

Abstract
Acetyl-CoA synthase (ACS) is a key enzyme in the Wood–Ljungdahl pathway of anaerobic CO2 fixation, which has long been proposed to operate by a novel mechanism involving a series of protein-bound organometallic (Ni–CO, methyl–Ni, and acetyl–Ni) intermediates. Here we report the first direct structural evidence of the proposed metal–carbon bond. We describe the preparation of the highly active metal-replete enzyme and near-quantitative generation of the kinetically competent carbonylated intermediate. This advance has allowed a combination of Ni and Fe K-edge X-ray absorption spectroscopy and extended X-ray absorption fine structure experiments along with density functional theory calculations. The data reveal that CO binds to the proximal Ni of the six-metal metallocenter at the active site and undergoes dramatic structural and electronic perturbation in forming this organometallic Ni–CO intermediate. This direct identification of a Ni–carbon bond in the catalytically competent CO-bound form of the A cluster of ACS provides definitive experimental structural evidence supporting the proposed organometallic mechanism of anaerobic acetyl-CoA synthesis.
Graphical Abstract
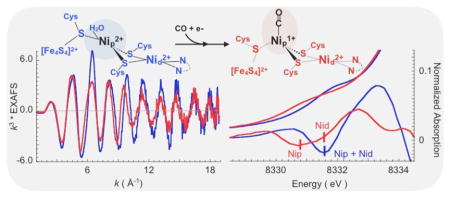
Thirty years ago, it was discovered that the nickel metalloenzyme CO dehydrogenase (CODH), involved in the conversion of CO2 to CO, also catalyzes an exchange reaction between CO and the carbonyl group of acetyl-CoA.1 The fact that CODH catalyzes this CO/acetyl-CoA exchange reaction demonstrated that this enzyme is solely responsible for binding the three components of acetyl-CoA (the carbonyl group, the methyl group, and CoA) and catalyzing C–C and C–S bond formation. Thus, the protein was renamed acetyl-CoA synthase, and it was proposed that the ACS-catalyzed reaction occurs through a sequence of enzyme-bound organometallic reactions.1 Later, the ACS protein was demonstrated to exist within a tightly associated two-protein complex with separate CODH and ACS active sites,2,3 so it became known as CODH/ACS.
Structural and spectroscopic studies of the two components of the CODH/ACS complex have demonstrated that both proteins contain unique Ni-based active sites, working harmoniously to reduce CO2 to CO (CODH) and functionalize CO to acetyl-CoA (ACS).4 X-ray crystallography revealed the active site of ACS, the so-called A cluster, to be a binuclear Ni center bridged through cysteine sulfur to a 4Fe–4S subcluster [Figure 1, Protein Data Bank (PDB) entry 1OAO].5–7 In this unusual metallocenter, the Ni distal to the FeS subcluster (Nid) is coordinated to a tetradentate Cys-Gly-Cys sequence in a square-planar N2S2 geometry, while the Ni proximal to the subcluster (Nip), which is coordinated by three cysteine thiolates, can adopt a square-planar or tetrahedral geometry, a structural change that may be coupled to a significant conformational change in the ACS polypeptide.6,8
Figure 1.

ACS active site determined by crystallography (PDB entry 1OAO, Moorella thermoacetica). “Closed” conformation (the access path from the CODH Ni–Fe center to the ACS active site is closed) showing Zn occupancy at the tetrahedral Nip site (left). “Open” conformation with Ni at the square-planar Nip site (right). The S–S–S–Ni dihedral angle demonstrates the change in geometry between the sites. A large difference in the Ni–metal distances is observed.
The proposed ACS reaction sequence (Figure 2, top panel), includes reductive activation of the active site Nip(II) center (species 1) to transiently generate Nip(I) (species 2), a reaction that is kinetically coupled to binding of CO to form the Nip(I)–CO intermediate (species 3), which is the intermediate characterized here. Then, addition of a methyl group generates methyl–Nip–CO species 4 that undergoes rapid reduction to form the more stable Nip(II) species 5. C–C bond formation then ensues to generate acetyl–Ni (species 6) followed by CoA-dependent thiolysis to generate acetyl-CoA and regenerate active species 2 for the next round of catalysis. This is a simplified diagram because pulse–chase kinetic studies demonstrate that ACS follows a random sequential mechanism (Figure 2, bottom panel) in which both methylated and carbonylated forms of ACS can serve as the first intermediate during the catalytic cycle.9 Because the Ni(I)–CO species is the primary subject of this paper, we illustrate only the upper “CO-first” branch (top panel).
Figure 2.

Organometallic mechanism of acetyl-CoA synthesis. All steps are reversible. See the text for details.
Two competing ACS mechanisms that differ in the oxidation state of the Nip have been proposed. The “paramagnetic mechanism” proposes that Nip(I) and Ni(I)–CO are central intermediates (Figure 2, top panel),10 while the “diamagnetic mechanism” (not shown) proposes a Ni(0) active catalyst with Ni(0)–CO and methyl–Ni(II) [without the paramagnetic methyl–Ni(III)] intermediates.11
The so-called “paramagnetic mechanism” proposes paramagnetic species as key intermediates, e.g., Ni(I) and Ni(I)–CO. The paramagnetic mechanism is supported by significant kinetic and spectroscopic evidence. For example, when ACS is exposed to CO under reducing conditions, the EPR active NiFeC species forms at catalytically relevant rates, and upon reaction with the methylated CFeSP, this species also decays at kinetically competent rates.12–14 Furthermore, stopped-flow infrared studies coupled with freeze quench EPR experiments show that reaction of ACS with CO generates the Ni(I)–CO intermediate and that this is the only carbonylated species that develops at catalytically competent rates.14 In addition, evidence that supports the proposed internal electron transfer step shown in Figure 2 has been found.15 Besides the NiFeC species, the Ni(I) intermediate to which CO binds has been trapped and identified by photolysis followed by EPR and infrared spectroscopic studies.16 This Ni(I) species exhibits an EPR spectrum with g values of 2.56, 2.10, and 2.01 and has an extremely low (1 kJ/mol) barrier for recombination with CO.16 Thus, when carbonylation is the first step in the ACS mechanism, the involvement of Ni(I) and the NiFeC species as catalytic intermediates is well-substantiated. On the basis of the random nature of the methylation and carbonylation steps in the ACS mechanism,9 the same Ni(I) catalyst that has been shown to form the Ni(I)–CO intermediate16 would be expected to be involved in methylation.
In the diamagnetic mechanism,17 the NiFeC species is viewed as an inhibited state and a Ni(0) state is proposed to be catalytically relevant.11,18 Gencic and Grahame suggested that the active intermediate is a spin-coupled [4Fe-4S]+ cluster linked to a Ni(I) site,19 evidence for which has been provided by Mossbauer spectroscopy.20 Regardless, a Ni(0) state in ACS has never been detected, and the spin-coupled species has not been observed to have any catalytic relevance.
Because CO and the methyl group bind in a random order to ACS,21 either the Ni–CO intermediate or the methyl–Ni intermediate can serve as the first intermediate in the ACS-catalyzed reaction, allowing one to trap and characterize either the metal–CO or the methyl–metal adduct. The methyl group is transferred to ACS from another organometallic intermediate, methyl–cob(III)alamin, bound to a corrinoid iron–sulfur protein (CFeSP).22,23
In 1985, when this organometallic hypothesis was proposed,1 it was not known that ACS activity (or CO binding) was associated with any metal other than Ni, an association based on the observation of an EPR signal (resulting from the species when the enzyme was incubated with CO) that exhibited hyperfine broadening from isotopic substitution with 61Ni and 13CO.24,25 The ACS active site was later shown to contain an iron–sulfur cluster on which significant unpaired spin density resides; thus, the Ni(I) designation assigned to species 2 in the NiFeC species is only a simplification, and formally, a fractional spin should be assigned to the Ni in that state.26 These results begged the question of whether CO binds to the Ni or Fe subcomponents of the ACS A cluster active site. That question has remained unresolved because the answer requires a method, such as X-ray crystallography, resonance Raman, or X-ray absorption spectroscopy (XAS), that can probe and identify the metal ion to which CO binds. Unfortunately, as described below, various issues, including the inability to obtain sufficiently large amounts of the active NiFeC species, prevented that definitive characterization. Thus, many papers have been published over the past 30 years in which the original organonickel hypothesis has been reproduced in different semblances, yet direct evidence of the nature of the metal–carbon bond has been lacking.
Besides the unusual nature of this bio-organometallic reaction sequence, CODH/ACS has attracted attention because it plays the key role(s) in the Wood–Ljungdahl pathway of autotrophic CO2 and CO fixation, a metabolic cycle that is important in the global carbon cycle as a predominant CO2 sink in anaerobic environments27 that is being exploited for the biotechnological conversion of waste and greenhouse gases into chemicals.28 It is the basis of energy metabolism in acetogenic bacteria, for autotrophic CO2 fixation in acetogenic bacteria and methanogens, and for acetate catabolism by sulfate-reducing bacteria and aceticlastic methanogens.27,29 This pathway has also been proposed to have fueled the primordial biochemical reactions required for the origin of life and to have developed from a prebiotic reaction sequence using H2 as the initial electron donor and CO2 as the initial electron acceptor.30 Thus, it is crucial to understand the mechanism by which this enzyme generates acetyl-CoA and in particular to define the nature of the proposed organometallic intermediates.
