

Abstract
Heparin, a member of the glycosaminoglycan family, is known to interact with more than 400 different types of proteins. For the past few decades, significant progress has been made to understand the molecular details involved in heparin-protein interactions. Based on the structural knowledge available from the FGF1-heparin interaction studies, we have designed a novel heparin-binding peptide (HBP) affinity tag that can be used for the simple, efficient, and cost-effective purification of recombinant proteins of interest. Purification of recombinant proteins using the HBP tag can be achieved using a simple sodium chloride gradient. In addition, owing to the high density of positive charges on the HBP tag, recombinant target proteins are preferably expressed in their soluble forms. Further, the purification of HBP-fused proteins/peptides can also be achieved in the presence of chemical denaturants like urea. Additionally, polyclonal antibodies raised against the affinity tag can be used to detect HBP-fused target proteins with high sensitivity. Given these properties, we believe that the HBP tag will be enthusiastically used by researchers for the purification of recombinant proteins.
Keywords: Heparin Sepharose, affinity chromatography, recombinant protein, fusion tag, resin
Introduction
This unit describes the use of novel heparin-binding peptide (HBP) as an affinity tag for purification of recombinant proteins from bacterial cell lysates. We constructed the expression vector using the standard pET22b™ vector backbone by inserting the nucleotide sequence that encodes for the affinity tag. A multiple cloning site (MCS) was restored downstream of the affinity tag with various combinations of restriction site(s) for directional cloning of the target genes/nucleotide sequences. Nucleotide sequence encoding the protein of choice can be amplified using PCR and ligated into the MCS region of the HBP expression vector. The HBP tag can be fused at either the N- or C- terminal end of the protein of interest. Recombinant proteins tagged with HBP can be purified to homogeneity using single–step heparin Sepharose™ affinity chromatography. Column-bound fusion protein can be collected under mild elution conditions consisting of a simple sodium chloride gradient.. After purification, the tag can be eliminated by subjecting the HBP fusion protein to specific restriction protease cleavage to obtain pure form of the protein of interest. Based on the criteria and the protocols for purification described, here in this unit, the novel HBP tag based affinity chromatography would be an ideal choice for purification of different types of recombinant target proteins. This purification technology/strategy can be adapted from a laboratory scale to an industrial scale for obtaining higher yields of purified proteins in a cost-effective manner. This aspect is critical for downstream processing in protein biotechnology. We believe that the HBP tag will also be a valuable avenue for the purification of small recombinant peptides. Currently, work is underway to further improve the heparin binding affinity of the HBP-tag through engineering mutations in the affinity tag to increase its protein solubilizing property. Increasing the solubilizing nature of the HBP tag will significantly benefit the biotechnology sector because several proteins of high biomedical importance are known to be expressed in E.coli as inclusion bodies. Further, we are planning to extend our research to exploit the HBP tag to selectively purify heparin from a mixture of different glycosaminoglycans. Lastly, efforts are currently underway to design and develop an exclusive resin to purify HBP-fused recombinant proteins in a cost-effective manner.
Strategic Planning
Design of the HBP tag
We had demonstrated the use of HBP tag for expression of diverse kinds of proteins, ranging from soluble intracellular proteins such as the C2A domain of synaptotagmin, S100A13 and also the C-terminal tail of the membrane protein, Albino-3 (Rajalinagam et al, 2005; Sivaraja et al, 2006; Marty et al, 2009). The HBP tag was rationally designed for optimal binding affinity based on the structural information available on various heparin-protein interactions (Beenken et al, 2009; Capila & Lindhart, 2002; Demepewolf et al, 2013). This affinity tag is 32 amino acids long consisting of the positively charged lysine in a defined repetitive manner interspersed with alanine and glutamine. Two tryptophan residues, one each at the N- and C-terminal ends of the HBP tag, were introduced to monitor the elution of HBP-fused recombinant protein(s) by UV-vis absorbance or fluorescence. In silico analysis of the HBP tag suggested that the positively charged side-chain group of lysines would be oriented to one side of the peptide, facilitating strong interaction(s) with the negatively charged heparin molecule(s). Circular dichroism spectroscopy data obtained on the HBP tag, in the presence and absence of heparin, revealed that the backbone conformation of the HBP tag transitions from an unfolded state(s) to a partial helical conformation upon binding to heparin (Morris et al, 2016). Isothermal titration calorimetry experiments showed that HBP has a strong binding affinity to heparin (Kd ∼ 190 nM, unpublished results).
Characteristics and detection of the HBP tag
We found that the expression yields of the HBP-fused recombinant target proteins in E.coli, were quite comparable with the yields obtained when the same target proteins were fused with glutathione S-transferase (GST). In fact, in some cases, the expression yields of the target proteins studied were marginally better than when fused to the GST tag. One other noteworthy feature is that HBP-fused target proteins were predominantly found in the soluble fraction rather than in the insoluble inclusion bodies. We believe that high content of positively charged residues in the HBP tag helps thwart the target proteins aggregating as inclusion bodies. Polyclonal antibodies raised against a N-terminal 15-amino segment of the HBP tag (QKAQKAQAKQAKQAC-) were very efficient and exhibited high sensitivity in the detection of recombinant HBP-fused target proteins present along with contaminating host (E.coli/yeast/mammalian cells) proteins. Western blot and ELISA results showed that polyclonal HBP antibodies can detect HBP-fused target proteins up to 5 ng – 10 ng quantity. In addition, multidimensional NMR spectroscopy revealed that the HBP tag does not interfere with the folding and activity of the fused recombinant target proteins (unpublished results).
