

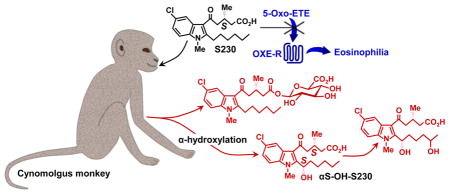
Abstract
We have developed a selective indole antagonist (230) targeting the OXE receptor for the potent eosinophil chemoattractant 5-oxo-ETE (5-oxo-6,8,11,14-eicosatetraenoic acid), that may be useful for the treatment of eosinophilic diseases such as asthma. In previous studies we identified ω2-oxidation of the hexyl side chain of racemic 230 as a major metabolic route in monkeys, but also obtained evidence for another pathway that appeared to involve hydroxylation of the hexyl side chain close to the indole. The present study was designed to investigate the metabolism of the active S-enantiomer of 230 (S230) and to identify the novel hydroxy metabolite and its chirality. Following oral administration, S230 rapidly appeared in the blood along with metabolites formed by a novel and highly stereospecific α-hydroxylation pathway, resulting in the formation of αS-hydroxy-S230. The chirality of α-hydroxy-S230 was determined by the total synthesis of the relevant diastereomers. Of the four possible diastereomers of α-hydroxy-230 only αS-hydroxy-S230 has significant OXE receptor antagonist activity and only this diastereomer was found in significant amounts in blood following oral administration of S230. Other novel metabolites of S230 identified in plasma by LC-MS/MS were αS,ω2-dihydroxy-S230 and glucuronides of S230 and ω2-hydroxy-S230. Thus the alkyl side chain of S230, which is essential for its antagonist activity, is also the major target of the metabolic enzymes that terminate its antagonist activity. Modification of this side chain might result in the development of related antagonists with improved metabolic stability and efficacy.
Keywords: Eicosanoids, 5-Lipoxygenase products, Inflammation, Granulocytes, Drug metabolism, Chiral analysis
Graphical abstract
1. Introduction
5-Oxo-6,8,11,14-eicosatetraenoic acid (5-oxo-ETE) is a product of the 5-lipoxygenase pathway, along with leukotrienes B4 and C4. It is formed by the NADP+-dependent oxidation of 5S-hydroxy-6,8,11,14-eicosatetraenoic acid (5S-HETE) by 5-hydroxyeicosanoid dehydrogenase [1], which is highly expressed in a variety of both inflammatory and structural cells [2]. The formation of 5-oxo-ETE is regulated by the availability of the cofactor NADP+, which is normally present at very low intracellular concentrations, but is dramatically elevated under conditions of oxidative stress, activation of the respiratory burst, and cell death [3]. 5-Oxo-ETE is a potent activator of human and monkey [4] eosinophils and is the only 5-lipoxygenase product with potent chemoattractant effects for these cells [5]. It acts via the selective OXE receptor, which is highly expressed on eosinophils and to a lesser extent on neutrophils and macrophages [6]. Intradermal injection of 5-oxo-ETE in humans results in the infiltration of eosinophils into the skin and this response is enhanced in asthmatic subjects [7].
The potent chemoattractant effects of 5-oxo-ETE on eosinophils, suggest that it may be an important proinflammatory mediator in eosinophilic disorders such as asthma and allergic rhinitis. For this reason, we sought to develop selective antagonists against the human OXE receptor, using calcium mobilization in human neutrophils as a screening test. We tested a series of indoles containing two side chains, one mimicking carbons 1–5 of 5-oxo-ETE and the other, carbons 15–20 of 5-oxo-ETE. We found that an indole containing a hexyl group in the 2-position and a 5-oxovalerate group in the 3-position selectively blocked 5-oxo-ETE-induced calcium mobilization. Modification of this compound led to the identification of the chiral compound 230 (Fig. 1) as a potent and selective OXE receptor antagonist [8], with the antagonist activity residing almost exclusively in the S-enantiomer (S230) [9].
Figure 1.

Inhibition of 5-oxo-ETE-induced eosinophil activation by S230.
To determine whether S230 may be useful therapeutically it will have to be tested in an animal model. Orthologs of the OXE receptor exist in many species but unfortunately not in rodents. We initially investigated the possibility that cats, which have an OXE receptor ortholog and are quite susceptible to asthma, might be a suitable experimental model for this purpose. Although 5-oxo-ETE is an extremely potent activator of feline eosinophils, 230 is only a weak antagonist in this species, presumably because of significant differences between the human and feline receptors [10]. We therefore plan to test 230 in monkeys. To do this it is first important to understand its metabolism and to establish whether adequate blood levels can be achieved. In an initial study we investigated the pharmacokinetics and metabolism of racemic 230, as at the time that the study was initiated we did not have a procedure for the synthesis of sufficient amounts of the pure S-enantiomer. We found that 230 is a potent OXE receptor antagonist in vitro in monkeys and that it rapidly appears in the blood, along with a number of metabolites, following oral administration [4]. Chiral analysis of 230 recovered from the blood revealed that the S-enantiomer was depleted more rapidly than the R-enantiomer. Although this study was not designed to follow the time courses of the metabolites, ω1- and ω2- oxidation products were identified by comparison with authentic standards in a blood sample taken 4 h after administration of 230. A number of other unidentified metabolites were also detected, including a pair of isomers that exhibited distinct chromatographic behaviour and may have been formed due to hydroxylation of the hexyl side chain α or possibly β to the indole moiety (see Fig. 1 for designation of the different carbons of the hexyl group).
The objective of the current study was to investigate the pharmacokinetics and metabolism of the active S-enantiomer of 230, which was made possible by a novel synthetic procedure that we recently developed [11]. We particularly wanted to conclusively identify the putative α- or β-hydroxy metabolite referred to above and to examine its antagonist activity, as hydroxylation of 5-oxo-ETE in the equivalent (ω6) position does not abolish its agonist activity [2].
2. Materials and Methods
2.1. Animal experiments
Three female cynomolgus monkeys weighing between 4.9 and 5.3 kg and housed at INRS-Institut Armand-Frappier, Laval, Quebec, were used for these studies. All experiments were performed in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the local institutional animal care committee.
S230, synthesized as described previously[11], was dissolved in EtOH at a concentration of 75 mg/mL and the appropriate volume added to 10 volumes of 20 mM NaHCO3 and the mixture vortexed. The resulting suspension (4.4 mL/kg) was administered to three monkeys by oral gavage at a dose of 30 mg/kg. Blood samples were collected in heparinized tubes just prior to gavage (1 mL) and 1, 4, 8, 12, and 24 h after gavage (4.5 to 5 ml for each time point, depending on the weight of the monkey). After centrifugation, plasma samples were frozen and stored at −80 °C.
2.2. Analysis of S230 and its metabolites by RP-HPLC
Solvents used for chromatographic analysis were purchased from Fisher Scientific, Markham, ON, Canada. Plasma samples were thawed and diluted by the addition of MeOH (2 volumes), followed by addition of the internal standard (1 μg 5-(5-chloro-1-methyl-2-(5-methylhexyl)-1H-indol-3-yl)-3-methyl-5-oxopentanoate; identical to 230 except that it contains an isoheptyl group instead of a hexyl group; synthesized as previously described [4]). After storing overnight at −80 °C the samples were thawed and centrifuged. The concentration of MeOH was adjusted to 30% by the addition of water and each sample was loaded onto a Sep-Pak C18 cartridge (Waters Corporation, Milford Massachusetts) [12], which was washed with 30% MeOH prior to the elution of indole metabolites with 100% MeOH. After evaporation of the solvent using a rotary evaporator the residue was dissolved in 30% MeOH and analyzed by precolumn extraction/RP-HPLC [13]. The precolumn was a C18 SecurityGuard cartridge (3.2 × 8 mm ID) from Phenomenex, Torrance, CA, whereas the analytical column was a Kinetex C18 column (5 μm particle size; 4.6 mm × 250 mm; Phenomenex;) with a linear gradient between 35 and 66% MeCN containing 0.02% HOAc over 62 min at a flow rate of 1 ml/min and a column temperature of 35 °C. Fractions corresponding to peaks of interest were collected and combined for subsequent analysis by LC-MS/MS.