Spectroscopic and kinetic studies have focused on identifying and characterizing the intermediates in the ACS-catalyzed reaction. For example, CO binds in an end-on fashion to one of the metal sites in the A cluster forming an organometallic EPR detectable metal–carbonyl species in which the unpaired electron is delocalized over the Fe and Ni centers and the CO subcomponent (labeled the NiFeC species).13,14,26,31–33 As described below, the rate constant for formation of the NiFeC species and its electrochemical properties are consistent with this species being a catalytic intermediate in acetyl-CoA synthesis;13,14,21,32,34,35 however, although it has been generally accepted that organometallic species involving CO, methyl, and acetyl groups accumulate at Nip, several problems have precluded direct identification of any of the metal–carbon bonds proposed to form during the ACS reaction cycle. CODH/ACS, the only form in which ACS is naturally found, contains a low proportion of the binickel center at the active site, with the Nip subcomponent depleted in Ni,36 and often substituted with other metals (Cu and Zn).6,37 Furthermore, the presence of two unique Ni–Fe clusters and additional Fe–S clusters in CODH/ACS makes spectroscopic studies, aimed at understanding binding of CO to the ACS subcomponent, extremely challenging. Thus, it has not been possible to prepare high levels of the fully reconstituted A cluster or to produce sufficient amounts of the carbonylated state, hamstringing any structural characterization of these intermediates. These problems have left the proposal of the organometallic intermediates without a firm structural basis and with a limited understanding of their electronic structure.
We have utilized a previously developed expression system for ACS alone in Escherichia coli35 and in some of the preparations obtained with a nearly fully loaded A cluster, with high levels of the NiFeC species, according to the nearly stoichiometric development of the EPR signal (termed the “NiFeC signal”) associated with the active state of ACS.12–14,21 We achieved spin percentages between 30 and 65% and utilized the protein preparation with a 65% spin percentage to perform high-resolution Fe and Ni K-edge XAS and EXAFS measurements on the as-isolated protein (AOX), its dithionite-treated form (traditionally and operationally called ARED), and the CO-bound form (with a strong NiFeC signal, ANiFeC), formed by treating ACS with dithionite and CO. These geometric and electronic structure determination techniques were combined with DFT calculations to unambiguously identify and characterize the organometallic Nip–CO bond in ANiFeC. Because XAS is an element-specific technique with exceptional precision (±0.02 Å) in measuring metal–ligand bond distances, the Fe and Ni sites can be individually probed at high resolution. Furthermore, any change in the Fe or Ni K-edge spectrum is due to a direct perturbation of the geometric and electronic structures at the respective site(s) in ACS. A combination of spectroscopic and DFT computational studies on ACS and model compounds38–41 have shown that the bisamidate coordination sphere around Nid stabilizes this site as a diamagnetic square-planar Ni(II) complex under all catalytic conditions;4 thus, the changes in the Ni K-edge XAS data overwhelmingly represent changes at the Nip site.
MATERIALS AND METHODS
Protein Expression and Purification, Metal Analysis, and EPR Spectroscopy
The α-subunit was prepared, purified, and Ni-reconstituted under strictly anaerobic conditions at <1 ppm O2 in a manner similar to that described previously but with several modifications.21 The pETACSMTHT plasmid was modified to have a TEV protease cleavage site just before the C-terminal His tag. Upon isolation of the pure protein, the His tag was removed using 1 mM sodium citrate instead of the commonly used EDTA in the protease reaction mixture to prevent any damage to the precious A cluster. Metal reconstitution was achieved by incubating the enzyme with a 6-fold excess of nickel(II) chloride at 45 °C for 3 days. Excess nickel was removed from the solution by passing the protein (<1 mL) twice through an anaerobic 8.3 mL PD-10 desalting column. The volume of protein applied to the column was limited to 1 mL to achieve adequate separation. The metal content of the samples was analyzed by ICP-OES at the Center for Applied Isotope Studies at the University of Georgia (Athens, GA). Concentrations were determined using the Rose Bengal protein assay using a standard curve prepared against dry ACS. Enzyme activity was determined by performing the acetyl-CoA/CO exchange reaction at 55 °C in 1 mL of 270 mM MES buffer (pH 6.1) containing 2 mM Ti(III) citrate, 0.2 mM methyl viologen, 0.2 mM acetyl-CoA, and 0.03–0.04 μCi of [1-14C]acetyl-CoA under an atmosphere of CO, and the data were analyzed as described previously.42
EPR samples were prepared in a manner similar to that of the CO-treated XAS samples as described below and transferred to EPR tubes. EPR spectra were recorded at 110 K under nonsaturating conditions. Spin quantification was performed by comparison with a 1 mM copper(II) standard.
XAS Sample Preparation
The reconstituted enzyme was buffer exchanged into 50 mM potassium phosphate (pH 7.5) with 100 μM DTT. As a glassing agent and to minimize ice crystal formation, glycerol was added to a final concentration of 30%. The as-isolated sample was produced directly from this initial preparation. The rest of the preparation was reduced by addition of concentrated sodium dithionite to a final concentration of 3 mM. The dithionite-reduced sample was made of this preparation. Some of this protein preparation was purged for 15 min with ultra-high-purity carbon monoxide that was further purified using an oxygen trap. This CO-purged enzyme was used to prepare the CO-ligated sample. Protein concentrations of the actual samples were determined to be 1.06 mM for ACS as isolated, 1.04 mM for the dithionite-reduced form, and 1.07 mM for the CO-bound samples using the Rose Bengal protein assay.
The solution samples (~80 μL) were transferred into 1 mm delrin XAS cells with 70 μm Kapton tape windows. The solution samples were immediately frozen after preparation and stored under liquid N2. Each sample holder afforded six fresh sample positions (1 mm × 4 mm beam) for EXAFS measurements. Three each were used to obtain the Ni and Fe XAS data.
EPR Spin Quantitation and Application to EXAFS
The Nip site in ACS is labile and binds weakly relative to the Nid site as evidenced by XRD data that show the site occupied by Zn and Cu. The loading of the Nip site is of special concern here, because free Ni in solution would dilute the signal from the ACS site. The presence of free Ni in ACS was evident in some preparations of ACS tested by EXAFS, which showed some differences in the rising-edge region between AOX and ARED, indicating the reduction of free Ni in solution. This was further validated by the lack of a statistically significant Ni–CO signal in the corresponding ANiFeC EXAFS data for these samples. This analysis is predicated on the relative standard redox potentials for free Ni(II)/(0) (−0.257 V, pH-independent) and dithionite (−0.46 V at pH 7).43 Therefore, dithionite is strong enough to reduce free Ni(II) to Ni(0).
EPR spectroscopy was used for spin quantitation of ANiFeC, which revealed 0.65 spin per monomer compared to that of a reference copper complex. The data presented here were measured on a sample with 1.75 Ni atoms per monomer of ACS. The similarity of the XAS data for AOX and ARED presented in Figure 4 shows that no free Ni is present in the sample used in this analysis. Because the spectroscopic features of Ni(0) are so dramatically different from those of Ni(II), the reduction of even a small amount of free Ni would significantly alter the Ni K-edge XANES. Assuming the tightly bound Nid site is fully occupied, we found the Nip site occupies 0.75 site and thus would lead to at most 0.75 spin in EPR if 100% of the Nip sites were bound by CO. Thus, the observed 0.65 spin indicates that ~87% of the Nip sites are CO-bound, which is expected to result in a 37% Ni–C contribution to the EXAFS data. This is approximated to 0.4 Ni–C in the FEFF fits.
Figure 4.

XAS results for oxidized and dithionite-reduced states of ACS. (A) Normalized Ni K-edge XAS data for AOX (black) and ARED (blue). The inset shows the expanded pre-edge region (inset, top) and the corresponding second derivatives (inset, bottom) for the pre-edge spectra. (B) Comparison of the non-phase-shift-corrected Fourier transforms and their corresponding EXAFS data (inset) for AOX (black) and ARED (blue).
X-ray Absorption Spectroscopic Measurements
X-ray absorption spectra were recorded at the Stanford Synchrotron Radiation Lightsource (SSRL) on focused 16-pole, 2 T wiggler side-station beamline 9-3 under standard ring conditions of 3 GeV and ~500 mA. A Si(220) double-crystal monochromator was used for energy selection. A Rh-coated harmonic rejection mirror was used to reject components of higher harmonics. During data collection, the samples were maintained at a constant temperature of ~10 K using an Oxford Instruments CF 1208 liquid helium cryostat. A 100-element Ge monolith fluorescence detector from Canberra Industries was employed for the measurement of fluorescence data.
Fe K-Edge Measurements
Fe K-edge XAS data were measured on the as-isolated form (AOX), the dithionite-reduced form (ARED), and the dithionite-reduced form subjected to CO treatment (ANiFeC) in the fluorescence mode by using soller slits equipped with a Mn filter in front of the Ge detector and using an Fe foil for energy calibration. The first inflection point of the foil spectrum was fixed at 7111.2 eV. The samples were monitored for photoreduction and showed no signs of change during the course of data collection. Fe K-edge EXAFS data were measured up to k = 17 Å−1. The data presented here are eight-scan average spectra, collected over three fresh sample positions for each of the samples.
Ni K-Edge Measurements
Ni K-edge XAS data were measured on AOX, ARED, and ANiFeC samples in fluorescence mode by using soller slits equipped with a Co filter in front of the Ge detector and using a Ni foil for energy calibration. The first inflection point of the foil spectrum was fixed at 8331.6 eV. The samples were monitored for photoreduction, and no sign of photodamage or Ni–CO bond cleavage was observed during the course of data collection. Ni K-edge EXAFS data were measured up to k = 19 Å−1. The data presented here are 12-scan average spectra, collected over three fresh sample positions for each of the samples.
