

Abstract
We herein report a Pd(II)-catalyzed β-C(sp3)–H (hetero)arylation of a variety of ketones using a commercially available 2,2-dimethyl aminooxyacetic acid auxiliary. Facile installation and removal of the auxiliary as well as its superior scope for both ketones and (hetero)aryl iodides overcome the significant limitations of the previously reported β-C(sp3)–H arylation of ketones. The ready availability of ketones renders this reaction a broadly useful method for alkyl-(hetero)aryl coupling involving both primary and secondary alkyls.
Graphical abstract
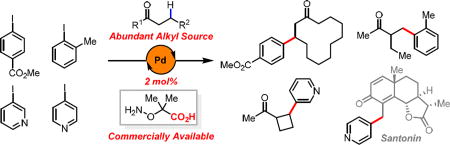
The availability and synthetic versatility of ketones have prompted chemists to further exploit their synthetic utility by functionalizing β-C(sp3)–H bonds with transition metal catalysts since the 1970s.1 Despite the early reports of oxime directed stoichiometric β-C(sp3)–H palladation and subsequent acetoxylation and halogenation,1 as well as the recent seminal contributions in rendering these processes catalytic,2,3 the oxime directed β-C(sp3)–H functionalization has largely been limited to primary C–H bonds. Recently, we have developed the Pd(II)-catalyzed β-C(sp3)–H arylation of ketones using a transient amino acid directing group.4 Extensive modification of a β-amino acid directing group has allowed for the methylene C–H arylation of a handful of ketones,5 however there exist two critical limitations: only methyl ketones are reactive; α-substituents or γ-substituents are not tolerated. In general, all the aforementioned β-C(sp3)–H functionalization reactions of ketones share the following fatal limitations in scope for both ketones and aryl coupling partners: (A) functionalization of methylene C(sp3)–H bonds is highly limited; (B) many common functional groups (i.e., ester, amine, ether, acetal etc.) are not compatible; (C) heteroarylation is inaccessible. Very recently, we reported a practical aminooxyacetic acid auxiliary for β-C(sp3)–H iodination.6 The weakly coordinating carboxyl motif in this bidentate directing group was found to be crucial for assembling the reactive palladium complex. Herein, we report a new aminooxyacetic acid auxiliary that successfully addressed all the limitations of β-C(sp3)–H arylation of ketones. Notably, the majority of the substrates employed are not compatible with previously reported methods. Late-stage diversification of Santonin through β-C(sp3)–H (hetero)arylation was also demonstrated. In light of the availability of diverse ketones and the versatile reactivity of carbonyl groups, this reaction provides an alternative method for alkylating (hetero)aryl iodides without using the conventional alkyl organometallic reagents.7
Based on our previous investigation of β-C(sp3)–H iodination of ketones using an aminooxyacetic acid auxiliary,6 we began our study by conducting β-methylene C–H arylation of 2-pentanone in the presence of Pd(OAc)2 (10 mol%), methyl 4-iodobenzoate (2.0 equiv), and AgTFA (2.0 equiv) in HFIP at 120 °C (Table 1). We were pleased to observe that using the optimal directing group (DG1) for β-C(sp3)–H iodination6 afforded the desired arylated product in 10% yield. However, the yield failed to be further improved after an extensive screening of conditions with DG1. Thus, we turned our attention to modifying DG1. We proposed that increasing the steric bulk of the aminooxyacetic acid carbon center by installing two methyl groups at its 2-position would favor the conformation necessary for bidentate binding to palladium, enhancing the coordinating ability of the directing group through Thorpe-Ingold effect and potentially promoting methylene C–H activation. Indeed, the yield drastically increased to 80% with this 2,2-dimethyl aminooxyacetic acid auxiliary (DG2). However, one iso-propyl substituent at the same position of aminooxyacetic acid (DG3) was proven ineffective presumably due to diminished Thorpe-Ingold effect, where both gem-substituents are required for the optimal directing group bite angle compression. In order to gain more insight into the directing group effect on reactivity, we also converted the acid moiety to the corresponding acidic amide (1a–4) and non-acidic amide (1a–5). However, only trace amounts of the products were detected in both cases (DG4 and DG5), which further highlights the unique property of the weakly coordinating carboxyl directing motif.
Table 1.

