

Abstract
Prader-Willi syndrome (PWS) is a genetic neurodevelopmental disorder that presents with hypotonia and respiratory distress in neonates. The Necdin-deficient mouse is the only model that reproduces the respiratory phenotype of PWS (central apnea and blunted response to respiratory challenges). Here, we report that Necdin deletion disturbs the migration of serotonin (5-HT) neuronal precursors, leading to altered global serotonergic neuroarchitecture and increased spontaneous firing of 5-HT neurons. We show an increased expression and activity of 5-HT Transporter (SERT/Slc6a4) in 5-HT neurons leading to an increase of 5-HT uptake. In Necdin-KO pups, the genetic deletion of Slc6a4 or treatment with Fluoxetine, a 5-HT reuptake inhibitor, restored normal breathing. Unexpectedly, Fluoxetine administration was associated with respiratory side effects in wild-type animals. Overall, our results demonstrate that an increase of SERT activity is sufficient to cause the apneas in Necdin-KO pups, and that fluoxetine may offer therapeutic benefits to PWS patients with respiratory complications.
Research organism: Mouse
eLife digest
Prader-Willi syndrome results from the disruption of a cluster of neighboring genes, including one called Necdin. Symptoms begin in early infancy and worsen with age. Affected children tend to develop an insatiable appetite, which often leads to obesity. They also experience serious problems with their breathing. Chest infections, high altitude and intense physical activity can be dangerous for children with Prader-Willi syndrome. This is because a slight shortage of oxygen may trigger breathing difficulties that could prove fatal.
The brain cells that produce a chemical messenger called serotonin help to control breathing. Several lines of evidence suggest that loss of Necdin may trigger breathing difficulties in Prader-Willi syndrome via effects on the serotonin system. First, serotonin neurons produce the Necdin protein. Second, laboratory mice that lack the gene for Necdin have abnormally shaped serotonin neurons. Third, these mice show breathing difficulties like those of individuals with Prader-Willi syndrome. But while this implies a connection between serotonin, Necdin and breathing difficulties, it falls short of establishing a causal link.
Matarazzo et al. now reveal an increase in the quantity and activity of a protein called the serotonin transporter in mutant mice that lacked the gene for Necdin compared to normal mice. Serotonin transporter proteins mop up the serotonin that neurons release when they signal to one another. Neurons in the mutant mice take up more serotonin than their counterparts in normal mice; this means they have less serotonin available for signaling. This may make it harder for the mutant mice to regulate their breathing.
Drugs called selective serotonin-reuptake inhibitors (or SSRIs for short) can block the serotonin transporter. These drugs, which include Fluoxetine (also called Prozac), are antidepressants. Matarazzo et al. show that SSRIs temporarily restore normal breathing in young mice that lack the gene for Necdin. However, these drugs have harmful long-term effects on breathing in non-mutant mice. Further studies should test whether short-term use of SSRIs could offer immediate relief for breathing difficulties in infants and children with Prader-Willi syndrome.
Introduction
Respiration is a complex function controlled in large part by raphe serotonergic (5-HT) neurons (Teran et al., 2014). Central 5-HT depletion induces severe apneas during the early postnatal period (Barrett et al., 2016; Trowbridge et al., 2011) and serotonopathy is implicated in the genesis of breathing disorders in human pathologies including neurodevelopmental diseases such as Sudden Infant Death Syndrome (Duncan et al., 2010; Hilaire et al., 2010; Kinney et al., 2011; Paterson et al., 2009), Rett syndrome (Abdala et al., 2010; Toward et al., 2013) and Prader-Willi Syndrome (PWS) (Zanella et al., 2008). However, the cellular and molecular events that underlie serotonopathy, and the causal link between serotonopathy and respiratory dysfunction in these pathologies are poorly understood.
PWS (prevalence 1/20000) is characterized by a combination of endocrine, metabolic, cognitive and behavioural/psychiatric symptoms (OMIM #176270). Its associated respiratory disturbances (Miller and Wagner, 2013; Nixon and Brouillette, 2002; Tan and Urquhart, 2017) are highly disruptive to the daily life of patients and represent the most common cause of death (73% of infants and 26% of adults) (Butler et al., 2017). They include both obstructive (Festen et al., 2006; Pavone et al., 2015) and central sleep apneas {Festen et al., 2006 #1495; Sedky et al., 2014), and blunted responses to hypercapnia/hypoxia possibly due to a lack of chemoreceptor sensitivity (Arens et al., 1996; Gozal et al., 1994; Schlüter et al., 1997; Gillett and Perez, 2016). Central apneas are present at birth (Zanella et al., 2008) and are prevalent throughout infancy while obstructive sleep apneas are more frequent in adolescents (Cohen et al., 2014).