XAS Data Reduction
Data presented in this study were processed by fitting a second-order polynomial to the pre-edge region and subtracting this from the entire spectrum as background in PySpline.44 A four-region spline with orders of 2, 3, 3, and 3 was used for both the Ni and Fe data sets to model the smoothly decaying postedge region. Theoretical EXAFS signals χ(k) were calculated by using FEFF (macintosh version 7)45,46 on the active site structure derived from the crystal structure of ACS (Protein Data Bank entry 1RU3).7 For the Fe EXAFS, the Fe atom bridging to Nip via a cysteine thiolate was chosen as the absorbing atom to generate phase and amplitude parameters. For the Ni EXAFS, structural models were generated for the different putative forms of Nip and Nid sites. The molecule editing software Avogadro was used to modify the structural parameters obtained from the crystal structure. DFT geometry-optimized structures were also used to generate phase and amplitude parameters for the CO-bound form of ACS (ANiFeC). Theoretical models were fit to the data using EXAFSPAK.47 The structural parameters varied during the fitting process were the bond distance (R) and bond variance σ2, which is related to the Debye–Waller factor resulting from thermal motion, and static disorder of the absorbing and scattering atoms. The nonstructural parameter ΔE0 (the energy at which k = 0) was also allowed to vary but was restricted to a common value for every component in a given fit. Coordination numbers were systematically varied in the course of the fit but were fixed within a given fit.
Statistical Evaluation of EXAFS Fits
The presence of a Ni–C component was statistically tested as follows. Two fits were performed: one without any Ni–CO components and one assuming a 0.4 Ni–CO contribution to the EXAFS data (Table 2). The reduced χ2 error values (which normalize the error function for the additional fit parameters, i.e., arising from the inclusion of single and multiple scattering components for the Ni–CO intermediate) are listed and are lower for the fit that includes the Ni–CO components and indicate that fit 2 is statistically better. A Hamilton F test was performed with a probability hypothesis of >90%.48 In addition to the statistical evaluation, a strong indicator of the presence of a Ni–C component is the behavior of the Ni–N σ2 value. In the fit without the Ni–C component (fit 1), the σ2 for the Ni–N component is 2-fold higher than in fit 2, which includes the Ni–CO components (Table 2). The Ni–N σ2 of ANiFeC in fit 2 is similar to that in AOX and ARED, consistent with the idea that bonding in Nid does not change significantly upon reduction and CO binding.
Table 2.
Ni K-Edge EXAFS Curve-Fitting Results for ANiFeC
| ANiFeC | coordination/path | R (Å)a | σ2 (Å2)b | ΔE0 (eV) | Fc |
|---|---|---|---|---|---|
| fit 1 | 1 Ni–N | 1.90 | 570 | 3.8 | 0.246 |
| 2.0 Ni–S | 2.21 | 353 | |||
| 0.5 Ni–S | 2.34 | 112 | |||
| 0.5 Ni–Fe/Ni | 2.78 | 606 | |||
| 1 Ni–C | 3.20 | 95 | |||
| fit 2 | 0.4 Ni–C | 1.77 | 323 | 1.5 | 0.231 |
| 0.4 Ni–O | /2.97 | /323 | |||
| 0.8 Ni–C–O | /2.97 | /323 | |||
| 1 Ni–N | 1.90 | 291 | |||
| 2.0 Ni–S | 2.21 | 351 | |||
| 0.5 Ni–S | 2.34 | 171 | |||
| 0.5 Ni–Fe/Ni | 2.79 | 710 | |||
| 1 Ni–C | 3.19 | 100 |
The estimated standard deviations for the distances are on the order of ±0.02 Å.
The σ2 values are multiplied by 105.
A slash indicates that the σ2 value was linked to the previous path.
The error is given by Σ[(χobsd − χcalcd)2k6]/(Ndata − Nvar).
The S02 factor was set at 1.
Density Functional Theory Calculations
Gradient-corrected spin unrestricted DFT calculations were performed using the ORCA49,50 package (ORCA version 3.03).51 The BP86 local functional and the following basis sets were employed for the calculations: Ahlrich’s all electron triple-ζ basis set TZVPP with three polarization functions on Ni, TZVP on all other atoms.52 Tight convergence criteria were imposed on all calculations. Calculations were performed in a dielectric continuum using the conductor-like screening model (COSMO)53 with a dielectric constant of 7. Starting guess geometries were obtained from the 1RU3 crystal structure and modified appropriately for the AOX, ARED, and ANiFeC states. The Cα atoms of six protein amino acid ligands to the cluster, marked with a star in Figure 6, for the sake of clarity were fixed for all calculations. These include the four cysteinates bound to the Fe–S clusters and one each from the phenylalanine and serine residues adjacent to the cysteinates bound to Nip and Nid. Tetrahedral and square-planar coordination was explored for the Nip site in all states. For AOX, a three- and four-coordinate Nip site was tested. A water molecule was added at the Nip open site in AOX to geometry optimize a four-coordinate Nip site. For AOX, optimizations without a fourth ligand led to unlikely perturbation of the active site; for example, the Nip site coordinated to the closed sulfide of the Fe–S cluster formed a Ni–S–Fe bond. This is consistent with previous reports in which a geometric constraint on the Ni–sulfide distance had to be imposed to theoretically evaluate the ARED state.54 The four-coordinate structure was optimized to a square-planar geometry, irrespective of the starting geometry (see Figure 6). Although it is likely that the four-coordinate Nip in AOX would lose the water ligand upon reduction, DFT optimizations of ARED with a four- or three-coordinate site invariably led to Ni–sulfide ligation (as described above and as observed previously).54 A representative input file is included in the Supporting Information, showing strategies employed for the convergence of pathological systems such as Fe–S clusters (the pathology being the large number of SCF convergence cycles, increasing the number of Fock matrices for the DIIS extrapolation and increasing the rebuilding frequency of the full Fock matrices). All calculations were checked for proper spin convergence of the [4Fe-4S]2+ cluster and to achieve the true broken symmetry ground state. Time-dependent DFT (TD-DFT) calculations were performed with the electronic structure program ORCA to calculate the energies and intensities of the Ni K pre-edges. The tight convergence criterion was imposed on all calculations. The same functional and basis sets that were used for the geometry optimizations were used for the TD-DFT calculations. The calculated energies and intensities were Gaussian-broadened with half-widths of 1.5 eV to account for core-hole lifetime and instrument resolution. The calculated pre-edge energy positions were linearly scaled to match the experimental spectra. This is generally the case with core level TD-DFT calculations because DFT does not describe core potentials accurately, resulting in the core levels having energies that are too high relative to those of the valence levels.
Figure 6.

DFT-calculated structures of oxidized and CO-bound forms of ACS. DFT geometry-optimized structures of AOX (left) and ANiFeC (right). The Cα atoms marked with an asterisk have been fixed to the crystal structure positions. The Nip site is square-planar in AOX (Ni–S–S–S dihedral angle of 5°) and tetrahedral in ANiFeC (Ni–S–S–S dihedral angle of 55°). Ni–Fe: 2.87 Å (AOX) and 3.93 Å (ANiFeC).
RESULTS AND DISCUSSION
Generation of ACS Containing High Levels of Nip Occupancy, Activity, and Spin Concentration
As described above, with slight modification of a previously published method,21 recombinantly expressed ACS was prepared, purified, and Ni-reconstituted to yield a nearly metal-replete sample exhibiting high levels of activity and spin concentrations. Generating a highly active sample is mandatory for detection of the organometallic intermediates because the bound CO is a relatively low intensity scatterer and because, even if the A cluster is 100% loaded in both Nip and Nid sites and no extraneous Ni is present, the maximal amount of Nip–CO will be 50% of the total Ni. Thus, our goal was to obtain a sample of ACS containing more than ~60% bound carbonyl, as assayed by the EPR spin intensity of the NiFeC signal. Generating such a sample has been an ongoing effort of this laboratory for decades.
The CO-treated enzyme used in the XAS experiments exhibited a large amount of FeNiC EPR signal intensity (0.65 spin/mol). This is much higher than the spin intensities reported for XAS samples reported earlier (Table S2), which were in the range of 0.06–0.25 spin/mol. Our high spin intensity value is congruent with the metal content, which was determined to be 4.2 Fe, 1.8 Ni, 0.05 Zn, and 0.01 Cu per monomeric unit of ACS, with all values having a 2–4% standard deviation. Furthermore, the ACS specific activity, measured by the acetyl-CoA/CO exchange reaction, was very high, i.e., 4.2 ± 0.2 units/mg at 55 °C and pH 6.1. As shown in Table S2, previous EXAFS experiments were performed with M. thermoacetica ACS samples exhibiting >10-fold lower specific activities (0.18–0.36 μmol min−1 mg−1). The highest previously reported value for an XAS sample was that of Carboxydothermus hydrogenoformans ACS, 2.4 μmol min−1 mg−1 (measured at 70 °C). The specific activity of 4.2 units/mg for a sample with 0.65 spin of NiFeC signal is consistent with that (3.2 units/mg with 0.5 spin per ACS monomer) exhibited by recent samples from our laboratory using a similar ACS preparation method.15 As described above (Materials and Methods), the observed 0.65 spin indicates that ~87% of the Nip sites are CO-bound, which is expected to result in a 37% Ni–C contribution to the EXAFS data. This is approximated to 0.4 Ni–C in the FEFF fits (below).