Design of the clone to overexpress HBP-fused target protein(s)
Yields of the final protein product depends on the optimization of different conditions starting from cloning all the way through protein purification. We have constructed a recombinant HBP-pET22b™ expression vector which in principle can be extended to other classes of expression vectors compatible with different expression hosts. The HBP tag in pET22b™ was cloned downstream of the ribosome binding site followed by the MCS consisting of a wide range of restriction sites for convenient directional cloning. The stop codon is located downstream of the MCS to facilitate termination of the translation process (Figure. 1). In our studies, all HBP-fused target protein-encoding DNA inserts contain a thrombin cleavage site between the HBP tag and target protein of interest. Users will have the provision of cloning the DNA insert, encoding for a specific polypeptide, in-frame with the HBP tag using any of the available restriction site(s) on the vector. However, one may introduce other well-known restriction protease sites such as factor Xa, enterokinase and TEV protease. The amino acid sequence of the HBP tag does not contain methionine, asparagine or glycine. Therefore, users will have an option to incorporate restriction site(s) that are amenable to chemical cleavage using specific reagents such as cyanogen bromide and hydroxylamine to enable the removal of the HBP-tag from the pure recombinant target protein
Figure 1.
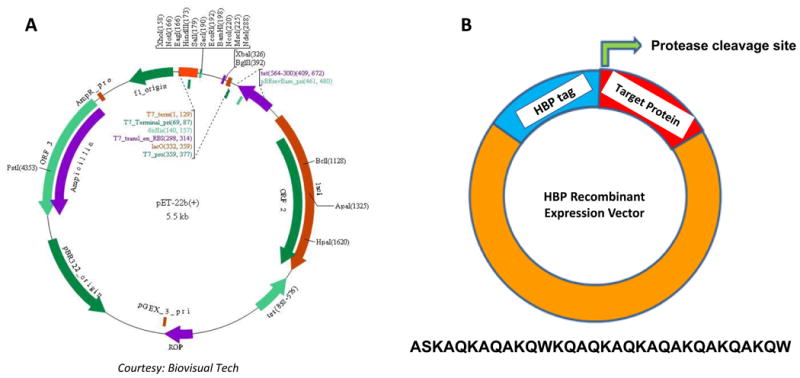
Construction of recombinant HBP Vector. Panel A, shows the pET22b vector map with restriction sites of the MCS region highlighted (courtesy from Biovisual Tech.), and Panel B, cartoon depicting the broad details of the affinity tag, cleavge site and target protein site available on the vector containing the constructed HBP recombinant expression vector. At the bottom, amino acid sequence corresponding to HBP tag is provided in single letter code.
Overexpression and Purification of HBP fused recombinant protein(s)
A flow chart depicting the steps involved in the purification of the target recombinant protein(s) using the HBP tag is shown in Figure 2. Optimization of the expression levels of the target protein is critical to render the entire purification process efficient and cost-effective. Some fusion proteins are known to be inherently toxic to the host cell and this aspect consequently impacts their expression levels. In fact, detection of the expressed target protein becomes challenging in the background of other contaminating proteins produced by the host cells. This problem was circumvented by using sensitive immunoblot detection for unambiguous identification of HBP-tagged proteins. The bacterial cell pellet collected was subjected to disruption using ultra-sonication or French press and the whole cell lysate was centrifuged at high speeds to collect the clear supernatant. SDS PAGE analysis indicated that most of the overexpressed HBP-fused target protein(s) was in the soluble fraction. The supernatant was loaded onto a heparin Sepharose column followed by wash and elution steps using either a linear or a step-gradient of sodium chloride. In all cases, the HBP-fused target proteins were eluted at NaCl concentrations greater than or equal to 500 mM. The fraction(s) containing the HBP-fused target protein was subjected to desalting and concentration process to equilibrate the target protein to the desired buffer and salt concentration. Subsequently, the HBP-tag was cleaved from the target protein by selective restriction protease (thrombin) treatment and the cleaved mixture was reloaded back onto the heparin Sepharose column to separate the HBP-tag from the protease (thrombin). The target protein was eluted in the wash buffer. Both the HBP-tag and thrombin, due to their strong binding affinity to heparin, were subsequently eluted at high salt concentration (> 500 mM NaC). The homogeneity of the target protein was verified from the SDS PAGE gels, stained separately with either Coomassie blue or silver staining or western blotting. The target protein was freeze-dried and stored at - 80° C for future use. The heparin Sepharose column was subsequently regenerated following the manufacturer's protocol.
Figure 2.

Flow-chart describing the steps involved in the purification of HBP-fused recombinant target protein.
In short, we have provided basic protocols for using HBP tag as an efficient purification tool for overexpression and isolation of protein of interest. In basic protocol 1 we =describie the steps required for performing bacterial overexpression which will be followed with basic protocol 2 wherein the details pertaining to the protein purification of HBP fused target proteins will be discussed. This unit will be concluded with the final support protocol describing the details for cleaving the affinity tag to obtain pure tag free homogenous recombinant protein.