The concentrations of 230 and its major metabolites were determined by measuring the ratios of their peak areas with that of the internal standard. The identities of the metabolites that were quantitated were confirmed by LC/MS/MS in all cases, as well as by cochromatography with authentic standards when these were available (230, ω2-OH-230, ω2-oxo-230, and αS-OH-S230). As we did not have standards for the glucuronide metabolites or αS,ω2-diOH-S230 we assumed that their extinction coefficients were identical to those of their precursors. The HPLC conditions were as described above except that a methanol gradient was used: 60 to 80% MeOH in H2O with 0.02% HOAc over 60 min at a temperature of 35 oC and a flow rate of 1 ml/min. The properties used for the quantitation of each compound were as follows: α,ω2-di-OH-S230 (tR, 13.9 min; λ, 311 nm; ε, 12,830), ω2-OH-S230 glucuronide (tR, 15.3 min; λ, 305 nm; ε, 12,710), ω2-oxo-S230 (tR, 16.6 min; λ, 305 nm; ε, 12,710), ω2-OH-S230 (tR, 19.3 min; λ, 305 nm; ε, 12,710), α-OH-S230 (tR, 35.1 min; λ, 311 nm; ε, 12,830), S230 glucuronide (tR, 38.6, min; λ, 305 nm; ε, 12,710), S230 (tR, 43.7 min; λ, 305 nm; ε, 12,710), internal standard (1 μg 5-(5-chloro-1-methyl-2-(5-methylhexyl)-1H-indol-3-yl)-3-methyl-5-oxopentanoate (tR, 50.4 min; λ, 305 nm; ε, 12,710).
2.3. Chiral HPLC
Chiral HPLC was carried out using a Lux Cellulose-2 column (5 μm particle size; 250 × 4.6 mm; Phenomenex). The mobile phases used are described in the legends to the relevant figures. A flow rate of 1 mL/min and a column temperature of 45 °C were used throughout.
2.4. Identification of S230 metabolites by LC-MS/MS
Fractions corresponding to the HPLC peaks shown in Fig. 5A were collected, with identical fractions from different time points being pooled. These fractions were then analyzed by LC/MS/MS using a model 1100 HPLC system (Agilent Technologies, Santa Clara, CA) connected to an LTQ Velos Orbitrap high resolution mass spectrometer via a heated electrospray ionization source (Thermo Scientific, San Jose, CA). Chromatographic separation was performed using a Phenomenex Kinetex C18 column (2.6 μm particle size; 50 mm × 2.1 mm) at the flow rate of 0.3 mL/min and a column temperature of 25 °C. The mobile phase was a gradient between solvents A (0.02% HOAc in H2O) and B (0.02% HOAc in MeCN) as follows: 0 min, 30% B; 1 min, 30% B; 20 min, 65% B. Samples were injected in a volume of 10 μL and analyzed by negative electrospray ionization (ESI) mode using the following settings: capillary temperature, 350°C; source heater temperature, 300 °C; sheath gas flow, 20; auxiliary gas flow, 10; capillary voltage, −3.0 kV. The MS settings were: S lens RF level, 60%; automatic gain control (AGC) target, 1 × 106 ions; mass range, m/z 250 to m/z 700; resolution, 100,000. Multiple levels of MSn analysis with data dependent acquisition (DDA) mode were used for the identification and elucidation of antagonist metabolites. In DDA mode, the selection of the precursor ion for MS2 analysis was based on the chlorine isotope pattern and/or isolation of the top three most intense ions from the full MS scan for fragmentation with dynamic exclusion disabled; activation type, collision-induced dissociation (CID); signal threshold, 5000; normalized collision energy, 35; isolation width, 2 Da; activation time, 30 s. MS3 used parent and product mass lists to trigger MS3 for selected ions and was performed with CID as the activation type; minimal signal threshold, 5000; isolation width, 2 Da; activation time, 30 s; normalized collision energy, 45.
Figure 5. Identification of plasma metabolites of S230 by RP-HPLC, UV absorbance and mass spectrometry.

A: Plasma was prepared from a blood sample taken from a cynomolgus monkey 8 h after oral administration of S230 (30 mg/kg). After solid-phase extraction the sample was analyzed by RP-HPLC using a Kinetex ODS-silica column with a gradient between 35 and 66% MeCN in H2O over 62 min. B: UV spectra of 230 and its major plasma metabolites formed by oxidation of the hexyl side chain. C: UV spectra of glucuronide metabolites a and c of 230. D: MS2 fragmentation of the [M-H]− ion at m/z 408 for plasma metabolite b. E: MS3 fragmentation of the ion at m/z 292 for metabolite b from panel D. F: MS2 fragmentation of the [M-H]− ion at m/z 552 for metabolite c. G: MS3 fragmentation of the ion at m/z 376 from metabolite c shown in panel F. H: MS2 fragmentation of the [M-H]− ion at m/z 568 for metabolite a. I: MS3 fragmentation of the ion at m/z 392 from metabolite a shown in panel G. J: MS2 fragmentation of synthetic ω2-OH-230 for comparison with the spectrum shown in panel I. K: MS2 fragmentation of 230 for comparison with the spectrum shown in panel G. * Ion formed due to a McLafferty rearrangement.
2.5. Evaluation of potencies of α-hydroxy-230 diastereomers in human neutrophils
The antagonist effects of α-hydroxy-230 diastereomers on 5-oxo-ETE-induced calcium mobilization in human neutrophils were examined as described previously [8]. Neutrophils prepared by dextran sedimentation and centrifugation over Ficoll-Paque (GE Healthcare, Mississauga, ON, Canada) were loaded with indo-1 AM (ThermoFisher Scientific, Waltham, MA) and placed in a thermostatted cuvette at 37 °C. After stabilization of baseline fluorescence, the α-hydroxy compound was added. 5-Oxo-ETE (10 nM; synthesized as previously described [14]) was added 2 min later followed 1 min later by digitonin (final concentration 0.1%). Fluorescence was measured using a Cary Eclipse spectrofluorometer (Agilent Technologies, Santa Clara, CA).
2.6. Chemistry
All reagents and solvents used for chemical synthesis were purchased from Sigma-Aldrich, Saint Louis, MO. All reactions were carried out under an argon atmosphere using dried, nitrogen-purged glassware and dry solvents. Reaction progress was monitored using Merck TLC Silica gel 60 F254 plates. The plates were visualized using 254 nm UV light, iodine chamber and anisaldehyde dip where appropriate, followed by gentle warming. 1H NMR and 13C NMR spectra were recorded on a BRUKER AMX 400 MHz spectrometer at rt in CDCl3, using TMS as an internal standard. The following abbreviations are used for the description of NMR spectra: s- singlet; d- doublet; t- triplet; q- quartet; dd- doublet of doublets and m- multiplet. High resolution mass spectrometry (HRMS) was performed using an AccuTOF mass spectrometer with positive ion ESI mode and DART as an ion source. The purity of all tested compounds was determined to be >95% by a combination of HPLC, NMR and HRMS.