The [4Fe-4S] Subcomponent of the A Cluster Is Similar in Its Oxidized, Dithionite-Reduced, and CO-Treated Forms
Electronic and geometric characterization of the [4Fe-4S] part of the A cluster was performed on the AOX (as-isolated), ARED (dithionite-treated), and ANiFeC (dithionite- and CO-treated) states using Fe K-edge and EXAFS studies. The XAS data (Figure 3A) for these forms are very similar to data published for [4Fe-4S]2+ clusters,55,56 with no significant shift in the rising-edge energy positions among the three states. This similarity in the edge data is mirrored in the Fe K-edge EXAFS data (Figure 3B) and the comparison of their corresponding fits (Figure S1 and Table S1). All data sets are consistent with four Fe–S components at 2.28 Å and three Fe–Fe components at 2.7 Å, further confirming the [4Fe-4S]2+ nature of the [4Fe-4S] cluster in all three states. Similarity in the EXAFS data also indicates that (a) CO does not bind at an Fe site and (b) no structural distortion of the cluster occurs in the process of CO binding. No evidence of the presence of a Fe–Nip contribution to the EXAFS data was found, which is expected because only one of the Fe atoms is bridged to the Nip center (via the thiolate sulfur). Thus, these spectroscopic results reveal that the [4Fe-4S]2+ site in the A cluster remains in the same oxidation state, with minimal change in the electronic structure upon reduction of AOX to ARED and conversion of ARED to ANiFeC upon treatment with CO. These data also indicate that the A cluster is near fully replete with Ni, because, as indicated earlier by Mossbauer and EPR studies of recombinant ACS,18 a Nip-deficient A cluster results in reduction of the [4Fe-4S] cluster by dithionite.
Figure 3.

Normalized Fe-edge XAS and EXAFS data for the three forms of ACS. (A) Normalized Fe K-edge XAS data for AOX (black), ARED (blue), and ANiFeC (red) states of ACS. The inset shows the expanded pre-edge region (inset, top) and the corresponding second derivatives (inset, bottom) for the pre-edge spectra. (B) Comparison of the non-phase-shift-corrected Fourier transforms and their corresponding EXAFS data (inset).
Geometric and Electronic Structures of the Oxidized and Dithionite-Reduced States of the A Cluster
The Ni centers in the AOX and ARED A cluster were studied using Ni K-edge XAS (Figure 4A) and EXAFS (Figure 4B). The weak pre-edge feature at 8331.9 eV that occurs before the onset of the rising edge (Figure 4A, inset) is attributed to the 1s → 3d transition indicating similar ligand-field strengths and oxidation states of the Ni centers in AOX and ARED. Furthermore, a comparison of the rising-edge region in both AOX and ARED shows a feature at ~8335 eV that is attributed to structural and electronic processes (four-body multiple scattering and formally forbidden shakedown transitions),57,58 which are enhanced for centrosymmetric square-planar systems but are attenuated when geometries diverge from the square-planar limit. The similarities in both energy position and intensity of this feature in AOX and ARED indicates that the structure and electronics at the Ni centers remain unperturbed upon dithionite treatment of AOX.
This similarity is also reflected in the nearly identical Ni K-edge EXAFS and Fourier transforms of AOX and ARED (Figure 4B). FEFF fits to the EXAFS data were performed to obtain quantitative local structure information. Because EXAFS data are an average of the Nip and Nid sites, where appropriate, the fits were performed with partial coordination numbers. The crystal structure (Figure 2) shows the Nid site to be square-planar with NiN2S2 ligation, while Nip can assume either a trigonal (NiS3) or a square-planar (NiS3O) coordination.6,8 The best fit to AOX was obtained with one Ni–N group at 1.90 Å, 2.5 Ni–S groups at 2.18 Å, and 0.5 Ni–Fe/Ni group at 2.95 Å (Figure S2A and Table 1). Note that EXAFS cannot differentiate between Fe and Ni backscattering contributions, especially when the contributions are weak and the backscatteres are similar distances from the absorbing atom. Therefore, the contribution at 2.95 Å is labeled as 0.5 Ni–Fe/Ni, suggesting that it could arise from a Ni or Fe backscatterer. Hereafter, this contribution is termed the Ni–metal contribution to emphasize the limitation of EXAFS. Contrary to previous results,7 no evidence of a weak Ni–O interaction (coordinating water molecule) was found; however, it is anticipated that any weak Ni–O interaction (contributing 0.5 Ni–O group to the total EXAFS), if present, would be obfuscated by the intense 2.5 Ni–S group contributions at 2.18 Å. The EXAFS fit for ARED (Figure S2B and Table 1) produces bond distances identical to those in AOX, suggesting nearly identical geometries for the Ni centers in these states. The somewhat lower Ni–metal coordination (Table 1), which indicates the difference in the Ni–Ni and Ni–Fe distance, is consistent with all previous EXAFS59–61 and X-ray crystallographic6 analysis of CODH/ACS and indicates that Nip can assume two conformations. Thus, the EXAFS data show a NipS3 with an average Ni–S bond length of 2.18 Å and a NidS2N2 with an average Ni–S bond length of 2.18 Å and a Ni–N bond length of 1.90 Å for both AOX and ARED. These values are in excellent agreement with the proposed geometries of Nip and Nid sites and agree well with the crystallography data (Figure 2).
Table 1.
Ni K-Edge EXAFS Curve-Fitting Results for AOX and ARED
| ACS | coordination/path | R (Å)a | σ2 (Å2)b | ΔE0 (eV) | Fc |
|---|---|---|---|---|---|
| AOX | 1 Ni–N | 1.90 | 224 | −2.64 | 0.36 |
| 2.5 Ni–S | 2.18 | 391 | |||
| 0.5 Ni–Fe/Ni | 2.95 | 578 | |||
| ARED | 1 Ni–N | 1.90 | 214 | −1.41 | 0.42 |
| 2.5 Ni–S | 2.18 | 351 | |||
| 0.5 Ni–Fe/Ni | 2.96 | 347 |
The estimated standard deviations for the distances are on the order of ±0.02 Å.
The σ2 values are multiplied by 105.
The error is given by Σ[(χobsd − χcalcd)2k6]/(Ndata − Nvar).
The S02 factor was set at 1.
In summary, the similarities among the Fe and Ni XAS and EXAFS data for AOX and ARED indicate that there is no observable structural or electronic change upon treatment of ACS with dithionite. This is consistent with previous studies showing that none of the metal sites in AOX can be reduced by dithionite alone,60 suggesting that the operational ARED designation for the dithionite-treated protein is incorrect. The XAS and EXAFS results also provide important information about the recalcitrance of the fully Ni loaded A cluster to reduction by dithionite (E0′ = −660 mV vs the normal hydrogen electrode62). The only published example of trapping and characterizing the one-electron-reduced “ARED” state to which CO binds involved photolysis of CO from the ANiFeC state.35 We refer to that state here as ARED*. This ARED* species is a metastable Ni(I) state that was shown by EPR and IR studies to rapidly recombine with CO in a temperature-dependent manner to regenerate ANiFeC. Thus, Figure 2 shows the equilibrium to heavily favor the Ni(II)–AOX form of the A cluster in the absence of CO, which kinetically traps and stabilizes the Ni(I) state as ANiFeC.35
The CO-Bound Intermediate on ACS Has a Nip–CO Bond
We used EPR spectroscopy to follow conversion of ARED to ANiFeC by treatment of ACS with CO and dithionite under anaerobic conditions. Normalization of the spectrum with protein and Ni concentrations showed a high level of accumulation (87% of available Nip sites) of the well-characterized NiFeC signal with g values of 2.074 and 2.028 (Figure S3). In comparing the Ni K-edge XAS spectra of ANiFeC (Figure 5A) to that of ARED (reproduced from Figure 4A), we find there are significant changes in the pre-edge, in the rising edge, and near the white-line maxima of the spectra. The rising-edge feature in ANiFeC occurs at 8335.2 eV and is attenuated relative to that in ARED, indicating that the Nip site deviates from a square-planar geometry in ARED to a tetrahedral geometry in ANiFeC (as Nid remains unchanged), The pre-edge feature in ANiFeC shifts to lower energies, and its second-derivative spectrum (Figure 5A, inset) shows the presence of two pre-edge features (at 8330.9 and 8331.7 eV). The energy of the higher-energy pre-edge feature (at 8331.7 eV) is similar to that of the single pre-edge feature present in ARED and AOX (see Figure 4A), suggesting that this feature in ANiFeC derives from Nid, which is unchanged in these three forms of the enzyme.57,63 Thus, the lower-energy pre-edge feature at 8330.9 eV is attributed to the 1s → 3d transition of Nip. Finally, the white-line region between 8340 and 8360 eV is different for ANiFeC and ARED. Such changes are due to a change in geometry around the Ni center and suggest a perturbation in site geometry upon CO binding. Thus, the Ni K-edge data of AOX, ARED, and ANiFeC unambiguously support CO binding at the Nip site.
Figure 5.

Comparison of XAS results for dithionite-reduced and CO-bound forms of ACS. (A) Normalized Ni K-edge XAS data for ARED (blue) and ANiFeC (red). The inset shows the expanded pre-edge region (inset, top) and the corresponding second derivatives (inset, bottom) for the pre-edge spectra. The pre-edge features are marked with gray bars in the second derivative. (B) Comparison of the nonphase-shift-corrected Fourier transforms and their corresponding EXAFS data (inset) for ARED (blue) and ANiFeC (red).