Basic Protocol 1
Cloning and expression of recombinant heparin-binding peptide (HBP) fusion proteins
Using gene-specific primers, nucleotide sequence (encoding the target protein) was PCR amplified and subjected to restriction digestion followed by directional cloning in the MCS region. The accuracy of cloning was verified by subjecting the plasmid to DNA sequencing. E.coli cells were transformed with the pET-22b-HBP plasmid and the transformed bacterial cells were selected by ampicillin resistance (Figure.3).
Figure 3.

Flow chart describing the steps of cloning. Steps involved in the cloning of gene of interest in for generating the pET22b-HBP recombinant construct.
High density shaker flask cultures, induced with IPTG (0.5-1 mM), usually result in significant intracellular expression of the fusion target protein. The protocol described herein is suitable for working with a culture of approximately 2L volume which could also be scaled to a pilot scale expression using fermenters/bioreactors. Optimal expression condition(s) for HBP fusion proteins involves aeration of the inoculated culture at 250 rpm (using an orbital shaker) at a temperature ranging between 25°C to 37°C until an optical density (OD) of 0.4 to 0.6 (at 600 nm) is reached. In our experience, the post-IPTG induction incubation time for the culture medium to reach the desired optical density is between 3 and 5 hours. These growth parameters can be adjusted to achieve maximum expression yields of the target protein(s).
Materials
Luria-Bertani (LB) medium pH 7.2
Antibiotic stock (ampicillin 100mg/mL)
Bacterial cell culture containing the HBP vector with desired gene of interest
1M IPTG
2L Erlenmeyer flasks
Temperature-controlled environmental shaker
UV-vis spectrophotometer
Refrigerated centrifuge, and centrifuge bottles (500 mL and 50 mL).
Bacterial overexpression of HBP fusion protein and detection
Prepare 2L of LB medium (500mL broth in 2L flasks - a total for four flasks) and sterilize the flask containing medium by autoclaving.
Cool all the media flasks to room temperature before inoculating the bacterial cell culture.
To the starter culture flask, add ampicillin (100 μg/mL), 1 mL of HBP recombinant vector glycerol stocks (frozen in -80°C freezer) and incubate the cells at 37°C with 250 rpm agitation for 14-16 hours until the bacterial culture inside the flasks reaches flocculent growth.
To 500 ml fresh, sterile LB broth in a 2 liter Erlenmeyer flask, transfer 25 mL of the overnight grown bacterial culture using a sterile falcon tube and also add appropriate amounts of antibiotic(s).
Allow cells to grow under the same growth conditions of 37°C and 250 rpm until the OD reaches 0.5.
Collect one milliliter of bacterial cells from the flask, centrifuge the sample and store the pellet in -20oC freezer as a pre-induction sample, for SDS-PAGE analysis.
To the rest of the bacterial cell culture, add 500 μL of 1 M (iso-propyl thio-β- D- galacto pyranoside) IPTG stock solution to achieve a final concentration of 1 mM.
Continue the growth of the bacterial cells for an additional 4 hours under the same incubation conditions.
Similar to pre-induction sample, collect 1 mL of post-induced sample before harvesting the cells for SDS-PAGE analysis.
Centrifuge the remaining volume of bacterial culture at 5000 × g for 15 minutes at 4°C in 500 mL centrifuge bottles.
Carefully discard the supernatant, consisting of LB broth, without disturbing the bacterial cell pellet.
To the bacterial cell pellet, add 50 mL of freshly prepared 1 × PBS (pH 7.2) and gently resuspend the cells using a glass rod and with intermittent mixing on a vortexer.
Transfer resuspended cells to a new conical tube (50 mL) and centrifuge again at 5000 × g for 15 minutes at 4°C.
-
Discard the clear 1 × PBS supernatant.
The buffer-rinsed bacterial cell pellet can be stored indefinitely in a -80°C freezer for future use.
-
Perform SDS-PAGE of the pre- and post-IPTG induced samples and visulaze the bands using Coomassie blue staining.
The resulting gel should show a prominent band corresponding to the molecular size of the HBP-fused target protein.
Under certain circumstances, recombinant proteins might express in very low amounts and therefore cannot be detected easily by regular Coomassie blue staining. In these cases, perform a western blot using polyclonal antibodies raised against HBP tag for specific detection of HBP fusion proteins at extremely low concentrations (5 - 10 nanograms/mL) shown in Figure 4.
Figure 4.
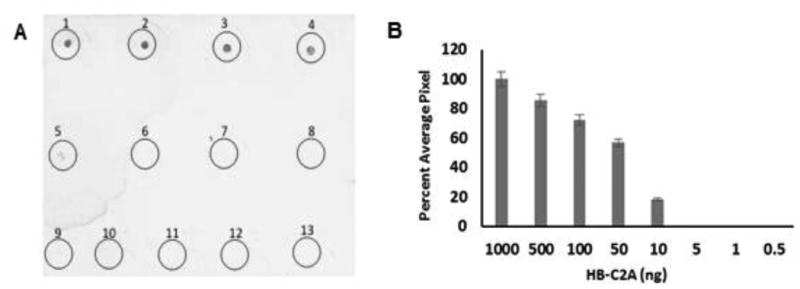
Detection of HBP fusion proteins. Panel A, shows the dot blot analysis of HBP fusion protein. The dot blots are labeled as Circle 1 to 8. 1 μg (circle 1); 500 ng (circle 2); 100 ng (circle 3); 50 ng (circle 4); 10 ng (circle 5); 5 ng (circle 6); 1 ng (circle 7); 0.5 ng (circle 8); Circles 9-13 are BSA: 1 μg (circle 9); 500 ng (circle 10); 100 ng (circle 11); 50 ng (circle 12); 10 ng (circle 13). Panel B, represents the bar graph depicting the densitometry scan of the dot blot with concentration of HBP-C2A on X-axis and percent average pixel on Y-axis. This figure was reproduced from Morris et al (Morris et al, 2016) with permission from the publisher.
