

Abstract
The cannabinoid CB1 receptor is abundant in the central nervous system and regulates neuronal transmission and other key physiological processes including those leading to pain, inflammation, memory, and feeding behavior. CB1 is activated by the endogenous ligands, arachidonoyl ethanolamine and 2-arachidonoyl glycerol, by various synthetic ligands (e.g., CP55940), and by Δ9-tetrahydrocannabinol, the psychoactive component of Cannabis sativa. These CB1 ligands are orthosteric and transduce downstream signals by binding CB1 and primarily inducing Gi coupling, but Gs and β-arrestin coupling are also possible. Recently, allosteric modulators for CB1 were discovered that bind to topographically distinct sites and can noncompetitively impact the potency and efficacy of orthosteric compounds. These offer the exciting potential for mechanistic analyses and for developing therapeutics. Yet, it is critical to elucidate whether a compound is a positive allosteric modulator or a negative allosteric modulator of orthosteric ligand-induced CB1 profiles to understand pathway specificity and ameliorate diseases. In this chapter, we present equilibrium and kinetic binding analysis to reveal the impact of allosteric modulators on CB1. Also described are activities consistent with CB1 activation (or inactivation) and include cellular internalization of CB1 and downstream signaling patterns. Since many CB1 allosteric modulators do not enhance G protein coupling, it is critical to distinguish CB1 activation and biased signaling patterns via β-arrestin from CB1 inactivation. These strategies can illuminate pathway specificity and are valuable for the fine-tuning of CB1 function.
1. INTRODUCTION
1.1 Cannabinoid Receptor System
The cannabinoid receptor 1 (CB1) binds the main active ingredient of Cannabis sativa (marijuana), Δ9-tetrahydrocannabinol (THC), to mediate its well-described psychoactive effects (Bhattacharyya et al., 2010; Little, Compton, Johnson, Melvin, & Martin, 1988; Prescott, Gold, & Martin, 1992). Regulation of CB1 functions has enormous potential to treat disorders of feeding behavior (Berry & Mechoulam, 2002; May, Leach, Sexton, & Christopoulos, 2007; Van Gaal et al., 2005), neuroinflammation (Saito, Rezende, & Teixeira, 2012; Walter & Stella, 2004), and pain relief (Russo, 2008). Modulation of CB1 has been traditionally achieved through orthosteric ligands, such as THC (Howlett et al., 2002; Pertwee et al., 2010), which bind to sites where endogenous cannabinoids bind. For instance, although orthosteric CB1 agonists such as Marinol® and Cesamet® are FDA-approved for limited applications, such as chemotherapy-induced nausea in cancer patients (Walsh, Nelson, & Mahmoud, 2003), these drugs have undesirable psychoactive side effects. Additionally, inverse agonists of the CB1 receptor such as rimonabant and taranabant were once touted as novel anti-obesity agents because of their anorectic and lipogenesis-decreasing properties (Ravinet Trillou et al., 2003); however, they may have also caused psychiatric side effects including depression and anxiety that terminated their clinical use (Rothman & Baumann, 2009). Therefore, while CB1 has strong potential as a therapeutic target, it has been underused and CB1 ligands with alternative mechanisms of action need to be developed.
CB1 is a G protein-coupled receptor (GPCR) localized in the membranes of presynaptic nerve terminals, and its activation directly inhibits neurotransmitter release (Mackie & Hille, 1992). Endogenous cannabinoids such as arachidonoyl ethanolamide and 2-arachidonoyl glycerol are synthesized from lipid molecules on demand as a result of an influx of calcium into the cell (Di Marzo et al., 1994; Stella, Schweitzer, & Piomelli, 1997). Upon activation, CB1 primarily binds to the intracellular Gi protein (Gi often refers to Gi/o proteins), which inhibits the enzyme adenylate cyclase and, thus, inhibits the production of cyclic adenosine monophosphate (cAMP) (Howlett & Fleming, 1984). However, CB1 can couple to other intracellular proteins including the Gs protein (Glass & Felder, 1997), and β-arrestin isoforms (Ahn, Mahmoud, Shim, & Kendall, 2013; Turu & Hunyady, 2010). The binding of different intracellular proteins leads to different physiological responses, and it would be advantageous to design drugs that selectively activate different signaling pathways. For example, the FDA-approved adrenergic receptor β-blocker carvedilol (Warne et al., 2008) and the agonist isoetharine (Liu, Horst, Katritch, Stevens, & Wuthrich, 2012) have different patterns of signaling in G protein and β-arrestin pathways, indicating that specific therapeutic outcomes can be achieved via different ligands for the same receptor.
1.2 Allosteric Modulation of the CB1 Receptor
In the past, the majority of GPCR-based drug discovery programs focused on the development of orthosteric molecules that compete with endogenous ligands. The recent development of more sophisticated functional reporter-based assays has made it possible to identify active ligands that bind to topographically distinct sites on the receptor, namely, allosteric sites. Allosteric ligands exert their effects by modifying the receptor conformation, leading to a change in the binding and/or functional properties and/or efficacies of orthosteric ligands (Fig. 1) (Langmead & Christopoulos, 2006; May et al., 2007; Wootten, Christopoulos, & Sexton, 2013). Compared to orthosteric ligands, allosteric ligands possess the following critical advantages for drug discovery:
Fig. 1.
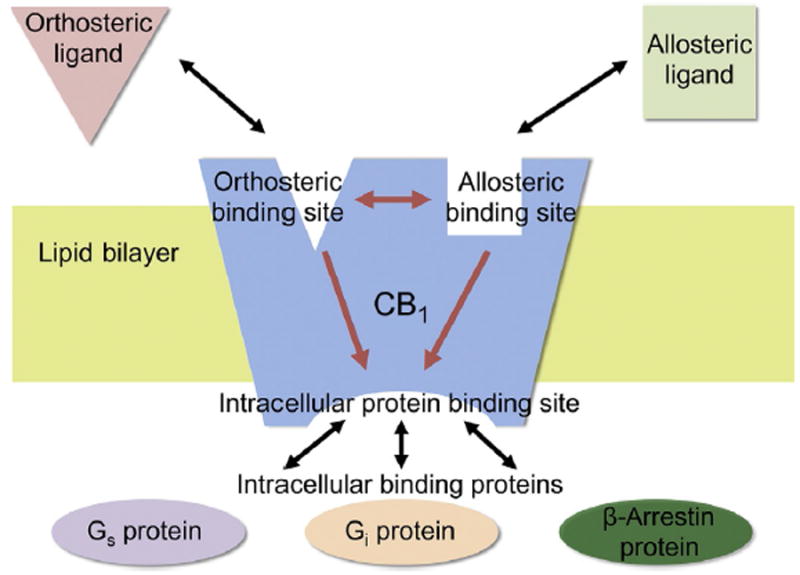
Cooperation between the GPCRs orthosteric and allosteric binding sites to induce intracellular protein coupling. Schematic illustration showing how binding of the allosteric modulator to an allosteric binding site distinct from the orthosteric binding site may influence binding of the orthosteric ligand to its respective site and how both ligands may impact binding of intracellular proteins to their respective binding sites. β-Arrestin binding can involve different isoforms not indicated and other G protein isoforms may also be impacted. Further, some ago-allosteric modulators can function without an orthosteric compound. Adapted from Fig. 1 in Conn, P. J., Christopoulos, A., & Lindsley, C. W. (2009). Allosteric modulators of GPCRS: A novel approach for the treatment of CNS disorders. Nature Reviews. Drug Discovery, 8(1), 41–54.
increased receptor subtype selectivity due to their binding to structurally less-conserved allosteric sites, and therefore, off-target side effects may be reduced (Christopoulos, 2002; Conn, Christopoulos, & Lindsley, 2009);
possess a “ceiling effect” on orthosteric ligand function such that increase in concentration beyond saturation of the allosteric receptor binding site does not increase the magnitude of the allosteric effect (avoidance of overdosing) (May et al., 2007);
lead to pathway-specific modulation (biased signaling) so that “on-target” side effects from untoward interference may be reduced (DeWire & Violin, 2011; Kenakin & Miller, 2010; May et al., 2007); and
while systemically administered classical ligands modulate all target receptors in all accessible tissues of the body, an allosteric modulator theoretically impacts CB1 signaling only in tissues where the endogenous ligand is present. Given that endocannabinoids are produced and released “on demand,” this feature is particularly relevant for CB1 allosteric modulators, as it would theoretically allow for further fine-tuning (Burford et al., 2013; Burford, Watson, Bertekap, & Alt, 2011; Conn et al., 2009).
