

Abstract
Hematopoietic stem cell transplantation (HSCT)-associated thrombotic microangiopathy (TA-TMA) is an understudied complication of HSCT that significantly affects transplant-related morbidity and mortality. Over the past several decades, the cause of TA-TMA has remained unknown, limiting treatment options to non-specific therapies adapted from other diseases. Recent prospective studies dedicated to the study of TA-TMA have provided new insights into the pathogenesis of, and genetic susceptibility to TA-TMA, raising awareness of this important transplant complication and allowing for the identification of potentially novel therapeutic targets. Specifically, many patients with TA-TMA develop multi-organ tissue injury through endothelial damage mediated by the activation of the complement pathway, leading to rational therapeutic strategies including complement blockade. This new knowledge has the potential to favorably influence clinical practice and change the standard of care for how patients with TA-TMA are managed. In this review, we summarize novel approaches to the recognition and management of TA-TMA, using case examples to illustrate key clinical points that hopefully lead to improved short and long-term outcomes for these complex HSCT patients, who remain at significant risk for treatment-related morbidity and mortality.
Keywords: Thrombotic microangiopathy, TA-TMA, Complement, Eculizumab, Hematopoietic stem cell transplant
1. Introduction
Hematopoietic stem cell transplantation-associated thrombotic microangiopathy (TA-TMA) is caused by systemic vascular endothelial injury triggered by the transplantation process [1,2]. TMA affects multiple organs and occurs in about 30% of hematopoietic stem cell transplant (HSCT) recipients. Half of patients who develop TA-TMA (~15% of all transplant patients) will present with severe disease and these cases have mortality rates in excess of 80%. Recently, it has been shown that complement system activation in patients with TA-TMA is a very poor prognostic sign and implicates complement dysregulation as a key pathway in the pathogenesis of TA-TMA and its disease phenotype [3]. The original diagnostic criteria for TA-TMA used hematologic and kidney injury markers that can only detect advanced disease and therefore often delay TA-TMA diagnosis in complex transplant patients [4–7]. This delay in diagnosis significantly impacts the institution of early therapeutic measures. We have shown that HSCT patients with TA-TMA respond to a complement blocking agent (eculizumab), but therapy is often started late after multi-organ injury has already occurred [8,9]. We also observed that patients who received complement-blocking therapy early in their disease course had better responses and outcomes. There is a significant unmet medical need to recognize TA-TMA early in the disease presentation or even to proactively identify patients who are at high-risk for a severe TA-TMA phenotype prior to the start of transplantation. In the past several years, significant progress has been made in advancing our understanding of TA-TMA, guiding favorable changes in clinical practice. The objectives of this review are to summarize new advances in TA-TMA diagnosis, pathophysiology and treatment while using case studies to provide practical suggestions for clinical management.
2. Refined TA-TMA diagnostic criteria
2.1. Laboratory testing
TA-TMA is a systemic disorder that occurs when endothelial injury in patients treated with HSCT presents with microangiopathic hemolytic anemia and platelet consumption, resulting in damage to the microcirculation. This endothelial injury affects multiple organs, both over the short- and long-term after transplant. Some patients exhibit signs of TA-TMA in one particular organ, such as the kidney, lung or bowel, while others present with multi-organ involvement that can result in severe, life-threatening, acute organ failure. It is not yet understood why certain patients have a predisposition to injury of particular organs, making it important to comprehensively assess the most commonly affected organs (kidney, bowel, heart/lung) in patients with clinical concern for TA-TMA [7,10].
The diagnosis of TA-TMA requires a high index of suspicion, especially since systemic signs of TA-TMA are often mistaken for other transplant-related blood test abnormalities, particularly prior to donor cell engraftment when cytopenia is expected as part of the transplant course. In patients who are awaiting donor cell engraftment and are still transfusion dependent it is very important to assess transfusion needs. Hemolytic or consumptive processes are suspected when transfusions are required several times per week, out of proportion to what is expected before engraftment, and hemoglobin and platelet decreases are significant and acute without evidence of blood loss. It is also important to note that in case of severe TA-TMA, schistocytosis might be absent due to high vascular permeability and red blood cell extravasation into the tissues. Accordingly, it has been observed that elevated blood lactate dehydrogenase (LDH), proteinuria, and hypertension are the earliest signs of TA-TMA and should trigger more comprehensive evaluation in HSCT patients [3,11,12].
We recently proposed refined TA-TMA diagnostic and risk criteria that included proteinuria, hypertension, and elevated markers of the activated terminal complement complex (sC5b-9) that were not part of previous diagnostic criteria but were indicative of disease phenotype and clinical outcomes [3]. Taking into consideration the challenges of identifying TA-TMA in the complex transplant patient, we observed that patients exhibiting at least five of the seven diagnostic criteria listed in Table 1 were very likely to have multi-organ TA-TMA and should be further evaluated with organ-specific tests. While haptoglobin was not included in these new updated diagnostic criteria, haptoglobin serves as an important prognostic marker of overall inflammatory status. In our prospective study we showed that patients with a severe TA-TMA phenotype resulting in multi-organ failure or death had elevated haptoglobin levels at TA-TMA diagnosis. Haptoglobin as a non-specific inflammatory marker likely reflects overall tissue inflammation and may take a prolonged time to drop below normal in patients with severe TA-TMA.
Table 1.
TA-TMA diagnostic criteria.
| The diagnosis of TA-TMA maybe be established:
| |
|---|---|
| A. Microangiopathy diagnosed on tissue biopsy | |
| or | |
| B. Laboratory and clinical markers indicating TMA
| |
| Laboratory or clinical marker | Description |
| 1Lactate dehydrogenase (LDH) | Elevated above the upper limit of normal for age |
| 2Proteinuria | Random urine protein/random urine creatinine ratio ≥2 mg/mg |
| 3Hypertension | <18 years of age: a blood pressure at the 95th percentile value for age, sex and height. ≥18 years of age: a blood pressure ≥140/90 mmHg. |
| 4De novo thrombocytopenia | Thrombocytopenia with a platelet count <50 × 109/L or a ≥ 50% decrease in the platelet count |
| 5De novo anemia | A hemoglobin below the lower limit of normal for age or anemia requiring transfusion support |
| 6Evidence of microangiopathy | The presence of schistocytes in the peripheral blood or histologic evidence of microangiopathy on a tissue specimen |
| 7Terminal complement activation | Elevated serum concentration of sC5b-9 above upper normal laboratory limit |
present: consider diagnosis of TA-TMA. Monitor very closely.
at TMA diagnosis indicate high features associated with poor outcome. This table is modified from previously published by Jodele et al. [7].