2.7. X-ray Single Crystal Diffractometry
Single crystals suitable for X-ray diffraction were mounted on a Mitegen micromesh mount with the help of a trace of mineral oil. Data were collected on a Rigaku R-axis curved image plate diffractometer equipped with a MicroMax002+ high-intensity copper X-ray source with confocal optics and examined with Cu Kα radiation (λ = 1.54184 Å). Data were collected using the d*TREK option of CrystalClear (Rigaku, The Woodlands, TX). All data sets were processed using HKL3000 [15] and were corrected for absorption and scaled using Scalepack [15]. The space group was assigned using XPREP from the Shexltl suite of programs (SHELXTL (2003) Bruker Advanced X-ray Solutions, Bruker AXS Inc., Madison, Wisconsin: USA) and the structures were solved by direct methods with SHELXS [16] and refined by full matrix least squares against F2 with all reflections using SHELXL-2014 [17] using the graphical user interface ShelXle [18]. H atoms attached to carbon and oxygen atoms were positioned geometrically and constrained to ride on their parent atoms, with carbon hydrogen bond distances of 0.95 Å for aromatic and alkene C-H, and 0.99 and 0.98 Å for aliphatic CH2 and CH3 moieties. Uiso(H) values were set to 1.5 times times Ueq(C/O) for methyl and hydroxyl hydrogen atoms, and to 1.2 times Ueq(C) for CH and CH2 moieties. Complete structures in CIF format have been deposited with the Cambridge Crystallographic Database. CCDC 1527527 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
2.8. 1-(5-chloro-1-methyl-1H-indol-2-yl)hexan-1-ol (3)
To a stirred solution of 5-chloro-1-methyl-1H-indole-2-carbaldehyde (1) (1.675 g, 8.651 mmol) in THF (25 mL) was added pentyl magnesium bromide (2) (9 mL, 2.0 M solution in Et2O) at −78°C dropwise over a period of 5 min. The reaction mixture was stirred at −78 °C for 20 min and then at rt for 15 min. Aqueous NH4Cl (20 mL) was added and the organic layer was extracted with EtOAc (3 × 25 mL). The combined organic extracts were washed with brine (50 mL), dried over Na2SO4, and the solvents were evaporated to obtain the crude mixture. Silica gel (from VWR International, Radnor, PA) column chromatography (15% EtOAc/hexane) afforded the product as a yellow solid (2.089 g, 91%). HRMS (ESI) m/z calcd for [C15H20ClNO + H]+, 266.1311; found 266.1616. 1H NMR (400 MHz, CDCl3): δ 7.53 (d, J = 1.6 Hz, 1H), 7.21 (d, J = 8.7 Hz, 1H), 7.15 (dd, J = 8.7 Hz, 1.9 Hz, 1H), 6.37 (S, 1H), 4.84 – 4.80 (m, 1H), 3.80 (s, 3H), 2.00 – 1.94 (m, 2H), 1.62 – 1.34 (m, 6H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (CDCl3): δ 143.4, 136.4, 128.1, 125.1, 122.0, 120.0, 110.1, 98.6, 67.3, 36.2, 31.7, 30.3, 25.8, 22.6, 14.0.
2.9. 2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indole (4)
To a stirred solution of 1-(5-chloro-1-methyl-1H-indol-2-yl)hexan-1-ol (3) in CH2Cl2 (8 ml) was added TBDMSCl (200 mg, 1.327 mmol) followed by imidazole (150 mg, 2.203 mmol) and the reaction mixture was stirred at rt for 14 h. H2O (8 mL) was added and the organic layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were washed with aqueous NaHCO3 (20 mL), dried over Na2SO4, concentrated to dryness, and purified using silica gel column chromatography (5% EtOAc in hexane) to obtain the product as a pale-yellow viscous oil (290 mg, 87%). HRMS (ESI) m/z calcd for [C21H34ClNOSi + H]+, 380.2176; found 380.2048. 1H NMR (400 MHz, CDCl3): δ 7.49 (d, J = 1.8 Hz, 1H), 7.19 (d, J = 8.7 Hz, 1H), 7.12 (dd, J = 8.7 Hz, 1.9 Hz, 1H), 6.22 (S, 1H), 4.83 (t, J = 7.0 Hz, 1H), 3.80 (s, 3H), 1.93 – 1.84 (m, 1H), 1.79 – 1.71 (m, 1H), 1.46 – 1.26 (m, 6H), 0.88 – 0.84 (m, 12H), 0.00 (s, 3H), −0.18 (s, 3H). 13C NMR (CDCl3): δ 143.7, 136.6, 128.4, 125.0, 121.3, 119.7, 109.8, 99.5, 70.1, 38.1, 31.5, 31.0, 25.8 (3C), 25.7, 22.6, 18.1, 14.0, −4.8, −5.2.
2.10. Methyl 5-(2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoate (6)
To a 100 mL round bottom flask containing 2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indole (4) (127 mg, 0.334 mmol) in CH2Cl2 (16 mL) was added methyl 5-chloro-3-methyl-5-oxopentanoate (5) (80 mg, 0.448 mmol) in CH2Cl2 (1 mL). The solution was cooled to 0°C, and Me2AlCl (0.43 mL, 1.0 M solution in hexanes) was added slowly over a period of 5 minutes. The reaction mixture was stirred at 0 °C for 1 h and then at rt for 2 h. Aqueous NaHCO3 solution (10 mL) was then added to quench the reaction. The aqueous layer was extracted with EtOAc (3 × 8 mL), and the combined organic extracts were washed with brine (20 mL), dried over Na2SO4, the volatiles removed under reduced pressure, and the resulting crude product was purified using silica gel column chromatography (2% EtOAc/hexane) to afford compound 6 as a pair of diastereomers (67 mg, 38%). HRMS (ESI) m/z calcd for [C28H44ClNO4Si + H]+, 522.2806; found 522.2985.
2.11. 5-(5-chloro-2-(1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (α-OH-230)
To a stirred solution of methyl 5-(2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoate (6) (10 mg, 0.0191 mmol) in CH3CN (5 mL) was added HF.Pyridine (0.1 ml, 30% pyridine, 70% HF) at 0 °C. The reaction mixture was allowed to warm to rt and stirred for 30 min. Aqueous NaHCO3 solution (5 mL) was added, the aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were dried over Na2SO4. Evaporation of the solvents in vacuo gave the crude product as a pale yellow solid that was dissolved in THF (2.5 mL) and H2O (0.6 mL). To this solution was added LiOH.H2O (32 mg, 0.763 mmol) and MeOH (0.1 mL). The reaction mixture was stirred at rt for 14 h. H2O (5 mL) was added and the aqueous layer was extracted with EtOAc (3 × 5 mL), and the organic extract dried over Na2SO4, concentrated to dryness, and the product purified using silica gel column chromatography (4% MeOH/CH2Cl2) to isolate the product as a pair of diastereomers (pale-yellow viscous oil, 6.1 mg, 80% over two steps). HRMS (ESI) m/z calcd for [C21H28ClNO4 + H]+, 394.1785; found 394.1883.
2.12 (S)-1-(5-chloro-1-methyl-1H-indol-2-yl)hexan-1-ol (7)
To a stirred solution of (S)-BINOL (503 mg, 1.757 mmol) in tBuOMe (52 mL) was added Ti(O-iPr)4 (3.5 mL, 11.945 mmol) dropwise at rt, and the resulting red solution was stirred for 30 min. In another flask, containing a solution of BDMAEE (5.3 mL, 27.813) in tBuOMe (52 mL) was added freshly prepared pentyl magnesium bromide (27.094 mmol in 20 mL THF) at 0 °C. The resulting cloudy suspension was stirred at 0 °C for 35 min. The mixture from the former flask was transferred slowly into the latter at −15 °C. The reaction mixture was allowed to warm to rt and then stirred for 1 h. After that, 5-chloro-1-methyl-1H-indole-2-carbaldehyde (1) (1.553 g, 8.020 mmol) in THF (20 mL) was added dropwise at −15 °C over a period of 5 min. The reaction mixture was allowed to warm to rt and stirred for 36 h. The reaction was quenched by adding aqueous NH4Cl solution (60 mL). The two layers were separated, the aqueous layer was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with brine (60 mL), dried over Na2SO4, filtered, and the solvents removed under reduced pressure. The brown viscous crude product was purified using silica gel column chromatography (10% EtOAc/hexane) to afford the product as a yellow solid (1.027 g, 48%). (See the experimental of the racemic compound 2 for the spectral properties). Using chiral HPLC, the %S enantiomer was determined to be 97.1. Recrystallization using hexane-methanol improved the enantiomeric purity to 100%.
2.13. (S)-2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indole (8)
The procedure was identical to that described for 2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indole (4).