|
Conditions: 1a–x (0.1 mmol, 1.0 equiv), methyl 4-iodobenzoate (2.0 equiv), Pd(OAc)2 (10 mol%), AgTFA (2.0 equiv), HFIP (1.0 mL), 120 °C, under air, 20 h. Ar1 = 4-CF3(C6F4), Ar2 = 3,5-CH3(C6H3).
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
The product was observed as an E/Z mixture.
With the optimal directing group and reaction conditions established, we set out to explore the scope of various ketones (Table 2). It is worth noting that the installation of the auxiliary for all the ketones was simple and chromatography free. We started the scope investigation with various methyl alkyl ketones (2a–f). 2-Pentanone and 2-decanone were selectively arylated at the β-position in good yields (2a, 2b). Arylation of benzylic and homobenzylic methylene C–H bonds gave the products in 52% and 65% yields respectively (2c, 2d). A phthaloyl-protected amino group at δ-position was well tolerated (2e). Sterically hindered α-substituted ketone was arylated smoothly at 80 °C with great diastereoselectivity (2f). Notably, such ketone is incompatible with the transient directing group strategy because the imine formation is retarded by steric hindrance. Besides methyl ketones, this method is also applicable to other ketones (2g–j). Symmetrical 4-heptanone was arylated in 82% yield with mono- and di-arylation (2g). For an unsymmetrical ketone 1h, selective mono-arylation on the less hindered side was achieved when a bulky iso-propyl group was presented at the β-position on the other side (2h). 2-Alkyl substituted cyclohexanones, such as 2-ethylcyclohexanone and 2-propyl-2-estercyclohexanone, were selectively arylated in good yields with moderate diastereoselectivity (2i, 2j). Apart from the arylation of acyclic methylene C–H bonds, the method is also feasible for cyclic methylene C–H bonds of 3-, 4-, 6-, and 12-membered rings (2k–p). Selective mono-arylation of methyl cyclohexyl ketone was achieved in 72% yield (2k). A methoxy group on the cyclohexyl ring was tolerated, giving the mono-arylated product in 67% yield (2l). Selective di-arylation of methyl cyclobutyl ketone was obtained with cis and trans isomers (2m). The trans isomer of product 2m was formed presumably due to epimerization at the α-position to avoid steric clashing between the two aryl groups and the directing group after C(sp3)–H di-arylation. A phthaloyl-protected amino substituent at the α-position of methyl cyclobutyl ketone was compatible, providing the arylated product in a high yield (2n). Inferior reactivity was observed for methyl cyclopropyl ketone and only the mono-arylated product was afforded (2o). Notably, the first example of methylene C–H activation of macrocyclic ketones, which are widely present in a plethora of biologically active natural products and clinic drugs,8 has been realized in an excellent yield (2p). Given the limited scope of primary C–H arylation of ketones using O-methyl oxime and transient amino acid directing group,2c,3b,4a we tested our newly developed method for β-methylene C–H arylation on β-primary ketones. As expected, the reaction gave uncontrollable mixtures of both primary and methylene C–H arylation products. Then we found that lowering the reaction temperature and replacing the HFIP solvent with PhCF3 can afford exclusive primary C–H arylation in good to excellent yields (2q–s). Notably, ketones containing α-ester and benzyl protected hydroxyl groups were arylated at β-methyl groups (2t, 2u). The lower yield of 2u can be attributed to the coordination of the protected hydroxyl group of the substrate to the palladium center to form a tridentate intermediate, disfavoring the formation of the bidentate reactive species that can undergo C–H activation. In addition to methyl ketones, ketones with n-butyl, ester and benzyl protected hydroxyl groups were well tolerated (2v–x). 2-Methylcyclohexanone and its derivatives containing acetal, cyclic ether, and ester functionalities were selectively arylated at 2-methyl groups in good to excellent yields (2y-ab).
Table 2.