Interestingly, despite the clear spectral changes that occur upon CO binding, the Ni K-edge inflection point (typically indicative of a change in oxidation state) does not shift significantly among AOX, ARED, and ANiFeC. The reason for these small changes in the Ni-edge spectra upon conversion to ANiFeC is that reduction can occur at only Nip, which represents only half of the probed Ni sites. In addition, the presence of five other heavy atom backscatterers in the A cluster tends to broaden edge features and obfuscate changes due to Zeff. Furthermore, ENDOR, EPR, and Mössbauer studies show that the unpaired electron in the Ni–CO state is delocalized among the Ni, CO, and [4Fe4S] cluster.18,26,31,33 Thus, although the bond is between Ni and C and we formally identify the species as Nip(I)–CO, only part of the unpaired electron spin density is on Nip.26 The lack of edge energy shifts in either Fe or Ni K-edge XAS data, the similarity of the Fe and Ni EXAFS, the results of the DFT and TD-DFT calculations (below), and the lack of any significant changes in the UV–visible spectrum upon conversion of AOX to ANiFeC all demonstrate that the average effect of reduction on each of the Fe atoms in the [4Fe-4S]2+ cluster or on the Nip site is small and further support the idea that ANiFeC is a highly delocalized spin system. This is also supported by the Mulliken population analysis of the valence orbitals (see Table S3 and the DFT section) involved in the Ni K-pre-edge transitions.
The Ni and Fe XAS and EXAFS data are relevant to a controversy about the intermediacy of ANiFeC (summarized in ref 4) versus a two-electron-reduced species formally identified as Ni(0)11,64 in the ACS catalytic cycle. The small differences in the Ni-edge and EXAFS spectra among AOX, ARED, and ANiFeC support one-electron reduction to form a highly delocalized ANiFeC rather than a formally two-electron reduced A cluster with a Ni(0) or Ni(I)/[4Fe-4S]+ electronic state. Furthermore, a Ni(0) state in ACS has never been reported, and two-electron reduction of Ni(II) to Ni(0) in the highly electropositive environment formed by Nid(II) and [4Fe-4S]2+ would be extremely difficult given that the redox potential for reductive activation of ACS to the Nip(I)–CO state is already very negative, approximately −500 to −540 mV.65,66
The Ni K-edge EXAFS data (and the non-phase-shift-corrected Fourier transforms) for the ARED and ANiFeC states exhibit large differences in the phase and amplitude parameters (Figure 5B). In the FT data of ANiFeC, both the first-shell (R′ between 1.2 and 1.8 Å) intensity and the second-shell (R′ between 1.9 and 3.0 Å) R′ values decrease, resulting in two main features compared to a single feature in the second shell of ARED. FEFF fits were performed on the EXAFS data for ANiFeC with several structural models, including that obtained from DFT geometry optimization. The best fits to the data were consistent with a first shell composed of 0.4 Ni–C group at 1.77 Å, 1 Ni–N group at 1.89 Å, 2 Ni–S groups at 2.22 Å, and 0.5 Ni–S group at 2.33 Å. A 0.5 Ni–metal component was found at 2.78 Å (Figure S4 and Table 2). It is important to note that a Ni–C–O multiple-scattering contribution had to be included in the fit to obtain reasonable Debye–Waller factors. This suggests a linear (or nearly linear) Ni–C–O arrangement upon CO binding, because a bent CO would not significantly contribute to the EXAFS data. This is supported by the DFT geometry optimizations performed on ANiFeC, which shows an Fe–C–O angle of 177° (see the next section and Figure 6). Together, the Ni K-edge and EXAFS data unambiguously show an organometallic Ni–CO bond in ANiFeC and provide the first structural evidence of binding of CO to Nip. Furthermore, although the data are consistent with the assumption that backbone amide(N)–Nid interaction remains virtually unchanged between ARED and ANiFeC (no change in the Ni–N component), a significant elongation of the Ni–S distances is observed. The single 2.5 Ni–S group contributions at 2.18 Å in ARED can now be separated into a shorter contribution from 2.0 Ni–S groups at 2.22 Å and a longer contribution from 0.5 Ni–S group at 2.33 Å in ANiFeC. This indicates that CO binding results in elongation of the Ni–S contribution in general and in particular at the Nip site. The EXAFS data also indicate that the Ni–metal (see above) bond is ~0.2 Å shorter than that in the AOX and ARED states.
Ni K-edge XANES and EXAFS have previously been exploited to study the geometric structure of the active sites of CODH and ACS. While EXAFS data have been quantitatively fit using FEFF (multiple-scattering methodology), the published XANES analysis has been very qualitative, mostly devoted to estimating the deviation of the Ni active site from a square-planar limit (and toward a tetrahedral limit) or in following the variability in the edge shape and energy position depending on ligand type.67 Furthermore, EXAFS analyses60,61,68 published prior to the crystal structures described in 2002 and 20035,6 were based on ACS containing a single Ni center at the ACS active site and therefore (as later revealed) had largely incorrect interpretations of the Ni centers. For example, a 1998 study reveals that upon removal of the “labile” site (later equated to Nip) with phenanthroline, the rising-edge feature (attributable to the “nonlabile” site and hence equated to Nid) becomes less intense,60 which is contrary to the finding that the Nid site is square-planar in nature. There are also significant limitations to the EXAFS studies of the ACS active site published after the discovery that the active site has two Ni centers. For example, Svetlitchnyi et al. performed EXAFS analysis of a sample of only as-isolated ACS; however, the data are fairly marginal (reliable only to k ~ 11 Å−1).7 Gu et al. performed a more comprehensive EXAFS study;59 however, their XANES data for the as-isolated protein resemble those of the phenathroline-treated protein,60 indicating significant removal of the Nip site. Because the Ni K-edge EXAFS figure in the paper is incorrect (the Fe K-edge EXAFS data are shown in the Ni K-edge EXAFS figure), a direct comparison of the two postcrystal structure EXAFS data is not possible. More importantly, the study does not show a significant change in the near-edge, pre-edge, or Fourier transforms upon CO treatment, indicating that an insignificant amount of the Ni–CO species was present in the sample. A more recent conference proceeding69 shows low-resolution Ni K-edge EXAFS data (k ~ 12 Å−1) on partially loaded as-isolated and reduced ACS, which are quantitatively different from those of Cramer et al.59 To complicate the analysis further, an EXAFS study of ACS containing Cu at the proximal center (i.e., CupNid) shows a dramatically short Cu–Ni distance (2.67 Å) compared to the longer Ni–Ni distance observed in other studies (~2.9 Å).37 These earlier Ni K-edge and EXAFS investigations performed on ACS have all yielded incorrect and at best incomplete information about the active site of ACS. Despite this, all the EXAFS studies are qualitatively consistent showing both Ni–S and Ni–N/O coordination and a longer distance Ni–metal interaction (see Table S2), properties that are consistent with the significantly higher quality theoretically validated EXAFS data presented here.
DFT Structural Correlation of AOX, ARED, and ANiFeC
Quantum chemical assessment70 of the geometries of AOX, ARED, and ANiFeC states was performed starting with the geometrical parameters obtained from the crystal structure of AOX (PDB entry 1RU3) (details of the various geometries and A cluster states are given in the Supporting Information).7 For ANiFeC, the CO was initially positioned near the Nip site and assumed not to bind to the Fe–S cluster or the Nid site, consistent with the XAS data presented in the previous sections. For AOX, optimizations without a fourth ligand led to unlikely perturbation of the Fe–S cluster and Ni–sulfide ligation. The four-coordinate structure was optimized to a square-planar geometry, irrespective of starting geometry (Figure 6). Although it is likely that the four-coordinate Nip in AOX would lose the water ligand upon reduction, DFT optimizations on ARED with either a four- or three-coordinate site invariably led to Ni–sulfide ligation as observed for the three-coordinate AOX. This is consistent with previous reports, in which a geometric constraint on the Ni–sulfide distance had to be imposed to theoretically evaluate the ARED state.54 Because of the similarity of the AOX and ARED spectra, which indicate similar geometric and electronic structures for these two states, we compared geometry-optimized structures for AOX and ANiFeC.
Geometry optimization on ANiFeC converged with CO bound at the Nip site in a tetrahedral geometry. This is consistent with the Ni K-edge rising-edge data, which showed that although the Nip site in AOX (and ARED) is consistent with a square-planar structure, the Nip site in ANiFeC is not. The calculated Ni–C distance of 1.75 Å is in good agreement with the EXAFS distance of 1.77 Å. The Ni–S distances for both the Nid and Nip sites increase in ANiFeC (Figure 6). The largest perturbation is in the Nip–S distance of the bridged Nip–S–Fe (from 2.18 to 2.36 Å), in good agreement with the EXAFS data, which show elongation of the Ni–S distance from 2.18 to 2.22 Å and splitting of the Ni–S contributions to include 0.5 Ni–S group at 2.33 Å. The comparison of the DFT structure with the EXAFS data confirms that the elongated Ni–S (obtained from the EXAFS data) is the Nip–S(Cys) of the bridging Nip–S–Fe in ACS. Reduction followed by CO binding (AOX to ANiFeC) leads to elongation of the Ni–Fe bond by 1.1 Å, while the Ni–Ni bond is shortened by 0.38 Å, consistent with the EXAFS results, which show a shortening of the Ni–metal bond. In summary, the experimental and theoretical data reveal a tetrahedral Nip site with a short Nip–C bond, longer Ni–S distances, especially the Nip–S(Cys) bond, and a shorter distance Nip–Nid interaction.
The structural models developed here were used to evaluate the Ni K-pre-edge data presented in Figure 5A, which show two pre-edge features for ANiFeC occurring at energies slightly lower than that of the single pre-edge feature in AOX. Ni 1s → 3d TD-DFT calculations (including both Nip and Nid sites)71 were performed on AOX and ANiFeC using the geometry-optimized structures (Figure 7). The calculations show a single peak in AOX at 8328.7 eV and two peaks separated by ~0.8 eV (8328.2 and 8329.0 eV) for ANiFeC, which is in excellent agreement with the experimental pre-edge energy separation. Individual 1s → 3d calculations from the Nip and Nid sites show that while both contribute at the same energy for AOX, they contribute dominantly to the 8328.2 and 8329.0 eV features, respectively. The lower pre-edge energy position for Nip in ANiFeC indicates a decrease in its ligand-field strength relative to that of AOX, which is consistent with a partially reduced Nip that has significantly elongated Ni–S distances. Both of these factors effectively offset the increase in ligand field due to the formation of the Ni–CO bond and lower the pre-edge energy position. This large structural and electronic perturbation is consistent with the idea that the Nip site is highly labile and flexible and can assume drastically different electronic configurations during catalysis.