2.14. (S)-5-(5-chloro-2-((S)-1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (αS-OH-S230)
To a stirred solution of (S)-2-(1-((tert-butyldimethylsilyl)oxy)hexyl)-5-chloro-1-methyl-1H-indole (8) (47 mg, 0.124 mmol) in CH2Cl2 (8 mL) was added methyl (R)-5-chloro-3-methyl-5-oxopentanoate (27 mg, 0.151 mmol) in CH2Cl2 (1.1 mL). The solution was cooled to 0 °C, and Me2AlCl (0.14 mL, 1.0 M solution in hexanes) was added over 15 min. The reaction mixture was then stirred at 0 °C for 1 h and then at rt for a further 24 h. Aqueous NaHCO3 (5 mL) was added and the aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic extracts were washed with brine (10 mL), dried over Na2SO4, concentrated to dryness, and the product was purified using silica gel column chromatography (2% EtOAc/hexane) to afford a yellow viscous oil containing the acylated product along with some inseparable impurities. The mixture, which was used as is without further purification, was dissolved in CH3CN (7 mL), followed by the addition of HF-Pyridine (0.18 mL, 30% pyridine, 70% HF) at 0 °C. The reaction mixture was stirred at rt for 30 min. Aqueous NaHCO3 (4 mL) was added and the aqueous layer was extracted with EtOAc (3 × 4 mL) and the organic extract dried over Na2SO4. After removal of the solvents in vacuo the crude product was passed through a short silica gel column and the combined filtrate was concentrated to obtain a yellow viscous oil that was subjected to ester hydrolysis under basic conditions. The yellow viscous oil was dissolved in THF (2 mL) and H2O (0.5 mL). To the stirred solution was added LiOH.H2O (24 mg, 0.572 mmol) and a drop of MeOH. The reaction mixture was stirred at rt for 16 h. H2O (10 mL) was added and the aqueous layer was extracted with EtOAc (3 × 4 mL), and the organic extract dried over Na2SO4, concentrated to dryness, and the product was purified using silica gel column chromatography (10% MeOH/CH2Cl2) to obtain the final product as a yellow viscous oil, 8.6 mg, 18% over three steps). HRMS (ESI) m/z calcd for [C21H28ClNO4 + H]+, 394.1785; found 394.1892. The diasteromeric ratio was determined to be 81/19 using chiral HPLC.
2.15. (R)-1-(5-chloro-1-methyl-1H-indol-2-yl)hexan-1-ol (10)
The procedure was identical to that described for (S)-1-(5-chloro-1-methyl-1H-indol-2-yl)hexan-1-ol (7) except (R)-BINOL was used instead of (S)-BINOL.
2.16. (S)-5-(5-chloro-2-((R)-1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (αR-OH-S230)
The procedure was identical to that described for (S)-5-(5-chloro-2-((S)-1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (α(S)-OH-S230).
3. Results
3.1. Identification of α-hydroxy metabolites of 230
We previously detected two isomeric forms of a plasma metabolite of 230 (metabolites d and e in Fig. 2A) with retention times between those of 230 and ω2-OH-230. Mass spectral evidence suggested that these isomers might be formed by hydroxylation of the alkyl side chain α, or possibly β, to the indole. However, we were unable to conclusively identify these isomers because of the lack of an authentic synthetic standard for comparison. To address the hypothesis that metabolites d and e are two stereoisomers of α-OH-230 we prepared a mixture of the 4 possible diastereomers of this compound2 as shown in Fig. 3. The α-OH center was introduced using a Grignard reaction between 5-chloro-1-methyl-1H-indole-2-carbaldehyde (1) and pentyl magnesium bromide (2). Because the free α-OH group was not tolerated in a Friedel-Craft’s reaction to introduce the 5-oxo-valerate side chain, it was first protected by conversion to a silyl ether (OTBDMS) derivative. RP-HPLC of synthetic α-OH-230 gave two peaks, labeled x and y in Fig. 2B, which had retention times identical to those of plasma-derived d and e (Fig. 2C) when chromatographed under the same conditions. Furthermore, d and e cochromatographed with x and y when equal amounts of these substances were mixed together Fig. 2D). We previously showed [4] that d and e have identical mass spectra, with [M-H]− ions consistent with monohydroxy metabolites of 230 and MS2 fragmentation patterns showing a single major ion at m/z 292 corresponding to loss of the alkyl side chain, which was consistent with the MS3 fragmentation pattern of this ion. In the present study we examined the mass spectral fragmentation of x and y derived from authentic α-OH-230. Both x and y had [M-H]− ions at m/z 392.1633, compared to the theoretical value of m/z 392.1629 expected for α-OH-230 (mass accuracy, 1.0 ppm) and had identical MS2 spectra (Fig. 2E), which contained a single major ion at m/z 292 (loss of alkyl group and a smaller ion at m/z 274 (loss of H2O). The MS3 profile resulting from fragmentation of the ion at m/z 292 (Fig. 2F) displayed major ions at m/z 274 (loss of H2O), 248 (loss of CO2), 206 (loss of CH2=CH-CH2-CO2H due to a McLafferty rearrangement), and 180. Both the MS2 and MS3 spectra of x and y are identical to those that we previously reported for d and e [4], confirming the identities of these metabolites as α-OH-230 diastereomers.
Figure 2. Cochromatography of metabolites d and e from plasma with synthetic α-hydroxy-230 diastereomers.

A: Plasma from a monkey obtained 4 h after oral administration of racemic 230 (30 mg/kg) was extracted as described in Materials and Methods and an aliquot was analyzed by RP-HPLC on a Kinetex ODS-silica column using a 30 to 65% gradient of MeCN in H2O over 30 min. B–D: RP-HPLC of synthetic racemic α-hydroxy-230 (B), metabolites d and e from plasma (C) and a mixture of plasma metabolites and synthetic racemic α-hydroxy-230 (D) using a 35 to 56% gradient of MeCN in H2O over 42 min. E: MS2 fragmentation of the [M-H]− ion at m/z 392 of synthetic α-OH-230 (peak y). F: MS3 fragmentation of the ion at m/z 292 shown in panel E. * Ion formed due to a McLafferty rearrangement.
Figure 3.

Procedure for the synthesis of 5-(5-chloro-2-(1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (α-OH-230).
As it contains two chiral carbons, we would expect synthetic α-OH-230 to be a 1:1:1:1 mixture of four diastereomers. As pairs of enantiomers are unlikely to be separated by RP- or NP- HPLC, we would assume that product x is a mixture of either the αS-OH,S-Me and αR-OH,R-Me diastereomers or alternatively the αS-OH,R-Me and αR-OH,S-Me diastereomers, and vice versa for product y. To investigate their antagonist potencies, we purified all four diastereomers by a combination of NP- and chiral HPLC. NP-HPLC of α-OH-230 gave 2 peaks, x and y, in the same elution order as with RP-HPLC (Fig. 4A). Product x was separated into two diastereomers, x1 and x2, by chiral HPLC (Fig. 4B). Chiral HPLC also separated product y into two diastereomers, y1 and y2 (Fig. 4C). The effects of the above four diastereomers on 5-oxo-ETE-induced calcium mobilization are shown in Fig. 4D. Only y1 had appreciable antagonist activity with an IC50 of 1.6 ± 0.3 μM, over 100 times higher than that of S230 (IC50, 13 ± 2 nM in these experiments). The other three α-OH-230 diastereomers had only very weak effects, inhibiting calcium mobilization by between 4 and 9% at a concentration of 10 μM.
Figure 4. Separation and antagonist activity of diastereomers of synthetic racemic α-OH-230.

A: NP-HPLC of synthetic α-OH-230 was performed as described in Materials and Methods. B: Peak x from panel A was subjected to chiral-HPLC on a Cellulose-2 column as described in Materials and Methods with a mobile phase of hexane/EtOH/HOAc (85:15:0.1). C: Peak y from panel E was subjected to chiral-HPLC on a Cellulose-2 column with a mobile phase of hexane/EtOH/HOAc (93:7:0.1). D: The effects of S230 and diastereomers x1 (▲), x2 (△), y1 (●) and y2 (○), purified as shown in panels A to C, on 5-oxo-ETE-induced calcium mobilization in human neutrophils were determined as described in Materials and Methods.