|
Conditions: 1 (0.1 mmol, 1.0 equiv), methyl 4-iodobenzoate (2.0 equiv), Pd(OAc)2 (10 mol%), AgTFA (2.0 equiv), HFIP (1.0 mL), 120 °C, under air, 20 h. See SI for work up procedures.
Isolated yields are provided and most of the products were isolated as an E/Z mixture.
The reaction temperature is 80 °C.
The numbers in parentheses indicate the ratio of mono:di.
The d.r. for the di-arylated product is 1:1.
The d.r. for the arylated product is 2:1.
The reaction temperature is 100 °C.
The d.r. for the arylated product is 2.4:1.
The ratio of cis:trans is 1:1.1.
The solvent is PhCF3 instead of HFIP, the reaction temperature is 80 °C.
AgOAc (2.0 equiv) was used instead of AgTFA.
Next, we examined the scope of aryl iodides (Table 3). The corresponding β-arylated products were obtained in good to excellent yields with iodobenzene and a variety of para-substituted aryl iodides (3a–l). For strongly coordinating groups (i.e., nitro and cyano) containing aryl iodides (3i, 3j), using HFIP instead of PhCF3 as a solvent was required to secure high yields. Notably, the free hydroxyl group of phenol remained intact throughout the reaction (3l). Different substituents at 3- and 3,5-positions of aryl iodides were successfully employed to give the arylated products in excellent yields (3m–s). Moreover, the C(sp3)–H arylation with sterically demanding ortho-substituted aryl iodides proceeded smoothly in good yields (3t–x). It is worth noting that the use of HFIP as a solvent was crucial for effectively coupling of bulky aryl iodides with C(sp3)–H bonds (3t, 3w, 3x). The use of aryl bromide or aryl chloride failed to give any desired arylation product under current conditions.
Table 3.

|
Conditions: 1s (0.1 mmol, 1.0 equiv), aryl iodide (2.0 equiv), Pd(OAc)2 (10 mol%), AgTFA (2.0 equiv), PhCF3 (0.5 mL), 80 °C, under air, 20 h. See SI for work up procedures.
Isolated yields are provided and all products were isolated as an E/Z mixture.
HFIP (1.0 mL) instead of PhCF3.
The numbers in parentheses indicate the ratio of mono:di.
HFIP (1.0 mL) instead of PhCF3 and the reaction temperature is 100 °C.
Heteroarylation of β-methylene C–H bonds could provide a useful method for alkylating medicinally important heteroaryls.9 However, the strong coordinating ability of heterocycles can often outcompete substrates for binding to palladium or simply deactivate the catalyst. As a consequence, there have been no successful examples of primary or methylene C–H heteroarylation of ketones. With our highly efficient directing group in hand, we tested the commonly used heteroaryl iodides with cyclobutyl ketone as the model substrate. Notably, heteroarylated cyclobutanes widely exist in natural products and drug molecules;10 and are inaccessible through conventional Michael addition (Table 4). For less strongly coordinating 2-substituted pyridines, moderate to good yields of heteroarylated products could be obtained (4a–h). More importantly, strongly coordinating pyridines and quinolines were also successfully coupled albeit with moderate yields (4i–l). Notably, the selective di-heteroarylation products were formed in good yields for weakly coordinating indole, thiophene, and furan heteroaryl iodides (4m–p). It is noteworthy that the epimerization at the α-position may occur during the reaction, giving structurally interesting trans-products (4d, 4i, 4j, 4o). We reason that the observed epimerization of (hetero)arylated cyclobutane products may be affected by two factors: (A) di-arylation products tend to epimerize to avoid the clash between directing group and two aryl groups and form thermodynamically more stable isomers; and (B) strongly basic aryl groups (pyridine and thiophene) may serve as a base to promote epimerization. The heteroarylation of acyclic substrates 1a and 1s also proceeded to give the desired products 4q and 4r respectively.
Table 4.