PWS is caused by the loss of paternal expression of several genes of the 15q11-q13 region, including NECDIN. Necdin protein is a member of the Mage family, with proposed functions in differentiation (Andrieu et al., 2003; Takazaki et al., 2002), migration (Kuwajima et al., 2010; Miller et al., 2009; Tennese et al., 2008), neurite growth (Liu et al., 2009; Tennese et al., 2008), axonal extension, arborization and fasciculation (Pagliardini et al., 2005), and cell survival (Aebischer et al., 2011; Andrieu et al., 2006; Kuwako et al., 2005; Tennese et al., 2008). Among several mouse models of PWS, only those with Necdin deletion, Necdin (Ndn)-KO mouse models (Ndntm1-Stw [Gérard et al., 1999] and Ndntm1-Mus [Muscatelli et al., 2000]), present breathing deficits. Newborns Ndn-KO showed severe arhythmia, apnea, and blunted responses to respiratory challenges that frequently result in early postnatal lethality (Ren et al., 2003; Zanella et al., 2008). This dyspnoeic phenotype is recapitulated in brainstem slices that contain the Inspiratory Rhythm Generator (IRG), which display an irregular inspiratory rhythm and apneas (Ren et al., 2003; Zanella et al., 2008). Interestingly, 5-HT application, as well as other neuromodulators that are commonly co-released by medullary 5-HT neurons, such as substance P and thyrotropin-releasing hormone (Hodges and Richerson, 2008; Holtman and Speck, 1994; Kachidian et al., 1991; Ptak et al., 2009), stabilized the in vitro inspiratory rhythm (Pagliardini et al., 2005; Zanella et al., 2008).
A role for serotonergic transmission in the genesis of respiratory dysfunction in the Necdin-KO model is supported by neuroanatomical studies: Pagliardini and colleagues report abnormal morphology and orientation of axonal fibers that contain large 5-HT/Substance P varicosities in the developing Ndntm1-Stw-KO medulla (Pagliardini et al., 2005; Pagliardini et al., 2008). Similarly, we have also previously found that 5-HT fibers contained ‘swollen 5-HT varicosities’ in the Ndntm1-Mus-KO model, and that Necdin is expressed in virtually all 5-HT neurons (Zanella et al., 2008).
These findings suggest a potential role for abnormalities in 5-HT metabolism and release as a potential mediator of respiratory dysfunction in the Necdin-KO model of PWS, but fall short of proving causality. Here, we demonstrate a causal link between the perturbed development of the 5-HT system in Ndntm1-Mus-KO mice (referred to hereafter as Ndn-KO) and their observed respiratory phenotype (central apnea and hypercapnia). Our data implicate increased activity of serotonin transporter (SERT) as a key mediator of central apnea in this model, and that its inhibition restores normal breathing in Ndn-KO mice.
Results and discussion
Lack of Necdin affects the development and function of 5-HT neurons
Pet-EYFP mice expressing YFP under Pet1-promoter control, an early marker of developing 5-HT neurons (Hawthorne et al., 2010), were used to show that Necdin is expressed from E10.5 in early post-mitotic 5-HT precursors and later on in all 5-HT neurons until adulthood (Figure 1A, Figure 1—figure supplement 1A–I).
Figure 1. Necdin expression in 5-HT neurons and alterations of 5-HT neuronal development and activity in Ndn-KO mice.
