

Abstract
The first example of palladium(II)-catalyzed β-C(sp3)–H iodination of a wide range of ketones using a commercially available aminooxyacetic acid auxiliary has been achieved. This L, X-type directing group overcomes the limitations of the transient directing group approach for C(sp3)–H functionalization of ketones. Practical advantages of this method include simple installation of the auxiliary without chromatography, exceptional tolerance of α-functional groups, as well as alkenes and alkynes, and rapid access to diverse sterically hindered quaternary centers.
Graphical abstract
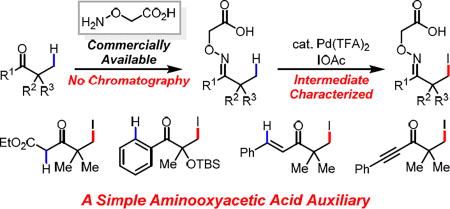
Ketones are ubiquitous in organic chemistry, serving as invaluable bulk chemicals, synthetic building blocks, and natural products. While the reactivity of the α-acidic C–H bonds has been explored extensively in asymmetric enolate chemistry,1 the direct functionalization of inert β-C(sp3)–H bonds could further expand the synthetic utility of ketones by providing new synthetic disconnections. Although stoichiometric oxime-directed β-C(sp3)–H palladation and subsequent acetoxylation and iodination have been reported since pioneering works by Shaw, Sutherland, Baldwin and others,2 the development of corresponding catalytic reactions has remained limited in scope and efficiency despite recent advances.3–5 Recently, the β-C(sp3)–H arylation of ketones has also been achieved using a catalytic transient amino acid directing group.6 However, this strategy is not compatible with many synthetically desirable transformations as the free transient directing group often interferes with the coupling reagents and catalysts. Compared to the C(sp3)–H functionalization of carboxylic acids and amines, the β-C(sp3)–H functionalization of ketones using either covalent or transient directing groups has been highly limited in terms of scope of substrates as well as transformations.7 Herein, we report a practical directing group that enables the β-iodination of ketones. The combination of the imino-carboxylic acid chelation established in our previous transient directing group in concert with the stability of an oxime linkage successfully overcomes the limitations of previous directing groups, presumably generating a highly active Pd precursor. Consequently, The substrate scope herein reported is significantly broader than previously reported C(sp3)–H functionalizations of ketones.
The use of amino acid transient directing groups for the β-C(sp3)–H functionalization of ketones is appealing from the viewpoint of step-economy. However, the free amino acid used is often incompatible with a range of oxidants or coupling reagents preventing the development of a diverse range of transformations. Since the bidentate imino-carboxyl chelation has been shown to be effective in directing C(sp3)–H activation,6 we proposed to replace the imine by a more stable oxime8 motif to render the linkage irreversible (Scheme 1). Importantly, L, X-type (i.e. neutral, anionic) directing groups have a fundamental advantage over the L, L (i.e. neutral, neutral) counterparts as shown in Scheme 2. Using L, L-type directing groups, a C–H bond must displace an acetate to form a thermodynamically unfavorable cationic Pd(II) species in order to achieve C–H insertion. (Scheme 2. Eq 2). Furthermore, the weakly coordinating nature of the carboxylic acid is also beneficial for the reactivity of the C–H insertion intermediate compared to other highly stable bidentate complexes.
Scheme 1.

Transient and Covalent Directing Groups based on Amino acids
Scheme 2.

