

Abstract
The life of an organism requires the assistance of an unlikely process: programmed cell death. Indeed, both development and the maintenance of homeostasis results in the production of superfluous cells, that must eventually be disposed of. Furthermore, programmed cell death can also represent a defense mechanism, for example by depriving pathogens of a replication niche. The responsibility of handling these dead cells falls on phagocytes of the immune system, who surveil their surroundings for dying or dead cells and efficiently clear them in a quiescent manner. This process, termed efferocytosis, depends on cooperation between both the phagocyte and the dying cell. In this review, we explore different types of programmed cell death and their impact on innate immune responses.
The importance of programmed cell death
“Death is so terribly final, while life is full of possibilities.”
- Tyrion Lannister
We have long recognized the importance of phagocytosis (from the Greek, meaning “the process of cellular devouring”) in the clearance of invading pathogens, but its more understated role is the sensing, recognition, and removal of cellular corpses. As over 50 billion cells undergo programmed cell death in the human body each day, it is imperative that their clearance occurs efficiently and silently. This evolutionarily conserved process, termed efferocytosis, is critical to maintain developmental and immune homeostasis and has been described in Drosophila melanogaster, Caenorhabditis elegans, and vertebrates [1, 2]. The magnitude of this efferocytotic effort is emphasized by the fact that uncleared dead cells are rarely observed, even in tissues with high rates of cellular turnover such as the thymus.
As the goal of efferocytosis is the quiet removal of cellular corpses, one could theorize that part of the program of programmed cell death would be the packaging of dying cells into immunologically inert pieces. While immunotolerance is one of the defining characteristics of apoptosis, not all modes of programmed cell death intrinsically induce tolerance. Necroptosis and pyroptosis both result in cell rupture, releasing their intracellular, immunostimulatory contents and elicit a robust inflammatory immune response that can be beneficial in the elimination of pathogens but is also linked to autoimmune and autoinflammatory disorders [3].
The immune system has evolved to recognize, respond, and remember danger, whether it be in the form of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [4, 5]. Though biologically beneficial and immunologically inert while contained safely within a cell, some intracellular components can transform into DAMPs upon release. DAMPs can be nuclear or cytosolic proteins, including high mobility group box 1 (HMGB1), S100 family of calcium-binding proteins, interleukin 1 (IL-1) family members, transcription factor A mitochondrial (TFAM), and histones, or non-protein molecules, such as ATP, uric acid, DNA, RNA, and mitochondrial DNA [6]. The effects of DAMPs are mediated by engagement of pattern recognition receptors (PRRs), including toll-like receptors (TLRs), cytosolic sensors (NOD-like receptors, DAI, RIG-I-like receptors, AIM2-like receptors), and the advanced glycosylation end product-specific receptor (RAGE) [7].
While lysis during cell death is known to release high quantities of HMGB1 compared to apoptotic death, it is also important to note that different forms of death can directly affect the biochemical status of these DAMPs. For example, the oxidative state of HMGB1 seems to determine the immunogenicity of this DAMP, as caspase-dependent oxidation of Cys106 on HMGB1 destroyed its ability to function as an immunostimulatory molecule [8]. The innate recognition of DAMPs also influences the maturation of antigen-presenting cells, ultimately shaping the adaptive immune response. Uptake of ruptured dead cells by dendritic cells results in antigen presentation to and activation of both CD4+ and CD8+ T cells, while engulfment of apoptotic cells results in only CD8+ T cell activation, thus removing the “help” from the CD8+ T cell response and rendering them susceptible to apoptosis following re-exposure to antigen [9]. DAMPs are also both a trigger for the production of autoantibodies by B cells and the maintenance of said autoantibodies, which are supported by the continuous exposure to autoantigens. The uncontrolled release of DAMPs (as well as autoantibodies to common DAMPs, such as self dsDNA and HMGB1) are highly associated with many inflammatory-related diseases, such as systemic-lupus erythematosus (SLE), rheumatoid arthritis, sepsis, atherosclerosis, and diabetes [10]. Exploring the mechanisms of DAMP release is vital to understanding their functions, both advantageous and deleterious, in pathological settings (Figure 1).
Figure 1. Different types of programmed cell death elicit different immune responses during efferocytosis.
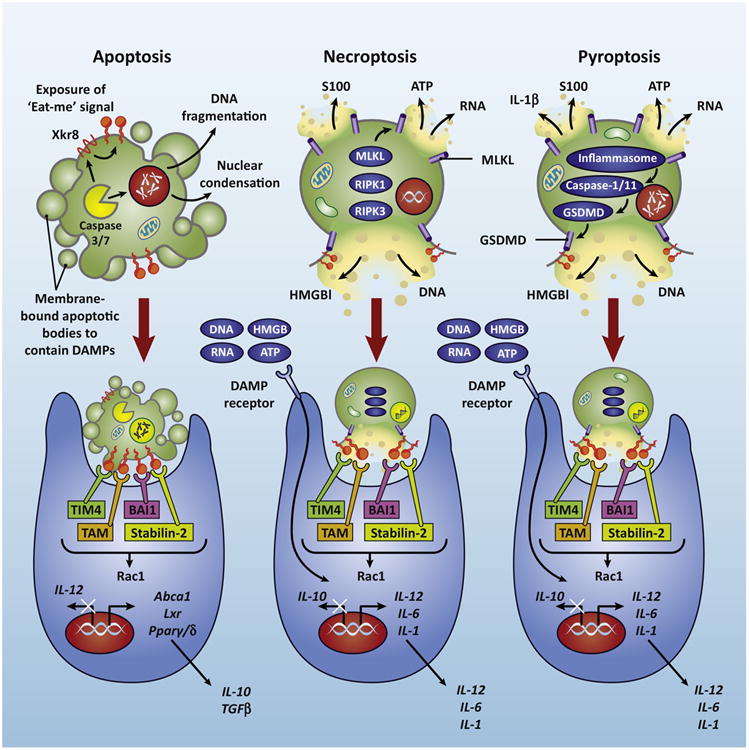
Caspase-mediated apoptosis results in DNA fragmentation, membrane blebbing, and caspase-dependent exposure of “eat-me” signals”. Phagocytes employ receptors that specifically recognize “eat-me” signals to facilitate engulfment of dying cells and a tolerant immune response. Necroptotic (mediated by RIPK1-RIPK3-MLKL) and pyroptosis (mediated by inflammatory caspases and GSDMD) results in membrane rupture and the release of DAMPs, like DNA, RNA, HMGB1, ATP, and IL-1β. These DAMPs are sensed by PRRs on the phagocyte, resulting in inflammation.