In several studies, proteinuria was more informative as a TA-TMA marker of kidney injury as compared to elevations of serum creatinine, especially in patients with prolonged illness and significant muscle mass loss, in whom serum creatinine often overestimates kidney function [13–17]. Proteinuria can be assessed by simple urinalysis (random urine protein of ≥30 mg/dL), but in patients receiving high volume hydration or with significant capillary leak, random urine protein/urine creatinine ratios are often more informative, as ratios of ≥2 mg/mg have been strongly associated with a diagnosis of TA-TMA in prospective studies [3].
2.2. Histopathologic tissue evaluation
While the tissue diagnosis of TA-TMA remains the gold standard, biopsy poses several challenges. It is often difficult to obtain tissue samples from HSCT patients who are critically ill or have a high risk for bleeding due to thrombocytopenia and/or hypertension. Historically, the kidney has been the preferred organ in which to diagnose a microangiopathic process. But in patients with systemic TA-TMA, renal biopsy may not always provide a diagnosis. This is because TA-TMA may selectively affect different organs, and any available tissue should be examined for vascular injury.
The histology of the kidney in TA-TMA usually shows classic microangiopathic features, indistinguishable from histologic signs seen in other microangiopathies such as atypical hemolytic uremic syndrome (aHUS) or thrombotic thrombocytopenic purpura (TTP) and usually includes glomeruli with thickened capillary walls, occluded small and medium-sized vessel lumens, red blood cell fragments trapped in the mesangial matrix, and renal arterioles and glomerular capillaries with separation of the endothelial cell layer from the vessel wall [18,19].
Gastrointestinal (GI) tract biopsies may provide very valuable information about systemic vascular injury [20–23]. These procedures are of lower risk compared to renal biopsies and are more commonly performed in HSCT recipients to look for evidence of graft versus host disease (GVHD). Because GVHD, a disease affecting epithelial cells, is most often the indication for a GI biopsy, the endothelial cells and the vascular compartment are often not carefully reviewed. We acknowledge that acquisition of an adequate vascular sample in GI biopsies is operator dependent, but TA-TMA can be suspected even by examining small caliber vessels. El-Bietar et al. proposed eight histologic criteria for the diagnosis of intestinal TA-TMA: mucosal hemorrhages, loss of glands, intraluminal schistocytes, intraluminal fibrin, intraluminal microthrombi, endothelial swelling, endothelial separation from the membrane, and total denudation of mucosa (Table 2) [24]. In addition, tissue samples should be examined for hemosiderin deposits indicating longstanding mucosal hemorrhages and providing a differential diagnosis distinct from procedure-related mucosal injury.
Table 2.
Diagnostic panel of intestinal TA-TMA.
| Clinical signs |
|
| Radiologic signs |
|
| GI endoscopy |
|
| Histologic signs on biopsy |
|
Hemosiderin deposits would indicate old bleeding and not procedure related hemorrhage.
This table is modified from previously published by Jodele et al. [7].
The evidence of mucosal denudation in HSCT patients with a clinical diagnosis of GI GVHD is often labeled as stage IV GVHD without more detailed evaluation as to what caused the mucosal surface denudation. As listed below in the clinical cases, it is very common for patients with intestinal bleeding that is usually staged as severe GVHD to present with ischemic colitis due to vascular injury resulting in damaging and sloughing of the bowel mucosa. It is certainly possible for intestinal GVHD and intestinal TA-TMA to coexist, especially in patients with severe symptomatology that is often refractory to GVHD-targeted therapy. Some investigators proposed a concept that patients presenting with therapy-refractory intestinal GVHD have both epithelial (bowel mucosa) and endothelial (vascular component) injury and both conditions – GVHD and TA-TMA – have to be treated to achieve a favorable clinical response [25,26].
In patients with systemic TA-TMA who require partial bowel resection for ischemic injury, larger bowel vessels should be carefully examined as they may show more severe vascular injury as compared to smaller submucosal vessels. Particular attention should be paid to the histology of vascular endothelial cells and the presence of fibrin deposition in the lumen, as microthromboses, contrary to what is often thought, is not an essential feature to diagnose a vascular microangiopathic process (Fig. 1).
Fig. 1.
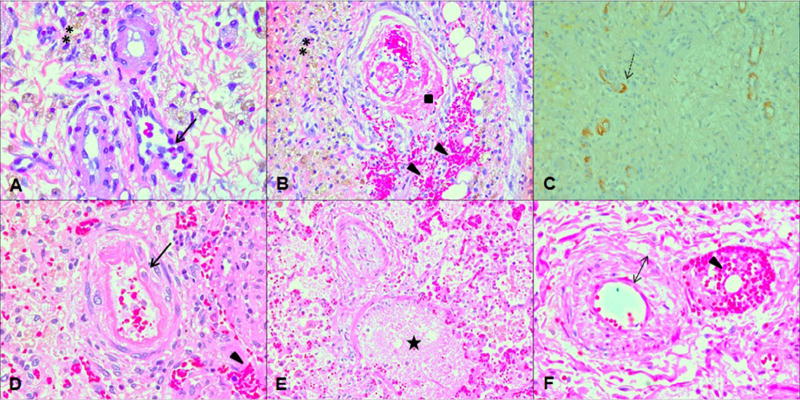
Histologic features of TA-TMA. (A) and (B) show histology from small bowel resection specimen displaying histopathologic changes of transplantation-associated thrombotic microangiopathy (TA-TMA) (H&E ×40). (A) This demonstrates severe acute microangiopathic vascular injury involving submucosal small bowel arterioles. Black arrows show endothelial cells “floating” in the vascular lumen after complete separation from the basement membrane and interstitial hemorrhages (triangle). (B) This shows organizing thrombus occluding submucosal small bowel arteriole with extensive hemorrhages into surrounding tissues (triangle), double stars mark hemosiderin deposits indicating old hemorrhagic areas. (C) C4d staining (×20) of small bowel tissue section shows diffuse positive staining in the degenerating submucosal vessels (dotted arrow) with microangiopathic changes indicating complement deposits in TMA affected vessels. (D), (E) and (F) show lung histology from autopsy specimen displaying histopathologic changes of TA-TMA and pulmonary arterial hypertension (H&E ×40). (A) This demonstrates severe acute microangiopathic vascular injury involving pulmonary arterioles. Black arrows show endothelial cells “floating” in the vascular lumen after complete separation from the basement membrane and interstitial hemorrhages (triangle) (B) lung arterioles affected by TA-TMA that are filled with fibrin debris trapping cells (star), (C) lung arterioles with proliferation of vascular wall (solid double headed arrow) with near obliteration of vascular lumen and red cell extravasation into the vessel wall (triangle).