Rational Design of a Powerful Directing Group
To test this design principle, we chose β-C(sp3)–H iodination as the model reaction for two reasons: First, C(sp3)–H iodination of ketones remains an unsolved problem except for a single example of stoichiometric iodination reaction of oxime reported by Sutherland in 1984.2b Second, β-iodoketones are extremely versatile synthons, yet the synthesis of β-iodoketones is a well-known challenge, especially for sterically hindered α-quaternary ketones.9 Therefore, the model substrate 1a-1 was readily prepared by the condensation between pinacolone and commercially available aminooxyacetic acid under mild conditions in high yield without chromatography (See SI for details). Guided by our previous study of oxazoline- and acidic amide-directed β-C(sp3)–H iodination of carboxylic acid substrates,5a,10 we found that treating model substrate 1a-1 with 1 equiv I2, PhI(OAc)2 and 10 mol% Pd(OAc)2 in 1,4-dioxane gave the desired iodination product in 70% yield. The use of other solvents gave significantly lower yields. Replacing Pd(OAc)2 with Pd(TFA)2 improved the yield to 88% (Table 1). Importantly, reducing catalyst loading to 5 mol% did not result in a noticeable decrease in yield, although slightly lower yields were obtained with a few demanding substrates containing coordinating functional groups. The observed high reactivity prompted us to investigate the coordination mode of the directing group and the impact of the structures on the reactivity. The corresponding L, L-type directing group bearing the methyl ester (DG2) gave poor yield which confirms the superiority of the L, X-type auxiliary (Scheme 2. Eq 1–2). Enhancing the bidentate coordination (due to the Thorp-Ingold effect) led to drastic increase in di- and tri-iodination products (2a–3), which is likely due to slow dissociation from Pd. Considering the well-established efficiency of the acidic amide directing group,11 we also converted the carboxylic acid into amides (DG4 and DG5). Although DG4 has comparable reactivity, the stronger binding affinity led to predominant formation of the tri-iodinated products. As expected, the less acidic amide DG5 is not as reactive as indicated by fewer turnover numbers (TONs), presumably due to its inability to adopt L, X-type coordination.
Table 1.

|
Conditions: Substrate (0.1 mmol, 1.0 equiv), Pd(TFA)2 (10 mol%), I2 (1.0 equiv), PhI(OAc)2 (1.0 equiv), 1,4-dioxane (1.25 mL), 40 °C, under air, 20 h. Ar1 = 4-CF3(C6F4), Ar2 = 3,5-CH3(C6H3).
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
The numbers in parenthesis indicate the ratio of mono:di or mono:di:tri (if applicable).
Pd(TFA)2 (5 mol%).
With the optimal directing group and reaction conditions establised, we evaluated the scope of C(sp3)–H iodination. It is worth highlighting the practical advantage of this directing group: preparation of a wide range of substrates was performed using the commercially available aminooxyacetic acid without chromatography. As shown in Table 2, a variety of alkyl substituted ketones were β-iodinated in good-to-excellent yields (2a–d). Note that the quaternary centers in products 2a–c and 2e–h are inaccessible via classic enolate alkylation methods as alkylation of the α-methyl group is kinetically favored;1 all of these products were formed in good-to-excellent yields. The bicyclic natural product Fenchone was iodinated with excellent mono-selectivity despite the presence of three α-methyl groups (2d). Notably, aromatic moieties trans to the carboxyl directing moiety were compatible with iodination conditions and no C(sp2)–H iodination was detected (2e–g, 2o). Ketone derived from Gemfibrozil was iodinated at β-methyl group and aromatic ring (2h). Various aromatic rings cis to the carboxyl directing moiety remains intact during the iodination reaction (2i–k, 2p–r). Remarkably, no α-iodination was observed in the presence of acidic α-hydrogen,12 only β-C(sp3)–H iodinated products were obtained (2k–m). Again, the quaternary centers in these products cannot be formed using enolate alkylation, which prefers the even more acidic and less hindered α-positions.1 Although halogens at β- or more distal positions should be tolerated as we can obtain di-iodination products from C(sp3)–H iodination of mono-iodination products, the halogenated substrates are not compatible with oxime condensation step due to the nucleophilicity of alkoxyamine. Finally, a variety of functional groups at the α-position, including nitriles, esters, TBS-protected hydroxyl, alkenyl, and alkynyl groups, were tolerated (2l–r), exhibiting significantly broader substrate scope than previously reported oxime-directed C(sp3)–H functionalizations. The selective di-iodination can be achieved by either extending the reaction time or adding more iodinating reagent. The auxiliary was removed by treating the β-iodinated product with 4 M HCl in 1,4-dioxane under 100 °C for 2 h, providing versatile β-iodinated ketone (see SI).
Table 2.