(A) Scheme adapted from (Hawthorne et al., 2010) representing expression profiles of Necdin (green), Pet1 (blue) and 5-HT (red) throughout embryonic development of 5-HT neurons as soon as the progenitors become post-mitotic and start their radial migration by successive waves between E11.5 and E13.5. (B–C) 5-HT immunolabelling of brainstem sagittal sections of WT and Ndn-KO at E16.5. (B) 5-HT nuclei: B4 to B9 (left panels) or B1-B2 (right panels) are abnormal in Ndn-KO compared to WT. (C) Quantification of 5-HT neurons in the B1-B2 raphe nuclei (WT: 1836 (1751, 1878), n = 8; Ndn-KO: 1312 (1234, 1384), n = 7; MW, p=0.0003). (D–E) (D) Brainstem coronal sections of Pet-EYFP neurons from E11.5 WT and Ndn-KO mice illustrating radial migration from the ventricular zone (V) to the pial surface. (E) Quantification of nonlinear migration by measuring the α angle (10 cells/mouse) between the ventricular process and a virtual axis crossing the two opposing points from which neurites extend from the soma: (Angle (°): WT: 1.1(0.5, 3.5), n = 4; Ndn-KO: 18(2.8, 93.5), n = 3; MW, p<0.0001). (F–H) Confocal time-lapse analyses of cell migration of Pet-EYFP and Pet-EYFP/Ndn-KO neurons. (F) Plots representing the coordinates of individual cell bodies over time illustrate different cell migration patterns in WT (n = 4) and Ndn-KO (n = 3) Pet-EYFP neurons (11 cells/mouse). (G) Tortuosity index was increased by 52% in Ndn-KO compared to WT mice (WT: 1.08(1.01, 1.26); Ndn-KO: 1.65(1.36, 1.93); MW, p=0.0005). (H) Velocity was decreased by 37% in Ndn-KO compared to WT (Velocity (µm.s−1): WT: 2.50 10−3 (2.00, 2.93); Ndn-KO: 1.57 10−3 (1.09, 2.00); MW, p<0.0001). (I–K) Current clamp recordings of Pet-EYFP neurons (2 cells/slice) in WT (n = 3) and Ndn-KO (n = 3) brain slices. (I) Spontaneous discharge pattern of Pet-EYFP neurons; (J–K) Firing rate (J) and resting membrane potential (K) in Ndn-KO cells and aged-matched WT controls. Frequency (Hz): WT: 2.50(1.20, 2.50); Ndn-KO (4.60(4.00, 7.90); MW, p=0.0025; Voltage (mV): WT: −44.37(-46.25, –43.76); Ndn-KO: −42.64(-43.03, –42.55); MW test, p=0.0002.

Figure 1—figure supplement 1. Necdin expression compared with Pet-1 and 5-HT expression throughout embryonic development and alteration of 5-HT projections in Ndn-KO embryos.

Figure 1—video 1. Two-photon timelapse video showing somal translocation on organotypic slice cultures of Pet-EYFP neurons in WT embryos (E12.5).
Figure 1—video 2. Two-photon timelapse video showing somal translocation on organotypic slice cultures of Pet-EYFP neurons in Ndn-KO embryos (E12.5).
We then assessed whether Necdin deficiency could induce alterations of 5-HT neuronal development. In wild-type mice rostral hindbrain 5-HT neurons project to the mesencephalon at E12.5, and we observed a decrease in those ascending 5-HT projections in Ndn-KO embryos (Figure 1—figure supplement 1J), confirming previous work (Pagliardini et al., 2008). At E16.5, when the 5-HT raphe nuclei reach their mature configuration, we observed misplaced 5-HT neurons in Ndn-KO embryos (Figure 1B), with ~30% reduction in the total number of 5-HT neurons in the B1-B2 caudal raphe nuclei at birth (Figure 1C).
Our observations suggested a defect in 5-HT neuronal migration; which was tested using the Pet-EYFP model. In E10.5 WT embryos, Pet-EYFP neurons displayed typical bipolar morphology with oval-shaped somata aligned with two primitive processes attached to the ventricular and pial surfaces, required for somal translocation and involved in migration processes (Hawthorne et al., 2010) (Figure 1D). In contrast cells were not correctly aligned and process orientation was significantly disturbed in Pet-EYFP/Ndn-KO embryos (Figure 1D–E). Cell migration was also defective in organotypic slice cultures prepared from E12.5 embryos. Two-photon time-lapse imaging indicated that migratory behavior, based on somal translocation, was altered in Ndn-KO mice (Figure 1F–H, Figure 1—video 1 and 2) with tracked cells exhibiting increased tortuosity (Figure 1G) and decreased velocity (Figure 1H) of their growth trajectories. Interestingly, a comparable migration defect has been described in primary cultures of Ndntm1-Stw-KO cortical neurons (Bush and Wevrick, 2010) Here we revealed an alteration of cell migration of 5-HT precursors leading to misplaced 5-HT raphe nuclei in Ndn-KO mice.