Recently, several CB1 allosteric modulators have been discovered. These novel CB1 ligands bind the receptor at sites topologically distinct from the orthosteric binding site (Fay & Farrens, 2012; Janero & Thakur, 2016; Shore et al., 2014; Stornaiuolo et al., 2015) and modulate the CB1 activity by acting as rheostats to alter the affinity and/or the efficacy of an orthosteric ligand in either a positive (positive allosteric modulator; PAM) or negative (negative allosteric modulator; NAM) manner depending on the functional property under investigation. For CB1, this includes: ORG27569, ORG27759, and ORG29647 (Price et al., 2005), PSNCBAM-1 (Horswill et al., 2007), RTI-371 (Navarro, Howard, Pollard, & Carroll, 2009), lipoxin A4 (Pamplona et al., 2012), and GAT211 and its enantiomer GAT229 (Laprairie, Bagher, & Denovan-Wright, 2017; Laprairie, Kulkarni, et al., 2017). Some of these structures are shown in Fig. 2. Pepcans (Bauer et al., 2012), cannabidiol (Laprairie, Bagher, Kelly, & Denovan-Wright, 2015), pregnenolone (Vallée et al., 2014), and GW405833 (Dhopeshwarkar, Murataeva, Makriyannis, Straiker, & Mackie, 2017) are also possible allosteric modulators of CB1. The therapeutic usefulness of these CB1 allosteric modulators is emerging.
Fig. 2.
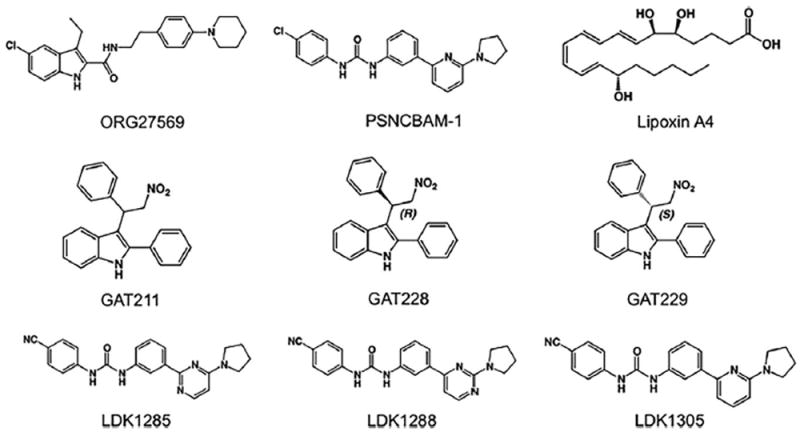
Structures of some known CB1 allosteric modulators. ORG27569 and PSNCBAM-1 are synthetic allosteric modulators (Ahn, Mahmoud, & Kendall, 2012; Horswill et al., 2007; Price et al., 2005), lipoxin A4 is an endogenous allosteric modulator (Pamplona et al., 2012), and GAT211 consists of a racemic mix of GAT228, the allosteric agonist R-(+)-enantiomer, and GAT229, the allosteric modulator S-(−)-enantiomer (Laprairie, Kulkarni, et al., 2017). LDK1285, LDK1288, and LDK1305, three positive allo-steric modulators derived from PSNCBAM-1, are referred to as 8d, 7d, and 29, respectively in Khurana et al. (2017) and German et al. (2014).
2. METHODS OF ALLOSTERIC LIGAND BINDING ANALYSIS
2.1 Impact of CB1 Allosteric Ligands on Orthosteric Ligand Affinity
To ascertain if a compound is an allosteric modulator, and if it promotes the active state of CB1 (a PAM) or the inactive state of CB1 (a NAM), a binding analysis is often used (Ahn, Mahmoud, Samala, Lu, & Kendall, 2013; Baillie et al., 2013; German et al., 2014; Khajehali et al., 2015; Khurana et al., 2014, 2017; Mahmoud et al., 2013; Price et al., 2005). For example, Ross and colleagues used equilibrium binding and kinetic binding assays to evaluate ORG27569 and related compounds and found that they elicit a conformational change that increases agonist affinity for the orthosteric site, although probe dependence was also observed (Baillie et al., 2013; Khajehali et al., 2015; Price et al., 2005). It should be noted that some functional assays show that ORG27569 and several derivatives do not induce Gi protein coupling with CB1 (Ahn et al., 2012; Ahn, Mahmoud, Samala, et al., 2013; Baillie et al., 2013; Khurana et al., 2014; Mahmoud et al., 2013; Price et al., 2005; Qiao et al., 2016) unlike many orthosteric agonists that do (Howlett & Fleming, 1984), but these compounds do induce effects consistent with coupling to the β-arrestin isoforms 1 and 2 (Ahn et al., 2012; Ahn, Mahmoud, Samala, et al., 2013; Ahn, Mahmoud, Shim, et al., 2013; Khurana et al., 2014).
Kendall and colleagues used equilibrium binding analysis to evaluate the affinity change in the presence of the allosteric modulator ORG27569 (Ahn et al., 2012). The basal activity of CB1 (R′) indicates that it is poised for conversion to either inactive (R) or more fully activated (R*) forms. T210 was identified on the same face of the helix as R214 of the DRY motif on TM3 (D’Antona, Ahn, & Kendall, 2006), and its substitution with Ile or Ala generates constitutively active or inactive receptor mutants, respectively, and provides powerful reagents to characterize these conformational states (D’Antona, Ahn, & Kendall, 2006; D’Antona, Ahn, Wang, et al., 2006). The T210I receptor exhibited enhanced agonist and diminished inverse agonist affinity for a variety of ligands, consistent with a shift toward the active form. Decreased thermal stability of the T210I receptor, higher levels of coupling activity, and increased internalization of the T210I receptor were also observed, consistent with constitutive activity. In contrast, the T210A receptor exhibited the opposite profile as predicted for an inactive receptor. Indeed, these receptors provide a window to different GPCR conformational states with minimal amino acid manipulation.
T210I, wild-type (WT) CB1, and T210A receptors reveal propensities of different ligands to promote R* vs R forms of CB1 regardless of effector coupling (Fig. 3). Taking advantage of the T210A, WT, and T210I CB1 receptors that cover a spectrum of receptor states, the effect of ORG27569 on agonist affinity has been elucidated. Saturation binding experiments were performed for CP55940 in the absence and presence of ORG27569. As we have shown (Ahn et al., 2012), the T210I receptor has the greatest affinity for CP55940, with a Kd of 0.31nM, followed by the WT (Kd = 2.15nM) and T210A receptors (Kd = 7.82nM). In the presence of 10 μM ORG27569, however, the three receptors exhibited an enhanced and similar Kd value for CP55940 (0.29nM, 0.42nM, and 0.30nM for the WT, T210I, and T210A, respectively) that corresponds to the high affinity observed with the T210I receptor. For WT CB1, ORG27569 enhances the affinity for the agonist, CP55940, by about sevenfold and decreases the affinity of the inverse agonist, SR141716A, by about eightfold as expected for an allosteric modulator that promotes a receptor active state. This suggests that ORG27569 induces a CB1 state (R*) characterized by enhanced agonist affinity, and separately, analysis with SR141716A shows it induces decreased inverse agonist affinity, consistent with induction of an “active” conformation.
Fig. 3.