|
Conditions: Substrate (0.1 mmol, 1.0 equiv), Pd(TFA)2 (10 mol%), I2 (1.0 equiv), PhI(OAc)2 (1.0 equiv), 1,4-dioxane (1.25 mL), 40 °C, under air, 20 h.
Isolated yield.
The numbers in parenthesis indicate the ratio of mono:di.
After the reaction, the carboxylic acid is converted to corresponding methyl ester (T1) and 3,5-dimethylamide (T2) for convenient chromatographic purification. See SI for work up procedures.
The reaction time is 3 h.
The reaction temperature is 80 °C.
1.0 mmol scale.
This new auxiliary offers a number of practical advantages over previously developed imine directing groups.2–5 Most importantly, imino-carboxyl directing group displayed superior reactivity over O-methyl oxime directing group with various substrates containing phenyl group, strained rings, alkenes, and other functional groups (Scheme 3).
Scheme 3.

Incompatible Ketone Substrates with O-Methyl Oxime Directing Group
Previous attempts to isolate the C–H insertion intermediate with our previous transient amino acid directing groups were not successful due to the instability of the imine linkage. With the more stable oxime linkage, we were pleased to observe the formation of the C–H insertion intermediate by stirring the substrate 1a with 1.2 equivalents of Pd(OAc)2 in HFIP at 80 °C. We were able to isolate a stable complex by trapping with PPh3 in 81% yield (Scheme 4). The structure was confirmed by X-ray crystallography. This intermediate provides the first direct evidence for the L, X-coordination mode of the oxime-carboxylic acid directing group, confirming the unique role of the carboxyl group as a directing moiety. Both inter- and intramolecular kinetic isotope effects (kH/kD = 2.7 and kH/kD = 5.0 respectively) indicate that the C–H activation step is the rate-limiting step (see SI).
Scheme 4.