Apoptosis
“Give me a good, clean death.”
- Tyrion Lannister
From a developmental and physiological perspective, apoptosis (from the Greek meaning “falling off”), a genetically programmed form of cellular suicide, is the most common form of cell death. Apoptosis results in the discreet partitioning of cells into plasma membrane-enclosed “apoptotic bodies” containing intracellular components. These compartments are a morphological characteristic of apoptosis, but also a strategic mechanism to contain possible DAMPs and prevent unwanted immune responses [11].
Apoptosis is largely coordinated by a family of cysteine proteases called caspases. Caspases are broadly grouped into initiator caspases (caspase-2, -8, and -9), executioner caspases (caspase-3, -6, and -7), and inflammatory caspases (human caspase-1, -4, and -5; rodent caspase-1 and –11) [12, 13]. Initiator caspases gain activity through dimerization (“induced proximity model”), resulting in autocatalytic cleavage and stabilization of a dimer capable of cleaving numerous targets, including executioner caspases [13]. Executioner caspases themselves achieve activation via this cleavage, which results in a conformational change that brings the dimer's two active sites together to create a functional mature protease. Once active, executioner caspases cleave and activate other executioner caspases, leading to a rapid feedback loop to facilitate apoptosis [14]. Caspases have a limited repertoire of specificity, primarily cleaving at aspartate-terminal sequence residues, though the Drosophila melanogaster caspase homologue, DRONC, has also been shown to cleave after glutamate residues [12].
1.1.1.1.1 The intrinsic or mitochondrial pathway is the most common mechanism of apoptosis, resulting in mitochondrial outer membrane permeabilization (MOMP) to release cytochrome c and other sequestered proteins present in the intermembrane space of mitochondria [15]. This pathway is activated by a variety of signals, notably developmental signals (hormones) and other stress-inducing stimuli (cytoskeletal disruption, DNA damage, accumulation of unfolded proteins, hypoxia) [13]. These signals converge on mitochondria, where the balancing act between pro-apoptotic and anti-apoptotic members of the BCL2 family control cell fate [13, 15] by controlling the release of cytochrome c. Once it reaches the cytosol, cytochrome c complexes with caspase-9 and the adaptor APAF-1 to form the apoptosome, leading to the activation of the downstream executioner caspases, caspase-3 and caspase-7 [16].
1.1.1.1.2 Apoptosis can also be triggered via the extrinsic pathway. Ligands that engage extracellular death receptors (DR), such as tumor necrosis factor (TNF),TNF-related apoptosis-inducing ligand (TRAIL), orCD95-ligand (CD95-L, Fas-L), induce the recruitment ofpro-caspase-8 to adaptor proteins FADD or TRADD localized to the cytoplasmic tail of the engaged DR. These proteins form the death-inducing signaling complex (DISC), at which homodimerized caspase-8 activates caspase-3 and caspase-7 [14]. Crosstalk between extrinsic and the intrinsic pathways exists, where caspase-8 activity cleaves and activates pro-apoptotic BCL2 family proteins to trigger mitochondrial apoptosis, amplifying the cell death cycle [13, 15].
While the development of a functional organism requires the proper execution of apoptosis, perhaps the most astounding aspect is that this task is accomplished in the absence of immune activation. The prevailing hypothesis states that apoptosis results in the inconspicuous packaging of potentially immunogenic cellular material into tolerogenic pieces, allowing for programmed cell death to occur without overt inflammation [11, 17]. The containment of DAMPs, such as DNA, RNA, HMGB1, nucleotides, and S100 proteins, within plasma membrane blebs or the intact apoptotic cell allows dying cells to escape detection by innate immune sensors [18]. This is supported by demonstrations that the effectiveness of anti-tumor therapies seems to rely on their ability to induce non-apoptotic cell death in which intracellular contents are released [19, 20].
Programmed necrosis
“Chaos isn't a pit, chaos is a ladder.”
– Petyr “Littlefinger” Baelish
Necrosis has long been considered as a form of accidental and uncontrolled cell death, however, as described in relatively recent studies, it is now clear that cells can undergo programmed necrosis, also called necroptosis. This form of cell death was named for its morphological similarity to the unprogrammed, lytic process of necrosis: necroptotic cells swell and burst, releasing their intracellular contents. This morphology stands in clear contrast to that of apoptosis (discussed above), and has led to the idea that necroptosis-mediated DAMP release renders this cell death program uniquely inflammatory [21].
Mechanistically, necroptosis is defined by the activation of two members of the receptor interacting protein kinase (RIPK) family, RIPK1 and RIPK3. These kinases interact via specialized domains termed RIP homotypic interaction motifs (RHIM) to form oligomeric structures within the cytosol called “necrosomes” or, sometimes, “ripoptosomes.” Within necrosomes, reciprocal interactions between RIPK1 and RIPK3 lead to activation of the latter, which in turn phosphorylates a pseudokinase called MLKL [22]. Once phosphorylated, MLKL translocates to cell membranes where it binds to phosphatidylinositol phosphates to affect cellular ion homeostasis, leading to osmotic cell membrane rupture [23-25].