3.2. Identification of plasma metabolites of S230 in cynomolgus monkeys
To investigate the chirality of metabolites d and e and the time course for their formation we conducted in vivo experiments in cynomolgus monkeys. Because nearly all of the antagonist activity of 230 resides in the S enantiomer, we focused on S230, which was administered to three monkeys by oral gavage at a dose of 30 mg/kg. Blood samples were obtained after 1, 4, 8, 12, and 24 h and S230 and its metabolites were extracted from plasma and analyzed by RP-HPLC. Fig. 5A shows the profile of S230 metabolites in plasma after 8 h (note that a shallower gradient was used for this chromatogram compared to that shown in Fig. 2A). A substantial amount of e was formed from S230, but in contrast to racemic 230, very little d was detected. Since the chiral methyl group of the substrate is in the S configuration, this suggests that the α-hydroxylation reaction is stereospecific, with the alkyl hydroxyl group being almost exclusively in either the R or S configuration. Metabolite e should therefore be either αS-OH-S230 or αR-OH-S230. These diastereomers should be separable by RP-HPLC, but authentic standards would be required to distinguish between them.
In addition to the two ω2-oxidized metabolites that we had previously identified [4] and α-OH-S230, we observed several other major metabolites of S230, including a and b, which were considerably more polar than ω2-OH-S230 (Fig. 5A). A minor product (c) was also observed after 8 h, but was much more prominent 1 h after administration of S230 (inset to Fig. 5A). The polar metabolite b has a UV spectrum almost identical to that of metabolite e, with a maximum at 312 nm, compared to about 305 nm for 230 and its ω2-oxidation products (Fig. 5B), suggesting that it might also have an alkyl hydroxyl group α to the indole. The mass spectrum of b revealed an [M-H]− ion at m/z 408.1571, compared to the theoretical value of m/z 408.1578 expected for a dihydroxy metabolite of 230 (mass accuracy, 1.7 ppm). The MS2 fragmentation pattern of b (Fig. 5D) is very similar to that of authentic α-OH-230 (Fig. 2E) except that the ion formed by loss of H2O was observed at m/z 390 instead of 374, consistent with the presence of an additional hydroxyl group. The MS3 fragmentation profile of the major ion in Fig. 5D at m/z 292 (Fig. 5E) formed by loss of the alkyl side chain was virtually identical to that shown in Fig. 2F for α-OH-230. From these data we conclude that b is a hydroxy metabolite of α-OH-230, most likely α,ω2-diOH-230.
Two additional major metabolites of S230 were also detected: c, which was prominent only at 1 h, and a, which was detected at all time points investigated. The UV spectra of a and c (Fig. 5C) are virtually identical to that of S230, indicating that they do not have a hydroxyl group on the alkyl side chain α to the indole. The mass spectrum of metabolite c had an [M-H]− ion at m/z 552.1957, compared to the theoretical value of m/z 552.2000 expected for 230 glucuronide (mass accuracy, 7.8 ppm). The MS2 fragmentation pattern for c had a single major ion at m/z 376 due to the characteristic loss of m/z 176 from a glucuronide (Fig. 5F). The MS3 spectrum of the latter ion (Fig. 5G) was virtually identical to the MS2 spectrum of 230 (Fig. 5K) with major ions at 358 (loss of H2O), 332 (loss of CO2), 316 (loss of CH2C(OH)2 due to a McLafferty rearrangement), 290 (loss of CH2=CH-CH2-CO2H due to a different McLafferty rearrangement), and 248 (loss of the acyl side chain).
Metabolite a was also identified as a glucuronide. It had an [M-H]− ion at m/z 568.1950, identical to the theoretical value expected for a glucuronide of a monohydroxy metabolite of 230. The MS2 spectrum of a (Fig. 5H) had a single ion at m/z 392, consistent with the loss of glucuronide (m/z 176). The MS3 fragmentation pattern of this ion was virtually identical to that of authentic ω2-OH-230 (Fig. 5J), prepared as previously described [4], with ions at m/z 374 (loss of H2O), 348 (loss of CO2), 332 (loss of CH2C(OH)2 due to a McLafferty rearrangement), 330 (loss of H2O and CO2), 306 (loss of CH2=CH-CH2-CO2H due to a different McLafferty rearrangement), 264 (loss of acyl side chain), and 246 (loss of the acyl side chain + H2O). These results indicate that a is the glucuronide derivative of an ω-hydroxy metabolite of S230, probably ω2-OH-S230 glucuronide.
3.3. Pharmacokinetics of S230 and its metabolites
S230 and its metabolites were quantitated by RP-HPLC using an isoheptyl analog of S230, which has a longer tR than S230, as an internal standard. The identities of all compounds quantitated were confirmed by LC-MS/MS as well as by cochromatography when the authentic standards were available.
S230 rapidly appeared in the blood, reaching a maximal concentration of 32 μM within 1 h and then declining to about 11% of this level by 4 h and to 5% by 8 h (Fig. 6A). Although they are quite high, these concentrations are lower than those that we previously reported for racemic 230 [4], also shown in Fig. 6A for comparison. In contrast, a biphasic pattern was observed with α-OH-S230, which reached maximal levels by 1 h, declined, and then rose again at 8 h (Fig. 6B). As would be expected, the concentration of the dihydroxy metabolite of S230 rose more slowly, reaching a maximum by 8 h and then declining. ω2-OH-S230 increased to maximal levels by 4 h and then declined, whereas the maximal level of its oxidation product ω2-oxo-S230 was observed at 8 h (Fig. 6C). The glucuronide metabolites of S230 both peaked at 1 h and then declined (Fig. 6D). In contrast to S230 glucuronide, which dropped to very low levels by 4 h, the time course for ω2-OH-S230 glucuronide was biphasic, with a second maximum being observed at 8 h.
Figure 6. Levels of S230 and its major metabolites in plasma.

Plasma was prepared from blood taken from cynomolgus monkeys at various times after oral administration of S230 (30 mg/kg). After solid-phase extraction, components were analyzed as illustrated in Fig. 5 except that a gradient of MeOH (60 to 80% over 60 min) in H2O was used (see Materials and Methods for further details). Plasma levels are shown for A: S230 (▲) and racemic 230 (△; data from ref 4), B: α-OH-S230 (●) and α,ω2-diOH-S230 (○), C: ω2-OH-S230 (●) and ω2-oxo-S230, and D: S230 glucuronide (●; Gluc) and ω2-OH-S230 glucuronide (○; ω2-OH-G). The scales for the Y-axes in panels B, C, and D are identical.
3.4. Chiral synthesis of α-hydroxy diastereomers of S230
To determine the chirality of the α-hydroxyl group in metabolite e we synthesized both αS-OH-S230 and αR-OH-S230 using a previously reported method for the stereospecific addition of Grignard reagents to aromatic aldehydes [19]. Accordingly, (S)-BINOL-mediated enantioselective addition of pentyl magnesium bromide (2) to the prochiral aldehyde 1 yielded the α-hydroxy compound, 97.1% of which was the S enantiomer 7 (Fig. 7). The small amount of R-enantiomer was completely removed after recrystallization as demonstrated by chiral HPLC (Fig. 8B) using conditions that completely separate the R and S enantiomers (Fig. 8A). The absolute configuration of 7 was confirmed using single crystal x-ray diffractometry (Fig. 8C). The α-OH group of 7 was protected using TBDMS to obtain 8. Friedel-Craft’s acylation of 8 using the chiral acyl side chain 9 [11], followed by removal of the TBDMS with HF-pyridine and ester hydrolysis under basic conditions, afforded the desired compound αS-OH-S230 with a chiral purity of 78% as determined by chiral HPLC.
Figure 7.

Procedure for the synthesis of (S)-5-(5-chloro-2-((S)-1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (α(S)-OH-S230).
Figure 8. Chiral HPLC and X-ray crystal structure of 7.

Chiral HPLC of the racemic version of 7 (A) and crystallized synthetic 7 (S-enantiomer) (B) on a Cellulose-2 column using hexane/MeOH/HOAc (98:2:0.1) as the mobile phase at a flow rate of 1 ml/min and a column temperature of 45 °C. C: X-ray crystal structure of 7. Thermal ellipsoids are set at the 50% probability level.