Lung tissue affected by TA-TMA may show histologic changes similar to what is seen in patients with pulmonary hypertension, in addition to microangiopathic vascular injury (Fig. 1). Different stages and severities of vascular injury may be present in the same sample ranging from acute microangiopathic injury with endothelial cells “floating” in the vascular lumen and red cell fragments extravasated into surrounding tissues to the thickening of all three layers of the vascular wall or even nearly complete obliteration of the vascular lumen, indicative of pulmonary hypertension. Intra-arteriolar fibrin microthrombi with focal areas of organization are quite common in long standing TA-TMA and indicate chronic and progressive endothelial injury. Patients with lung TA-TMA usually have significant interstitial lung hemorrhages explaining acute hemoglobin drops without evidence of airway bleeding as seen in diffuse alveolar hemorrhage after transplantation [27–32]. In lung thromboembolic events unrelated to microangiopathy, arteriolar endothelium is intact, there are no interstitial hemorrhages and thrombi are detected in larger arteries without any evidence of systemic injury in smaller arterioles.
2.3. Echocardiographic studies
Pericardial effusion has been strongly associated with TA-TMA in HSCT patients [33,34]. Pericardial effusion can be suspected on chest X-ray or may be detected on echocardiographic screening even in asymptomatic patients. Patients with pericardial effusions should be closely monitored as they can acutely progress to cardiac tamponade. In patients with pericardial effusion and a history of hematologic malignancy but without evidence of systemic signs of TA-TMA, local malignancy relapse should be included in the differential diagnosis. Patients with polyserositis (pericardial, pleural effusions and ascites), especially those without any evidence of skin or GI GVHD, should be evaluated for TA-TMA.
Echocardiographic evidence of increased right ventricular pressure indicative of elevated pulmonary artery pressure or pulmonary hypertension has also been strongly associated with TA-TMA in prospective studies. Dandoy et al. showed that pulmonary vascular resistance increases as early as day +7 after HSCT in patients who later develop hematologic or tissue evidence of TMA, indicating that vascular injury occurs very early after HSCT and currently available laboratory studies are not sensitive enough to diagnose early disease [30]. Routine echocardiographic screening of HSCT patients at day+7 and day +30 was very useful in diagnosing early pathology such as elevated pulmonary pressures and pericardial effusion even in asymptomatic patients, guiding more targeted monitoring and interventions. Hypoxemia was the most significant clinical symptom associated with elevated pulmonary pressures in patients with TA-TMA.
Echocardiographic evaluation remains an expensive modality for evaluation of pulmonary hypertension associated with TA-TMA and blood biomarkers indicative of endothelial injury are being investigated.
3. Clinical presentation
Recognition of TA-TMA in complex HSCT patients requires a high index of suspicion and prospective monitoring using a routine “TMA panel” can be expanded to include more specific tests associated with TA-TMA if warranted by clinical suspicion. LDH, urinalysis, and careful blood pressure measurement should be routinely monitored in all HSCT recipients. Persistent evidence of elevated LDH, proteinuria, and hypertension out of proportion to expected steroid or calcineurin inhibitor therapy should trigger more detailed evaluation for TA-TMA including complement studies and organ function assessment. Certain clinical events should prompt clinicians to consider TA-TMA in the differential diagnosis, as illustrated in the following clinical cases.
3.1. Case 1
A six year old African-American female received an autologous HSCT for neuroblastoma using carboplatin, etoposide, and melphalan (day −7 to the day −4). On day +2 after stem cell infusion the patient developed fever to 39 °C, severe hypertension with PRES (posterior reversible encephalopathy syndrome) requiring a nicardipine infusion, followed by acute kidney injury requiring hemodialysis on day +3. Despite hypertension, the patient was presumed to have a serious bacterial infection and was started on broad-spectrum antibiotics. Prior to becoming anuric, she was noted to have a rising urine protein/creatinine ratio from a pre-transplant normal baseline of 0.1 mg/mg to 64 mg/mg (normal <0.2 mg/mg) on the day she started hemodialysis. The patient was red blood cell and platelet transfusion dependent, had rising LDH since day −4, and an elevated haptoglobin. There were no schistocytes on examination of the peripheral blood smear. On day +4, the patient developed respiratory failure requiring mechanical ventilation. On day +12 the patient was noted to have schistocytes in the blood and the haptoglobin dropped below normal. She developed severe abdominal distention, was not able to sustain adequate oxygenation on maximal ventilator support and passed away without receiving any TA-TMA targeted therapy. Autopsy showed signs of multi-organ TA-TMA with severe pulmonary and intestinal arteriolar endothelial injury and thromboses and no evidence of infection on tissue cultures.
This case illustrates a multi-organ presentation of acute TA-TMA that is often presumed to be a septic event with multi-organ failure after HSCT. It is very important to note that the patient was always hypertensive requiring a continuous nicardipine infusion, arguing against a clinical picture of septic shock. Even though in all circumstances of acute illness HSCT patients will be prescribed broad-spectrum antibiotics for the possibility of infection, other differential diagnoses should be considered. This case also illustrates the delayed appearance of hematologic TMA markers schistocytes and low haptoglobin, with persistent elevation of LDH, proteinuria, and hypertension being early markers of evolving TA-TMA. Hypoxemia in critically ill patients should also trigger consideration of echocardiography where in this case pulmonary failure was a result of acute arteriolar thrombosis and interstitial hemorrhaging that usually presents as acute pulmonary hypertension.