Necrosome assembly can be triggered by activation of RHIM containing proteins, of which there are four in the mammalian proteome: in addition to RIPK1 and RIPK3, the TLR adapter TRIF and the putative DNA sensor DAI encode RHIM domains, and their activation can therefore trigger necrosome assembly and necroptosis [26, 27]. RIPK1 activation can occur directly downstream of the same death receptors discussed above as activators of apoptosis, while TRIF is activated by ligation of TLR3 or TLR4 [28]. Activating ligands for DAI are less well-characterized; this sensor was initially described as a sensor of Z-form DNA, but recent work has shown that it can mediate necroptosis in response to infection by both DNA and RNA viruses [27]. Importantly, necroptosis is under tight negative control by the pro-apoptotic protease caspase-8, which acts in concert with its paralog cFLIP to suppress necrosome assembly [21]. Thus, necroptotic cell death in response to the stimuli enumerated above generally occurs only when caspase-8 is absent or inhibited. Notably, many pathogens encode caspase inhibitors, leading to the idea that necroptosis represents a “trap door” by which cell death can occur when the apoptotic pathway is subverted during infection.
As mentioned above, necroptosis is a lytic form of cell death, and early studies of necroptosis presumed that DAMP release would lead to potent inflammation in response to this form of cell death. This may be true in some cases, but two observations hint that the immune response triggered by necroptosis extends beyond simple DAMP sensing. First, it is worth noting that activation of the TLRs or death receptors, such as TNF receptor, lead to the activation of NF-κB, IRF, and MAPK pathways; these are among the most potent inflammatory transcription programs available to the cell. When TLR or death receptor ligation is combined with caspase inhibition, these same stimuli trigger “inflammatory” necroptosis. However, as pointed out in an elegant and heterodox study by Martin and colleagues, the inflammatory potential of a live cell producing TNF or TLR-dependent inflammatory cytokines greatly outstrips that of the DAMPs released by the death of that cell [29]. Thus, in some cases necroptosis may actually curtail inflammatory signaling through elimination of cells in which these programs are active. It may therefore be that elimination of infected cells by whatever means—and thereby elimination of the replicative niche of the pathogens they contain—is more important to the immune response than the DAMPs that accompany cell death [30].
Beyond these observations, recent work has pointed to roles for the necrosome that extend beyond the induction of cell death. Several studies have pointed to differences in the phenotype of mice lacking RIPK3 and those lacking MLKL [25, 31]. In both cases, necroptotic cell death is entirely absent, but relative to Mlkl-/- animals, Ripk3-/- mice display enhanced protection in models of ischemic injury and sterile shock, and increased susceptibility to infection with influenza and West Nile virus [32, 33]. Together, these findings imply that RIPK3 has roles in coordinating inflammation and immunity that extend beyond the activation of MLKL and necroptosis. Consistent with this idea, we recently found that RIPK1 and RIPK3 is required to coordinate neuroinflammation during West Nile virus encephalitis, and that this role is wholly independent of MLKL or the induction of necroptosis. Notably, mature neurons displayed a general resistance to necroptosis, a finding consistent with previous reports that this cell type is also resistant to apoptosis [34]. Because mature neurons are both essential and non-regenerating, it may be that they have evolved to avoid cell death as a defensive strategy; that they are, in effect, “too important” to die. It may therefore be the case that necroptotic signaling has been re-wired in these cells, such that infection-induced RIPK1-RIPK3 signaling restricts infection through transcriptional responses, not cell death.
Pyroptosis
“I take what is mine with fire and blood.”
—Daenerys Targaryen
Mechanisms of programmed cell death have evolved to promote inflammation by releasing intracellular DAMPs and cytokines extracellularly. This process, known as pyroptosis (from the Greek word pyro, meaning “fire”) is a lytic, regulated cell death mode that requires the enzymatic activity of inflammatory caspases [35]. Like necroptosis, pyroptosis involves plasma membrane pore formation and cellular swelling, ultimately leading to rupture. However, pyroptosis has characteristics reminiscent of apoptosis, including caspase-dependence, chromatin condensation, and DNA fragmentation [36]. The defining feature of pyroptosis is its dependence on the inflammatory caspases (human caspase-1/4/5; rodent caspase-1/11). Caspase-1 was first identified by its ability to convert pro-IL-1β and pro-IL-18 into their active forms [37]. Caspase-1 activation is achieved upon interaction with a cytoplasmic, multiprotein complex called the inflammasome.
Several different types of inflammasome can be produced depending upon the protein components involved and the cellular perturbations triggering their activation. The PRR component of inflammasomes senses dangers signals, and five cytoplasmic PRRs have been confirmed to form inflammasomes: NLRP1 (anthrax lethal toxin), NLRP3 (PAMPs, asbestos, silica, ATP, serum amyloid A, uric acid crystals), NLRC4 (flagellin, type III/IV secretion systems, sensed through the NAIP family members), AIM2 (dsDNA), and Pyrin (bacterial toxins) [38, 39]. These PRRs contains either a caspase activation and recruitment domain (CARD) or a pyrin domain (PYD). PPRs with a CARD (NLRP1 and NLRC4) can interact directly with pro-caspase-1 through its CARD and promote pro-caspase-1 activation. PRRs that contain only a PYD domain (NLRP3) must first recruit adapter apoptosis-associated speck-like protein containing a CARD (ASC), through PYD-PYD interactions, to recruit and activate caspase-1 [36]. Non-canonically, hexa-acylated LPS and lipid A bind directly to the CARD of caspase-4/5/11 to induce their oligomerization and caspase-1 activation [40, 41]. Caspase-11 can also mediate pyroptosis independently of caspase-1, though its control of cytokine processing and production is dependent on both the NLRP3 inflammasome and caspase-1. The mechanisms by which caspase-11 activates the NLRP3 inflammasome is still being debated [37, 42].