The α(R)-hydroxy compound, αR-OH-S230, was prepared in a similar fashion as shown in Fig. 9. The enantioselective addition of the Grignard reagent 2 to the aldehyde 1 in the presence of (R)-BINOL afforded the α-hydroxy compound, 97.3% of which was the R enantiomer 10. Compound 10 was then treated similarly to the S-enantiomer 7 to yield the final product αR-OH-S230 with a chiral purity of 72%.
Figure 9.

Procedure for the synthesis of (S)-5-(5-chloro-2-((R)-1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoic acid (α(R)-OH-S230).
3.5. Identification of metabolite e as αS-OH-S230
To determine the configuration of the α-hydroxy metabolite e we compared its chromatographic properties to those of the synthetic compounds αS-OH-S230 and αR-OH-S230. Initial studies revealed that with RP-HPLC the αR-OH diastereomer (labeled “RS” in Fig. 10A) has a shorter retention time than the αS-OH diastereomer (labeled “SS” in the figure). The tR of metabolite e under the same conditions was identical to that of αS-OH-S230 (Fig. 10B) and this was confirmed by cochromatography of e with a mixture of the two synthetic α-hydroxy compounds (Fig. 10C). There appeared to be a very small peak (d) corresponding to the elution position of αR-OH-S230 (Fig. 10B). We measured the area of this peak at all time points and in all cases it amounted to about 3% of the area of metabolite e (Fig. 10G).
Figure 10. Cochromatography of metabolite e with αS-OH-S230.

A–C: Synthetic α-OH-S230 diastereomers and metabolite e isolated from plasma were analyzed by RP-HPLC using a Kinetex ODS column with a mobile phase consisting of a gradient between 45 and 55% MeCN in H2O over 30 min with a flow rate of 1 ml/min and a column temperature of 35 °C. A: a mixture of synthetic αS-OH-S230 (SS) and αR-OH-S230 (RS). B: Metabolite e purified from plasma from a monkey following administration of 230 (30 mg/kg). The arrow shows the elution position of αR-OH-S230 (RS), run separately. C: Cochromatography metabolite E with a mixture of the synthetic SS and RS isomers of α-OH-S230. D–F: Chiral HPLC of the above compounds on a Cellulose-2 column with a mobile phase of hexane/EtOH/HOAc (93:7:0.1) and a flow rate and column temperature of 1 ml/min and 45 °C, respectively. D: a mixture of synthetic αS-OH-S230 (SS) and αR-OH-S230 (RS); E: Metabolite e purified from plasma. F: Cochromatography metabolite e with a mixture of the synthetic SS and RS isomers of α-OH-S230. The elution positions of αR-OH-R230 (RR), and αS-OH-R230 (SR), run separately, are shown by arrows. G: Levels of metabolites d and e in plasma following the administration of S230 (30 mg/kg) by oral gavage.
Similar experiments were performed using chiral HPLC. In this case, the tR of αS-OH-S230 was shorter than that of the corresponding αR-OH diastereomer (Fig. 10D). The tR of e (Fig. 10E) was identical to that of αS-OH-S230 and the two compounds cochromatographed with one another (Fig. 10F). Furthermore, we analyzed metabolite e, purified by RP-HPLC using an acetonitrile gradient, by chiral HPLC at all time points investigated and did not detect any other diastereomers (data not shown). Therefore, we can conclude that e is identical to αS-OH-S230. Metabolite d derived from S230 is therefore probably identical to αR-OH-S230, although the amount detected is too small to be certain of this.
4. Discussion
In our previous studies we identified racemic 230 as a potent 5-oxo-ETE antagonist [8] that rapidly appears in the blood following oral administration to monkeys [4]. By preparing a series of potential metabolites of 230 by total chemical synthesis we identified ω2-oxidation of the hexyl side chain as a major metabolic pathway for this compound. However, we also observed a number of additional metabolites, including a pair of isomeric compounds that displayed a bathochromic shift in their UV spectra compared to 230 (305 nm → 311 nm), suggestive of a modification close to the indole chromophore. This was supported by mass spectral evidence showing that these metabolites possess an additional hydroxyl group in the alkyl side chain α or possibly β to the indole, but we were unable to confirm their identities due to the lack of authentic standards.
For the reasons discussed above we hypothesized that the isomeric metabolites d and e absorbing at 311 nm are a mixture of isomers of α-OH-230, which has 2 chiral carbons and therefore 4 possible diastereomers. We initially developed a procedure for the synthesis of a mixture of all four diastereomers. RP-HPLC of this mixture gave two peaks that cochromatographed with our novel metabolites and had identical MS2 and MS3 spectra, confirming the presence of the hydroxyl group on the alkyl side chain α to the indole.
We expected that addition of a hydroxyl group at the ω-end of the hexyl group of 230 would reduce antagonist activity, consistent with the loss of agonist activity following ω1-hydroxylation of 5-oxo-ETE [20], and this proved to be the case [4]. However, it is more difficult to predict the effect of addition of a hydroxyl group α to the indole, which would be equivalent to the ω6-position of 5-oxo-ETE. 5-Oxo-ETE can be hydroxylated in the ω6-position by 15-lipoxygenase, resulting in the formation of 5-oxo-15S-HETE [21], which, although not as potent as 5-oxo-ETE, is an OXE receptor agonist [5, 22]. In contrast, 5-oxo-ETE can be hydroxylated in the ω9-position by platelet 12-lipoxygenase to give 5-oxo-12S-HETE, which was the first OXE receptor antagonist to be recognized [23]. It was therefore important to examine the antagonist activities of the four synthetic α-OH-230 diastereomers, which were purified by a combination of normal-phase and chiral HPLC. Interestingly, only one of these diastereomers, subsequently identified as αS-OH-S230 (Table 1), displayed significant antagonist activity (IC50, 1.6 μM), being about 100 times less potent than S230 in inhibiting 5-oxo-ETE-induced calcium mobilization. This is similar to the IC50 for ω2-hydroxy-230 (1.4 μM) and a bit higher than that for ω1-hydroxy-230 (0.27 μM) [4].
Table 1.
Synthetic and biological α-hydroxy metabolites of 230. The IC50 values were determined from the data shown in Fig. 3D. The IC50 value for S230 in these experiments was 13 ± 2 nM.
| Synthetic diastereomer | Structure | Plasma metabolite | IC50 (μM) |
|---|---|---|---|
| x1 | αS-OH-R230 | ≫10 | |
| x2 | αR-OH-S230 | d (?) | ≫10 |
| y1 | αS-OH-S230 | e | 1.6 ± 0.3 |
| y2 | αR-OH-R230 | ≫10 |
Because an S-methyl group is required for appreciable antagonist activity it could be assumed that the active α-hydroxy metabolite of 230 has an S-methyl group. Of the three inactive diastereomers two would have an R-methyl group and would therefore be expected to have little or no activity. However, the remaining inactive diastereomer would have an S-methyl group and either an R or an S α-hydroxyl group, indicating that only one of these configurations is compatible with antagonist activity. The impact of the chirality of the α-hydroxyl group could possibly be due to an effect on the orientation of the alkyl group, which we have shown to be very important for interaction with the OXE receptor [24]. There could also be hydrogen bonding between the α-hydroxyl group and the oxo group, which would likely be affected by its chirality.
For further studies on the pharmacokinetics and metabolism of 230 we focused on the active S-enantiomer (S230), which is over 400 times more potent than the R-enantiomer (R230) [8, 9]. To provide the gram-amounts of antagonist that would be required for this and subsequent in vivo studies we developed a novel procedure using the chiral reagent (S)-Tol-BINAP for the synthesis of the chiral acyl side chain of S230 [11]. This permitted the synthesis of substantial amounts of this compound with high (>98%) enantiomeric purity.
The pharmacokinetic profile of S230 is quite similar to that which we previously observed for racemic 230 [4] except that the plasma concentrations were lower at all time points, peaking at 32 μM after 1 h, compared to 87 μM for racemic 230. This could be explained, at least in part, by differences in the rates of metabolism or intestinal uptake between S230 and R230, consistent with our finding of higher plasma levels of the R enantiomer following oral administration of racemic 230 [4].