3.2. Case 2
A two year old Caucasian boy with hemophagocytic lymphohistiocytosis (HLH) received a matched unrelated donor HSCT using a reduced intensity regimen with alemtuzumab, fludarabine, and melphalan. The post-transplant course was complicated by stage III acute skin GVHD that responded to therapy. He had persistent elevation of LDH (1.5 times the upper normal limit), intermittent schistocytosis, persistent proteinuria >30 mg/dL, low cystatin C-estimated glomerular filtration rate, and hypertension requiring three medications for blood pressure control. He had no documented infections. On day +150 he presented to the emergency department with significant hypoxemia. Lungs were clear on chest X-ray. CT scan of the chest showed clear lungs and increased vascular markings. The patient was scheduled for diagnostic bronchoalveolar lavage. Upon transitioning to the intensive care after the bronchoscopy, he acutely developed respiratory failure requiring re-intubation and mechanical ventilation, but was not able to achieve adequate oxygenation. Echocardiography showed a right ventricular pressure 92% of systemic pressure, indicating severe pulmonary hypertension. The patient passed away before being able to undergo emergency atrial septostomy. Autopsy showed histologic lung vasculature changes compatible with TA-TMA and pulmonary hypertension.
This case illustrates pulmonary hypertension that developed due to long standing thrombotic microangiopathy. Acute hypoxemia in HSCT patients with TA-TMA not otherwise explained by acute viral or bacterial illness should be evaluated with echocardiography for evidence of pulmonary hypertension. Patients with persistent symptoms of TA-TMA who otherwise look well, but have ongoing pulmonary vascular injury, can succumb to acute cardiorespiratory failure after hypoxemic triggers such as anesthesia, acute respiratory viral illness, or due to mechanical pressure changes while undergoing procedures such as photopheresis for GVHD. HSCT patients with evidence of elevated right ventricular pressure should be monitored very closely and evaluated by a cardiologist, since symptoms may progress very rapidly to death.
3.3. Case 3
A six year old Caucasian female received a matched unrelated HSCT for HLH using a reduced intensity regimen with alemtuzumab, fludarabine, and melphalan. About 50 days after transplant she developed bloody diarrhea. GI endoscopy showed mucosal hemorrhages with only a few epithelial cells demonstrating apoptosis. The patient was diagnosed with intestinal GVHD based on clinical presentation and was treated with steroids and infliximab without improvement in the intestinal bleeding. She continued to have rectal bleeding without voluminous diarrhea and had severe abdominal pain and evidence of clinical ileus. There was no evidence of skin or liver GVHD. Chart review revealed persistent elevation of LDH and intermittent appearance of schistocytes since completing transplant chemotherapy. A random urine protein/creatinine ratio was 8 mg/mg (>2 mg/mg is nephrotic range) and she required four medications for hypertension control. Serum creatinine remained normal (0.3 mg/dL). The patient was cushingoid from prolonged steroid use. Blood sC5b-9 levels were above normal. There were no pulmonary symptoms and echocardiographic studies were normal. Repeated GI endoscopy was compatible with TA-TMA without evidence of GVHD. The patient was started on eculizumab therapy while GVHD medications were weaned. Hematologic TA-TMA signs and GI bleeding was controlled using pharmacokinetically guided eculizumab therapy but the patient continued to exhibit symptoms of clinical ileus and was diagnosed with a small bowel stricture by CT enterography. The patient was maintained on parenteral nutrition and bowel rest until resolution of active symptoms and until the small bowel strictures could be resected. She is doing well two years post transplantation without TMA or GI symptoms.
This case illustrates a typical presentation of intestinal TA-TMA manifesting as ischemic colitis with rectal bleeding and severe pain. While intestinal symptoms after HSCT are most often thought to be a consequence of GVHD, microangiopathic bowel injury should be considered in the differential diagnosis when patients continue with bloody stools and abdominal pain in the absence of the voluminous diarrhea typical for GVHD. TA-TMA causes ischemic bowel injury resulting in clinical ileus, bloody stool and severe pain refractory to narcotics. In this particular case, our patient did not have obvious histologic evidence of acute GVHD and even on bowel areas unaffected by ischemia, there were no apoptotic bodies indicative of GVHD. More commonly, HSCT patients may have both intestinal GVHD and intestinal TA-TMA together presenting with bloody high output diarrhea. We are now learning that patients with both epithelial (GVHD) and endothelial (TMA) components involved present with what may be called “steroid refractory GVHD” and clinical remission is not achieved unless both pathologies, GVHD and TMA, are treated. Despite debates as to whether TA-TMA is a separate entity in these patients or just a companion for severe GVHD, both clinical complications should be recognized and treated for clinical success. GI specimens from patients with refractory intestinal GVHD should be thoroughly reviewed for vascular injury, especially in cases of completely denuded mucosa indicating ischemic bowel injury.
4. Pathogenesis
Endothelial injury after HSCT is multifactorial resulting from the transplant conditioning regimen, infections, GVHD, medications, and likely inherited tissue sensitivity to the injury. Endothelial dysfunction also plays an important role in the pathogenesis of steroid refractoriness in GVHD [35,36]. Multiple endothelial injury pathways have been proposed in transplant patients, but for this review, we will focus on the role of complement activation because complement modifying therapy is currently the most promising and clinically available treatment option in TA-TMA.
We recently demonstrated that HSCT patients with TA-TMA have a high incidence of genetic variants in complement genes predisposing them to develop TMA with inferior survival after transplant [37]. Examining 17 genes involved in regulation of the complement pathway, we identified variants in 65% of HSCT recipients with TMA as compared with only 9% in those without TMA (p < 0.01). Furthermore, the incidence of TA-TMA was higher in patients with gene variants detected, regardless of race. However, the incidence of TA-TMA and the number of gene variants were higher in non-Caucasian transplant recipients, the majority of whom were African Americans, compared with Caucasian recipients. Importantly, the higher number of variants observed in non-Caucasian HSCT recipients was associated with a more severe TA-TMA phenotype and higher mortality, suggesting that complement gene variants likely modify susceptibility to TA-TMA after HSCT and determine disease severity in patients exposed to the transplantation process and its associated endothelial insults.