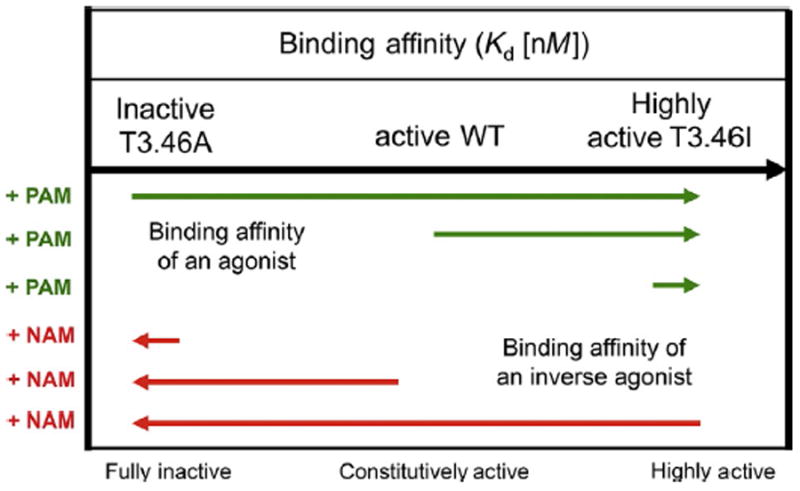
Allosteric modulator impact on ligand binding affinity to the CB1 receptor. In the presence of a PAM, the respective agonist binding affinities (green) for the fully inactive T3.46A mutant receptor or the constitutively active WT receptor are enhanced to match the affinity for the fully constitutively active mutant T3.46I (Ahn et al., 2012). The increase in binding affinity is shown schematically by the length and direction of the arrows. Hypothetically, in the presence of a NAM, the inverse agonist (red) would experience the opposite effect by having diminished respective binding affinities for the fully active T3.46I mutant and constitutively active WT receptor, which are similar to that of the fully inactive T3.46A mutant. The top arrow depicts the increasing agonist binding and functional activity of the three CB1 receptors.
2.2 Equilibrium Binding Assays With Allosteric Modulators
The degree of binding of the allosteric modulator is typically dependent on the affinity of the orthosteric ligand for the receptor. Thus, to elucidate how an allosteric modulator impacts orthosteric ligand binding, it is necessary to first examine how the orthosteric ligand independently binds to the protein. This involves saturation binding assays to determine the equilibrium dissociation constant, Kd, of the orthosteric ligand to the receptor and competition binding assays involving a labeled and unlabeled orthosteric compound, to determine Ki. IC50 can be converted to the inhibition equilibrium constant, Ki, using the Cheng–Prusoff equation (Cheng & Prusoff, 1973) shown in Eq. (1):
| (1) |
where [A] is the concentration of the competitive ligand and Ki is dependent on Kd, which was determined from saturation binding assays. Equilibrium binding assays with allosteric modulators show the strength of binding to the receptor, and the impact that the allosteric modulator has on the binding of orthosteric ligands. They reveal the cooperativity between molecules binding at two distinct sites, that is, if binding of the allosteric modulator enhances or decreases the binding of the orthosteric ligand. When an allosteric modulator is the test compound, it will not compete with the orthosteric compound (usually radiolabeled) on the target protein. Instead of using the one-site model to determine the Ki, the allosteric ternary complex model is used, which describes the interaction of an allosteric modulator binding to a GPCR in the presence of an orthosteric ligand (Christopoulos & Kenakin, 2002; De Lean, Stadel, & Lefkowitz, 1980). This model is applied to find the fraction of specific binding as shown in Eq. (2) (Price et al., 2005):
| (2) |
where Y is the specific binding fraction of the radiolabeled ligand, [A] and [B] are the concentrations of the orthosteric ligand and allosteric modulator, respectively, KA and KB are the equilibrium dissociation constants of the orthosteric ligand and allosteric modulator, respectively, and a is the magnitude of the cooperativity binding factor. This value expresses how the allosteric modulator affects orthosteric ligand binding (Fig. 4). An α value greater than 1 indicates that the compound is a PAM and improves the binding of the orthosteric ligand. An α value less than 1 indicates that the compound is a NAM by inducing less favorable orthosteric ligand binding. When α=1, the compound is a silent allosteric modulator (SAM) because it has no effect on the binding of the orthosteric ligand. Of the parameters shown in Eq. (2), [A] and KA are fixed constants, with the latter value is determined by saturation binding assays for the orthosteric agonist. The values of α and KB are established by nonlinear regression (Price et al., 2005).
Fig. 4.
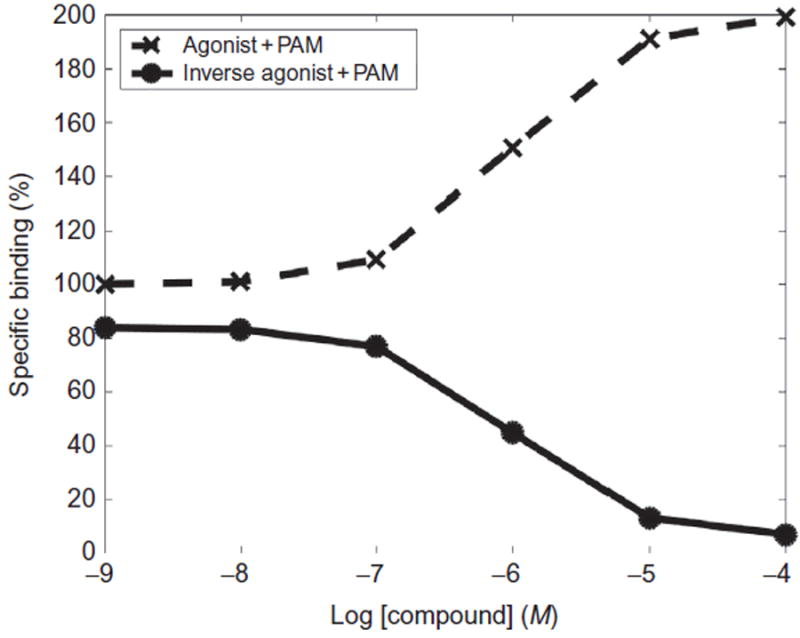
Hypothetical dose–response curves of experimental assays showing agonist and inverse agonist binding in the presence of a PAM. Radiolabeled agonist (dashed line, crosses) and radiolabeled inverse agonist (solid line, circles) binding percentage measured in the presence of a PAM.
2.2.1 Equipment and Reagents for Equilibrium Binding
Needed equipment are 11 pairs of silanized test tubes, a vortexer, microcentrifuge, glass pestle tissue grinder, shaking water bath, Brandel cell harvester, refrigerated condensation trap, Whatman GF/C filter paper for 24 wells without deposit system (Brandel), GraphPad Prism software (for plotting and analysis), liquid scintillant, and liquid scintillation counter.
Chemicals, biological agents, and other reagents needed are membrane preparation of human embryonic kidney (HEK293) cells transfected with CB1 protein, cold compounds such as CP55940 (Tocris), a radiolabeled orthosteric tracer such as 3H-CP55940 (PerkinElmer), and allosteric modulators (e.g., LDK1285, LDK1288, LDK1305). Also needed are dimethyl sulfoxide (DMSO), TME buffer (25mM Tris–HCl, 5mM MgCl2, 1mM EDTA, pH 7.4), fatty acid-free bovine serum acid (BSA) (Sigma Aldrich), TME buffer+5% BSA, TME+0.2% BSA, TME+7% sucrose, and ScintiSafe™ 30% Cocktail (Fisher Chemical).
2.2.2 Protocol for Equilibrium Binding
Note: The following procedure is written for reactions that are read in a liquid scintillation counter. The procedure can be adapted for a microtiter plate reader.
Perform serial dilution of the test allosteric modulator compounds. Add DMSO to the compounds to create nine samples typically ranging in concentrations of 0.1μM to 1.0mM.
Create 1.0mM concentration of CP55940 (or other cold orthosteric compound for nonspecific binding) by mixing 2μL of 10mM of the stock CP55940 and 18μL of DMSO.
Add 2μL of each compound to each of the pairs of test tubes. To the first pair, add 2μL of DMSO—this is the control that represents total binding of the orthosteric compounds to the CB1 protein. In the second pair of test tubes, add 2μL of the cold 1.0mM CP55940 created in the step above—to represent the amount of nonspecific binding to the CB1 membrane preparation. In the remaining nine pairs of test tubes, add 2μL of the test allosteric modulators in concentrations increasing from 0.1μM to 1.0mM.
In a separate test tube, add TME+0.2% BSA buffer. This amount depends on the activity level of the radioactive CP55940 compound.