Analysis of the plasma metabolites of S230 by RP-HPLC revealed a number of peaks that we had not previously identified (a, b, and c), and only one significant peak (e) for α-OH-S230 in contrast to the two peaks (d and e) observed with racemic 230. Since e is derived from S230 it could be either αS-OH-S230 or αR-OH-S230, which should be separable by RP-HPLC, as they are not mirror images of one another. This suggests that the α-hydroxylation reaction is stereospecific. To test this hypothesis, and to determine whether the α-hydroxy metabolite of S230 in plasma is the one with antagonist activity, we prepared both of the above diastereomers using (S)-BINOL and (R)-BINOL and confirmed the absolute chirality of the products by X-ray diffractometry. Comparison of the chromatographic properties of the resulting authentic standards clearly indicated that both metabolite e derived from S230 and the α-hydroxy-230 diastereoisomer with antagonist activity are identical to αS-OH-S230. It is interesting that this highly stereoselective hydroxylation reaction results in the formation of the only diastereomer with significant antagonist activity.
The site at which the α-hydroxylation reaction occurs in the monkey is not clear. In our previous studies we did not detect α-OH-230 following incubation of racemic 230 with monkey liver microsomes in the presence of NADPH, which instead converted this compound almost exclusively to ω2- and to a much lesser extent, ω1- oxidation products [4]. We have subsequently examined microsomes from other sources, including kidney, intestine, and lungs but did not detect α-hydroxylation activity in the presence of NADPH (data not shown). Neither did we detect such products as a result of incubating 230 with either plasma or intact leukocytes. One possibility might be that this reaction is catalyzed by bacteria in the gastrointestinal tract.
Although benzylic hydroxylation is quite well known [25, 26] and is an important step in the formation of noradrenaline, there are few examples in the literature of α-hydroxylation of alkylindoles. The bacterial enzyme indolyl-3-alkane α-hydroxylase, isolated from Pseudomonas, has been shown to catalyze α-hydroxylation of the side chain in the 3-position of tryptophan [27, 28]. This is not a direct hydroxylation, but rather proceeds by the initial abstraction of 2 hydrogens, one on the indole nitrogen, and the other on the side chain carbon adjacent to the indole ring. This results in the formation of a 3-alkylidene indoline intermediate that undergoes the 1,4 addition of H2O to give the α-hydroxy product [29]. However, 2-alkylindoles such as S230 would not be a substrate for such a series of reactions.
In addition to αS-OH-S230 we identified several other novel metabolites of S230. One of these (metabolite b) was formed by a combination of α- and ω-oxidation (Fig. 11). Although we cannot be certain of the location of the second hydroxyl group, it is highly likely that it is in the ω2-position, because ω2-hydroxylation is a major pathway for the metabolism of S230. The resulting product, αS,ω2-dihydroxy-S230 could be formed from either αS-OH-S230 or ω2-OH-S230.
Figure 11. In vivo metabolism of S230 in monkeys.

Metabolites shown in red were identified for the first time in the present study (i.e. the two α-OH metabolites and the two glucuronides). The broken arrows indicate that αS,ω2-diOH-S230 could potentially be formed from either ω2-OH-S230 or αS-OH-S230. The glucuronate in ω2-OH-S230 glucuronide could be attached to either the carboxyl group or the hydroxyl group. * It is highly likely that the hydroxyl group is in the ω2-position in a and b, but this has not been conclusively proven.
We also detected glucuronide metabolites of S230 and hydroxy-S230, presumably ω2-OH-S230. The S230 metabolite would clearly be an acyl glucuronide, as this compound does not have a free hydroxyl group. In contrast, ω2-OH-S230 could be converted to either an acyl or an ether glucuronide, or to both. At all time points later than 1 h, the concentrations of ω2-OH-S230 glucuronide(s) were considerably higher than those of S230 glucuronide, even when the plasma concentrations of S230 were much higher than its ω2-hydroxy metabolite, which might be consistent with the glucuronide being in a different (i.e. ether) position, resulting in a more stable metabolite.
Overall, the combined concentrations of S230 metabolites remained between 2 and 3 μM over the first 8 h and then slowly declined, in contrast to S230, which declined fairly rapidly after reaching high initial levels by 1 h. By 8 h, the combined amounts of S230 metabolites exceeded that of S230 itself. Nevertheless, its concentration at this time was still greater than 1.5μM, over 100 times higher than its IC50 (13 nM) for inhibition of 5-oxo-ETE-induced calcium mobilization in vitro. Multiple dosing with S230 (e.g. every 8 h) may be required to achieve therapeutic levels over a period of 24 h in future in vivo experiments.
In conclusion, we have identified a novel stereoselective α-hydroxylation pathway for the metabolism of a 2-alkylindole antagonist in primates. The αS-hydroxy product of this pathway still retains some antagonist activity, albeit considerably reduced, in contrast to the corresponding αR-hydroxy compound, which is virtually inactive. Thus S230 is metabolized in vivo by a combination of α-hydroxylation, ω2-hydroxylation and glucuronidation. In spite of its metabolism by these pathways, plasma levels of S230 well in excess of its in vitro IC50 value were maintained for 8 h. These results raise the possibility that either S230 itself or a related compound modified to resist hydroxylation, might be a useful therapeutic agent in eosinophilic disorders such as asthma. Studies are currently underway in primate models of allergic disease to test this hypothesis.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (WSP: Grants MOP-6254 and PP2-133388), the American Asthma Foundation (JR: Grant 12-0049), and the National Heart, Lung, and Blood Institute (JR: Grant R01HL081873) and by AmorChem (Montreal, QC). The Meakins-Christie Laboratories-MUHC-RI are supported in part by a Centre grant from Le Fond de la Recherche en Santé du Québec as well as by the J. T. Costello Memorial Research Fund. JR also wishes to acknowledge the National Science Foundation for the AMX-360 (Grant CHE-90-13145) and Bruker 400 MHz (Grant CHE-03-42251) NMR instruments. DV and IS were supported by the Natural Sciences and Engineering Research Council of Canada (Grant RGPIN/435814-2103). IS also wishes to acknowledge the Centre for Biological Applications of Mass Spectrometry at Concordia University for PhD scholarship funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Abbreviations
- 5-HETE
5S-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid
- 5-oxo-ETE
5-oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid
- 230
5-(5-chloro-2-hexyl-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoate
- S230
(S)-5-(5-chloro-2-hexyl-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoate
- αS-OH-S230
(S)-5-(5-chloro-2-((S)-1-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoate
- ω2-OH-S230
(3S)-5-(5-chloro-2-(5-hydroxyhexyl)-1-methyl-1H-indol-3-yl)-3-methyl-5-oxopentanoate
- BDMAEE
bis[2-(N,N-dimethylamino)ethyl] ether
- DART
direct analysis in real time
- HF
Hydrofluoric acid
- HRMS
high-resolution mass spectra
- MeOH
methanol
- (S)-BINOL
(S)-(−)-1,1’-bi(2-naphthol)
- TBDMSCl
tert-butyldimethylchlorosilane
- THF
tetrahydrofuran
- TMS
tetramethylsilane
Footnotes
WSP and JR hold a patent on OXE receptor antagonists related to compounds 230 and 264 (Powell WS and Rokach J, 5-Oxo-ETE receptor antagonist compounds, US patent US-2014-0323535-A1 (2014)).
“α” is used to designate the carbon of the hexyl side chain adjacent to the indole moiety, whereas “ω2” is used to designate the penultimate carbon of the hexyl side chain.