Figure 7.
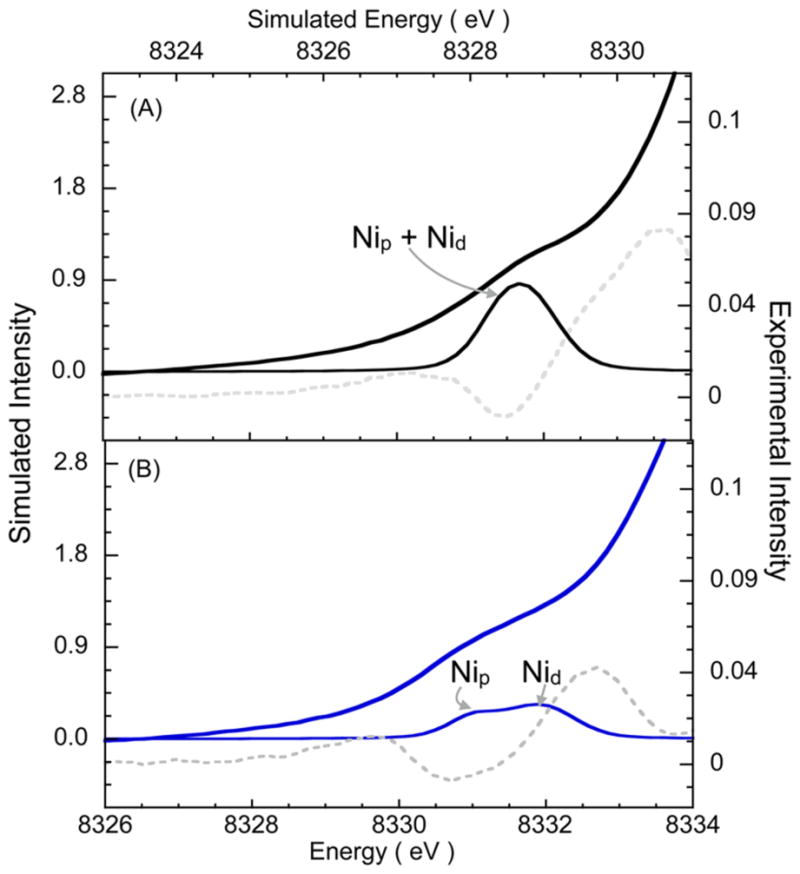
TD-DFT-calculated XAS spectra. TD-DFT-calculated 1s → 3d data compared to the Ni K-pre-edge data (—) and their second derivatives (---) for (A) AOX and (B) ANiFeC using the structures depicted in Figure 6. The arrows indicate the dominant contribution of Nip and Nid sites to individual pre-edge features.
IMPLICATIONS
Our results convincingly demonstrate that biological acetate synthesis occurs through formation of an organometallic Ni–CO intermediate, thus validating a central tenet of the unique organometallic pathway used by a wide class of anaerobic bacteria in energy metabolism and carbon dioxide fixation. Acetyl-CoA synthase (ACS), the key enzyme in anaerobic CO and CO2 fixation, catalyzes the production of acetyl-CoA from CO, a methyl group and CoA as part of the Wood–Ljungdahl pathway.4 Various lines of evidence support the concept that this pathway involves an organometallic reaction sequence in which the CO, methyl, and CoA components of acetyl-CoA bind sequentially to a metal center in the A cluster of ACS (Figure 2). Because of the novelty of Ni in the enzyme and the lack of any structural information about the complexity of this cluster (later shown to be a dinickel unit bridged to a [4Fe-4S] cluster), it was proposed three decades ago that acetyl-CoA assembly took place at Ni;1 however, direct structural evidence of a nickel–carbon bond or, in fact, any type of metal–carbon bond on ACS has been lacking. The Ni K-edge XAS results provide this unambiguous structural evidence demonstrating that the carbon of CO binds to Nip within the A cluster of ACS.
This Nip(I)–CO (ANiFeC) species is shown as intermediate 3 in Figure 2. It is formed from intermediate species 1 (AOX) through another intermediate 2, described here as Ared*, a highly labile paramagnetic Ni(I) state that has so far been observed only by photolysis of the Nip(I)–CO state.35 The EPR spectrum of Ared* is significantly different from that of the ANiFeC species in that it exhibits g values (2.56, 2.10, and 2.01) similar to those of Ni(I) model compounds,72 suggestive of a Ni(I) state with little electron delocalization into the other subcomponents of the A cluster. The data presented here support previous results that indicate that Nip(II) is not reduced by DTT, dithionite, or Ti(III) citrate alone; however, in the presence of CO, the reduction of AOX is coupled to formation of ANiFeC, described formally as a Ni(I)–CO species. A combination of EXAFS and DFT results shows that when CO binds to ACS, Nip converts from a square-planar NiS3O to tetrahedral NiS3CO site accompanied by a 0.16 Å lengthening of the Fe–S bond for the bridging Nip–S(Cys)–Fe bond and 0.04 Å elongation of the Nip–S(Cys) bridged to Nid. Nid retains a N2S2 square-planar geometry but experiences a 0.04 Å lengthening of the Nid–S bond upon reduction and CO binding to Nip. This, along with the short Ni–Ni distance predicted by DFT (Figure 6), suggests a subtle structural/electronic role of Nid in forming ANiFeC. One might ask if a simple Ni–CO model complex or peptide may have structural, spectroscopic, and/or catalytic properties similar to those of the A cluster containing five other thiol-associated metal ions embedded within a large protein.
While ANiFeC and Ared* are one-electron reduced species relative to AOX, our results indicate that the species traditionally called ARED actually is equivalent to AOX. This is in contrast to “free” [4Fe-4S]2+ clusters such as that present in the Fe protein in nitrogenase,55,56 which undergo reduction in the presence of dithionite accompanied by significant changes in the UV–visible spectra and in the Fe K-edge XAS and EXAFS. The only time that the [4Fe-4S] cluster undergoes reduction is in the Ni-depleted protein, so we propose that this might be the species more properly designated as ARED. Thus, ARED, defined as the state of the A cluster that undergoes reduction in the absence of CO to generate a reduced [4Fe-4S] cluster, is not part of the ACS catalytic cycle.
Upon reaction of ANiFeC with the methylated CFeSP, a putative methyl–Ni(III) species, 4, is formed, which is expected to be a highly oxidizing and unstable intermediate that would undergo immediate one-electron reduction to generate 5, requiring a net one-electron uptake and positing a net input of two electrons between 1 and 5. The second electron is obtained from internal electron transfer, but the actual site for docking of that one electron has not been identified.15 Nucleophilic attack by CoA would then lead to reductive elimination of the acetyl group as acetyl-CoA to regenerate 2. Acetyl-CoA formation from a metal–carbonyl species is analogous to some well-known industrial processes involving carbonylation chemistry, including the Monsanto process, hydroformylation, and the Reppe process.73 The intermediate steps in the Monsanto process for formation of acetic acid from methanol and CO are nearly identical to those in the catalytic mechanism of ACS in that both use organometallic mechanisms that feature low-valent metal centers [e.g., Rh(I) vs Ni(I)] to react with CO to form a metal–carbonyl bond (M–CO) or with a methyl donor to generate a methyl–metal bond (M–CH3) that undergoes carbonyl insertion (methyl migration) and reaction with a nucleophile to generate an activated form of acetate (acetyl iodide instead of acetyl-CoA). Thus, it is interesting to consider how the ACS active site is poised to catalyze C–C and C–S bond formation, while CODH, which also contains a Ni ion bridged to a [4Fe-4S] cluster, proficiently catalyzes CO oxidation and/or CO2 reduction.
Supplementary Material
Acknowledgments
Portions of this research were performed at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393).
Funding
This work was supported by Grant R37-GM39451 (S.W.R.).
ABBREVIATIONS
- ACS
acetyl-CoA synthase
- ACDS
acetyl-CoA decarbonylase/synthase
- CODH
CO dehydrogenase
- CFeSP
corrinoid iron–sulfur protein
- DFT
density functional theory
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- Nip
Ni atom proximal to the Fe4S4 subcluster
- Nid
Ni atom distal to the Fe4S4 subcluster
- TD
time-dependent
- XAS
X-ray absorption spectroscopy
- XANES
X-ray near-edge spectroscopy
Footnotes
Author Contributions
M.C. and L.J.G. contributed equally to this work. R.S., S.W.R., L.G., and M.C. designed and performed the research. R.S., L.G., and M.C. analyzed the data.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00983.