To this same test tube, add the desired amount of the radioactive compound. Wrap the top of the test tube in parafilm and vortex to mix the solution.
Pipette 148μL of the mixed radioactive solution into each of the 11 pairs of test tubes. Vortex each test tube.
In a 3-mL glass pestle tissue grinder, add 1175μL of TME+7% sucrose buffer.
Pipette 75μL CB1 membrane preparation from cells expressing CB1 into the 3-mL glass pestle tissue grinder with the buffer.
Homogenize the CB1 containing membrane preparation.
Pipette 50μL of the membrane preparation from cells expressing CB1 into each of the 11 pairs of test tubes. Vortex each test tube.
Incubate at 30°C for 1h in a Precision™ shaking water bath.
Stop the reaction by adding 300μL TME+5% BSA to each test tube.
Place filter paper in the Brandel cell harvester. Rinse the test tubes twice with 2mL of 1× TME buffer followed by rinsing twice with 4mL of 1× TME buffer.
Collect the filter paper and put each piece in a separate scintillation vial, one corresponding to each test tube.
Fill each vial with 4mL of ScintiSafe scintillation fluid. Vortex each vial. Let it for at least 8h.
Measure the radioactivity of each vial with the liquid scintillation counter.
2.3 Kinetic Binding Assays With Allosteric Modulators
The rates of association and dissociation of ligands (often labeled with radioactivity) are useful to determine when an experiment has reached equilibrium, to determine the cooperativity of binding, and to validate test compounds as allosteric modulators. First, the kinetics of the orthosteric ligand are determined. In association binding kinetic assays, the radiolabeled ligand is incubated with the membrane for times typically ranging from 2 to 20min. In dissociation binding kinetics assays (Fig. 5), the radiolabeled ligand is incubated with the membranes for a full hour to ensure full association before the competing orthosteric ligand is added at times ranging from 2 to 20min. Data are analyzed with the one-phase association model to obtain the observed association rate, kobs, and the dissociation rate, koff. Using these two values and the concentration of the orthosteric ligand, we can determine the association rate constant, kon, as shown in Eq. (3):
| (3) |
Fig. 5.
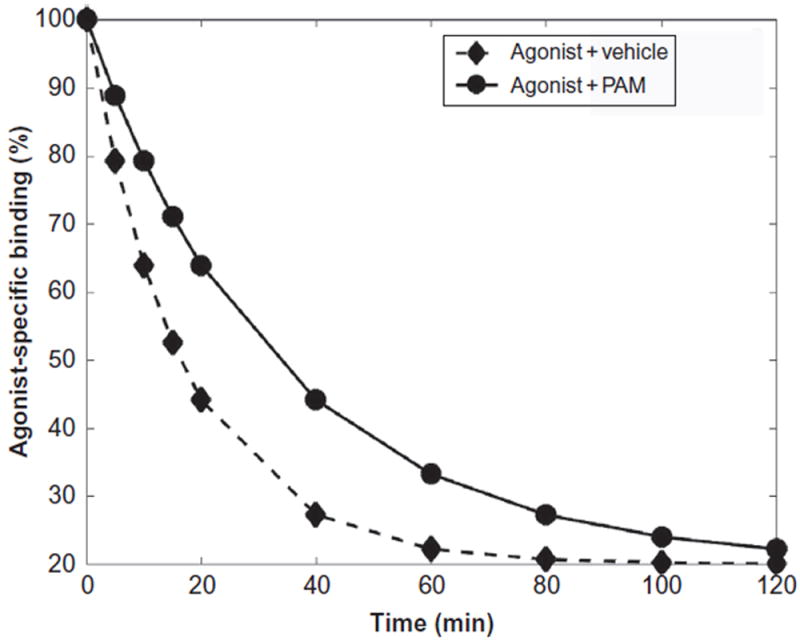
Hypothetical curves of experimental kinetic dissociation assays showing a PAM in the presence of an agonist (circles) effect with respect to the vehicle in the presence of an agonist (diamonds) on the binding of the agonist with respect to time.
The observed association and dissociation rates of the orthosteric ligand are dependent on its binding affinity for the receptor, as shown in Eq. (4):
| (4) |
The dissociation constant (Kd) is inversely proportional to the kon and directly proportional to the koff.
Allosteric modulators can impact the kinetics of dissociation of the orthosteric ligand in ways that competitive orthosteric ligands cannot. Both orthosteric ligands and allosteric modulators affect the association kinetics because orthosteric ligands and the radiolabeled ligand compete for the same binding site, and binding of an allosteric modulator induces a conformational change that will impact the orthosteric site. The rate of radiolabeled ligand dissociation, however, only changes as a result of conformational changes, such as those caused by the allosteric modulator (Christopoulos, 2001), and is not affected by the presence of a radiolabeled ligand. Therefore, dissociation kinetic binding assays are especially useful in examining if a ligand is an allosteric modulator or another orthosteric ligand. ORG2769, for example, was shown to be an allosteric modulator as opposed to an agonist because it affected the kinetics of CP55940 binding by decreasing the rate of dissociation of CP55940 alone from CB1 (Price et al., 2005). The procedure for the dissociation binding assays in the presence of the allosteric modulator follows that of the equilibrium binding assays except the reactions are incubated for different periods of time and a highly concentrated solution of the unlabeled ligand needs to be added to prevent the radiolabeled ligand from reassociating with the proteins following disassociation (Christopoulos, 2001).
3. G-PROTEIN COUPLING
CB1 predominantly couples to the Gi protein, which, upon activation, undergoes an exchange of guanosine diphosphate (GDP) by guanosine triphosphate (GTP). G protein binding assays determine the extent to which a PAM induces CB1 coupling to the Gi protein. To evaluate the extent of G protein coupling, as a result of the impact of an allosteric modulator, stimulation of 35S-guanine 5′-3-O-(thio)triphosphate (GTPγS) binding (Ahn et al., 2012; Ahn, Mahmoud, Samala, et al., 2013; Baillie et al., 2013; Khurana et al., 2014; Mahmoud et al., 2013; Price et al., 2005; Qiao et al., 2016) or the level of cAMP accumulation is monitored (Baillie et al., 2013; Cawston et al., 2013; Khajehali et al., 2015; Laprairie, Kulkarni, et al., 2017) using cells or inhibition of electrically stimulated contractions in a vas deferens assay from mice is evaluated (Laprairie, Kulkarni, et al., 2017; Price et al., 2005). This has been carried out for ORG27569, PSNCBAM-1, and several analogues of these that function as PAMs with respect to agonist binding, yet most show inhibition of the extent of G protein coupling and some specifically Gi coupling (Ahn et al., 2012; Ahn, Mahmoud, Samala, et al., 2013; Baillie et al., 2013; Khurana et al., 2014, 2017; Mahmoud et al., 2013; Qiao et al., 2016).
Here we describe the protocol for using a GTPγS assay, in which GTPγS acts as a substitute for GTP and binds to the Gi protein during activation (Howlett & Fleming, 1984). The radiolabeled component is the GTPγS, and GDP is added to promote the exchange. Unlabeled agonists and allosteric modulators are used in this assay. The amount of G protein exchange is determined by measuring the amount of radiolabeled 35S-GTPγS bound. The degree to which a test compound stimulates (or inhibits) GTPγS binding is measured by the EC50 values, or the concentration of ligand needed to produce 50% of the maximum biological response and/or the Emax value which corresponds to the level of coupling achieved.
GTPγS assays clarify the extent of receptor coupling in the presence of the allosteric modulator and may indicate if the compound is a PAM or a NAM. One example is ORG2769, which showed positive cooperativity with the agonist CP55940 and increased its binding. For ORG2769 to be considered a PAM (as opposed to a NAM), it is expected that the compound would enhance the potency and efficacy of G protein binding because PAMs increase the binding affinity of agonists (likely by a conformation that corresponds to the active state of the receptor) to the target. Counterintuitively, GTPγS assays showed that, in the presence of the agonist WIN55212-2 (Baillie et al., 2013) or CP55940 (Ahn et al., 2012; Price et al., 2005), ORG2769 did not affect the potency of G protein binding and decreased its efficacy, which suggests that ORG2769 inhibits the activation of CB1 even in the presence of an agonist. Subsequent studies indicated that ORG2769 induced internalization of CB1 and signaled in a β-arrestin-dependent manner, which suggested that ORG2769 is a biased PAM (Ahn et al., 2012; Ahn, Mahmoud, Shim, et al., 2013). Thus, the GTPγS assays provided insight into the function and selective signaling of this allosteric modulator.
