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lacy PE, Kostianovsky M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–39. doi: 10.2337/diab.16.1.35. PMID:5333500 [DOI] [PubMed] [Google Scholar]
- [14].Le TN, Elsea SH, Romero R, Chaiworapongsa T, Francis GL. Prolactin receptor gene polymorphisms are associated with gestational diabetes. Genet Test Mol Biomarkers. 2013;17:567–571. doi: 10.1089/gtmb.2013.0009. PMID:23651351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pasek RC, Gannon M. Advancements and challenges in generating accurate animal models of gestational diabetes mellitus. Am J Physiol Endocrinol Metab. 2013;305:E1327–1338. doi: 10.1152/ajpendo.00425.2013. PMID:24085033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Perkins J, Dunn JP, Jagasia SM. Perspectives in gestational diabetes mellitus: A review of screening, diagnosis, and treatment. Clinical diabetes: a publication of the American Diabetes Association. 2007;25:57–62. doi: 10.2337/diaclin.25.2.57. [DOI] [Google Scholar]
- [17].Rachfal AW, Brigstock DR. Structural and functional properties of CCN proteins. Vitam Horm. 2005;70:69–103. doi: 10.1016/S0083-6729(05)70003-0. PMID:15727802 [DOI] [PubMed] [Google Scholar]
- [18].Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab: TEM. 2010;21:151–158. doi: 10.1016/j.tem.2009.11.001. PMID:20015659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Riley KG, Pasek RC, Maulis MF, Dunn JC, Bolus WR, Kendall PL, Hasty AH, Gannon M. Macrophages are essential for CTGF-mediated adult beta-cell proliferation after injury. Mol Metab. 2015;4:584–591. doi: 10.1016/j.molmet.2015.05.002. PMID:26266091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Riley KG, Pasek RC, Maulis MF, Peek J, Thorel F, Brigstock DR, Herrera PL, Gannon M. Connective tissue growth factor modulates adult beta-cell maturity and proliferation to promote beta-cell regeneration in mice. Diabetes. 2015;64:1284–1298. doi: 10.2337/db14-1195. PMID:25392241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shibata A, Ludvigsen CW Jr., Naber SP, McDaniel ML, Lacy PE. Standardization of a digestion-filtration method for isolation of pancreatic islets. Diabetes. 1976;25:667–672. doi: 10.2337/diab.25.8.667. PMID:182606 [DOI] [PubMed] [Google Scholar]
- [22].Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. PMID:9230352 [DOI] [PubMed] [Google Scholar]
- [23].Staels W, Heremans Y, Leuckx G, Van Gassen N, Salinno C, De Groef S, Cools M, Keshet E, Dor Y, Heimberg H, and De Leu N. Conditional islet hypovascularisation does not preclude beta cell expansion during pregnancy in mice. Diabetologia. 2017;60:1051–1056. doi: 10.1007/s00125-017-4243-1. PMID:28299380 [DOI] [PubMed] [Google Scholar]
- [24].Yuchi Y, Cai Y, Legein B, De Groef S, Leuckx G, Coppens V, Van Overmeire E, Staels W, De Leu N, Martens G, et al.. Estrogen Receptor alpha Regulates beta-Cell Formation During Pancreas Development and Following Injury. Diabetes. 2015;64:3218–3228. doi: 10.2337/db14-1798. PMID:26015547 [DOI] [PubMed] [Google Scholar]
- [25].Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, Gannon M. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes. 2010;59:143–152. doi: 10.2337/db09-0050. PMID:19833884 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.