In contrast to other microangiopathies like aHUS where pathogenic complement gene variants are thought to be causal of disease, we believe that some of the identified genetic variants in HSCT recipients may not have biological importance in the course of normal life, but under the stress of the transplantation process, with significant vascular injury caused by intense radiation, chemotherapy, infections, and GVHD prophylaxis, susceptible individuals will develop TA-TMA via complement-mediated endothelial injury (Fig. 2) [38,39].
Fig. 2.
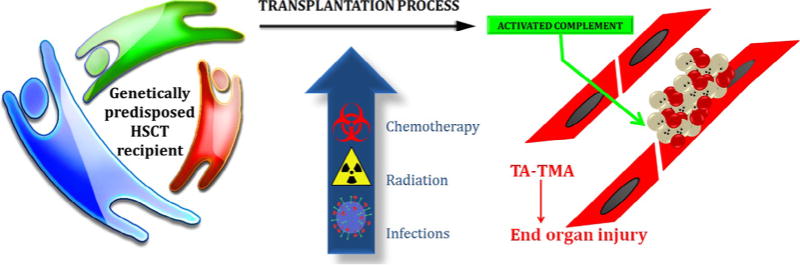
TA-TMA pathogenesis. Transplant recipient’s genotype will determine subject at risk to develop TA-TMA under adequate stressors during HSCT like chemotherapy, radiation, infections, and medications. Complement system will be activated by the stressor in susceptible individuals leading to vascular endothelial injury due to activated terminal complement complex resulting in end organ injury.
Accordingly, activated terminal complement as measured in the blood by elevated sC5b-9 (soluble terminal complement complex) is a poor prognostic marker of the TA-TMA phenotype and indicates the need for complement modifying therapy. About 70% of HSCT patients with a diagnosis of TA-TMA had elevations in sC5b-9 above normal (>244 ng/dL). Elevated blood sC5b-9 levels in conjunction with proteinuria are associated with the most severe TA-TMA phenotype after HSCT resulting in dismal outcomes in untreated patients [3]. Patients with higher sC5b-9 elevations required more intensive complement blocking therapy to achieve clinical response, likely reflecting higher degree of tissue injury by terminal complement [9].
5. Therapy
TA-TMA management can be supportive or disease targeted. In our prospective study, patients with only hematologic signs of microangiopathy without proteinuria or elevated sC5b-9 fully recovered from TA-TMA without treatment, while those with evidence of proteinuria and elevated sC5b-9 levels had a very poor survival of <20% indicating a high-risk group of patients that can benefit from targeted medical interventions using complement modifying drugs [3].
Factors such as viral or bacterial infections that can exacerbate TA-TMA should be adequately treated. Patients with persistent TA-TMA should be screened for invasive fungal disease as one of the causes of microangiopathic vascular changes. Proper hypertension management is essential to allow vascular healing, as severe hypertension by itself can trigger TMA. Selection of antihypertensive therapies should target the underlying pathophysiological mechanisms of the elevated blood pressure [40–43].
Multiple literature sources report calcineurin inhibitors and sirolimus as important causal factors for TA-TMA, although discontinuation of these agents in patients at high-risk for GVHD remains debatable [44]. Often, calcineurin inhibitors are discontinued due to severe acute kidney injury, hypertension, or PRES. In other situations, clinical decisions regarding these agents must balance the risks of developing acute GVHD. Patients with TA-TMA who are at high-risk for GVHD and have well-controlled blood pressures and adequate kidney function can be treated with eculizumab without stopping calcineurin inhibitors. In patients with active GVHD and impaired organ function, less organ toxic options like photopheresis may be considered in conjunction with targeted TA-TMA therapy.
The most promising currently available targeted therapy for TA-TMA is the complement blocking agent eculizumab [8,45–47]. We demonstrated improved outcomes in patients with high-risk TA-TMA treated with eculizumab (n = 18) who historically have dismal outcome as compared with patients with the same high risk TA-TMA features from our prospective observational study who did not receive complement blocking therapy (n = 11) (1 year overall survival 56% vs 9%, p = 0.003) [9]. In Fig. 3 we display updated outcome data in now 30 treated patients with high-risk TA-TMA supporting the initial observations of improved survival in patients receiving eculizumab (1 year overall survival 62% vs 9%, p = 0.0007). All these high risk patients had proteinuria, activated terminal complement, and multi-organ impairment (Table 3). Patients received a median of 14 eculizumab doses (range 2–38). All survivors were able to safely discontinue eculizumab therapy without TA-TMA relapse. It is important to note that HSCT patients have variable eculizumab clearance and require personalized drug dosing based on pharmacokinetics to achieve adequate complement blockade and clinical response. Patients should be carefully selected for therapy, with early treatment offered to those with evidence of activated terminal complement pathway (elevated blood sC5b-9) and proteinuria. Normal blood levels of sC5b-9 should not discourage consideration for complement blocking therapy in patient meeting other diagnostic parameters of TA-TMA, since complement mediated injury can still be present at the vascular endothelial level and functional assays to measure complement deposition on vascular endothelium are currently under development.
Fig. 3.
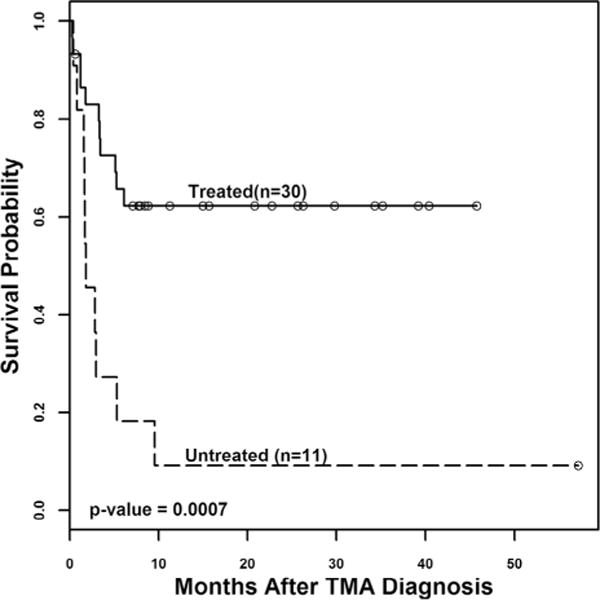
Survival in HSCT recipients with high-risk TA-TMA. Survival curves for patients with high-risk TA-TMA who were treated with the terminal complement blocker eculizumab (“Treated”, n = 30) and historical control subjects from prospective observational TA-TMA study with the same high-risk TMA features who did not receive eculizumab (“Untreated”, n = 11) were calculated using Kaplan–Meier and log rank tests starting at TA-TMA diagnosis to assess statistical significance. Patients with high-risk TA-TMA who received eculizumab therapy had better survival than untreated patients (62% versus 9% at 1 year from TA-TMA diagnosis, p = 0.0007).