The acquisition of specific firing properties is considered a critical marker of 5-HT neuronal and circuit maturation (Rood et al., 2014). Using visually guided patch-clamp recordings on brain slices (P15), we demonstrated a significant increase of spontaneous firing in Pet-EYFP/Ndn-KO cells (Figure 1I–K) suggesting a decreased availability of extracellular 5-HT (Maejima et al., 2013). Overall, our results show that Necdin is responsible for the normal migration of 5-HT precursor neurons during development and exerts effects on their electrophysiological properties in post-natal life.
Lack of Necdin increases the expression and activity of serotonin transporter
We hypothesised that reduced availability of extracellular 5HT could have contributed to the excessive electrophysiological activity we observed in Pet-EYFP neurons in Ndn-KO animals and examined potential mechanisms through which extracellular 5-HT could be reduced. We compared the distributions of 5-HT- immunoreactive enLarged Punctiform Axonal stainings (5-HT LPAs, previously named ‘swollen large varicosities’ [Pagliardini et al., 2005; Zanella et al., 2008]) in Ndn-KO and WT mice. In all regions analyzed we found significantly more 5-HT LPAs in Ndn-KO mice (Figure 2A–B). These 5-HT LPAs could result from (1) an increase of 5-HT synthesis and/or (2) a decrease in 5-HT degradation and/or (3) an increase of 5-HT reuptake. HPLC analyses showed a similar level of L-Trp and 5-HT in Ndn-KO compared with WT mice, but a significant increase of 5HIAA product in mutants (the ratio of 5HIAA/5-HT also being increased: Figure 2—figure supplement 1A–D). Noticeably, transcript levels of Tryptophan hydroxylase 2, the enzyme that converts L-Trp to 5-HT, were similar in Ndn-KO and WT mice (Figure 2—figure supplement 1E). These results suggest that the increase in 5-HT LPAs found in Ndn-KO brainstems probably result from an accumulation of intracellular 5-HT due to an increased 5-HT reuptake, since there is no increase of 5-HT synthesis but, on the contrary, an increase of 5-HT degradation.
Figure 2. Large punctiform axonal 5-HT staining (5-HT LPAs) results from an increase in SERT expression and activity in Ndn-KO mice.
(A–B) (A) Axonal 5-HT immunoreactivity illustrating 5-HT LPAs in the raphe of WT, Ndn-KO, Slc6a4-KO and Ndn/Slc6a4-DKO neonates (P1). (B) 5-HT LPAs were counted for all different genotypes (n = 3/genotype) in the raphe nuclei (B1–B2, B3, B7, B9), cortex and hippocampus.: Raphe B1-B2: WT: 2.2 ± 0.4; Ndn-KO: 11.8 ± 0.8, p=0.003; Raphe B3: WT: 2.6 ± 0.8; Ndn-KO: 9.3 ± 1.6, p=0.01; Raphe B7: WT: 6.5 ± 1.2; Ndn-KO: 15.5 ± 2.6, p=0.07; Raphe B9: WT: 5.1 ± 2.72; Ndn-KO: 14.6 ± 2.9, p=0.0001; Cortex: WT: 5.9 ± 2.0; Ndn-KO: 13.4 ± 0.8, p=0.01; Hippocampus: WT: 4.5 ± 1.2; Ndn-KO: 11.8 ± 2.5, p=0.01. p-values determined by two-way ANOVA followed by Bonferroni post-hoc test. DKO: double KO. Bar graphs represent mean ±SEM. (C–D) (C) Western blot analysis of SERT protein expression in brainstem collected from WT, Ndn-KO and Slc6a4-KO (negative control) neonates (P1). (D) Quantification of SERT expression normalized to β3 tubulin expression: WT: 0.45 (0.29, 0.56), n = 5; Ndn- KO: 1.11 (0.75, 1.68), n = 4; MW, p=0.016). Scatter dot plots, report Q2 (Q1, Q3). (E–H) Real time and single living cell analyses of SERT uptake activity using the fluorescent substrate ASP+, a fluorescent substrate of SERT. (E) Kinetic experiment recordings of accumulation of ASP+ over