4. CELLULAR INTERNALIZATION OF CB1
Since binding assays and G protein coupling assays can give apparently conflicting results (Ahn et al., 2012; Ahn, Mahmoud, Samala, et al., 2013; Khurana et al., 2014, 2017; Mahmoud et al., 2013; Price et al., 2005; Qiao et al., 2016), it is helpful to evaluate allosteric modulators by other means to determine if CB1 functions as if it is activated or inactivated by the modulator. For example, following prolonged activation, GPCRs typically become desensitized and become internalized in cells (Appleyard, Patterson, Jin, & Chavkin, 1997; Jin et al., 1999; Kovoor, Nappey, Kieffer, & Chavkin, 1997; Krupnick & Benovic, 1998; Zhang et al., 1997). Since the WT CB1 localizes mainly to intracellular vesicles in various cell lines in the absence of ligand (Ahn, Nishiyama, Mierke, & Kendall, 2009; Kenakin, 1995; Leterrier, Bonnard, Carrel, Rossier, & Lenkei, 2004) consistent with its partial constitutive activity, Kendall and colleagues have used the previously characterized inactive T210A mutant receptor, which is exclusively expressed at the cell surface (Ahn et al., 2012; D’Antona, Ahn, & Kendall, 2006). Alternatively, treatment of the WT CB1 with the inverse agonist SR141716A produces a more inactive form of the receptor and brings it to the cell surface, but SR141716A must then be removed thoroughly without cell loss prior to subsequent treatment. The extent of internalization can be assessed by colocalization of the receptors with the late endosome marker, LAMP-1 (Ahn et al., 2012, 2009; Kenakin, 1995). Confocal microscopy shows that CB1 T210A expressing HEK293 cells treated with CP55940 plus ORG27569, or ORG27569 alone, internalized the receptor. When treated with both CP55940 and ORG27569, the CB1 T210A receptors are internalized more readily than CP55940 treatment alone (Ahn et al., 2012). These results are consistent with activation of the receptor by this allosteric modulator and showed receptor internalization as seen following CP55940 agonist treatment alone (Daigle, Kwok, & Mackie, 2008; Hsieh, Brown, Derleth, & Mackie, 1999).
Furthermore, a different primary role of the two β-arrestin isoforms was observed. While suppressing β-arrestin 1 expression levels with isoform-specific siRNA, no effect on CB1 T210A internalization was observed induced by CP55940 or that induced by ORG27569. Yet, internalization was dramatically attenuated in β-arrestin 2 siRNA-transfected cells, in both cases, suggesting that the CB1 receptor undergoes β-arrestin 2-mediated internalization following both CP55940 treatment and ORG27569 treatment. Thus, β-arrestin 2 but not β-arrestin 1 appears to play a critical role in receptor internalization regardless of the extent of G protein coupling induced by different compounds. β-Arrestin 1, however, appears to be mediate signaling (see Section 5), indicating a differential role of the two β-arrestin isoforms in CB1 signaling and internalization (Ahn, Mahmoud, Shim, et al., 2013).
4.1 Protocol for Cellular Internalization of CB1
HEK293 cells expressing CB1-GFP receptors (carrying the T210A mutation or CB1 WT treated with SR141716A then washed thoroughly) are seeded onto 35-mm glass-bottomed dishes precoated with poly-d-lysine. They are treated with ligands (orthosteric and/or allosteric and/or vehicle) for various lengths of time (typically 5min to 3h) then washed three times with phosphate-buffered saline (PBS) followed by fixation with 4% paraformal-dehyde for 10min at room temperature.
Cells are mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and visualized using a Leica TCS SP2 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Images are collected from at least three independently transfected cell dishes and processed for presentation using Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA). Quantification of colocalization is performed using ImageJ software (National Institutes of Health, Bethesda, MD) with the JACoP plugin.
5. ANALYSIS OF KINASE PHOSPHORYLATION
5.1 ERK1/2 Phosphorylation
As shown in Fig. 6, both the Gi and β-arrestin proteins cause downfield signaling that typically results in the phosphorylation of extracellular signal-regulated kinase isoforms 1 and 2 (ERK1/2) (for review, see Turu & Hunyady, 2010). However, the use of pertussis toxin (PTX) precludes CB1 from coupling with the Gi protein. Thus, by using immunoblotting procedures, we can determine if ERK1/2 phosphorylation is a result of activating endogenous Gi protein. Alternatively, transfection with specific β-arrestin siRNA greatly reduces the expression of endogenous β-arrestin isoforms 1 or 2, and it can be determined if β-arrestins contribute to G protein-independent ERK1/2 activation (see Fig. 7). This is a comparable procedure for orthosteric and allosteric modulators. Strikingly, the reduced expression of β-arrestin 1 nearly abolished ORG27569-induced ERK1/2 phosphorylation (but not that by CP55940), whereas cotransfection with β-arrestin 2 siRNA did not alter patterns of ERK1/2 phosphorylation (induced by either ligand) compared to those observed by control siRNA transfection. Analogues of ORG27569 are typically comparable (Ahn et al., 2012; Ahn, Mahmoud, Samala, et al., 2013; Ahn, Mahmoud, Shim, et al., 2013; Khurana et al., 2014; Mahmoud et al., 2013; Qiao et al., 2016), but other results have been observed (Gamage, Anderson, & Abood, 2016).
Fig. 6.
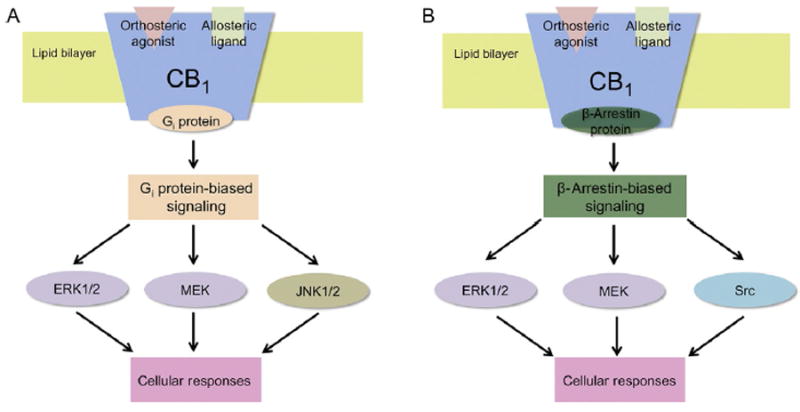
Schematic illustration showing some Gi protein and β-arrestin-mediated signaling resulting from PAM binding to CB1 and subsequent Gi protein and β-arrestin recruitment. Gi protein (orange) binding to CB1 results in cellular responses such as phosphorylation of ERK1/2, MEK, or JNK1/2 (A). β-Arrestin (green) binding to CB1, on the other hand, can typically result in the cellular responses including phosphorylation of ERK1/2, MEK, or Src (B). The proteins that are shown here can either be phosphorylated as a result of Gi protein-mediated signaling (brown), β-arrestin-mediated signaling (blue), or both (purple).
Fig. 7.