Table 3.
Demographics and disease characteristics for HSCT patients with high risk TA-TMA.
| Treated with eculizumab n = 30 |
Not treated n = 11 |
p-Value | |
|---|---|---|---|
| Age | 5.3 (3.6–11.1) | 5.2 (1.8–11.8) | 0.453 |
| Male | 17 (56.7%) | 7 (63.6%) | 0.74 |
| Race | 0.28 | ||
| Caucasian | 22 (73.3%) | 6 (54.5%) | |
| Non-Caucasian | 8 (26.7%) | 5 (45.5%) | |
| Diagnosis | 1 | ||
| Bone marrow failure | 9 (30%) | 3 (27.3%) | |
| Immune deficiency | 13 (43.3%) | 6 (54.5%) | |
| Malignancy | 4 (13.3%) | 1 (9.1%) | |
| Benign hematology | 4 (13.3%) | 1 (9.1%) | |
| Donor type | 0.27 | ||
| Related | 5 (16.7%) | 4 (36.4%) | |
| Unrelated | 20 (66.7%) | 7 (63.6%) | |
| Autologous | 5 (16.7%) | 0 (0%) | |
| Stem cell source | 0.33 | ||
| Bone marrow | 24 (80%) | 7 (63.6%) | |
| PBSCs | 5 (16.7%) | 3 (27.3%) | |
| Cord blood | 1 (3.3%) | 1 (9.1%) | |
| HLA match | 1 | ||
| Matched | 16/25 (64%) | 7 (63.6%) | |
| Mismatched | 9/25 (36%) | 4 (36.4%) | |
| Conditioning regimen | 0.49 | ||
| Myeloablative | 18 (60%) | 5 (45.5%) | |
| Reduced intensity | 12 (40%) | 6 (54.5%) | |
| Calcineurin inhibitor of GVDH prophylaxis | 30 (100%) | 11 (30%) | 1 |
| GVHD (3–4) | 15/25 (60%) | 6 (54.5%) | 1 |
| TMA diagnosis (days after HSCT) | 28 (13.8–48.5) | 26 (17–38.5) | 0.96 |
| sC5b-9 (normal <244 ng/mL) at TA-TMA diagnosis | 334.5 (280.8–420.2) | 458.5 (324.2–708.4) | 0.08 |
| Cystatin C based GFR(mg/mL) at TA-TMA diagnosis | 45.5 (25.5–72) | 45 (34.5–56) | 0.94 |
| Urine random protein/creatinine ratio at TA-TMA diagnosis | 8.3 (4.9–13.5) | 2.6 (1.5–3.3) | <0.0001 |
| Renal replacement therapy | 11 (36.7%) | 4 (36.4%) | 1 |
| Overall survival at 1 year after TA-TMA diagnosis | 62% | 9% | 0.0007 |
The first eculizumab dose should be based on the patient’s weight as currently recommended for aHUS. Subsequent induction doses and intervals are dictated by TA-TMA activity measured by the degree of sC5b-9 in the blood and can be calculated using the patient’s actual weight, the sC5b-9 level, and first eculizumab dose in mg as published by Jodele et al. [9]. If this calculator is not available, patients with sC5b-9 elevations above normal should receive the first 2–3 loading drug doses at least every 72 hours before receiving drug weekly. More frequent than weekly dosing might be required until the sC5b-9 normalizes. Total hemolytic complement activity (CH50) measurement in the blood can serve as an easily available and fast test to aid eculizumab dosing in HSCT patients, also after sC5b-9 normalizes. CH50 should be measured prior to starting eculizumab to obtain baseline reference values and to assure that patient does not have hypocomplementemia with low baseline CH50 in which case CH50 measurement might not be useful for drug dosing monitoring. CH50 should then be measured daily after starting eculizumab therapy until steady therapeutic drug level is established. CH50 should remain suppressed at <10% of the normal value if eculizumab drug concentration in blood is adequate to block terminal complement which correlates with a therapeutic eculizumab drug level >100 μg/mL during induction therapy. A rise in the CH50 above 10% of normal indicates that another eculizumab dose is needed. Eculizumab should be dosed no less than weekly for the first 4–6 weeks of therapy until TA-TMA is controlled and before spacing the dosing schedule and stopping the drug. In HSCT patients, complement blockade is only required as a temporary measure to abort the TA-TMA process, restore the complement pathway back into balance, and is not needed as lifelong therapy.
6. Conclusions and future directions
Significant advances have been made in our understanding of HSCT-associated TMA with the complement system playing a significant role in the pathogenesis and the selection of appropriate therapeutic options. New complement blocking agents are being developed for use in patients with thrombotic microangiopathy after HSCT. While TA-TMA may coincide with other significant transplant complications like GVHD, signs of thrombotic microangiopathy in critically ill HSCT patients should not be ignored or attributed to other clinical diagnoses since targeted TA-TMA therapy can abort the multi-organ injury process and improve long term outcomes. As new diagnostic, prognostic, and therapeutic strategies become available, it will be important to test their efficacy in well-designed, prospective, multi-center clinical trials.
Acknowledgments
None.
Footnotes
Authorship contributions
SJ and BLL wrote the paper and prepared figures. CED, KCM, JE, AN, and GW prepared organ specific sections of the paper. All authors reviewed and revised manuscript critically for important intellectual content and approved the final article version.