Basic Protocol 2
Protein Purification using Heparin Sepharose affinity chromatography
Purification of HBP fusion proteins can be successfully achieved by using well-established conditions for heparin Sepharose chromatography. Depending on the isoelectric point, solubility, and susceptibility of the target protein to cellular proteolytic cleavage, appropriate cell lysis buffer conditions may be chosen. Inclusion of commercially available protease inhibitor cocktail in the cell lysis buffer can prevent undesired degradation of the expressed recombinant target-HBP fusion proteins. We have not encountered problems of degradation of the protein affinity tag after lysis of bacterial cells overexpressing HBP-fused target proteins. Cell debris and other insoluble material are eliminated by high speed centrifugation (35, 000 ×g). Depending on the thermal stability of the target protein, it may be required to perform the heparin Sepharose affinity chromatography at specific temperature conditions. It is important to equilibrate the buffers and the heparin Sepharose column to the required cold temperatures if the purification of the target protein is to be performed under cold room conditions (4°C). The HBP-tagged target protein can be expected to elute at higher (> 500 mM NaCl) concentrations of sodium chloride. An advantage of using heparin Sepharose resin is that it can be reused multiple times by subjecting it to appropriate regeneration procedures recommended by the manufacturer immediately after every use. The heparin Sepharose column needs to be stored in either 20% ethanol or in buffers containing 0.1% w/v sodium azide to avoid bacterial growth.
Materials
Heparin Sepharose™ resin (GE Healthcare)
Sodium phosphate buffer
Step/linear gradient of sodium chloride (250mL gradient mixer with a starting NaCl concentration of 100mM)
Gradient mixer
Protease-inhibitor cocktail (Sigma)
Overexpressed bacterial cell pellet containing the HBP fusion protein (from Basic Protocol 1?)
Econo column from BioRad, Econo UV columns from BioRad with dimensions 1.6cm × 20 cm can be used to pack heparin Sepharose with a bed volume of ∼ 15 ml.Econo UV monitor
Low-flow peristaltic pump
Ultrasonicator or French cell press
Oakridge tubes
High speed refrigerated centrifuge (Beckman Coulter)
Millipore ultrafiltration centrifugal concentrating devices with appropriate molecular weight cutoffs.
Column packing, cell lysis and protein purification
NOTE: Unless otherwise mentioned, all the purification steps are preferably conducted at desired temperature.
Prepare 15 ml of a 50% slurry of heparin Sepharose in 20% ethanol and degas under vacuum (∼2mTorr for 15 minutes).
Pour the degassed slurry into a 1.6cm × 20 cm Econo UV column along the walls using a glass rod as a guide to avoid the formation of air pockets.
Allow the heparin Sepharose resin beads to settle by gravity flow for efficient packing. Wash the beads with equilibration buffer (starting buffer of purification, in most cases this buffer will be the same for cell lysis and for the first wash step) with a constant flow rate of 1-2 ml/min for at least 5 column volumes (CV) of buffer.
Resuspend a frozen bacterial pellet from a 2L culture in 30 mL of ice-cold equilibration/lysis buffer.
Lyse the resuspended cellsby ultrasonication (amplitude # 15 watts output) with an alternate cycles of 10 seconds in the ON and the OFF.
To achieve higher efficiency of cell lysis, pass post-sonicated sample through a French Pressure Cell at 30,000 psi for 3 cycles.
Centrifuge the lysed cell mixture at 35,000 × g for 30 minutes at 4°C to separate the insoluble material from the soluble cellular components.
Carefully decant the supernatant/cell lysate into a fresh Falcon tube without disturbing the insoluble pellet.
Collect a small aliquot of supernatant for SDS-PAGE analysis.
Load the cleared supernatant onto the pre-equilibrated heparin Sepharose column at a uniform flow rate of 1-2 mL/minute.
Care should be taken such that the amount of fusion protein loaded should not exceed the binding capacity of the column.
-
11
Wash the column with 2 column volumes of wash buffer/equilibration buffer followed with gradient of sodium chloride to remove the bacterial contaminants.
Most of the bacterial proteins will be eliminated at lower concentrations of sodium chloride and pure HBP fusion protein usually elutes at 500mM NaCl (in case of HBP-C2A). But if the fusion partner has high positive charge, it might bind to heparin Sepharose column more tightly and will be eluted at higher concentrations of sodium chloride. From our studies, we have observed that c-Alb3, a positively charged protein (pI ∼ 9.3) bound to heparin Sepharose column and eluted at 1M NaCl concentration.
-
12
Subject the collected fractions to SDS-PAGE to check for the presence of the HBP fusion protein (Figure. 5).
-
13
Desalt and concentrate the column-eluted HBP fusion protein using an ultrafiltration centrifugal device with molecular weight cutoff suitable the size of fusion protein. Estimate the concentration of the obtained fusion protein by measuring the absorbance at 280nm..