Gasdermin D (GSDMD) was recently identified as a critical effector of pyroptosis and IL-1β release downstream of inflammatory caspases. Of note, pro-IL1β processing was not inhibited in GSDMD-deficient cells, indicating that IL-1β secretion is pyroptosis-dependent [42, 43]. Mechanistically, Caspase-1/4/5/11 cleave both mouse and human GSDMD at conserved residues, which allows GSDMD to bind lipids in the cell membrane and oligomerize into pore structures, resulting in cell rupture [44-46]. Like other types of lytic cell death, pyroptosis results in the extracellular release of DAMPs. HMGB1 has been shown to be released in a caspase-1-dependent manner [47], and IL-1α, which is active in both its pro- and processed forms, is released upon permeabilization of the plasma membrane [48]. Similarly, secreted IL-1β itself has been proposed as a potential DAMP and is associated with inflammation [49]. Further experimentation is needed to determine the role that pyroptotic cell death, apart from cytokine secretion, plays in inflammasomopathies and the potential for modulation of pyroptosis as an effective immunotherapeutic strategy.
Notably, GSDMD is not the only member of the Gasdermin family, and recent work has demonstrated that other family members share the property of pore formation upon cleavage-mediated activation. However, while GSDMD has been clearly defined as a substrate of the inflammatory caspases, proteases responsible for cleaving other members of the Gasdermin family remain elusive. These findings have led to a proposal to redefine pyroptosis as “Gasdermin-mediated programmed necrosis,” with inflammasome/GSDMD-dependent cell death as just one of a potentially related family of cell death modalities [50].
Other types of cell death
Ferroptosis
“I pay the iron price.”
– Balon Greyjoy
Ferroptosis (from the Latin ferrum, meaning <xps:span class=“ceCheck”>‘</xps:span>iron<xps:span class=“ceCheck”>’</xps:span>) is a cell death pathway distinct from apoptosis and necrosis. Ferroptosis requires iron and the generation of radicals in the form of reactive oxygen species (ROS) and lipid peroxide species and may have evolved as an ancient defense against the buildup of toxic radicals within a cell [51]. Discovered following small-molecule screens for cell death-inducing compounds, ferroptosis can be initiated by erastin and RSL3, both of which require the presence of iron, ROS, and oncogenic MEK activity and result in a toxic, oxidative state in the cell [52]. Morphologically, ferroptosis exhibits condensed, ruptured mitochondria, and rounding-up, yet lacks many of the defining characteristics of other programmed death, such as blebbing, lysis, or DNA fragmentation. However, it has been reported that ferroptosis results in the release of DAMPs, such as HMGB1 and IL-33, though further studies are required to delineate this pathway [53].
Autophagy
“If you think this has a happy ending, you haven't been paying attention.”
- Ramsay Bolton
The essence of autophagy is a pro-survival strategy in response to stress, including environmental damage, chemical fluctuations, metabolic deprivation, or pathogen exposure, allowing cells to preserve biosynthetic and energetic function by cannibalizing intracellular substrates for de novo protein and ATP synthesis [54]. Under nutrient rich conditions, autophagy is inhibited by the mTOR complex, but during nutrient depletion, the pre-initiation complex becomes activated as the mTOR complex is simultaneously inhibited [55]. The now active pre-initiation complex then activates the Class III PI3K complex, composed of core components Beclin1, VPS34 (the class III PI3K), and VPS15, to initiate vesicle nucleation [56]. The Class III PI3K complex generates PI(3)P which serves as a recruitment signal for two ubiquitination-like, conjugation systems: the ATG5-12-16L and LC3-PE conjugation systems. Collectively, these two conjugation systems mediate the elongation, stabilization, ultimate closure, and maturation of the autophagosomes. LC3 (or ATG8), is cleaved to its cytosolic form, LC3-I, exposing a carboxyl terminal glycine, which is subsequently lipidated with phosphatidylethanolamine (PE) to its membrane-bound form, LC3-II. Recent evidence suggest LC3-II is crucial for the targeting of autophagosomes to lysosomes and ultimately successful autophagy [57, 58].
We now recognize that the autophagy machinery is linked to a myriad of biological processes, like apoptosis, necroptosis, mitophagy, xenophagy, and LC3-associated phagocytosis (LAP, below) that function beyond the realm of nutrient stress and survival [57]. It has been reported that cells can die via an autophagy-dependent mechanism. Originally described as “autophagic cell death”, the only criteria for this phenomenon was the presence of excessive autophagosomes in dying cells. Whether autophagy played a causative role in death or was merely an attempt to prevent said death was not known. Recent studies, however, have described an autophagy-dependent, non-apoptotic form of cell death termed autosis. During autosis, autophagy-triggering peptides, starvation, or ischemia can induce autosis requiring both autophagic machinery and activity of Na+,K+-ATPase. Morphologically, autosis is characterized by numerous autophagosomes, plasma membrane rupture, enhanced cell substrate adhesion, focal ballooning of perinuclear space, and disruption of endoplasmic reticulum [59]. The role that autotic cell death plays in inflammation has yet to be described, though its lytic nature indicates that it is pro-inflammatory.
Efferocytosis
“If a man paints a target on his chest, he should expect that sooner or later someone will loose an arrow on him.”