References
- 1.Elsallabi O, Bhatt VR, Dhakal P, Foster KW, Tendulkar KK. Hematopoietic stem cell transplant-associated thrombotic microangiopathy. Clin Appl Thromb Hemost. 2016;22:12–20. doi: 10.1177/1076029615598221. [DOI] [PubMed] [Google Scholar]
- 2.Keir L, Coward RJ. Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy. Pediatr Nephrol. 2011;26:523–33. doi: 10.1007/s00467-010-1637-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, et al. Refined diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a prospective study in children and young adults. Blood. 2014;124(4):645–53. doi: 10.1182/blood-2014-03-564997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho VT, Cutler C, Carter S, Martin P, Adams R, Horowitz M, et al. Blood and marrow transplant clinical trials network toxicity committee consensus summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11:571–5. doi: 10.1016/j.bbmt.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Ruutu T, Barosi G, Benjamin RJ, Clark RE, George JN, Gratwohl A, et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica. 2007;92:95–100. doi: 10.3324/haematol.10699. [DOI] [PubMed] [Google Scholar]
- 6.Cho BS, Yahng SA, Lee SE, Eom KS, Kim YJ, Kim HJ, et al. Validation of recently proposed consensus criteria for thrombotic microangiopathy after allogeneic hematopoietic stem-cell transplantation. Transplantation. 2010;90:918–26. doi: 10.1097/TP.0b013e3181f24e8d. [DOI] [PubMed] [Google Scholar]
- 7.Jodele S, Laskin BL, Dandoy CE, Myers KC, El-Bietar J, Davies SM, et al. A new paradigm: diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 2014;29(3):191–204. doi: 10.1016/j.blre.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jodele S, Fukuda T, Vinks A, Mizuno K, Laskin BL, Goebel J, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2013;20(4):518–25. doi: 10.1016/j.bbmt.2013.12.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jodele S, Fukuda T, Mizuno K, Vinks AA, Laskin BL, Goebel J, et al. Variable eculizumab clearance requires pharmacodynamic monitoring to optimize therapy for thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22(2):307–15. doi: 10.1016/j.bbmt.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uderzo C, Bonanomi S, Busca A, Renoldi M, Ferrari P, Iacobelli M, et al. Risk factors and severe outcome in thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Transplantation. 2006;82:638–44. doi: 10.1097/01.tp.0000230373.82376.46. [DOI] [PubMed] [Google Scholar]
- 11.Glezerman IG, Jhaveri KD, Watson TH, Edwards AM, Papadopoulos EB, Young JW, et al. Chronic kidney disease, thrombotic microangiopathy, and hypertension following T cell-depleted hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2010;16:976–84. doi: 10.1016/j.bbmt.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmeister PA, Hingorani SR, Storer BE, Baker KS, Sanders JE. Hypertension in long-term survivors of pediatric hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2010;16:515–24. doi: 10.1016/j.bbmt.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nehus EJ, Laskin BL, Kathman TI, Bissler JJ. Performance of cystatin C-based equations in a pediatric cohort at high risk of kidney injury. Pediatr Nephrol. 2013;28:453–61. doi: 10.1007/s00467-012-2341-3. [DOI] [PubMed] [Google Scholar]
- 14.Laskin BL, Goebel J, Davies SM, Khoury JC, Bleesing JJ, Mehta PA, et al. Early clinical indicators of transplant-associated thrombotic microangiopathy in pediatric neuroblastoma patients undergoing auto-SCT. Bone Marrow Transplant. 2011;46:682–9. doi: 10.1038/bmt.2010.182. [DOI] [PubMed] [Google Scholar]
- 15.Hingorani S, Gooley T, Pao E, Sandmaier B, McDonald G. Urinary cytokines after HCT: evidence for renal inflammation in the pathogenesis of proteinuria and kidney disease. Bone Marrow Transplant. 2014;49:403–9. doi: 10.1038/bmt.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura Y, Yujiri T, Ando T, Hisano S, Tanizawa Y. Nephrotic syndrome associated with thrombotic microangiopathy following allogeneic stem-cell transplantation for myelodysplastic syndrome. Br J Haematol. 2007;136:857–9. doi: 10.1111/j.1365-2141.2007.06515.x. author reply 9–60. [DOI] [PubMed] [Google Scholar]
- 17.Hingorani SR, Seidel K, Lindner A, Aneja T, Schoch G, McDonald G. Albuminuria in hematopoietic cell transplantation patients: prevalence, clinical associations, and impact on survival. Biol Blood Marrow Transplant. 2008;14:1365–72. doi: 10.1016/j.bbmt.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang A, Hingorani S, Kowalewska J, Flowers ME, Aneja T, Smith KD, et al. Spectrum of renal pathology in hematopoietic cell transplantation: a series of 20 patients and review of the literature. Clin J Am Soc Nephrol. 2007;2:1014–23. doi: 10.2215/CJN.01700407. [DOI] [PubMed] [Google Scholar]
- 19.Siami K, Kojouri K, Swisher KK, Selby GB, George JN, Laszik ZG. Thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation: an autopsy study. Transplantation. 2008;85:22–8. doi: 10.1097/01.tp.0000297998.33418.7e. [DOI] [PubMed] [Google Scholar]
- 20.Narimatsu H, Kami M, Hara S, Matsumura T, Miyakoshi S, Kusumi E, et al. Intestinal thrombotic microangiopathy following reduced-intensity umbilical cord blood transplantation. Bone Marrow Transplant. 2005;36:517–23. doi: 10.1038/sj.bmt.1705099. [DOI] [PubMed] [Google Scholar]
- 21.Nishida T, Hamaguchi M, Hirabayashi N, Haneda M, Terakura S, Atsuta Y, et al. Intestinal thrombotic microangiopathy after allogeneic bone marrow transplantation: a clinical imitator of acute enteric graft-versus-host disease. Bone Marrow Transplant. 2004;33:1143–50. doi: 10.1038/sj.bmt.1704512. [DOI] [PubMed] [Google Scholar]
- 22.Yamada-Fujiwara M, Miyamura K, Fujiwara T, Tohmiya Y, Endo K, Onishi Y, et al. Diagnosis of intestinal graft-versus-host disease and thrombotic microangiopathy after allogeneic stem cell transplantation. Tohoku J Exp Med. 2012;227:31–7. doi: 10.1620/tjem.227.31. [DOI] [PubMed] [Google Scholar]
- 23.Hewamana S, Austen B, Murray J, Johnson S, Wilson K. Intestinal perforation secondary to haematopoietic stem cell transplant associated thrombotic microangiopathy. Eur J Haematol. 2009;83:277. doi: 10.1111/j.1600-0609.2009.01267.x. [DOI] [PubMed] [Google Scholar]