Figure 5.

Affinity chromatography for purification of HBP-fused recombinant proteins. SDS-PAGE analysis of the purified proteins: Lane-1 shows the broad range protein marker and lanes 2 and 3 show the bands corresponding to HBP fusion protein and the cleaved protein of interest. This figure was reproduced from Morris et al (Morris et al, 2016) with permission from the publisher.
Alternate protocol 1
Purification of HBP-fusion proteins under denaturation conditions
Unlike other commonly used purification tags, such as glutathione S-transferase (GST), maltose binding protein (MBP) and others, HBP-fused target proteins can also purified under denaturing conditions in the presence of 8 M urea. Circular dichroism spectroscopy analysis revealed that the HBP tag in 10 mM phosphate buffer (pH 7.2) containing 100 mM sodium chloride is a random coil. In addition, the HBP tag binds strongly to heparin in the unfolded state(s). The presence of the denaturant, 8M urea, does not affect the binding affinity of the HBP-fused target protein to heparin Sepharose. However, it is recommended that before attempting to purify HBP-fused target protein under denaturing conditions, care should be taken to ensure that the target protein exhibits reversible denaturant-induced unfolding. In contrast, we found that HBP-fused target proteins fail to show binding to heparin Sepharose when guanidinium hydrochloride is included in the elution buffer. Guanidinium hydrochloride is an ionic denaturant that interferes with the electrostatic interaction(s) between the HBP-tag and the heparin Sepharose resin.
Materials (see Basic Protocol 2)
Materials used for purification of the HBP-fused proteins under native and denaturing conditions are the same, with the exception of 8M urea.
Follow Basic Protocol 2, steps 1 through 6 the insoluble component consisting of the inclusion bodies will be obtained. .
The insoluble pellet will be dissolved in the buffer containing 8M urea.
Centrifuge the urea-solubilized pellet at 35,000 × g for 30 minutes at 4°C. Carefully transfer the cleared into a fresh Falcon tube.
Load the supernatant onto a pre-equilibrated heparin Sepharose column with 8M urea solution at a constant flow rate of 1-2 mL/min.
Perform wash steps using a decreasing gradient of urea concentration until the urea is completely eliminated and the bound HBP fusion protein from heparin Sepharose column is eluted in the increasing NaCl gradient with no urea in the buffers. An alternate method of elution can also be employed in which the urea concentration is descreasing and the NaCl concentration will be simultaneously increasing.
Analyze all the fractions collected by SDS-PAGE to detect the presence of HBP fusion protein.
Support protocol 3
ON/OFF column cleavage of HB fusion tag from the target protein
Cleavage using proteases (such as thrombin, factor Xa, enterokinase or TEV protease) for the removal of HBP tag can be successfully accomplished, but restriction protease cleavage conditions need to be optimized for each individual HBP fusion protein to achieve maximum efficiency of cleavage using extremely low amounts of restriction protease. Common parameters adjusted for obtaining an optimal cleavage include choice of cleavage buffer, addition of reducing agents or mild denaturation, pH, temperature, length of incubation and addition of divalent cations, like Ca2+ to the buffer.
Materials
HBP fusion protein solution
bovine thrombin 1U/μl
0.2M PMSF solution
constant temperature water bath
OFF-column cleavage
For performing cleavage in solution (OFF column), add restriction protease thrombin to the HBP– fused target protein solution at a ratio of 1U of thrombin to 250 μg of HBP-fused target protein.
Incubate the HBP fusion protein-protease mixture at 4°C or 37°C, depending upon the stability of the fusion partner, for 14 – 24 hours with continuous mixing on a rotator.
Terminate the cleavage reaction by adding 0.2mM PMSF solution.
Reload the cleavage mixture onto the heparin Sepharose resin.
Collect the unbound sample in sodium phosphate buffer containing 100mM NaCl, pH 7.2 consisting of the protein of interest and regenerate the column to remove the bound HBP tag and the restriction protease thrombin.
Subject the purified protein product to desalting and concentration using a Millipore ultrafiltration concentrator(s) with the appropriate molecular weight cut-off suited to the size of the protein of interest.
ON-column cleavage
-
7
To perform ON column cleavage, add restriction protease thrombin in larger amounts at a ratio of 1U / 125 μg of fusion protein for achieving a higher cleavage efficiency in a shorter time interval.
Reagents and Solutions
Stock Solutions
Ampicillin, 100 mg/ml
100 mg of ampicillin in 1 mL of water. Filter sterilize the solution and store in -20° C freezer for 1 month.
Isopropyl-1-thio- B-D-galactopyranoside (IPTG), 1 M
238 mg of IPTG in 1 ml of water. Store at -20 °C for 1 month.
Growth medium
LB medium
Dissolve 25 grams of LB ready-made powder in 1 liter of water. Dispense into flasks covered with aluminum foil s and autoclave with liquid cycle – 15 minutes at 121.5°C at 15 lbs pressure. Must be used immediately.
Equilibration/Lysis/Wash buffer
10 mM sodium phosphate buffer containing 100 mM NaCl; pH 7.2. Can be stored for 1 month at 4°C.
Elution buffer
Equilibration buffer with increasing gradient of NaCl concentration. Can be stored for 1 month at 4°C.