- Tyrion Lannister
Efferocytosis (from the Latin meaning “to take to the grave”) is the process by which dying cells are cleared by phagocytes, and is a carefully orchestrated process that requires active participation from both the dying cells and phagocytes. Both professional (macrophages, dendritic cells) and non-professional (epithelial cells) phagocytes are recruited toward areas of cell death (“Find-me” signals), recognize and engage cellular corpses (“Eat-me” signals), and internalize dying cells for degradation and processing [60]. Apoptotic cells actively “advertise” their presence to other phagocytes to facilitate their own clearance, releasing “find-me” signals, such as ATP, UTP, sphingosine-1-phosphate (S1P), lysophosphatidylcholine (LPC), and CX3CL1 (fractalkine), which are sensed by phagocytes via cognate receptors (P2Y2, S1PRs, G2A, and CXCR3, respectively) [61]. While the release of “find-me” signals is often caspase-dependent, and an important first step in efferocytosis, recent studies have demonstrated that their function, specifically in vivo, lies beyond their ability to attract phagocytic cells [62, 63]. Deficiency of a single “find-me” signal or its receptor often does not result in a defect in efferocytosis, indicating that other factors can regulate recruitment [64]. The ability of “find-me” signals to act more than locally is also unknown. The lipid S1P is present in the circulation at a concentration higher than that released by apoptotic cells, and the release of nucleotides from apoptotic cells is less than 2% of intracellular nucleotide levels and can be easily degraded by extracellular nucleotidases [65]. A recent study demonstrated that S1P signaling on phagocytes initiates an EPO-EPOR-dependent upregulation of phagocytic molecules, including Mer, MFGE8, CD36, and Gas6 [66]. Collectively, we now recognize that while “find-me” signals can act as a local chemoattractant, they also play a vital role in priming the phagocyte for engulfment. The ability of the immune system to distinguish living cells from dead cells is vital to development and the prevention of unwanted inflammation. This feat is accomplished by the display of “eat-me” signals on dying cells. The most well-characterized “eat-me” signal is phosphatidylserine (PS), a lipid confined to the inner leaflet in viable cells, but rapidly externalized by caspase-3-mediated activation of the scramblase Xkr8 [67, 68].
Conversely, the flippase ATP11C is inactivated by caspase-3, rendering PS externally exposed [69]. While the presence of PS on a cell is considered an insurmountable and evolutionarily-conserved signal of cellular demise, not all PS exposures are treated equally and not all PS exposure is a death sentence. Indeed, activated T cells expose PS on their surface, yet avoid uptake by phagocytes, possibly through the co-expression of “don't eat me” signals, such as CD47 [70, 71]. Similarly, constitutive PS exposure by mutant TMEM16F can result in rapid and reversible PS externalization, which does not result in phagocytic uptake, contrary to Xkr8-mediated irreversible PS externalization [72]. Recent work has demonstrated that PS exposure and even plasma membrane permeability are not irreversible events during programmed necrosis [23]. Furthermore, recent reports demonstrate that early exposure of PS on cells, that precedes blebbing or other signs of apoptotic execution, is sufficient to induce efferocytosis, a process that itself leads to the actual death of engulfed cell, possibly in an attempt to preemptively clear dying cells before secondary necrosis and release of DAMPs.
Phagocytes recognize exposed PS via a variety of PS-specific receptors, such as T cell immunoglobulin mucin receptor 4 (TIM4), brain-specific angiogenesis inhibitor 1 (BAI1), and stabilin-2, or bridging molecules, such as milk fat globule-EGF factor 8 (MFG-E8) and Gas6, which further link to integrins or Tryo3-Axl-Mer (TAM) receptors [73, 74]. PS can be found at low levels extracellularly on viable, activated cells, and it is thought that “don't eat-me” signals, such as CD31, CD47, and CD61, on PS-positive cells could negatively regulate phagocytosis to signal that this cell is not intended for clearance [60]. Finally, other molecules such as ICAM3, oxidized LDL-like molecules, calreticulin (CRT), and C1q bound serum proteins have been described to act as “eat-me” signals [60, 65, 75].
Phagocytes residing within different tissues preferentially express different PS receptors including BAI-1 (bone marrow, spleen, brain), TIM1-4 (kidney), and stabilin-2 (sinusoidal endothelial cells), suggesting that different tissues require specialized PS receptor mechanisms [3, 60, 62]. Engagement of PS receptors (or surface receptors engaged by bridging molecules) results in the activation of the Rho family of small GTPases, converging on evolutionarily conserved Rac1 activation, though the downstream molecules used by specific PS receptors can differ. Signaling via integrins or Mer by bridging molecules or membrane-bound BAI1 recruits the adaptor proteins ELMO1 and DOCK180 to the phagocytic cup to activate Rac1 [75]. Stabilin-2 requires the activity of adaptor protein, GULP, to activate the Rac1 pathway [76]. The signaling machinery downstream of TIM4, however, is currently unknown [1, 75].
Once engulfed, the dead cell is now cargo that must be properly degraded, digested, and processed. Rab5 and Rab7, small GTPases, are recruited to the phagosome and facilitate fusion to the lysosomal network [75]. The lysosomal milieu contains acidic proteases and nucleases that digests cellular corpses into their basic cellular components including fats, sterols, peptides, and nucleotides. One such enzyme, DNAse-II, is required for the degradation of DNA, and DNAse-II deficiency results in accumulation of undigested DNA fragments, polyarthritis, and joint inflammation [77]. LAP is a form of non-canonical autophagy, wherein an extracellular particle, such as a pathogen, immune complex, or dead cell, is sensed by TLR, FcR, and PS receptors, respectively, during phagocytosis and recruits some, but not all, of the autophagy machinery to the cargo-containing, single-membraned vesicle [1, 78]. This LC3-decorated, cargo-containing phagosome is termed the LAPosome, and successful LAP facilitates the rapid destruction of the cargo via fusion with the lysosomal pathway [58]. While LAP and canonical autophagy share some of the same molecular components, LAP is a process distinct from canonical autophagy in both molecular mechanism and function (Figure 2). Rubicon (RUN domain protein as Beclin-1 interacting and cysteine-rich containing) was recently described as a protein that is required for LAP but not canonical autophagy [58]. Rubicon is crucial for both the localization of and PI(3)P generation by the Class III PI3K complex, as well as the production of ROS via the NOX2 complex [58, 79].