COMPOUNDS
5S-HETE (PubChem CID: 5280733); 5-oxo-ETE (PubChem CID: 5283159); BINOL (PubChem CID: 11762); 5-chloro-1-methyl-1H-indole-2-carbaldehyde (PubChem CID: 23004695); pentyl magnesium bromide (PubChem CID: 121513990); methyl 5-chloro-3-methyl-5-oxopentanoate (PubChem CID: 10888500); Indo-1 AM (PubChem CID: 123918); TBDMSCl (PubChem CID: 28928); BDMAEE (PubChem CID: 18204); tBuOMe (PubChem CID: 15413)
Database link
Data for the crystal structure of compound 7 has been deposited with the Cambridge Crystallographic Data Centre (deposition number, CCDC 1527527).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Powell WS, Gravelle F, Gravel S. Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. J Biol Chem. 1992;267:19233–41. [PubMed] [Google Scholar]
- 2.Powell WS, Rokach J. The eosinophil chemoattractant 5-oxo-ETE and the OXE receptor. Prog Lipid Res. 2013;52:651–65. doi: 10.1016/j.plipres.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham FD, Erlemann KR, Gravel S, Rokach J, Powell WS. Oxidative stress-induced changes in pyridine nucleotides and chemoattractant 5-lipoxygenase products in aging neutrophils. Free Radic Biol Med. 2009;47:62–71. doi: 10.1016/j.freeradbiomed.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cossette C, Chourey S, Ye Q, Nagendra Reddy C, Gore V, Gravel S, et al. Pharmacokinetics and Metabolism of Selective Oxoeicosanoid (OXE) Receptor Antagonists and Their Effects on 5-Oxo-6,8,11,14-eicosatetraenoic Acid (5-Oxo-ETE)-Induced Granulocyte Activation in Monkeys. J Med Chem. 2016;59:10127–46. doi: 10.1021/acs.jmedchem.6b00895. [DOI] [PubMed] [Google Scholar]
- 5.Powell WS, Chung D, Gravel S. 5-Oxo-6,8,11,14-eicosatetraenoic acid is a potent stimulator of human eosinophil migration. J Immunol. 1995;154:4123–32. [PubMed] [Google Scholar]
- 6.Jones CE, Holden S, Tenaillon L, Bhatia U, Seuwen K, Tranter P, et al. Expression and characterization of a 5-oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid receptor highly expressed on human eosinophils and neutrophils. Mol Pharmacol. 2003;63:471–7. doi: 10.1124/mol.63.3.471. [DOI] [PubMed] [Google Scholar]
- 7.Muro S, Hamid Q, Olivenstein R, Taha R, Rokach J, Powell WS. 5-oxo-6,8,11,14-eicosatetraenoic acid induces the infiltration of granulocytes into human skin. J Allergy Clin Immunol. 2003;112:768–74. doi: 10.1016/s0091-6749(03)01888-8. [DOI] [PubMed] [Google Scholar]
- 8.Gore V, Gravel S, Cossette C, Patel P, Chourey S, Ye Q, et al. Inhibition of 5-oxo-6,8,11,14-eicosatetraenoic acid-induced activation of neutrophils and eosinophils by novel indole OXE receptor antagonists. J Med Chem. 2014;57:364–77. doi: 10.1021/jm401292m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel P, Reddy CN, Gore V, Chourey S, Ye Q, Ouedraogo YP, et al. Two Potent OXE-R Antagonists: Assignment of Stereochemistry. ACS Med Chem Lett. 2014;5:815–9. doi: 10.1021/ml500161v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cossette C, Gravel S, Reddy CN, Gore V, Chourey S, Ye Q, et al. Biosynthesis and actions of 5-oxoeicosatetraenoic acid (5-oxo-ETE) on feline granulocytes. Biochem Pharmacol. 2015;96:247–55. doi: 10.1016/j.bcp.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy CN, Ye QJ, Chourey S, Gravel S, Powell WS, Rokach J. Stereoselective synthesis of two highly potent 5-oxo-ETE receptor antagonists. Tetrahedron Lett. 2015;56:6896–9. [Google Scholar]
- 12.Powell WS. Rapid extraction of oxygenated metabolites of arachidonic acid from biological samples using octadecylsilyl silica. Prostaglandins. 1980;20:947–57. doi: 10.1016/0090-6980(80)90144-6. [DOI] [PubMed] [Google Scholar]
- 13.Powell WS. Precolumn extraction and reversed-phase high-pressure liquid chromatography of prostaglandins and leukotrienes. Anal Biochem. 1987;164:117–31. doi: 10.1016/0003-2697(87)90375-7. [DOI] [PubMed] [Google Scholar]
- 14.Khanapure SP, Shi XX, Powell WS, Rokach J. Total synthesis of a potent proinflammatory 5-oxo-ETE and its 6,7-dihydro biotransformation product. J Org Chem. 1998;63:337–42. [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–22. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 17.Sheldrick GM. Crystal structure refinement with SHELXL. Acta Crystallogr C Struct Chem. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubschle CB, Sheldrick GM, Dittrich B. ShelXle: a Qt graphical user interface for SHELXL. J Appl Crystallogr. 2011;44:1281–4. doi: 10.1107/S0021889811043202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Da CS, Wang JR, Yin XG, Fan XY, Liu Y, Yu SL. Highly Catalytic Asymmetric Addition of Deactivated Alkyl Grignard Reagents to Aldehydes. Org Lett. 2009;11:5578–81. doi: 10.1021/ol9020942. [DOI] [PubMed] [Google Scholar]
- 20.Powell WS, MacLeod RJ, Gravel S, Gravelle F, Bhakar A. Metabolism and biologic effects of 5-oxoeicosanoids on human neutrophils. J Immunol. 1996;156:336–42. [PubMed] [Google Scholar]
- 21.Schwenk U, Schröder JM. 5-Oxo-eicosanoids are potent eosinophil chemotactic factors - functional characterization and structural requirements. J Biol Chem. 1995;270:15029–36. doi: 10.1074/jbc.270.25.15029. [DOI] [PubMed] [Google Scholar]
- 22.O’Flaherty JT, Kuroki M, Nixon AB, Wijkander J, Yee E, Lee SL, et al. 5-Oxo-eicosatetraenoate is a broadly active, eosinophil- selective stimulus for human granulocytes. J Immunol. 1996;157:336–42. [PubMed] [Google Scholar]
- 23.Powell WS, Gravel S, Khanapure SP, Rokach J. Biological inactivation of 5-oxo-6,8,11,14-eicosatetraenoic acid by human platelets. Blood. 1999;93:1086–96. [PubMed] [Google Scholar]
- 24.Patel P, Cossette C, Anumolu JR, Gravel S, Lesimple A, Mamer OA, et al. Structural requirements for activation of the 5-oxo-6E,8Z, 11Z,14Z-eicosatetraenoic acid (5-oxo-ETE) receptor: identification of a mead acid metabolite with potent agonist activity. J Pharmacol Exp Ther. 2008;325:698–707. doi: 10.1124/jpet.107.134908. [DOI] [PubMed] [Google Scholar]
- 25.Lindeke B, Ericsson O, Jonsson A, Noren B, Stromberg S, Vangbo B. Biotransformation of Terodiline.3. Opposed Stereoselectivity in the Benzylic and Aromatic Hydroxylations in Rat-Liver Microsomes. Xenobiotica. 1987;17:1269–78. doi: 10.3109/00498258709047158. [DOI] [PubMed] [Google Scholar]
- 26.Shetty HU, Nelson WL. Chemical Aspects of Metoprolol Metabolism - Asymmetric-Synthesis and Absolute-Configuration of the 3-[4-(1-Hydroxy-2-Methoxyethyl)Phenoxy]-1-(Isopropylamino)-2-Propanols, the Diastereomeric Benzylic Hydroxylation Metabolites. Journal of Medicinal Chemistry. 1988;31:55–9. doi: 10.1021/jm00396a009. [DOI] [PubMed] [Google Scholar]
- 27.Tsai MD, Floss HG, Rosenfeld HJ, Roberts J. Stereochemistry and mechanism of reactions catalyzed by indolyl-3-alkane alpha-hydroxylase. J Biol Chem. 1979;254:6437–43. [PubMed] [Google Scholar]
- 28.Noda Y, Takai K, Tokuyama T, Narumiya S, Ushiro H, Hayaishi O. Tryptophan side chain oxidase from Pseudomonas. pH-dependent formation of alpha,beta-didehydro, beta-hydroxy, and beta-keto derivatives of N-acetyltryptophanam. J Biol Chem. 1978;253:4819–22. [PubMed] [Google Scholar]
- 29.Rosenfeld HJ, Watanabe KA, Roberts J. Mechanism of action of indolyl-3-alkane alpha-hydroxylase. J Biol Chem. 1977;252:6970–3. [PubMed] [Google Scholar]