time (5 min recording). Coefficient of Determination R2: WT = 0.97; Ndn KO = 0.99; Slc6a4-KO = 0.93. Mean velocity (v) of ASP+ accumulation obtained by linear regression analyses of the slopes: (AUF.s−1): WT: 0.36 ± 0.01, n = 18; Ndn-KO: 0.51 ± 0.01, n = 18, covariance (ANCOVA), p<0.0001. Non-specific accumulation of ASP+ fluorescence was evaluated in Slc6a4-KO neurons and found to be strongly low (0.02 ± 0.01 (n = 6 cells). (F) Saturation experiments using gradual concentration of APS+. Non-linear curve-fitting yielded a one-phase exponential association, with a Vmax (G) and Km (H): Vmax (AUF.s−1): WT: 0.45 ± 0.05, n = 64; Ndn-KO: 0.84 ± 0.12, n = 67; Slc6a4-KO: 0.01 ± 0.03, n = 37, p<0.0001; Km (µM): WT: 6.03 ± 1.60, n = 64; Ndn-KO: 12.03 ± 3.55, n = 67; Slc6a4-KO: 1.83 ± 1.60, n = 37, p<0.0001. AUF: arbitrary unit of fluorescence. p-values determined by K-W test, followed by Dunn post-hoc test. Bar graphs represent mean ±SEM. *p<0.05; **p<0.01; ***p<0.001. Scatter dot plots, report Q2 (Q1, Q3). **p<0.01; ***p<0.001.

Figure 2—figure supplement 1. 5-HT metabolism, Tph2 and Slc6a4 transcripts quantification in Ndn-KO mice.

Figure 2—figure supplement 2. ASP+ uptake in neurons of raphe primary cultures.


We hypothesised that overexpression of serotonin transporter (SERT) represents a plausible mechanism through which 5-HT could be accumulated in Ndn-KO mice, based on the observation that inactivation of Maged1, another member of the Mage gene family, leads to overexpression of SERT (encoded by the Slc6a4 gene) (Mouri et al., 2012). Indeed, we observed a 3.2 fold increase in SERT protein expression in the brainstems of Ndn-KO compared to WT pups (Figure 2C–D), while Slc6a4 transcript levels were similar (Figure 2—figure supplement 1F). This suggests post-transcriptional or post-translational dysregulation of Slc6a4/SERT in Ndn-KO. Subsequently, in 5-HT neurons of raphe primary cultures, we assessed SERT activity by live single cell uptake assay, using ASP+ (4(4-(dimethylamino)styryl)-N-methylpyridinium), a fluorescent substrate of SERT (Lau et al., 2015; Oz et al., 2010). Changes in the kinetics and saturation of ASP+ uptake were measured after 8 days in vitro culture in 5-HT neurons from neonatal (P0) WT, Ndn-KO, and Slc6a4-KO mice (Figure 2E–H, Figure 2—figure supplement 2A–B). As expected, cultures accumulated ASP+ over time in all conditions tested. However, kinetics experiments show that ASP+ accumulation was significantly faster (greater mean velocity v) in Ndn-KO compared to WT raphe neurons (Figure 2E). Saturation experiments using increasing concentrations of ASP+ confirmed that ASP+ uptake is a saturable process (Figure 2F) and showed a Vmax (Figure 2G) and KM (Figure 2H) significantly higher in Ndn-KO than in WT or Slc6a4-KO neurons. ASP+ uptake was ~2 fold increased in Ndn-KO while it was null in Slc6a4-KO cell cultures. We conclude that there is an increase of ASP+ uptake in Ndn-KO neurons, specifically dependent on SERT activity, suggesting a mechanism for 5-HT LPAs accumulation in vivo. To determine whether in vivo deletion of Slc6a4 could suppress the 5-HT LPAs in Ndn-KO, we compared the number of 5-HT LPAs in Ndn-KO, Slc6a4-KO and Ndn/Slc6a4-double KO (Ndn/Slc6a4-DKO) neonates in various brain structures. The number of 5-HT LPAs was similar in brains of Ndn/Slc6a4-DKO and WT mice (Figure 2A–B), indicating that the absence of Ndn is functionally compensated for by the lack of Slc6a4.