Cleavage buffer
Same as equilibration buffer. May be CaCl2 can be included in the buffer depending upon the conditions used.
Commentary
Background information
The HBP affinity tag-based protein purification procedure mainly relies on the electrostatic interaction between the positively charged HBP peptide fragment and the highly negatively charged heparin immobilized on the agarose bead. This type of interaction(s) is very well documented in different heparin-binding proteins ranging from growth factors to different extracellular matrix proteins.
A specific arrangement of basic amino acids in a particular pattern provide spatial distribution of positive charged amino acids that facilitate the strong non-covalent interaction with the heparin moiety. Such a specific binding can be exploited for purification of the recombinant proteins. Electrostatic-based interactions can be selectively disrupted by varying the ionic concentration of the buffer. Using this simple procedure, HBP tag-fused recombinant proteins can be eluted from the heparin Sepharose with a simple salt gradient.
Critical parameters
Chromatography conditions
The binding affinity of HBP-tagged fused target protein(s) to heparin Sepharose, at any pH, can be potentially influenced by the net charge on the affinity tag-fused target protein. The strength of the heparin-protein interaction will dictate the concentration of salt that will be required to elution HBP-tagged fused target protein(s). For example, a highly acidic target protein (pI < 4.0) can decrease the affinity of the HBP-fused target protein to the heparin Sepharose resin. In this context, it is recommended that a small scale purification be performed initially to determine the appropriate salt gradient to achieve the optimal purity of the HBP-fused target protein(s). In general, it is recommended to incubate the heparin Sepharose with cleared cell lysate (containing the overexpressed HBP tag-fused target protein) for at least 60 minutes (at 4°C) to facilitate tight binding and consequently achieve high yields of the HBP-fused target protein. Such optimization of elution conditions is critical to avoid wastage of time and resources during large batch/scale purification.
Optimization of expression
Optimization of expression conditions, through standardization of bacterial cell growth conditions, can result in significant gains in the final yield(s) of pure target recombinant proteins. The major parameters that affect the expression yields are growth temperature, agitation/aeration, time of induction, length of post-induction before harvesting, and the concentration of IPTG used for induction. Further, addition of protease inhibitor cocktail to the lysis buffer will significantly decrease degradation of HBP-fused target proteins, especially when the protein purification is carried out at room temperature.
Removal of HBP tag
Optimization of the restriction protease cleavage conditions to removethe HBP tag from the recombinant target protein is another parameter that needs to determined with different concentrations of the restriction protease and a low concentration of HBP-fused target protein.
Binding of HBP tag to heparin Sepharose resin
Extreme changes in the buffer pH and random temperature changes during purification may result in aggregation of protein on the column.Proper maintenance and storage of heparin Sepharose in bactericidal solvents will help retain the efficiency of binding of HBP-fused target protein.
Anticipated Results
The HBP affinity tag has advantages over other commonly used protein affinity tags. As the HBP-tag is a small positively charged polypeptide, the effective intracellular concentrations of the overexpressed HBP-fused target protein(s) can reach high concentrations without being trapped in unproductive inclusion bodies. Depending on the properties of the fusion protein, an appropriate bacterial host needs to be selected for obtaining maximum expression yields. This could be achieved by using expression bacterial hosts that are protease-deficient and have special codons for efficient incorporation such as, BL-21 (DE3) Star strains of E.coli. As the heparin-HBP tag interaction is highly specific to heparin Sepharose resin, very little or no loss of the HBP-tagged target protein can be expected during the initial wash. More than 95% of the contaminating bacterial host proteins should be eliminated in initial buffer washes, containing low salt concentration, and a highly pure HBP fusion protein will be eluted in high salt conditions. Pure recombinant target protein, free of the restriction protease (especially if the restriction protease used is thrombin), can be obtained by performing off-column cleavage and passing the cleaved mixture (containing the restriction protease, HBP-tag, and the target recombinant protein) through the heparin Sepharose column. The restriction protease and the HBP tag bind to the heparin Sepharose resin and the pure, tagless target protein is eluted in the flow-through.
Time Considerations
Typically, when the HBP affinity tag is used for purification, the large scale overexpression to the final purification of the target protein can be completed in ≤ 16 hours.
Breakdown of the experimental time involved for carrying out the different steps is as follows: bacterial transformation (∼ 2 hours), growth of transformants on plate (∼ 14 hours), growth of starter culture (∼14 hours), large scale bacterial overexpression (∼8 hours), preparation of bacterial cell lysate and purification using affinity column chromatography (∼4 hours), and desalting/concentration (∼1 hour) and freeze drying (∼2 hours).
Acknowledgments
This work was supported in part by grants from the National Institutes of Health [NCRR COBRE Grant 1 P20 RR15569 (TKSK), P30 GM103450 (TKSK)], the Department of Energy [(Grant DE-FG02-01ER15161 (to TKSK)], National Science Foundation [Grant IOS0843397 9TO (to TKSK)] and the Arkansas Biosciences Institute (to TKSK).
Footnotes
Conflict of Interest: Authors claim no conflict of interest.