Characterization of the Palladacycle
In summary, C(sp3)–H iodination of ketones was developed using a commercially available aminooxyacetic acid auxiliary. This reaction features facile installation and removal of the auxiliary. The substrate scope is significantly broader than the previously reported C–H functionalizations of ketones using various approaches. The characterization of the C–H insertion intermediate also provides the first direct evidence for the L, X-type coordination mode of the oxime-carboxylic acid directing groups.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support. R.-Y.Z. was supported by the Boehringer Ingelheim Fellowship. We thank T. G. Saint-Denis, K. Wu, K. Hong, H. Park, and Y. Wu for editorial assistance.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Mukaiyama T. Angew. Chem. Int. Ed. 2004;43:5590. doi: 10.1002/anie.200300641. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Vogel E, Nelson JV. J. Am. Chem. Soc. 1979;101:6120. [Google Scholar]; (c) Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496. [Google Scholar]; (d) Cowden CJ, Paterson I. Org. React. 1997;51:1. [Google Scholar]; (e) Cano R, Zakarian A, McGlacken GP. Angew. Chem. Int. Ed. 2017;56:2. doi: 10.1002/anie.201703079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For selected examples of stoichiometric palladation reactions: Constable AG, McDonald WS, Sawkins LC, Shaw BL. J. Chem. Soc. Chem, Commun. 1978:1061.Carr K, Sutherland JK. J. Chem. Soc. Chem, Commun. 1984:1227.Baldwin JE, Nájera C, Yus M. J. Chem. Soc. Chem, Commun. 1985:126.Baldwin JE, Jones RH, Nájera C, Yus M. Tetrahedron. 1985;41:699.
- 3.For early examples of Pd-catalyzed C(sp2)–H Halogenation: Fahey DR. J. Organomet. Chem. 1971;27:283.Andrienko OS, Goncharov VS, Raida VS. Russ. J. Org. Chem. 1996;32:89.
- 4.For advances of catalytic C(sp3)–H activation using oxime directing groups: Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542. doi: 10.1021/ja046831c.Thu H-Y, Yu W-Y, Che C-M. J. Am. Chem. Soc. 2006;128:9048. doi: 10.1021/ja062856v.Kang T, Kim Y, Lee D, Wang Z, Chang S. J. Am. Chem. Soc. 2014;136:4141. doi: 10.1021/ja501014b.Gao P, Guo W, Xue J, Zhao Y, Yuan Y, Xia Y, Shi Z. J. Am. Chem. Soc. 2015;137:12231. doi: 10.1021/jacs.5b06758.
- 5.For C(sp3)–H halogenation of carboxylic acid substrates: Giri R, Chen X, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884.Giri R, Wasa M, Brazzano SP, Yu J-Q. Org. Lett. 2006;8:5685. doi: 10.1021/ol0618858. (c) Two substrates have been reported. Wasa M, Yu J-Q. J. Am. Chem. Soc. 2008;130:14058. doi: 10.1021/ja807129e. (d) One substrate has been reported. Stowers KJ, Kubota A, Sanford MS. Chem. Sci. 2012;3:3192. doi: 10.1039/C2SC20800H.Rit RK, Yadav M, Ghosh RK, Shankar M, Sahoo AK. Org. Lett. 2014;16:5258. doi: 10.1021/ol502337b.Yang X, Sun Y, Sun T-Y, Rao Y. Chem. Commun. 2016;52:6423. doi: 10.1039/c6cc00234j.
- 6.(a) Zhang F-L, Hong K, Li T-J, Park H, Yu J-Q. Science. 2016;351:252. doi: 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang K, Li Q, Liu Y, Li G, Ge H. J. Am. Chem. Soc. 2016;138:12775. doi: 10.1021/jacs.6b08478. [DOI] [PubMed] [Google Scholar]
- 7.For selected reviews on Pd-catalyzed C(sp3)–H functionalizations: Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058.Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e.He J, Wasa M, Chan KSL, Shao Q, Yu J-Q. Chem. Rev. 2017;117:8754. doi: 10.1021/acs.chemrev.6b00622.
- 8.Kalia J, Raines RT. Angew. Chem., Int. Ed. 2008;47:7523. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roman BI, Kimpe ND, Stevens CV. Chem. Rev. 2010;110:5914. doi: 10.1021/cr900409h. [DOI] [PubMed] [Google Scholar]
- 10.For acidic amide-directed C(sp3)–H iodination: Zhu R-Y, Saint-Denis TG, Shao Y, He J, Sieber JD, Senanayake CH, Yu J-Q. J. Am. Chem. Soc. 2017;139:5724. doi: 10.1021/jacs.7b02196.
- 11.For selected examples: Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:3680. doi: 10.1021/ja1010866.Wasa M, Chan KSL, Zhang X-G, He J, Miura M, Yu J-Q. J. Am. Chem. Soc. 2012;134:18570. doi: 10.1021/ja309325e.Li S, Chen G, Feng C-G, Gong W, Yu J-Q. J. Am. Chem. Soc. 2014;136:5267. doi: 10.1021/ja501689j.Zhu R-Y, He J, Wang X-C, Yu J-Q. J. Am. Chem. Soc. 2014;136:13194. doi: 10.1021/ja508165a.Zhu R-Y, Tanaka K, Li G-C, He J, Fu H-Y, Li S-H, Yu J-Q. J. Am. Chem. Soc. 2015;137:7067. doi: 10.1021/jacs.5b04088.Wu Q-F, Shen P-X, He J, Wang X-B, Zhang F, Shao Q, Zhu R-Y, Mapelli C, Qiao JX, Poss MA, Yu J-Q. Science. 2017;355:499. doi: 10.1126/science.aal5175.
- 12.For selected exmaples of α-iodination for ketones: Bekaert A, Barberan O, Gervais M, Brion J-D. Tetrahedron Lett. 2000;41:2903.Jereb M, Stavber S, Zupan M. Synthesis. 2003:853.Wang Z, Yin G, Qin J, Gao M, Cao L, Wu A. Synthesis. 2008:3565.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.