Together, our data show that increased SERT expression in Ndn-KO mice underlies an increase of 5-HT reuptake, which accumulates in 5-HT LPAs. In the absence of any increase in 5-HT synthesis (and in fact increased 5-HT degradation), this sequence of events could be sufficient to cause a physiologically relevant decrease extracellular 5-HT.
Genetic ablation or pharmacological inhibition of SERT uptake restores normal breathing in Ndn-KO mice
As exogenous 5-HT application stabilized respiratory rhythm of Ndn-KO mice in vitro, (Zanella et al., 2008), we hypothesized that SERT dysregulation observed in Ndn-KO mice might underlie their respiratory phenotype. To further investigate this causal link, we compared breathing parameters in WT, Ndn-KO, Ndn/Slc6a4-DKO and in Ndn-KO pups treated with Fluoxetine, a selective 5-HT reuptake inhibitor (SSRI) used clinically to increase extracellular 5-HT (Figure 3A–B). First, we confirmed that respiratory deficits, quantified as the percentage of mice exhibiting apnea (Figure 3C), the number of apneas per hour (Figure 3D), or the accumulated apnea duration (Figure 3E), were significantly increased in Ndn-KO compared to WT mice. These deficits were suppressed by reducing SERT function either by constitutive genetic inactivation (Ndn/Slc6a4-DKO pups) or by 10 days of Fluoxetine treatment (P5-P15; 10 mg/kg/day) in Ndn-KO pups (Figure 3C–E). Other basic respiratory parameters (minute ventilation, frequency of breathing, tidal volume) were unchanged between all genotypes (Figure 3F–H). Therefore, our results show that increasing extracellular 5-HT is sufficient to suppress apneas in juvenile Ndn-KO mice.
Figure 3. Genetic ablation or pharmacologic inhibition of SERT suppresses apnea and rescues central chemoreflex in Ndn-KO mice.
(A) Workflow experiment of constant airflow whole body plethysmography performed in unanaesthetized, unrestrained WT, Ndn-KO, Ndn/Slc6a4-DKO and Ndn-KO+Fluox mice at the age of P15. Ndn-KO and WT animals (indicated here and in the figure as WT or Ndn-KO) have been pre-treated with 0.9% NaCl from age P5 to P15. Ndn-KO mice (indicated here and in the figure as Ndn-KO+Fluox) have been pre-treated with with Fluoxetine (10 mg/Kg/day) from age P5 to P15. (B) Plethysmographic recordings of WT, Ndn-KO, Ndn/Slc6a4-DKO and Ndn-KO+Fluox mice at the age of P15. (C–E) Quantification of apnea in P15 mice. (C) Proportion of apneic mice: WT: 2 of 8; Ndn-KO: 7 of 8; corresponding respectively to 25% and 87%; Chi2 test, p=0.01. Genetic ablation of Slc6a4or early Fluoxetine treatment normalized the number of Ndn-KO apneic mice: Ndn/Slc6a4 DKO: 2 of 8, 25%; Chi2 test, p>0.99, N.S.; Ndn-KO+Fluox: 2 of 8, 25%; Chi2 test, p>0.99, N.S. (D) Number of apnea in Ndn-KO compared to WT mice: WT: 0.0 (0.0, 1.5), n = 8; Ndn-KO: 3.8 (2.0, 8.0), n = 8; p=0.01. Genetic ablation of Slc6a4 or Fluoxetine treatment normalized the number of apnea of Ndn-KO mice to WT values: Ndn/Slc6a4-DKO: 0.0 (0.0, 2.0), n = 8; p>0.99, N.S.; Ndn-KO+Fluox: 0.0 (0.0, 2.2), n = 8; p>0.99, N.S. p-values determined by KW test followed by Dunn post-hoc test with comparison to WT. (E) Cumulative distribution of apnea (number of cumulated values) over apnea duration (msec) in WT, Ndn-KO, Ndn/Slc6a4-DKO and Ndn-KO treated by Fluoxetine. Compared to WT, Ndn-KO mice demonstrated a significant increase of cumulative apnea both in term of number and duration (Kolmogorov-Smirnov test, p=0.01). However, such increase was normalized to WT after genetic deletion of Slc6a4 or Fluoxetine treatment. (F–H) Basic breathing parameters: (F) Minute ventilation, VE (the total volume breathed over one min): WT: 24.5 (17.7, 32.7), n = 8; Ndn-KO: 17.5 (15.2, 18.7), n = 8; p=0.14, N.S.; Ndn/Slc6a4-DKO: 18.0 (17.0, 20.0), n = 8; p=0.25, N.S. and Ndn-KO+Fluox: 21.0 (18.2, 29.7), n = 8; p>0.99, N.S.. (G) Frequency of breathing, Rf (breaths/min): WT: 338 (312, 3867), n = 8; Ndn-KO: 296 (270, 352), n = 8; p=0.56, N.S.; Ndn/Slc6a4-DKO: 292 (2890, 305), n = 8; p=0.16, N.S. and Ndn-KO+Fluox: 329 (289, 388), n = 8; p>0.99, N.S. (H) Tidal Volume, VT (the volume flow per breath): WT: 0.07 (0.05, 0.09), n = 8; Ndn-KO: 0.06 (0.06, 0.06), n = 8; p=0.38, N.S.; Ndn/Slc6a4-DKO: 0.06 (0.05, 0.07), n = 8; p=0.51, N.S. and Ndn-KO+Fluox: 0.07 (0.06, 0.08), n = 8; p>0.99, N.S. p-values determined by K-W test followed by Dunn post-hoc test with comparison to WT.Scatter dots represent Q2 (Q1, Q3). N.S.: non-significant; *p<0.05.

Figure 3—figure supplement 1. Early life Fluoxetine treatment has only short-term positive effects on Ndn-KO apneas.

Figure 3—figure supplement 2. Early life treatment of Fluoxetine on respiratory apnea in wild-type mice.

Since Fluoxetine treatment in early life has positive effects on apneas, we next questioned the long-term consequences of this treatment. Novel cohorts of WT, Ndn-KO and Ndn-KO pups were treated as above with Fluoxetine or vehicle and then submitted to plethysmography 0, 15 and 45 days after treatment (DAT) (Figure 3—figure supplement 1A–B). The positive effect of Fluoxetine on respiratory function in Ndn-KO pups at the end of treatment were confirmed in this cohort, but did not persist at 15 and 45 DAT (Figure 3—figure supplement 1C–E). Other respiratory parameters (minute ventilation, frequency of breathing, tidal volume) measured at 45 DAT were unchanged between all genotypes (Figure 3—figure supplement 1F–H).
An altered ventilatory response to hypercapnia was previously observed in adult Ndn-KO mice (Zanella et al., 2008), so we next investigated whether this deficit is apparent in P0-P1 pups. We examined the chemoreflex of Ndn-KO and WT neonates by initially subjecting them to a moderate hypercapnia (5 min; 4% CO2) (Figure 4A–C). Under hypercapnic stress, WT but not Ndn-KO neonates progressively increased their respiratory frequency (Rf) (Figure 4D), leading to an increase in minute ventilation (volume breathed over 1 min,VE) (Figure 4F). In contrast, no significant effects of hypercapnia were detected on any respiratory variables in Ndn-KO pups and thus Ndn-KO pups appear relatively insensitive to hypercapnia. To determine whether altered central 5-HT transmission contributes to this effect, we performed electrophysiological recordings of rhythmic phrenic bursts using en bloc brainstem-spinal cord preparations from P0-P1 WT and Ndn-KO pups. During perfusion with physiological aCSF (pH 7.4), we found no significant difference in phrenic burst (PB) shape, amplitude or discharge frequency (PBf) between WT and Ndn-KO pups (Figure 4G–H). As expected, PBf in WT preparations progressively increased upon acidosis (pH = 7.1, Figure 4I,L). However, this effect was not observed in Ndn-KO preparations (Figure 4J,L).
Figure 4. Alteration of respiratory chemoreflex in Ndn-KO neonates is rescued by Fluoxetine.