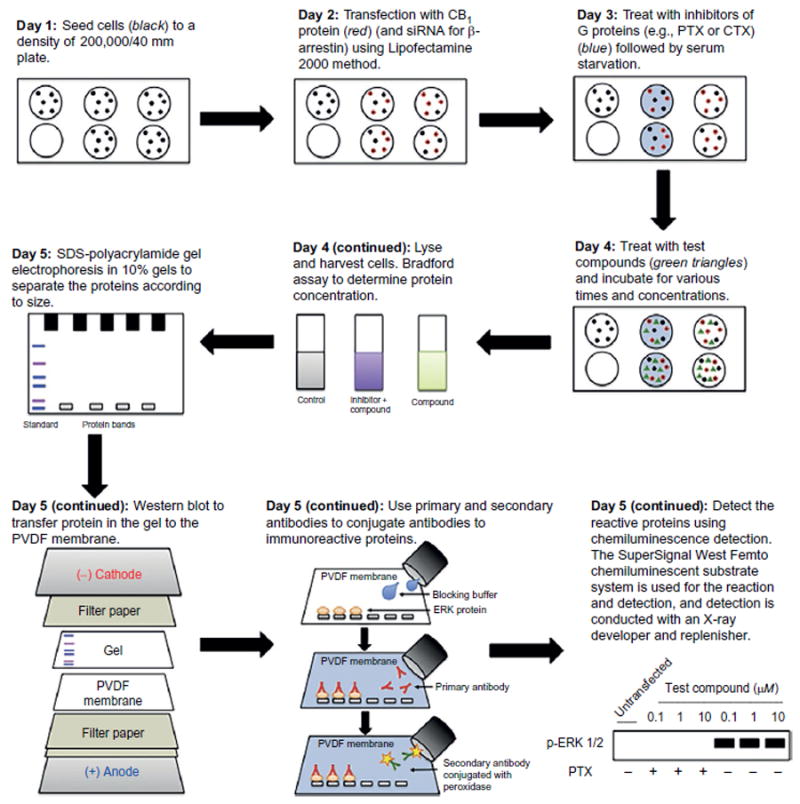
Illustration of the cell culture and immunoblotting procedures to examine the effect on ERK1/2 phosphorylation of treating cells expressing CB1 with test allosteric modulators. A comparable approach can be done with other kinases including a global phospho-kinase antibody array analysis to find which human kinases are phosphorylated as a result of treatment by the test compounds.
5.2 Phosphorylation of Other Kinases
To elucidate other phosphorylation patterns, Kendall and colleagues investigated the activation of other kinases resulting from the ORG27569-induced CB1-β-arrestin 1 interaction relative to the CB1 Gi protein interaction resulting from CP55940 alone. These include MEK1/2, Src, and Akt kinases. First, phosphorylation of MEK1/2, an upstream kinase (MAPKK) of MAPK in the MEK-ERK signaling pathway, was assessed to evaluate the effect of β-arrestins on its signaling. In control siRNA transfection, both CP55940 and ORG27569 produced a peak at 5min after treatment. The silencing of β-arrestin 1 abolished MEK1/2 phosphorylation upon treatment with ORG27569 but had no significant effect on CP55940-induced phosphorylation. Thus, we demonstrated that the MEK1/2-ERK1/2 pathway is β-arrestin 1-dependent upon ORG27569 treatment. Second, we assessed the activation of upstream kinases, Src and Akt. Src phosphorylation is a necessary step in the activation of various mitogenic signaling pathways activated by GPCRs. It has been shown that non-receptor tyrosine kinases from the Src family are recruited to GPCRs via β-arrestin interactions (Barlic et al., 2000; Imamura et al., 2001). Akt also plays a role in the β-arrestin-mediated ERK1/2 signaling pathway (Goel, Phillips-Mason, Raben, & Baldassare, 2002; Povsic, Kohout, & Lefkowitz, 2003). Interestingly, CP55940 failed to induce Src phosphorylation, whereas ORG27569 produced a peak of Src phosphorylation at 5min. Src phosphorylation only required β-arrestin 1 but not β-arrestin 2, suggesting that the Src pathway is only activated by ORG27569 and via β-arrestin 1 interaction. Both CP55940 and ORG27569 failed to alter Akt phosphorylation levels (Ahn, Mahmoud, Shim, et al., 2013). The procedure utilized is comparable to that described earlier in Section 5.1 and can be used to evaluate a variety of allosteric modulators.
To ensure that the effect of ORG27569 on the Src-MEK1/2-ERK1/2 signaling cascade is not limited to HEK293 cells, we tested the effect of ORG27569 on the activation of the signaling components using rat hippocampal neurons endogenously expressing the CB1 receptor. Consistent with the phosphorylation pattern in HEK293 cells, ORG27569 resulted in increased phosphorylation of ERK1/2, MEK1/2, and Src compared to basal levels, whereas Akt phosphorylation remained unaffected by CP55940 or ORG27569 (Ahn, Mahmoud, Shim, et al., 2013). This suggests that ORG27569 induces allosteric ligand biased signaling patterns and that these are dependent on β-arrestin 1. Nonetheless, cell and tissue type has suggested differences in other CB1 systems (Khajehali et al., 2015) and may involve other mechanisms when analyzed differently (Cawston et al., 2013; Gamage et al., 2016; Straiker, Mitjavila, Yin, Gibson, & Mackie, 2015). Moreover, other kinases can also be assessed including using global phosphorylation kinase array analysis.
Carrying out CB1-induced kinase phosphorylation assays and their sub-sequent analysis can be challenging. The time course for phosphorylation typically varies with test compound and one needs to examine that for different compounds. It should be noted that some commercially available assays are provided as end-point assays with 30–60min readouts and may not monitor other phosphorylation times. Also, the test compounds are best used at a variety of concentrations. If the test allosteric modulator is not an ago-agonist and is used with an orthosteric agonist, attention must be given to the relative concentrations of orthosteric and allosteric modulator employed and their relative affinity for CB1. Furthermore, if the orthosteric ligand is biased for coupling to the receptor differently than the allosteric ligand, additional competitions may be at play (e.g., in some instances orthosteric ligands induce CB1 Gi coupling, while the allosteric ligand might preclude G protein coupling and instead induce β-arrestin 1 binding). In addition, other factors which can influence subsequent cell lysis or phos-phorylation and its detection include the cell density employed, the effect of serum starvation on the signal, the stimulation time, the titer of the antibody used and antibody specificity, best practices in compound dispensing, and the relative hydrophobicity of the orthosteric and allosteric ligands used.
5.2.1 Reagents for Kinase Phosphorylation
Reagents used are HEK293 cells, Dulbecco’s modified Eagle’s medium (DMEM), six-well cell culture plates (Corning), Lipofectamine 2000 transfection kit (Invitrogen), PTX (Calbiochem), siRNA (QIAGEN), lysis buffer (R&D Systems), 1× TBS-T buffer (20mM Tris, 150mM NaCl, pH 7.6), Superblock T20 PBS solution (Thermo Scientific), rabbit anti-phospho-p44/42 primary antibody (Cell Signaling; for ERK1/2 phosphorylation) or other antibody reactive toward the phosphorylated kinase to be assessed, goat anti-rabbit IgG-conjugated peroxidase secondary antibody (Millipore), and SuperSignal West Femto Maximum Substrate (Thermo Scientific).
5.2.2 Protocol for Kinase Phosphorylation
For the kinase phosphorylation analysis, the procedure usually takes 5 days to complete.
Day 1: HEK293 cells are seeded to 50%–70% confluency in a six-well plate and maintained in DMEM with 10% fetal bovine serum and 0.35mg/mL glucose at 37°C in an incubator with 5% CO2.
Day 2: HEK293 cells were transfected with CB1 DNA alone or cotransfected with control siRNA and siRNA to β-arrestin 1 or 2 with Lipofectamine 2000. After transfection, cells are grown for 24h.
Day 3: The medium is changed to DMEM serum-free medium, cells are treated with PTX at 10ng/mL if this treatment is used, and cell growth is continued for 16h.
Day 4: Cells are treated with various concentrations of test compounds (antagonist, agonist, or allosteric modulator) for the specified time (typically 5–20min). The cells are solubilized and lysed in 120μL/well in lysis buffer. The protein concentration is determined by Bradford assay (Bradford, 1976).
Day 5: The protein, the amount determined from the Bradford assay, is loaded into 10% SDS-polyacrylamide gel electrophoresis run to completion. The protein in the gel is transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane blocked with Superblock T20 PBS solution at 4°C with gentle shaking overnight. The PVDF membrane is incubated for 1h with anti-phospho-p44/42 (ERK) primary antibody (1:4000) (or another primary antibody diluted appropriately) at room temperature. The membrane is washed with 1× TBS-T buffer and then incubated with secondary antibody (1:6000) conjugated with peroxidase for 1h at room temperature. After washing with 1× TBS-T buffer, the antibody-reactive components on the immunoblot are detected by using a SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) following the manufacture’s protocol and visualized with X-ray film.