Figure 2. Efferocytosis utilizes LC3-associated phagocytosis.
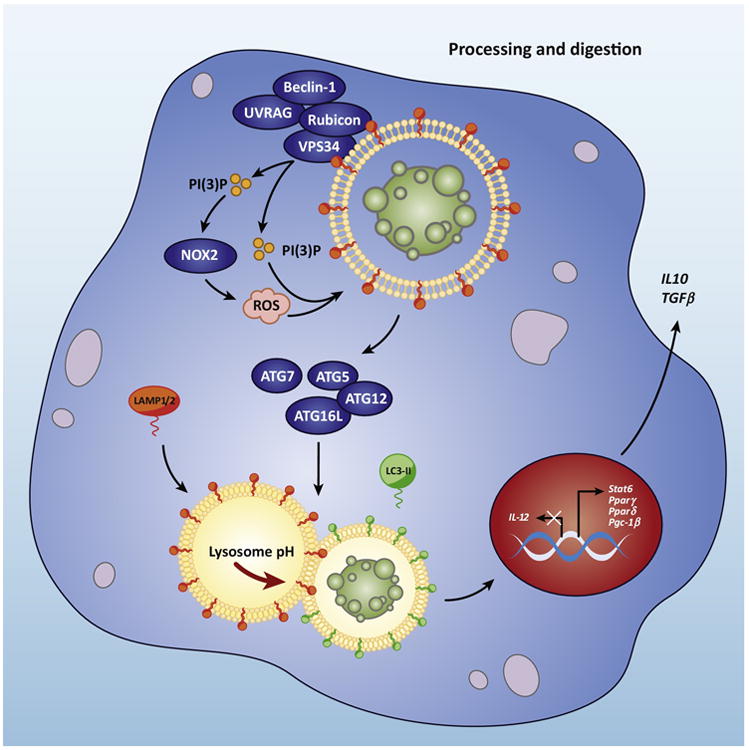
Upon engulfment of dying cells, components of the LC3-associated phagocytosis (LAP) pathway are recruitment to dead cell-containing LAPosome. The Class III PI3 K complex, composed of Beclin-1, VPS34, UVRAG, and Rubicon, is critical to the sustained and localized production of PI(3)P at the LAPosome. PI(3)P aids in the recruitment of the downstream autophagic/LAP machinery (such as ATG5, ATG12, ATG16L, ATG7), as well as the production of ROS by NOX2. Both ROS and PI(3)P are required for lipidation and translocation of LC3-II to the LAPosome. The antiinflammatory effects of efferocytosis are mediated by the activity of lipid and cholesterol sensors (Abca1, Lxr, Pparγ, Pparδ, and Pgc-1β), leading to the production of antiinflammatory mediators, IL-10 and TGFβ, while pro-inflammatory mediators, such as IL-12, are actively suppressed.
Importantly, LAP can have a profound effect on the degradation of and immune response to the engulfed material, including dying cells and the immunotolerance associated with uptake of apoptotic cells [1, 58, 78]. LAP-deficient (Rubicon- or NOX2-deficient), but not canonical autophagy-deficient (FIP200- or ATG14-deficient) macrophages present with a failure in phagosomal acidification and subsequent corpse degradation. Consequently, LAP-deficient macrophages produce increased levels of IL-1β and IL-6 when fed dying cells, and virtually no anti-inflammatory cytokines, like IL-10 [1, 2]. Animals with LAP deficiency, but not autophagy only-deficiency, develop age-related SLE-like disease, with increased serum levels of pro-inflammatory cytokines, autoantibodies, and kidney dysfunction. LAP exerts this protective effect by facilitating efficient clearance of dying cells [2]. Characterization of LAP as a critical regulator of inflammation in response to dying cells allows for the design of anti-inflammatory therapeutics that specifically target LAP, while maintaining the quality control mechanisms of canonical autophagy unaffected.
The engulfment of a cellular corpse essentially doubles a phagocyte's levels of cellular components. We now recognize that the mechanisms that mediate the metabolic stress of efferocytosis are intricately linked to its pursuant immune response. Efferocytosis leads to the activation of peroxisome proliferator-activated receptor γ/δ (PPARγ/δ) and liver × receptor (LXR) families, both important regulators of cellular lipid homeostasis [80, 81]. This results in the upregulation of other phagocytic machinery, such as TAM family, and basal cholesterol efflux machinery, such as ABCA1 (ATP-binding cassette subfamily A, member 1), to accommodate this increase in cholesterol levels [82]. Interestingly, PPARγ- and PPARδ-deficient macrophages show a defect in efferocytosis, signaling the interconnectedness between efferocytosis and metabolism [80]. Cholesterol homeostasis is a critical regulator of the non-immunogenic nature of apoptotic cell efferocytosis, characterized by production of anti-inflammatory cytokines, like TGFβ and IL-10, and active suppression of pro-inflammatory cytokines, such as TNFα, IL-1, and IL-12 [83, 84]. Agonists for both PPARγ and LXR have been shown to inhibit inflammatory responses [60, 80].