- 24.El-Bietar J, Warren M, Dandoy C, Myers KC, Lane A, Wallace G, et al. Histologic features of intestinal thrombotic microangiopathy in pediatric and young adult patients after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2015;21:1994–2001. doi: 10.1016/j.bbmt.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dietrich S, Falk CS, Benner A, Karamustafa S, Hahn E, Andrulis M, et al. Endothelial vulnerability and endothelial damage are associated with risk of graft-versus-host disease and response to steroid treatment. Biol Blood Marrow Transplant. 2013;19:22–7. doi: 10.1016/j.bbmt.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 26.Andrulis M, Dietrich S, Longerich T, Koschny R, Burian M, Schmitt-Graf A, et al. Loss of endothelial thrombomodulin predicts response to steroid therapy and survival in acute intestinal graft-versus-host disease. Haematologica. 2012;97:1674–7. doi: 10.3324/haematol.2011.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houtchens J, Martin D, Klinger JR. Diagnosis and management of pulmonary arterial hypertension. Pulm Med. 2011;2011:845864. doi: 10.1155/2011/845864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkowska-Ptasinska A, Sulikowska-Rowinska A, Pazik J, Komuda-Leszek E, Durlik M. Thrombotic nephropathy and pulmonary hypertension following autologous bone marrow transplantation in a patient with acute lymphoblastic leukemia: case report. Transplant Proc. 2006;38:295–6. doi: 10.1016/j.transproceed.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 29.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–13. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dandoy C, Davies SM, Hirsch R, Chima RS, Paff Z, Cash M, et al. Abnormal echocardiography seven days after stem cell transplant may be an early indicator of thrombotic microangiopathy. Biol Blood Marrow Transplant. 2015;21(1):113–8. doi: 10.1016/j.bbmt.2014.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jodele S, Hirsch R, Laskin B, Davies S, Witte D, Chima R. Pulmonary arterial hypertension in pediatric patients with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2013;19:202–7. doi: 10.1016/j.bbmt.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 32.Dandoy CE, Hirsch R, Chima R, Davies SM, Jodele S. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19:1546–56. doi: 10.1016/j.bbmt.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Lerner D, Dandoy C, Hirsch R, Laskin B, Davies SM, Jodele S. Pericardial effusion in pediatric SCT recipients with thrombotic microangiopathy. Bone Marrow Transplant. 2014;49(6):862–3. doi: 10.1038/bmt.2014.40. [DOI] [PubMed] [Google Scholar]
- 34.Yanagisawa R, Ishii E, Motoki N, Yamazaki S, Morita D, Sakashita K, et al. Pretransplant-corrected QT dispersion as a predictor of pericardial effusion after pediatric hematopoietic stem cell transplantation. Transpl Int. 2015;28:565–74. doi: 10.1111/tri.12532. [DOI] [PubMed] [Google Scholar]
- 35.Cooke KR, Jannin A, Ho V. The contribution of endothelial activation and injury to end-organ toxicity following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:23–32. doi: 10.1016/j.bbmt.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Carmona A, Diaz-Ricart M, Palomo M, Molina P, Pino M, Rovira M, et al. Distinct deleterious effects of cyclosporine and tacrolimus and combined tacrolimus-sirolimus on endothelial cells: protective effect of defibrotide. Biol Blood Marrow Transplant. 2013;19:1439–45. doi: 10.1016/j.bbmt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, Myers KC, et al. The genetic fingerprint of susceptibility for transplant associated thrombotic microangiopathy. Blood. 2016;127(8):989–96. doi: 10.1182/blood-2015-08-663435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of the alternative pathway and terminal complement activity at presentation confirms the clinical diagnosis of aHUS and differentiates aHUS from TTP. Blood. 2014;123(24):3733–8. doi: 10.1182/blood-2013-12-547067. [DOI] [PubMed] [Google Scholar]
- 39.Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25:55–64. doi: 10.1681/ASN.2013050453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114:555–76. [PubMed] [Google Scholar]
- 41.Moulder JE, Cohen EP, Fish BL. Captopril and losartan for mitigation of renal injury caused by single-dose total-body irradiation. Radiat Res. 2011;175:29–36. doi: 10.1667/RR2400.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hingorani S. Chronic kidney disease after liver, cardiac, lung, heart-lung, and hematopoietic stem cell transplant. Pediatr Nephrol. 2008;23:879–88. doi: 10.1007/s00467-008-0785-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wuhl E, Trivelli A, Picca S, Litwin M, Peco-Antic A, Zurowska A, et al. Strict blood-pressure control and progression of renal failure in children. N Engl J Med. 2009;361:1639–50. doi: 10.1056/NEJMoa0902066. [DOI] [PubMed] [Google Scholar]
- 44.Shayani S, Palmer J, Stiller T, Liu X, Thomas SH, Khuu T, et al. Thrombotic microangiopathy associated with sirolimus level after allogeneic hematopoietic cell transplantation with tacrolimus/sirolimus-based graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant. 2013;19:298–304. doi: 10.1016/j.bbmt.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhakal P, Giri S, Pathak R, Bhatt VR. Eculizumab in transplant-associated thrombotic microangiopathy. Clin Appl Thromb Hemost. 2015 doi: 10.1177/1076029615599439. [DOI] [PubMed] [Google Scholar]
- 46.de Fontbrune FS, Galambrun C, Sirvent A, Huynh A, Faguer S, Nguyen S, et al. Use of eculizumab in patients with allogeneic stem cell transplant-associated thrombotic microangiopathy: a study from the SFGM-TC. Transplantation. 2015;99(9):1953–9. doi: 10.1097/TP.0000000000000601. [DOI] [PubMed] [Google Scholar]
- 47.Kim SS, Patel M, Yum K, Keyzner A. Hematopoietic stem cell transplant-associated thrombotic microangiopathy: review of pharmacologic treatment options. Transfusion. 2015;55:452–8. doi: 10.1111/trf.12859. [DOI] [PubMed] [Google Scholar]