|
Conditions: 1m (0.1 mmol, 1.0 equiv), heteroaryl iodide (1.5 equiv), Pd(OAc)2 (10 mol%), AgTFA (2.0 equiv), HFIP (1.0 mL), 100 °C, under air, 20 h. T’ means no further methyl esterification, the acid form was isolated. See SI for work up procedures.
Isolated yields.
The ratio of cis:trans is 1:1.4.
The reaction temperature is 120 °C.
The ratio of cis:trans is 1:1.8.
The ratio of cis:trans is 1:2.5.
Heteroaryl iodide (2.0 equiv) were used and the reaction temperature is 80 °C.
AgOAc (2.0 equiv) was used instead of AgTFA.
The widespread presence of carbonyl groups in natural products and drug molecules also renders this reaction a useful method for late-stage diversification. Thus, direct C(sp3)–H (hetero)arylation of Santonin was performed with a variety of coupling partners to provide potentially useful analogues (Scheme 2).11
Scheme 2. The Late-Stage C(sp3)–H (Hetero)Arylation of Santonina,b.

aConditions: 1ac (0.1 mmol, 1.0 equiv), (hetero)aryl iodide (1.5 equiv), Pd(OAc)2 (10 mol%), AgTFA (2.0 equiv), HFIP (1.0 mL), 100 °C, under air, 20 h. T’ means no further methyl esterification, the acid form was isolated. See SI for work up procedures. bIsolated yields. cMethyl 4-iodobenzoate (2.0 equiv) was used and the reaction temperature is 80 °C. dThe reaction temperature is 120 °C.
To further showcase the synthetic utility of this method, the arylation of 1s was carried out in 2.0 mmol scale with reduced catalyst loading (2 mol%). The desired C(sp3)–H arylation product was obtained in 75% yield (Scheme 3). The facile removal of the auxiliary was demonstrated by treating the arylated product with 4 M HCl in 1,4-dioxane and water at 100 °C, providing β-arylated ketone in 85% yield (Scheme 4).
Scheme 3. The Scale Up Reaction with Reduced Pd Loadinga,b.

aConditions: 1s (2.0 mmol, 1.0 equiv), 4-iodotoluene aryl iodide (2.0 equiv), Pd(OAc)2 (2 mol%), AgTFA (2.0 equiv), HFIP (20 mL), 80 °C, under air, 20 h. See SI for work up procedures. bIsolated yields.
Scheme 4. The Removal of Auxiliarya,b.

aConditions: 2a (0.1 mmol, 1.0 equiv), HCl (4.0 M in dioxane) (1.25 mL), H2O (0.25 mL), 100 °C, under air, 20 h. See SI for work up procedures. b Isolated yields.
In summary, a simple and efficient directing group has been developed to enable β-methylene and primary C–H arylation of ketones. The α,α,-dimethyl groups in the aminooxyacetic acid auxiliary are crucial for enabling the reactivity. Broad substrate scope for ketones and (hetero)aryl iodides has been demonstrated, overcoming the significant limitations of previous β-C–H functionalization of ketones.
Supplementary Material
Scheme 1. Transition Metal-Catalyzed β-C(sp3)–H Arylation of Ketones.

Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support. R.-Y. Zhu is supported by the Boehringer Ingelheim Fellowship. We thank N. Chekshin for proofreading.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected examples of stoichiometric palladation and subsequent functionalization reactions: Constable AG, McDonald WS, Sawkins LC, Shaw BL. J. Chem. Soc. Chem, Commun. 1978:1061.Carr K, Sutherland JK. J. Chem. Soc. Chem, Commun. 1984:1227.Baldwin JE, Nájera C, Yus M. J. Chem. Soc. Chem, Commun. 1985:126.Baldwin JE, Jones RH, Nájera C, Yus M. Tetrahedron. 1985;41:699.
- 2.For oxime directed C(sp3)–H functionalizations with palladium catalyst: Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542. doi: 10.1021/ja046831c.Thu H-Y, Yu W-Y, Che C-M. J. Am. Chem. Soc. 2006;128:9048. doi: 10.1021/ja062856v.Peng J, Chen C, Xi C. Chem. Sci. 2016;7:1383. doi: 10.1039/c5sc03903g.Mu Y, Tan X, Zhang Y, Jing X, Shi Z. Org. Chem. Front. 2016;3:380.
- 3.For oxime directed C(sp3)–H functionalizations with iridium catalyst: Kang T, Kim Y, Lee D, Wang Z, Chang S. J. Am. Chem. Soc. 2014;136:4141. doi: 10.1021/ja501014b.Gao P, Guo W, Xue J, Zhao Y, Yuan Y, Xia Y, Shi Z. J. Am. Chem. Soc. 2015;137:12231. doi: 10.1021/jacs.5b06758.
- 4.(a) Zhang F-L, Hong K, Li T-J, Park H, Yu J-Q. Science. 2016;351:252. doi: 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang K, Li Q, Liu Y, Li G, Ge H. J. Am. Chem. Soc. 2016;138:12775. doi: 10.1021/jacs.6b08478. [DOI] [PubMed] [Google Scholar]
- 5.Hong K, Park H, Yu J-Q. ACS Catal. 2017;7:6938. doi: 10.1021/acscatal.7b02905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu R-Y, Liu L-Y, Yu J-Q. J. Am. Chem. Soc. 2017;139:12394. doi: 10.1021/jacs.7b06851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For selected reviews on alkyl-(hetero)aryl cross-coupling: Cherney AH, Kadunce NT, Reisman SE. Chem. Rev. 2015;115:9587. doi: 10.1021/acs.chemrev.5b00162.Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA. Acc. Chem. Res. 2016;49:1429. doi: 10.1021/acs.accounts.6b00214.
- 8.For selected examples: Parenty A, Moreau X, Campagne JM. Chem. Rev. 2006;106:911. doi: 10.1021/cr0301402.Driggers EM, Hale SP, Lee J, Terrett NK. Nat. Rev. Drug Discovery. 2008;7:608. doi: 10.1038/nrd2590.Mallinson J, Collins I. Future Med. Chem. 2012;4:1409. doi: 10.4155/fmc.12.93.
- 9.Vitaku E, Smith DT, Njardarson JT. J. Med. Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 10.For selected examples: Hansen TV, Stenstrøm Y. In: Naturally Occurring Cyclobutanes. In Organic Synthesis: Theory and Applications. Hudlicky T, editor. Vol. 5. Elsevier Science; Oxford U.K: 2001. pp. 1–38.Kurosawa K, Takahashi K, Tsuda EJ. Antibiot. 2001;54:541. doi: 10.7164/antibiotics.54.541.Chi YM, Nakamura M, Zhao XY, Yoshizawa T, Yan WM, Hashimoto F, Kinjo J, Nohara T. Chem. Pharm. Bull. 2005;53:1178. doi: 10.1248/cpb.53.1178.Dembitsky VM. J. Nat. Med. 2008;62:1. doi: 10.1007/s11418-007-0166-3.Sergeiko A, Poroikov VV, Lumir O, Hanu LO, Dembitsky VM. Open Med. Chem. J. 2008;2:26. doi: 10.2174/1874104500802010026.
- 11.For selected examples: Duplan V, Serba C, Garcia J, Valot G, Barluenga S, Hoerlé M, Cuendet M, Winssinger N. Org. Biomol. Chem. 2014;12:370. doi: 10.1039/c3ob42049c.Chinthakindi PK, Singh J, Gupta S, Nargotra A, Mahajan P, Kaul A, Ahmed Z, Koul S, Sangwan PL. Eur. J. Med. Chem. 2017;127:1047. doi: 10.1016/j.ejmech.2016.11.018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.