Why then does efferocytosis of necroptotic or pyroptotic cells not mediate the same tolerant effect as apoptotic cells, despite providing excess cholesterol and fatty acids that tend to trigger an anti-inflammatory program [1, 85]? A key to this puzzle lies in the nature of each type of cell death. During apoptosis, DAMPs, such as DNA and RNA, are neatly packaged into blebs and sequestered from their cognate PRRs within the cytoplasm of the phagocytosing cell. Thus, the developmentally and functionally normal process of apoptosis is likened to nucleic acid compartmentalization within the nucleus that presumably allowed for the evolution of viral RNA and DNA sensors, which are critical for host defense [86].
In contrast, lytic forms of cell death, such as necroptosis and pyroptosis, can be viewed as catastrophic events requiring immune attention. These lytic cellular deaths unleash DAMPs on to phagocyte PRRs, which may set in motion pro-inflammatory cytokine production that outweighs anti-inflammatory efferocytosis pathways. Necroptotic and pyroptotic cells engage PS receptors, but this signal is not enough to induce a tolerant response [1, 9]. Moreover, lytic cell death often results in an increased release of “find-me” signals, which can double as DAMPs [65].
Different forms of programmed lytic cell death have likely evolved to more finely tune the immune response to cells infected by pathogens or damaged by mechanical stress. The end result of both pyroptosis and necroptosis is plasma membrane permeabilization, but each of these cell death programs has additional layers of signaling that contribute to the response of surrounding cells. As stated above, necroptosis appears to alert the immune system of cells infected with pathogens equipped with caspase- or RHIM-inhibiting machinery [87]. In addition, the signaling molecules involved in necroptosis initiation, such as RIPK3, may help to coordinate the immune response to viruses apart from cellular rupture [33]. Similarly, pyroptosis alerts the immune system to a variety of viral and intracellular bacteria. IL-1β released during pyroptosis heightens the inflammatory environment during infection. In addition, it is now understood that pyroptotic cells trap intracellular bacteria within structures dubbed pore-induced intracellular traps or PITs. Following PIT formation, complement and scavenger receptor stimulation promotes the efferocytosis of PITs by neutrophils. Therefore, pyroptosis and necroptosis may represent two unique biological pathways in which the host counteracts pathogen evasion mechanisms [36].
Conclusions
“I learned how to die a long time ago.”
- Eddard Stark
The immune system is poised to recognize and respond to the plethora of dying cells it encounters over the lifetime of any organism, and will utilize different pathways depending on the type of dead cell it faces. Evolutionarily, the vast majority of cell death that an immune system must cope with is of an apoptotic nature, wherein dying cells are neatly packaged into easily digestible apoptotic blebs that protect organisms from overt immune activation. Safely sequestered within intact, viable cells reside potential immunostimulatory components, capable of triggering a chain reaction of inflammation but also potentially autoimmunity once released into the extracellular environment. While necessary for the survival, function, and identity of a living cell, components like DNA, RNA, histones, and ATP are viewed as immunological threats once sensed by the immune system. It is not merely their release that triggers an inflammatory response. These genetic programs of cell death can also actively transform DAMPs, altering their immunogenicity and dictating the effect of cell death on the phagocytes and the immune response.
Unlike necrosis, which occurs in the absence of a defined genetic program, lytic, programmed cell death, like necroptosis or pyroptosis, employs machinery specifically designed to puncture the cell membrane as part of its demise. This technique is an evolutionarily invaluable contribution to innate immunity, combining the killing of pathogen-infected cells with alerting the immune system through the release of DAMPs. Immunologically, however, this is a double-edged sword, as cell death provides the antigens necessary for peripheral tolerance, but excessive inflammation is detrimental and linked to many pathologies.
Efferocytosis is not a battle that occurs in a vacuum. The process and subsequent outcome is molded by the type of cell that dies, how it dies, where it dies, and who disposes of its corpse. All of these factors must be taken into account when assessing an immune response to dying cells. In vivo administration of apoptotic cells has been successfully used in graft versus host disease to mediate a tolerant response in inflamed tissues [88]. Therapeutics designed to enhance efferocytosis, could be effective in treating disorders linked to chronic inflammation. How a phagocyte translates efferocytosis into an appropriate immune response is an area of growing interest, and as we uncover more about this complex process, from both the side of the dying cell and the side of the phagocyte, potential mechanisms could arise as targets for drug therapy.
Trends.
Efferocytosis is the process employed by phagocytes wherein dying cells are recognized, engulfed, and digested in order to maintain developmental and immune homeostasis.
Dying cells actively participate in their own clearance, by recruiting and priming phagocytes for efferocytosis and displaying “eat-me” molecules that signal to phagocytes that should be removed from circulation.
Whereas the most common form of programmed cell death, apoptosis, is designed to be immunologically silent, other forms of programmed cell death, like necroptosis and pyroptosis, are lytic in nature and result in the release of damage-associated molecular patterns (DAMPs).
We now recognize that defects in efferocytosis, including defects in the proper processing of ingested cellular corpses, can lead to inflammatory and autoimmune disorders
Outstanding Questions.
Can therapeutics modulate inflammation by exploiting some of the efferocytotic machinery?
Could these therapies extend beyond autoimmune disorders into infectious disease conditions?
How does LC3-associated phagocytosis (LAP) mediate its anti-inflammatory effect during efferocytosis?
While the efferocytosis machinery is highly conserved across species, animals that lack an adaptive immune response do not display inflammation in response to uncleared dying cells – why?
Are there single nucleotide polymorphisms (SNPs) in LAP genes that are associated with inflammatory or autoimmune pathologies?
What is the evolutionary benefit to autosis, a cell death pathway originating from a cell survival mechanisms?
What is the contribution lytic programmed cell death to the development of human cancers?