References
- Arnau J, Lauritzen C, Petersen GE, Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr Purif. 2006;48(1):1–13. doi: 10.1016/j.pep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Batra S, Sahi N, Mikulcik K, Shockley H, Turner C, Laux Z, et al. Efficient and inexpensive method for purification of heparin binding proteins. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(24):2437–2442. doi: 10.1016/j.jchromb.2011.06.047. [DOI] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonyuen U, Promnares K, Junkree S, Day NP, Imwong M. Efficient in vitro refolding and functional characterization of recombinant human liver carboxylesterase (CES1) expressed in E. coli. Protein Expr Purif. 2015;107:68–75. doi: 10.1016/j.pep.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41(3):391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9(1):21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- Culley FJ, Fadlon EJ, Kirchem A, Williams TJ, Jose PJ, Pease JE. Proteoglycans are potent modulators of the biological responses of eosinophils to chemokines. Eur J Immunol. 2003;33(5):1302–1310. doi: 10.1002/eji.200323509. [DOI] [PubMed] [Google Scholar]
- de Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2(11):735–744. doi: 10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dempewolf C, Morris J, Chopra M, Jayanthi S, Kumar T, Li W. Identification of consensus glycosaminoglycan binding strings in proteins. 2013 International Conference on Information Science and Applications (ICISA) 2013:1, 24–26. [Google Scholar]
- Esko J, Linhardt R, Varki A, Cummings R. Essentials of Glycobiology. 2nd ed. Cold Spring Harbor Laboratory Press; New York: 2009. Proteins that Bind Sulfated Glycosaminoglycans. chapter 35. [PubMed] [Google Scholar]
- Esposito D, Chatterjee DK. Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol. 2006;17(4):353–358. doi: 10.1016/j.copbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Fang J, Dong Y, Salamat-Miller N, Middaugh CR. DB-PABP: a database of polyanion-binding proteins. Nucleic Acids Res. 2008;36(Database issue):D303–306. doi: 10.1093/nar/gkm784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Miralles N, Corchero JL, Kumar P, Cedano JA, Gupta KC, Villaverde A, et al. Biological activities of histidine-rich peptides; merging biotechnology and nanomedicine. Microb Cell Fact. 2011;10:101. doi: 10.1186/1475-2859-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken KL, Hiemstra HS, van Meijgaarden KE, Subronto Y, den Hartigh J, Ottenhoff TH, et al. Purification of his-tagged proteins by immobilized chelate affinity chromatography: the benefits from the use of organic solvent. Protein Expr Purif. 2000;18(1):95–99. doi: 10.1006/prep.1999.1162. [DOI] [PubMed] [Google Scholar]
- Fromm JR, Hileman RE, Caldwell EE, Weiler JM, Linhardt RJ. Pattern and spacing of basic amino acids in heparin binding sites. Arch Biochem Biophys. 1997;343(1):92–100. doi: 10.1006/abbi.1997.0147. [DOI] [PubMed] [Google Scholar]
- Fromm JR, Hileman RE, Caldwell EE, Weiler JM, Linhardt RJ. Pattern and spacing of basic amino acids in heparin binding sites. Arch Biochem Biophys. 1997;343(1):92–100. doi: 10.1006/abbi.1997.0147. [DOI] [PubMed] [Google Scholar]
- Gandhi N, Mancera R. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Fruitos E, Vazquez E, Diez-Gil C, Corchero JL, Seras-Franzoso J, Ratera I, et al. Bacterial inclusion bodies: making gold from waste. Trends Biotechnol. 2012;30(2):65–70. doi: 10.1016/j.tibtech.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182(2):319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Hunt I. From gene to protein: a review of new and enabling technologies for multi-parallel protein expression. Protein Expr Purif. 2005;40(1):1–22. doi: 10.1016/j.pep.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Launay G, Salza R, Multedo D, Thierry-Mieg N, Ricard-Blum S. MatrixDB, the extracellular matrix interaction database: updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2015;43(Database issue):D321–327. doi: 10.1093/nar/gku1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Comparison of affinity tags for protein purification. Protein Expr Purif. 2005;41(1):98–105. doi: 10.1016/j.pep.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Macinkovic IS, Abughren M, Mrkic I, Grozdanovic MM, Prodanovic R, Gavrovic-Jankulovic M. Employment of colorimetric enzyme assay for monitoring expression and solubility of GST fusion proteins targeted to inclusion bodies. J Biotechnol. 2013;168(4):506–510. doi: 10.1016/j.jbiotec.2013.09.019. [DOI] [PubMed] [Google Scholar]
- Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J Biol Chem. 1993;268(26):19228–19231. [PubMed] [Google Scholar]
- Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J Biol Chem. 1993;268(26):19228–19231. [PubMed] [Google Scholar]
- Martinez-Ceron MC, Targovnik AM, Urtasun N, Cascone O, Miranda MV, Camperi SA. Recombinant protein purification using complementary peptides as affinity tags. N Biotechnol. 2012;29(2):206–210. doi: 10.1016/j.nbt.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Marty NJ, Rajalingam D, Kight AD, Lewis NE, Fologea D, Kumar TK, et al. The membrane-binding motif of the chloroplast signal recognition particle receptor (cpFtsY) regulates GTPase activity. J Biol Chem. 2009;284(22):14891–14903. doi: 10.1074/jbc.M900775200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem Int Ed Engl. 2003;42(8):864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Mitsi M, Forsten-Williams K, Gopalakrishnan M, Nugent MA. A catalytic role of heparin within the extracellular matrix. J Biol Chem. 2008;283(50):34796–34807. doi: 10.1074/jbc.M806692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, Shet D, Prasanna C, Atreya H. High yield expression of proteins in E. coli for NMR studies. Adv Biosci Biotech. 2013;4:751–767. [Google Scholar]