6. PERSPECTIVES
Early thinking about GPCR function assumed the existence of just two GPCR states, the “off” (R) and “on” (R*) modes representing the inactive and active states of the receptor, respectively (De Lean et al., 1980). It is now clear that GPCRs can adopt multiple distinct conformations displaying different states of receptor activation, resulting in different signaling efficacies (Kenakin, 2003). Thus, the transition from inactive to active GPCR forms may include several distinct conformational substrates that can be differentially stabilized by different ligands—and this seems to be true for CB1.
Allosteric modulation of CB1 adds yet another variable and, depending on the allosteric and orthosteric ligands utilized, may adopt different activated (or inactivated) conformations. This may introduce several complicated and apparently confounding consequences (Conn et al., 2009; Khoury, Clément, & Laporte, 2014; Wang, Martin, Brenneman, Luttrell, & Maudsley, 2009) including bias for different coupling partners. Thus, while allosteric modulators of CB1 offer many advantages for mechanistic analysis and for drug development including impacting specific downstream pathways (Basavarajappa, 2007; Hudson, Hébert, & Kelly, 2009; Picone & Kendall, 2015; Smith, Sim-Selley, & Selley, 2010; Turu & Hunyady, 2010), their outcome is complex and must also be evaluated in vivo. While this chapter focused on in vitro analysis of some behaviors of allosteric modulators of CB1, it is anticipated that patterns will emerge and clear correlations will be made between in vitro and in vivo profiles.
Acknowledgments
This research was supported in part by the National Institutes of Health grant DA039942 (to D.A.K.). We thank all authors who have contributed to the elucidation of CB1 modulation although many were not included in this review. We also thank D.A.K.’s laboratory coworkers who have contributed to the knowledge of CB1 allosteric modulators especially Kwang H. Ahn, Leepakshi Khurana, Mariam M. Mahmoud, and Yu-Hsien Liao. The authors gratefully acknowledge Andrew Alt for helpful discussions.
ABBREVIATIONS
- BSA
fatty acid-free bovine serum acid
- CB1
cannabinoid receptor 1
- DMSO
dimethyl sulfoxide
- ERK1/2
extracellular signal-regulated kinase isoforms 1 and 2
- GDP
guanosine diphosphate
- GPCR
G protein-coupled receptor
- GTP
guanosine triphosphate
- GTPγS
[35S]-guanine 5′-3-O-(thio)triphosphate
- HEK293 cells
human embryonic kidney cells
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- PTX
pertussis toxin
- SAM
silent allosteric modulator
- SDS
sodium dodecyl sulfate
- WT
wild-type CB1
Footnotes
Conflict of Interest. The authors have no conflict of interest.
References
- Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. The Journal of Biological Chemistry. 2012;287(15):12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Samala S, Lu D, Kendall DA. Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. Journal of Neuro-chemistry. 2013;124:584–589. doi: 10.1111/jnc.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Shim J-Y, Kendall DA. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) The Journal of Biological Chemistry. 2013;288(14):9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Nishiyama A, Mierke DF, Kendall DA. Hydrophobic residues in helix 8 of cannabinoid receptor 1 are critical for structural and functional properties. Biochemistry. 2009;49(3):502–511. doi: 10.1021/bi901619r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard SM, Patterson TA, Jin W, Chavkin C. Agonist-induced phosphorylation of the κ-opioid receptor. Journal of Neurochemistry. 1997;69(6):2405–2412. doi: 10.1046/j.1471-4159.1997.69062405.x. [DOI] [PubMed] [Google Scholar]
- Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, et al. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Molecular Pharmacology. 2013;83(2):322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, et al. Regulation of tyrosine kinase activation and granule release through [beta]-arrestin by CXCR1. Nature Immunology. 2000;1(3):227–233. doi: 10.1038/79767. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS. The endocannabinoid signaling system: A potential target for next-generation therapeutics for alcoholism. Mini Reviews in Medicinal Chemistry. 2007;7(8):769–779. doi: 10.2174/138955707781387920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, et al. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. The Journal of Biological Chemistry. 2012;287(44):36944–36967. doi: 10.1074/jbc.M112.382481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry EM, Mechoulam R. Tetrahydrocannabinol and endocannabinoids in feeding and appetite. Pharmacology & Therapeutics. 2002;95(2):185–190. doi: 10.1016/s0163-7258(02)00257-7. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T, et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology. 2010;35(3):764–774. doi: 10.1038/npp.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72(1):248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O’Connell J, et al. Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(26):10830–10835. doi: 10.1073/pnas.1300393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochemical Pharmacology. 2011;81(6):691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M. Real-time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. British Journal of Pharmacology. 2013;170(4):893–907. doi: 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical Pharmacology. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. Quantification of allosteric interactions at G protein coupled receptors using radioligand binding assays. Current Protocols in Pharmacology. 2001;11(1.22):1.22.1–1.22.40. doi: 10.1002/0471141755.ph0122s52. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nature Reviews Drug Discovery. 2002;1(3):198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacological Reviews. 2002;54(2):323. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRS: A novel approach for the treatment of CNS disorders. Nature Reviews Drug Discovery. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kwok ML, Mackie K. Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. Journal of Neurochemistry. 2008;106(1):70–82. doi: 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antona AM, Ahn KH, Kendall DA. Mutations of CB1 T210 produce active and inactive receptor forms: Correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry. 2006;45(17):5606–5617. doi: 10.1021/bi060067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antona AM, Ahn KH, Wang L, Mierke DF, Lucas-Lenard J, Kendall DA. A cannabinoid receptor 1 mutation proximal to the dry motif results in constitutive activity and reveals intramolecular interactions involved in receptor activation. Brain Research. 2006;1108(1):1–11. doi: 10.1016/j.brainres.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. The Journal of Biological Chemistry. 1980;255(15):7108–7117. [PubMed] [Google Scholar]
- DeWire SM, Violin JD. Biased ligands for better cardiovascular drugs dissecting G-protein-coupled receptor pharmacology. Circulation Research. 2011;109(2):205–216. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar A, Murataeva N, Makriyannis A, Straiker A, Mackie K. Two Janus cannabinoids that are both CB2 agonists and CB1 antagonists. Journal of Pharmacology and Experimental Therapeutics. 2017;360(2):300–311. doi: 10.1124/jpet.116.236539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372(6507):686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Fay JF, Farrens DL. A key agonist-induced conformational change in the can-nabinoid receptor CB1 is blocked by the allosteric ligand Org 27569. The Journal of Biological Chemistry. 2012;287(40):33873–33882. doi: 10.1074/jbc.M112.352328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage TF, Anderson JC, Abood ME. CB1 allosteric modulator Org27569 is an antagonist/inverse agonist of ERK1/2 signaling. Cannabis and Cannabinoid Research. 2016;1(1):272–280. doi: 10.1089/can.2016.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, et al. Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: Structure–activity relationship studies on 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1) Journal of Medicinal Chemistry. 2014;57(18):7758–7769. doi: 10.1021/jm501042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments camp accumulation in striatal neurons: Evidence for a G(s) linkage to the CB1 receptor. Journal of Neuroscience. 1997;17(14):5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel R, Phillips-Mason PJ, Raben DM, Baldassare JJ. A-thrombin induces rapid and sustained Akt phosphorylation by β-arrestin 1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. The Journal of Biological Chemistry. 2002;277(21):18640–18648. doi: 10.1074/jbc.M108995200. [DOI] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, et al. Psncbam-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. British Journal of Pharmacology. 2007;152(5):805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacological Reviews. 2002;54(2):161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate-cyclase. Pharmacology of the response in neuro-blastoma cell-membranes. Molecular Pharmacology. 1984;26(3):532–538. [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. Journal of Neurochemistry. 1999;73(2):493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Hudson BD, Hébert TE, Kelly MEM. Ligand- and heterodimer-directed signaling of the CB1 cannabinoid receptor. Molecular Pharmacology. 2009;77(1):1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, et al. B-arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. The Journal of Biological Chemistry. 2001;276(47):43663–43667. doi: 10.1074/jbc.M105364200. [DOI] [PubMed] [Google Scholar]