Figures S1–S4, Tables S1–S4, references, and a sample DFT input file (PDF)
References
- 1.Ragsdale SW, Wood HG. Acetate biosynthesis by acetogenic bacteria: evidence that carbon monoxide dehydrogenase is the condensing enzyme that catalyzes the final steps of the synthesis. J Biol Chem. 1985;260:3970–3977. [PubMed] [Google Scholar]
- 2.Kumar M, Lu WP, Liu L, Ragsdale SW. Kinetic evidence that CO dehydrogenase catalyzes the oxidation of CO and the synthesis of acetyl-CoA at separate metal centers. J Am Chem Soc. 1993;115:11646–11647. [Google Scholar]
- 3.Xia J, Sinclair JF, Baldwin TO, Lindahl PA. Carbon monoxide dehydrogenase from Clostridium thermoaceticum: quaternary structure, stoichiometry of its SDS-induced dissociation, and characterization of the faster-migrating form. Biochemistry. 1996;35:1965–1971. doi: 10.1021/bi9511853. [DOI] [PubMed] [Google Scholar]
- 4.Can M, Armstrong FA, Ragsdale SW. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem Rev. 2014;114:4149–4174. doi: 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doukov TI, Iverson T, Seravalli J, Ragsdale SW, Drennan CL. A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 6.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open alpha subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat Struct Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 7.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Romer P, Huber R, Meyer O. A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc Natl Acad Sci U S A. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drennan CL, Doukov TI, Ragsdale SW. The Metalloclusters of Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase: A Story in Pictures. JBIC, J Biol Inorg Chem. 2004;9:511–515. doi: 10.1007/s00775-004-0563-y. [DOI] [PubMed] [Google Scholar]
- 9.Seravalli J, Ragsdale SW. Pulse-chase studies of the synthesis of acetyl-CoA by carbon monoxide dehydrogenase/acetyl-CoA synthase - Evidence for a random mechanism of methyl and carbonyl addition. J Biol Chem. 2008;283:8384–8394. doi: 10.1074/jbc.M709470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragsdale SW. Metals and their scaffolds to promote difficult enzymatic reactions. Chem Rev. 2006;106:3317–3337. doi: 10.1021/cr0503153. [DOI] [PubMed] [Google Scholar]
- 11.Lindahl PA. Acetyl-coenzyme A synthase: the case for a Nip (0)-based mechanism of catalysis. JBIC, J Biol Inorg Chem. 2004;9:516–524. doi: 10.1007/s00775-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 12.Seravalli J, Kumar M, Ragsdale SW. Rapid kinetic studies of acetyl-CoA synthesis: evidence supporting the catalytic intermediacy of a paramagnetic NiFeC species in the autotrophic Wood-Ljungdahl pathway. Biochemistry. 2002;41:1807–1819. doi: 10.1021/bi011687i. [DOI] [PubMed] [Google Scholar]
- 13.Gorst CM, Ragsdale SW. Characterization of the NiFeCO Complex of Carbon-Monoxide Dehydrogenase as a Catalytically Competent Intermediate in the Pathway of Acetyl-Coenzyme-a Synthesis. J Biol Chem. 1991;266:20687–20693. [PubMed] [Google Scholar]
- 14.George SJ, Seravalli J, Ragsdale SW. EPR and infrared spectroscopic evidence that a kinetically competent paramagnetic intermediate is formed when acetyl-coenzyme A synthase reacts with CO. J Am Chem Soc. 2005;127:13500–13501. doi: 10.1021/ja0528329. [DOI] [PubMed] [Google Scholar]
- 15.Bender G, Ragsdale SW. Evidence That Ferredoxin Interfaces with an Internal Redox Shuttle in Acetyl-CoA Synthase during Reductive Activation and Catalysis. Biochemistry. 2011;50:276–286. doi: 10.1021/bi101511r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender G, Stich TA, Yan L, Britt RD, Cramer SP, Ragsdale SW. Probing the catalytic mechanism of acetyl-CoA synthase by infrared and EPR characterization of the photolyzed Ni(I)-CO intermediate. Biochemistry. 2010;49:7516–7523. doi: 10.1021/bi1010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ragsdale SW. Nickel and the Carbon Cycle. J Inorg Biochem. 2007;101:1657–1666. doi: 10.1016/j.jinorgbio.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bramlett MR, Stubna A, Tan X, Surovtsev IV, Munck E, Lindahl PA. Mossbauer and EPR study of recombinant acetyl-CoA synthase from Moorella thermoacetica. Biochemistry. 2006;45:8674–8685. doi: 10.1021/bi060003+. [DOI] [PubMed] [Google Scholar]
- 19.Gencic S, Grahame DA. Two Separate One-Electron Steps in the Reductive Activation of the A Cluster in Subunit beta of the ACDS Complex in Methanosarcina thermophila. Biochemistry. 2008;47:5544–5555. doi: 10.1021/bi7024035. [DOI] [PubMed] [Google Scholar]
- 20.Tan X, Martinho M, Stubna A, Lindahl PA, Munck E. Mossbauer Evidence for an Exchange-Coupled {[Fe4S4]1+ Nip1+} A-Cluster in Isolated alpha Subunits of Acetyl-Coenzyme A Synthase/Carbon Monoxide Dehydrogenase. J Am Chem Soc. 2008;130:6712–6713. doi: 10.1021/ja801981h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seravalli J, Ragsdale SW. Pulse-Chase Studies of the Synthesis of Acetyl-CoA by Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase: Evidence for a Random Mechanism of Methyl and Carbonyl Addition. J Biol Chem. 2008;283:8384–8394. doi: 10.1074/jbc.M709470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu WP, Harder SR, Ragsdale SW. Controlled Potential Enzymology of Methyl Transfer-Reactions Involved in Acetyl-CoA Synthesis by Co Dehydrogenase and the Corrinoid Iron-Sulfur Protein from Clostridium thermoaceticum. J Biol Chem. 1990;265:3124–3133. [PubMed] [Google Scholar]
- 23.Tan XS, Sewell C, Lindahl PA. Stopped-Flow Kinetics of Methyl Group Transfer between the Corrinoid-Iron-Sulfur Protein and Acetyl-Coenzyme A Synthase from Clostridium thermoaceticum. J Am Chem Soc. 2002;124:6277–6284. doi: 10.1021/ja016676r. [DOI] [PubMed] [Google Scholar]
- 24.Ragsdale SW, Ljungdahl LG, DerVartanian DV. EPR evidence for nickel substrate interaction in carbon monoxide dehydrogenase from Clostridium thermoaceticum. Biochem Biophys Res Commun. 1982;108:658–663. doi: 10.1016/0006-291x(82)90880-4. [DOI] [PubMed] [Google Scholar]
- 25.Ragsdale SW, Ljungdahl LG, DerVartanian DV. 13C and 61Ni isotope substitution confirm the presence of a nickel(III)-carbon species in acetogenic CO dehydrogenases. Biochem Biophys Res Commun. 1983;115:658–665. doi: 10.1016/s0006-291x(83)80195-8. [DOI] [PubMed] [Google Scholar]
- 26.Ragsdale SW, Wood HG, Antholine WE. Evidence that an iron-nickel-carbon complex is formed by reaction of CO with the CO dehydrogenase from Clostridium thermoaceticum. Proc Natl Acad Sci U S A. 1985;82:6811–6814. doi: 10.1073/pnas.82.20.6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl Pathway of CO2 Fixation. Biochim Biophys Acta, Proteins Proteomics. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiel-Bengelsdorf B, Durre P. Pathway engineering and synthetic biology using acetogens. FEBS Lett. 2012;586:2191–2198. doi: 10.1016/j.febslet.2012.04.043. [DOI] [PubMed] [Google Scholar]
- 29.Ferry JG. Enzymology of one-carbon metabolism in methanogenic pathways. FEMS Microbiol Rev. 1999;23:13–38. doi: 10.1111/j.1574-6976.1999.tb00390.x. [DOI] [PubMed] [Google Scholar]
- 30.Martin W, Russell MJ. On the origin of biochemistry at an alkaline hydrothermal vent. Philos Trans R Soc, B. 2007;362:1887–1925. doi: 10.1098/rstb.2006.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan C, Gorst CM, Ragsdale SW, Hoffman BM. Characterization of the Ni-Fe-C complex formed by reaction of carbon monoxide with the carbon monoxide dehydrogenase from Clostridium thermoaceticum by Q-band ENDOR. Biochemistry. 1991;30:431–435. doi: 10.1021/bi00216a018. [DOI] [PubMed] [Google Scholar]
- 32.Lindahl PA, Münck E, Ragsdale SW. CO dehydrogenase from Clostridium thermoaceticum: EPR and electrochemical studies in CO2 and argon atmospheres. J Biol Chem. 1990;265:3873–3879. [PubMed] [Google Scholar]
- 33.Lindahl PA, Ragsdale SW, Münck E. Mössbauer studies of CO dehydrogenase from Clostridium thermoaceticum. J Biol Chem. 1990;265:3880–3888. [PubMed] [Google Scholar]
- 34.Doukov TI, Blasiak LC, Seravalli J, Ragsdale SW, Drennan CL. Xenon in and at the end of the tunnel of bifunctional Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase. Biochemistry. 2008;47:3474–3483. doi: 10.1021/bi702386t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bender G, Stich TA, Yan L, Britt RD, Cramer SP, Ragsdale SW. Infrared and EPR spectroscopic characterization of a Ni(I) species formed by photolysis of a catalytically competent Ni(I)-CO intermediate in the acetyl-CoA synthase reaction. Biochemistry. 2010;49:7516–7523. doi: 10.1021/bi1010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin WS, Lindahl PA. Low-Spin Quantitation of Nifec Epr Signal from Carbon-Monoxide Dehydrogenase Is Not Due to Damage Incurred during Protein-Purification. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1993;1161:317–322. doi: 10.1016/0167-4838(93)90231-f. [DOI] [PubMed] [Google Scholar]