- Morris J, Jayanthi S, Langston R, Daily A, Kight A, McNabb DS, et al. Heparin-binding peptide as a novel affinity tag for purification of recombinant proteins. Protein Expr Purif. 2016;126:93–103. doi: 10.1016/j.pep.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Park N, Ryu J, Jang S, Lee H. Metal ion affinity purification of proteins by genetically incorporating metal-chelating amino acids. Tetrahedron. 2012;68:4649–4654. [Google Scholar]
- Peysselon F, Ricard-Blum S. Heparin-protein interactions: from affinity and kinetics to biological roles. Application to an interaction network regulating angiogenesis. Matrix Biol. 2014;35:73–81. doi: 10.1016/j.matbio.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Pina AS, Lowe CR, Roque AC. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol Adv. 2014;32(2):366–381. doi: 10.1016/j.biotechadv.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin VH, Sharp JS, Li X, Wang L, Prestegard JH. Characterization of glycosaminoglycans by 15N NMR spectroscopy and in vivo isotopic labeling. Anal Chem. 2010;82(10):4078–4088. doi: 10.1021/ac1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam D, Graziani I, Prudovsky I, Yu C, Kumar TK. Relevance of partially structured states in the non-classical secretion of acidic fibroblast growth factor. Biochemistry. 2007;46(32):9225–9238. doi: 10.1021/bi7002586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam D, Kathir KM, Ananthamurthy K, Adams PD, Kumar TK. A method for the prevention of thrombin-induced degradation of recombinant proteins. Anal Biochem. 2008;375(2):361–363. doi: 10.1016/j.ab.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam D, Kumar TK, Yu C. The C2A domain of synaptotagmin exhibits a high binding affinity for copper: implications in the formation of the multiprotein FGF release complex. Biochemistry. 2005;44(44):14431–14442. doi: 10.1021/bi051387r. [DOI] [PubMed] [Google Scholar]
- Raman R, Sasisekharan V, Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem Biol. 2005;12(3):267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, et al. A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14(4):2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel M, Soler DF, Kermode JC, Harris RB. Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J Biol Chem. 1992;267(13):8857–8862. [PubMed] [Google Scholar]
- Someya S, Kakuta M, Morita M, Sumikoshi K, Cao W, Ge Z, et al. Prediction of carbohydrate-binding proteins from sequences using support vector machines. Adv Bioinformatics. 2010 doi: 10.1155/2010/289301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraja V, Kumar TK, Rajalingam D, Graziani I, Prudovsky I, Yu C. Copper binding affinity of S100A13, a key component of the FGF-1 nonclassical copper-dependent release complex. Biophys J. 2006;91(5):1832–1843. doi: 10.1529/biophysj.105.079988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60(5):523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- Torrent M, Nogues MV, Andreu D, Boix E. The “CPC clip motif”: a conserved structural signature for heparin-binding proteins. PLoS One. 2012;7(8):e42692. doi: 10.1371/journal.pone.0042692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler-Cross R, Sobel M, Marques D, Harris RB. Heparin binding domain peptides of antithrombin III: analysis by isothermal titration calorimetry and circular dichroism spectroscopy. Protein Sci. 1994;3(4):620–627. doi: 10.1002/pro.5560030410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Han J, Wang S, Li P. Expression and purification of recombinant human bone morphogenetic protein-7 in Escherichia coli. Prep Biochem Biotechnol. 2014;44(1):16–25. doi: 10.1080/10826068.2013.782043. [DOI] [PubMed] [Google Scholar]
- Westra DF, Welling GW, Koedijk DG, Scheffer AJ, The TH, Welling-Wester S. Immobilised metal-ion affinity chromatography purification of histidine-tagged recombinant proteins: a wash step with a low concentration of EDTA. J Chromatogr B Biomed Sci Appl. 2001;760(1):129–136. doi: 10.1016/s0378-4347(01)00261-4. [DOI] [PubMed] [Google Scholar]
- Wong H, Schotz MC. The lipase gene family. J Lipid Res. 2002;43(7):993–999. doi: 10.1194/jlr.r200007-jlr200. [DOI] [PubMed] [Google Scholar]
- Yu W, Hill JS. Mapping the heparin-binding domain of human hepatic lipase. Biochem Biophys Res Commun. 2006;343(2):659–665. doi: 10.1016/j.bbrc.2006.02.175. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jiang N, Jia B, Chang Z, Wei X, Zhou J, et al. A comparative study on the heparin-binding proteomes of Toxoplasma gondii and Plasmodium falciparum. Proteomics. 2014;14(15):1737–1745. doi: 10.1002/pmic.201400003. [DOI] [PubMed] [Google Scholar]
Key references
- Morris J, Jayanthi S, Langston R, Daily A, Kight A, McNabb DS, et al. Heparin-binding peptide as a novel affinity tag for purification of recombinant proteins. Protein Expr Purif. 2016;126:93–103. doi: 10.1016/j.pep.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Dempewolf C, Morris J, Chopra M, Jayanthi S, Kumar T, Li W. Identification of consensus glycosaminoglycan binding strings in proteins. 2013 International Conference on Information Science and Applications (ICISA) 2013:1–5. 24–26. [Google Scholar]