- Janero DR, Thakur GA. Leveraging allostery to improve G protein-coupled receptor (GPCR)-directed therapeutics: Cannabinoid receptor 1 as discovery target. Expert Opinion on Drug Discovery. 2016;11(12):1223–1237. doi: 10.1080/17460441.2016.1245289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, et al. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. Journal of Neuroscience. 1999;19(10):3773. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Pharmacological proteus? Trends in Pharmacological. Sciences. 1995;16(8):256–258. doi: 10.1016/s0165-6147(00)89037-9. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Ligand-selective receptor conformations revisited: The promise and the problem. Trends in Pharmacological Sciences. 2003;24(7):346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacological Reviews. 2010;62(2):265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K. Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Molecular Pharmacology. 2015;88(2):368. doi: 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- Khoury E, Clément S, Laporte SA. Allosteric and biased G protein-coupled-receptor signaling regulation: Potentials for new therapeutics. Frontiers in Endocrinology. 2014;5:68. doi: 10.3389/fendo.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Ali HI, Olszewska T, Ahn KH, Damaraju A, Kendall DA, et al. Optimization of chemical functionalities of indole-2-carboxamides to improve allosteric parameters for the cannabinoid receptor 1 (CB1) Journal of Medicinal Chemistry. 2014;57(7):3040–3052. doi: 10.1021/jm5000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Fu B-Q, Duddupudi AL, Liao Y-H, Immadi SS, Kendall DA, et al. Pyrimidinyl biphenylureas: Identification of new lead compounds as allosteric modulators of the cannabinoid receptor CB1. Journal of Medicinal Chemistry. 2017;60(3):1089–1104. doi: 10.1021/acs.jmedchem.6b01448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovoor A, Nappey V, Kieffer BL, Chavkin C. M and δ opioid receptors are differentially desensitized by the coexpression of β-adrenergic receptor kinase 2 and β-arrestin 2 in Xenopus oocytes. The Journal of Biological Chemistry. 1997;272(44):27605–27611. doi: 10.1074/jbc.272.44.27605. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annual Review of Pharmacology and Toxicology. 1998;38(1):289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. Allosteric agonists of 7TM receptors: Expanding the pharmacological toolbox. Trends in Pharmacological Sciences. 2006;27(9):475–481. doi: 10.1016/j.tips.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Denovan-Wright EM. Cannabinoid receptor ligand bias: Implications in the central nervous system. Current Opinion in Pharmacology. 2017;32:32–43. doi: 10.1016/j.coph.2016.10.005. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly MEM, Denovan-Wright EM. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. British Journal of Pharmacology. 2015;172(20):4790–4805. doi: 10.1111/bph.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Kulkarni PM, Deschamps JR, Kelly MEM, Janero DR, Cascio MG, et al. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chemical Neuroscience. 2017;8(6):1188–1203. doi: 10.1021/acschemneuro.6b00310. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive endocytic cycle of the CB1 cannabinoid receptor. The Journal of Biological Chemistry. 2004;279(34):36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR. Pharmacology and stereoselectivity of structurally novel cannabinoids in mice. Journal of Pharmacology and Experimental Therapeutics. 1988;247(3):1046–1051. [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335(6072):1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(9):3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud MM, Ali HI, Ahn KH, Damaraju A, Samala S, Pulipati VK, et al. Structure-activity relationship study of indole-2-carboxamides identifies a potent allosteric modulator for the cannabinoid receptor 1 (CB1) Journal of Medicinal Chemistry. 2013;56(20):7965–7975. doi: 10.1021/jm4009828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annual Review of Pharmacology and Toxicology. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB 1) receptor by RTI-371, a selective inhibitor of the dopamine transporter. British Journal of Pharmacology. 2009;156(7):1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona FA, Ferreira J, Menezes de Lima O, Duarte FS, Bento AF, Forner S, et al. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(51):21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacological Reviews. 2010;62(4):588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone RP, Kendall DA. Minireview: From the bench, toward the clinic: Therapeutic opportunities for cannabinoid receptor modulation. Molecular Endocrinology. 2015;29(6):801–813. doi: 10.1210/me.2015-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povsic TJ, Kohout TA, Lefkowitz RJ. B-arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis. The Journal of Biological Chemistry. 2003;278(51):51334–51339. doi: 10.1074/jbc.M309968200. [DOI] [PubMed] [Google Scholar]
- Prescott WR, Gold LH, Martin BR. Evidence for separate neuronal mechanisms for the discriminative stimulus and catalepsy induced by delta 9-THC in the rat. Psychopharmacology. 1992;107(1):117–124. doi: 10.1007/BF02244975. [DOI] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Molecular Pharmacology. 2005;68(5):1484. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Qiao C-J, Ali HI, Ahn KH, Kolluru S, Kendall DA, Lu D. Synthesis and biological evaluation of indole-2-carboxamides bearing photoactivatable functionalities as novel allosteric modulators for the cannabinoid CB1 receptor. European Journal of Medicinal Chemistry. 2016;121:517–529. doi: 10.1016/j.ejmech.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet Trillou C, Arnone M, Delgorge C, Gonalons N, Keane P, Maffrand JP, et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2003;284(2):R345–R353. doi: 10.1152/ajpregu.00545.2002. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH. Appetite suppressants, cardiac valve disease and combination pharmacotherapy. American Journal of Therapeutics. 2009;16(4):354–364. doi: 10.1097/MJT.0b013e31817fde95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo EB. Cannabinoids in the management of difficult to treat pain. Therapeutics and Clinical Risk Management. 2008;4(1):245–259. doi: 10.2147/tcrm.s1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito VM, Rezende RM, Teixeira AL. Cannabinoid modulation of neuroinflammatory disorders. Current Neuropharmacology. 2012;10(2):159–166. doi: 10.2174/157015912800604515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore DM, Baillie GL, Hurst DH, Navas F, 3rd, Seltzman HH, Marcu JP, et al. Allosteric modulation of a cannabinoid G protein-coupled receptor: Binding site elucidation and relationship to G protein signaling. The Journal of Biological Chemistry. 2014;289(9):5828–5845. doi: 10.1074/jbc.M113.478495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB(1) receptor-interacting proteins: Novel targets for central nervous system drug discovery? British Journal of Pharmacology. 2010;160(3):454–466. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388(6644):773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Stornaiuolo M, Bruno A, Botta L, Regina GL, Cosconati S, Silvestri R, et al. Endogenous vs exogenous allosteric modulators in GPCRs: A dispute for shuttling CB1 among different membrane microenvironments. Scientific Reports. 2015;5:15453. doi: 10.1038/srep15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mitjavila J, Yin D, Gibson A, Mackie K. Aiming for allosterism: Evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacological Research. 2015;99:370–376. doi: 10.1016/j.phrs.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. Journal of Molecular Endocrinology. 2010;44(2):75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- Vallée M, Vitiello S, Bellocchio L, Hébert-Chatelain E, Monlezun S, Martin-Garcia E, et al. Pregnenolone can protect the brain from cannabis intoxication. Science. 2014;343(6166):94–98. doi: 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S RIO-Europe Study Group. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365(9468):1389–1397. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- Walsh D, Nelson KA, Mahmoud FA. Established and potential therapeutic applications of cannabinoids in oncology. Support Care Cancer. 2003;11(3):137–143. doi: 10.1007/s00520-002-0387-7. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. British Journal of Pharmacology. 2004;141(5):775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Martin B, Brenneman R, Luttrell LM, Maudsley S. Allosteric modulators of G protein-coupled receptors: Future therapeutics for complex physiological disorders. Journal of Pharmacology and Experimental Therapeutics. 2009;331(2):340–348. doi: 10.1124/jpet.109.156380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454(7203):486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nature Reviews: Drug Discovery. 2013;12(8):630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Aber MJ, Giros B, Lefkowitz RJ. Molecular mechanisms of G protein-coupled receptor signaling: Role of G protein-coupled receptor kinases and arrestins in receptor desensitization and resensitization. Receptors & Channels. 1997;5(3–4):193–199. [PubMed] [Google Scholar]