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martinez J, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(42):17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez J, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Nagata S, et al. Autoimmunity and the clearance of dead cells. Cell. 2010;140(5):619–30. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R. Approaching the asymptote: 20 years later. Immunity. 2009;30(6):766–75. doi: 10.1016/j.immuni.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, et al. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9(4):451–8. doi: 10.4161/auto.23691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang D, et al. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kazama H, et al. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29(1):21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferguson TA, et al. Armed response: how dying cells influence T-cell functions. Immunol Rev. 2011;241(1):77–88. doi: 10.1111/j.1600-065X.2011.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jounai N, et al. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front Cell Infect Microbiol. 2012;2:168. doi: 10.3389/fcimb.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green DR, et al. Immunogenic and tolerogenic cell death. Nature reviews Immunology. 2009;9(5):353–63. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor RC, et al. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–41. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 13.Green DR. Means to an end: apoptosis and other cell death mechanisms. Cold Spring Harbor Laboratory Press; 2011. [Google Scholar]
- 14.Boatright KM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11(2):529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 15.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–32. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 16.Shiozaki EN, et al. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc Natl Acad Sci U S A. 2002;99(7):4197–202. doi: 10.1073/pnas.072544399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rock KL, et al. Innate and adaptive immune responses to cell death. Immunol Rev. 2011;243(1):191–205. doi: 10.1111/j.1600-065X.2011.01040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11(7):615–28. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauter B, et al. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191(3):423–34. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zelenay S, Reis e Sousa C. Adaptive immunity after cell death. Trends Immunol. 2013;34(7):329–35. doi: 10.1016/j.it.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez DA, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2015 doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong YN, et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169(2):286–300 e16. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dondelinger Y, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 25.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Upton JW, et al. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–7. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dillon CP, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kearney CJ, et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015;22(8):1313–27. doi: 10.1038/cdd.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaczmarek A, et al. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Rickard JA, et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife. 2014;3 doi: 10.7554/eLife.03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu Y, et al. MLKL inhibition attenuates hypoxia-ischemia induced neuronal damage in developing brain. Exp Neurol. 2016;279:223–31. doi: 10.1016/j.expneurol.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 33.Daniels BP, et al. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell. 2017;169(2):301–313 e11. doi: 10.1016/j.cell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kole AJ, et al. Mature neurons: equipped for survival. Cell Death Dis. 2013;4:e689. doi: 10.1038/cddis.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Vasconcelos NM, et al. Inflammasomes as polyvalent cell death platforms. Cell Mol Life Sci. 2016;73(11-12):2335–47. doi: 10.1007/s00018-016-2204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–42. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 38.Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213(6):617–29. doi: 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265(1):6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagar JA, et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 42.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 43.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 44.Aglietti RA, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113(28):7858–63. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–6. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 47.Lamkanfi M, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185(7):4385–92. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daniels MJ, Brough D. Unconventional Pathways of Secretion Contribute to Inflammation. Int J Mol Sci. 2017;18(1) doi: 10.3390/ijms18010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cullen SP, et al. Diverse Activators of the NLRP3 Inflammasome Promote IL-1beta Secretion by Triggering Necrosis. Cell Rep. 2015;11(10):1535–48. doi: 10.1016/j.celrep.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Shi J, et al. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42(4):245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165–76. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yagoda N, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–8. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin-Sanchez D, et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol. 2017;28(1):218–229. doi: 10.1681/ASN.2015121376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194(2):341–61. doi: 10.1534/genetics.112.149013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao K, Klionsky DJ. AMPK activates autophagy by phosphorylating ULK1. Circ Res. 2011;108(7):787–8. doi: 10.1161/RES.0b013e3182194c29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Itakura E, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–72. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levine B, et al. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez J, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Liu Y, et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–71. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poon IK, et al. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166–80. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–6. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5(1):a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chekeni FB, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467(7317):863–7. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Truman LA, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112(13):5026–36. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- 65.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207(9):1807–17. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo B, et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity. 2016;44(2):287–302. doi: 10.1016/j.immuni.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 68.Suzuki J, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341(6144):403–6. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 69.Segawa K, et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344(6188):1164–8. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 70.Fischer K, et al. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108(13):4094–101. doi: 10.1182/blood-2006-03-011742. [DOI] [PubMed] [Google Scholar]
- 71.Liu X, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21(10):1209–15. doi: 10.1038/nm.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Birge RB, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–78. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zizzo G, et al. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189(7):3508–20. doi: 10.4049/jimmunol.1200662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanayama R, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304(5674):1147–50. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 75.Martinez J. Prix Fixe: Efferocytosis as a Four-Course Meal. Curr Top Microbiol Immunol. 2015 doi: 10.1007/82_2015_467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park SY, et al. Requirement of adaptor protein GULP during stabilin-2-mediated cell corpse engulfment. J Biol Chem. 2008;283(16):10593–600. doi: 10.1074/jbc.M709105200. [DOI] [PubMed] [Google Scholar]
- 77.Kawane K, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443(7114):998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 78.Henault J, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang CS, et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe. 2012;11(3):264–76. doi: 10.1016/j.chom.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mukundan L, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15(11):1266–72. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roszer T, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186(1):621–31. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell. 2011;147(7):1442–5. doi: 10.1016/j.cell.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.N AG, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31(2):245–58. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim S, et al. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21(5):643–53. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 85.Kiss RS, et al. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006;16(22):2252–8. doi: 10.1016/j.cub.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 86.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38(5):870–80. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brault M, Oberst A. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol. 2017;95(2):131–136. doi: 10.1038/icb.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Korns D, et al. Modulation of macrophage efferocytosis in inflammation. Front Immunol. 2011;2:57. doi: 10.3389/fimmu.2011.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]