- 37.Seravalli J, Xiao Y, Gu W, Cramer SP, Antholine WE, Krymov V, Gerfen GJ, Ragsdale SW. Evidence That Ni-Ni Acetyl-CoA Synthase Is Active And That The Cu-Ni Enzyme Is Not. Biochemistry. 2004;43:3944–3955. doi: 10.1021/bi036194n. [DOI] [PubMed] [Google Scholar]
- 38.Schenker RP, Brunold TC. Computational studies on the A cluster of acetyl-coenzyme A synthase: geometric and electronic properties of the NiFeC species and mechanistic implications. J Am Chem Soc. 2003;125:13962–13963. doi: 10.1021/ja037893q. [DOI] [PubMed] [Google Scholar]
- 39.Harrop TC, Olmstead MM, Mascharak PK. Synthetic analogues of the active site of the A-cluster of acetyl coenzyme A synthase/CO dehydrogenase: syntheses, structures, and reactions with CO. Inorg Chem. 2006;45:3424–3436. doi: 10.1021/ic0520465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathrubootham V, Thomas J, Staples R, McCraken J, Shearer J, Hegg EL. Bisamidate and Mixed Amine/Amidate NiNi2S2 Complexes as Models for Nickel-Containing Acetyl Coenzyme A Synthase and Superoxide Dismutase: An Experimental and Computational Study. Inorg Chem. 2010;49:5393–5406. doi: 10.1021/ic9023053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsumoto T, Ito M, Kotera M, Tatsumi K. A dinuclear nickel complex modeling of the Nid(II)-Nip(I) state of the active site of acetyl CoA synthase. Dalton transactions. 2010;39:2995–2997. doi: 10.1039/b924915j. [DOI] [PubMed] [Google Scholar]
- 42.Raybuck SA, Bastian NR, Orme-Johnson WH, Walsh CT. Kinetic characterization of the carbon monoxide-acetyl-CoA (carbonyl group) exchange activity of the acetyl-CoA synthesizing CO dehydrogenase from Clostridium thermoaceticum. Biochemistry. 1988;27:7698–7702. doi: 10.1021/bi00420a019. [DOI] [PubMed] [Google Scholar]
- 43.Li CW, Yu JH, Liang YM, Chou YH, Park HJ, Choo KH, Chen SS. Ni removal from aqueous solutions by chemical reduction: Impact of pH and pe in the presence of citrate. J Hazard Mater. 2016;320:521–528. doi: 10.1016/j.jhazmat.2016.08.030. [DOI] [PubMed] [Google Scholar]
- 44.Tenderholt A, Hedman B, Hodgson KO. PySpline: A Modern, Cross-Platform Program for the Processing of Raw Averaged XAS Edge and EXAFS Data. In: Hedman B, Pianetta P, editors. X-ray Absorption Fine Structure: XAFS13. American Institute of Physics, Stanford University; 2007. pp. 105–107. [Google Scholar]
- 45.Rehr JJ, Mustre de Leon J, Zabinsky SI, Albers RC. Theoretical X-Ray Absorption Fine-Structure Standards. J Am Chem Soc. 1991;113:5135–5140. [Google Scholar]
- 46.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Multiple-Scattering Calculations of X-Ray-Absorption Spectra. Phys Rev B: Condens Matter Mater Phys. 1995;52:2995–3009. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 47.George GN. EXAFSPAK and EDG-FIT. Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center; Stanford, CA: 2000. [Google Scholar]
- 48.Hamilton W. Significance tests on the crystallographic R factor. Acta Crystallogr. 1965;18:502–510. [Google Scholar]
- 49.Neese F. ORCA: An Ab initio, DFT and Semiempirical Electronic Structure Package, version 2.6.35. University of Bonn; Bonn, Germany: 2008. [Google Scholar]
- 50.Neese F, Olbrich G. ORCA publication. Chem Phys Lett. 2002;362:170–178. [Google Scholar]
- 51.Neese F. ORCA: An ab initio, DFT and semiempirical SCF-MO package, version 3.0.3. University of Bonn; Bonn, Germany: 2016. [Google Scholar]
- 52.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 53.Sinnecker S, Rajendran A, Klamt A, Diedenhofen M, Neese F. Calculation of solvent shifts on electronic g-tensors with the conductor-like screening model (COSMO) and its self-consistent generalization to real solvents (direct COSMO-RS) J Phys Chem A. 2006;110:2235–2245. doi: 10.1021/jp056016z. [DOI] [PubMed] [Google Scholar]
- 54.Chmielowska A, Lodowski P, Jaworska M. Redox Potentials and Protonation of the A-Cluster from Acetyl-CoA Synthase. A Density Functional Theory Study. J Phys Chem A. 2013;117:12484–12496. doi: 10.1021/jp402616e. [DOI] [PubMed] [Google Scholar]
- 55.Musgrave KB, Angove HC, Burgess BK, Hedman B, Hodgson KO. All-Ferrous Titanium(III) Citrate Reduced Fe Protein of Nitrogenase: An XAS Study of Electronic and Metrical Structure. J Am Chem Soc. 1998;120:5325–5326. [Google Scholar]
- 56.Mitra D, George SJ, Guo Y, Kamali S, Keable S, Peters JW, Pelmenschikov V, Case DA, Cramer SP. Characterization of [4Fe-4S] Cluster Vibrations and Structure in Nitrogenase Fe Protein at Three Oxidation Levels via Combined NRVS, EXAFS, and DFT Analyses. J Am Chem Soc. 2013;135:2530–2543. doi: 10.1021/ja307027n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarangi R. X-ray absorption near-edge spectroscopy in bioinorganic chemistry: Application to M-O systems. Coord Chem Rev. 2013;257:459–472. doi: 10.1016/j.ccr.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DuBois JL, Mukherjee P, Stack TDP, Hedman B, Solomon EI, Hodgson KO. A Systematic K-edge X-ray Absorption Spectroscopic Study of Cu(III) Sites. J Am Chem Soc. 2000;122:5775–5787. [Google Scholar]
- 59.Gu W, Gencic S, Cramer SP, Grahame DA. The A-cluster in subunit beta of the acetyl-CoA decarbonylase/synthase complex from Methanosarcina thermophila: Ni and Fe K-edge XANES and EXAFS analyses. J Am Chem Soc. 2003;125:15343–15351. doi: 10.1021/ja036602a. [DOI] [PubMed] [Google Scholar]
- 60.Russell WK, Stalhandske CMV, Xia JQ, Scott RA, Lindahl PA. Spectroscopic, redox, and structural characterization of the Ni-labile and nonlabile forms of the acetyl-CoA synthase active. Site of carbon monoxide dehydrogenase. J Am Chem Soc. 1998;120:7502–7510. [Google Scholar]
- 61.Xia JQ, Dong J, Wang SK, Scott RA, Lindahl PA. EXAFS, EPR, and electronic absorption spectroscopic study of the alpha metallosubunit of CO dehydrogenase from Clostridium thermoaceticum. J Am Chem Soc. 1995;117:7065–7070. [Google Scholar]
- 62.Mayhew SG. The redox potential of dithionite and SO2− from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur J Biochem. 1978;85:535–547. doi: 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- 63.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 64.Lindahl PA, Graham DE. Acetyl-Coenzyme A Synthases and Nickel Containing Carbon Monoxide Dehydrogenases. In: Sigel A, Sigel H, Sigel RKO, editors. Nickel and its Surprising Impact in Nature. John Wiley and Sons; West Sussex, England: 2007. pp. 357–416. [Google Scholar]
- 65.Lu WP, Ragsdale SW. Reductive activation of the coenzyme A/acetyl-CoA isotopic exchange reaction catalyzed by carbon monoxide dehydrogenase from Clostridium thermoaceticum and its inhibition by nitrous oxide and carbon monoxide. J Biol Chem. 1991;266:3554–3564. [PubMed] [Google Scholar]
- 66.Bhaskar B, DeMoll E, Grahame DA. Redox-dependent acetyl transfer partial reaction of the acetyl-CoA Decarbonylase/Synthase complex: kinetics and mechanism. Biochemistry. 1998;37:14491–14499. doi: 10.1021/bi9812423. [DOI] [PubMed] [Google Scholar]
- 67.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Mascharak PK, Baidya N. X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases. Inorg Chem. 1991;30:920–928. [Google Scholar]
- 68.Cramer SP, Eidsness MK, Pan WH, Morton TA, Ragsdale SW, DerVartanian DV, Ljungdahl LG, Scott RA. X-ray absorption spectroscopic evidence for a unique nickel site in Clostridium thermoaceticum carbon monoxide dehydrogenase. Inorg Chem. 1987;26:2477–2479. [Google Scholar]
- 69.Schrapers P, Mebs S, Ilina Y, Warner DS, Wörmann C, Schuth N, Kositzki R, Dau H, Limberg C, Dobbek H, Haumann M. The binuclear nickel center in the A-cluster of acetyl-CoA synthase (ACS) and two biomimetic dinickel complexes studied by X-ray absorption and emission spectroscopy. J Phys: Conf Ser. 2016;712:012029. [Google Scholar]
- 70.Neese F. The ORCA program system. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2012;2:73–78. [Google Scholar]
- 71.DeBeer George S, Petrenko T, Neese F. Prediction of iron K-edge absorption spectra using time-dependent density functional theory. J Phys Chem A. 2008;112:12936–12943. doi: 10.1021/jp803174m. [DOI] [PubMed] [Google Scholar]
- 72.Manesis AC, Shafaat HS. Electrochemical, Spectroscopic, and Density Functional Theory Characterization of Redox Activity in Nickel-Substituted Azurin: A Model for Acetyl-CoA Synthase. Inorg Chem. 2015;54:7959–7967. doi: 10.1021/acs.inorgchem.5b01103. [DOI] [PubMed] [Google Scholar]
- 73.Appel A, Bercaw J, Bocarsly A, Dobbek H, DuBois D, Dupuis M, Ferry J, Fujita E, Hille R, Kenis P, Kerfeld C, Morris R, Peden C, Portis A, Ragsdale S, Rauchfuss T, Reek J, Seefeldt L, Thauer R, Waldrop G. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem Rev. 2013;113:6621–6658. doi: 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.