(A–F) Effect of hypercapnia on in vivo ventilatory parameters of WT and Ndn-KO neonates. (A) Workflow experiment of constant airflow whole body plethysmography performed in unanaesthetized, unrestrained WT, Ndn-KO neonates at P0-P1 when breathing either air or hypercapnic mixture containing 4% CO2 in air for 5 min. Data for analyses were collected in the last 5 min (air) or the last min (hypercapnia). (B–C) (B) Plethysmographic recordings of WT and Ndn-KO neonates when breathing air or (C) at 5th min upon hypercapnic respiratory challenge. (D) Respiratory frequency (Rf) in WT and Ndn-KO pups when subjected to hypercapnic stress: WT Air: 91 ± 8; WT hypercapnia: 163 ± 16; n = 8, p=0.004; Ndn-KO Air: 95 ± 12; Ndn-KO hypercapnia: 123 ± 11; n = 8, p=0.31, N.S. p-values determined by two-way-ANOVA test followed by Bonferroni post-hoc test. Bar graphs represent mean ±SEM; **p<0.01; N.S.: non-significant. (E) Tidal Volume (VT) (µl.g−1) in WT neonates: WT Air: 9 ± 0.7; WT hypercapnia: 12 ± 1; n = 8, p=0.12 N.S.; in Ndn-KO: Ndn-KO Air: 7.5 ± 0.9; Ndn-KO hypercapnia: 10.9 ± 1.1; n = 8, p=0.055. p-values determined by two-way ANOVA test followed by Bonferroni post-hoc test. (F) Minute Ventilation (VE) (ml.min−1.g−1) in WT neonates: WT Air: 0.8 ± 0.1; WT hypercapnia: 2.1 ± 0.1; n = 8, p=0.01; in Ndn-KO: Ndn-KO Air: 0.7 ± 0.1; Ndn-KO hypercapnia: 1.3 ± 0.2; n = 8, p=0.3 N.S. p-values determined by two-way ANOVA test followed by Bonferroni post-hoc test. Bar graphs represent mean ±SEM. *p<0.05. (G–H) Effect of Fluoxetine treatment on the resting phrenic burst frequency (PBf) and the PBf response to acidosis in Ndn-KO medulla preparations. (G) Electrophysiological recordings of PBf produced in vitro in WT and Ndn-KO en bloc brainstem-spinal cord preparations at P0-P1 when superfused first with neutral artificial cerebrospinal fluid (aCSF) (pH 7.4) and then acidified aCSF (pH 7.1). (H) Workflow experiment of the electrophysiological recordings on medullary preparations to assess central chemosensitivity in WT and treated with Fluoxetine (20 μM) or untreated Ndn-KO mice. (I–K) Examples of continuous electrophysiological recordings of rhythmic phrenic bursts produced in en bloc brainstem-spinal cord preparations of (I) one WT, (J) one Ndn-KO and (K) one Ndn-KO treated with Fluoxetine (20 µM) pup and superfused with first neutral aCSF (pH 7.4) (left column recordings) or acidified aCSF (pH 7.1) (right column recordings). (L) Quantifications of the resting PBf (c.min−1) of en bloc preparations superfused with neutral aCSF (pH 7.4) or acidified aCSF (pH 7.1) respectively of WT (WT (pH 7.4): 8.8 c.min−1 ±1.2; WT (pH 7.1) 12.8 ± 0.3 c.min−1; n = 12, p<0.001), Ndn-KO (Ndn-KO (pH 7.4): 9.5 ± 1.0; Ndn-KO (pH 7.1): 9.8 ± 0.3 c.min−1; n = 12, p=0.41, N.S.) and Ndn-KO treated with Fluoxetine: (Ndn-KO+Fluox (pH 7.4): 9.6 ± 0.3; Ndn-KO+Fluox (pH 7.1): 12.6 ± 0.7; n = 12, p=0.04). Noticeably, under neutral aCSF (pH 7.4) no difference was observed between WT, Ndn-KO and Ndn-KO+Fluox. However, in acidified aCSF (pH 7.1), Fluoxetine significantly increased the PBf of Ndn-KO preparations. p-values determined by two-way ANOVA test followed by Tukey post-hoc test. Bar graphs represent mean ±SEM. N.S: non-significant; *p<0.05.

Figure 4—figure supplement 1. Effect of pre-treatment with the 5-HT1A-R agonist 8OHDPAT on the resting PBf and the PBf response to acidosis in Ndn-KO en bloc brainstem-spinal cord preparations of P0-P1 pups.
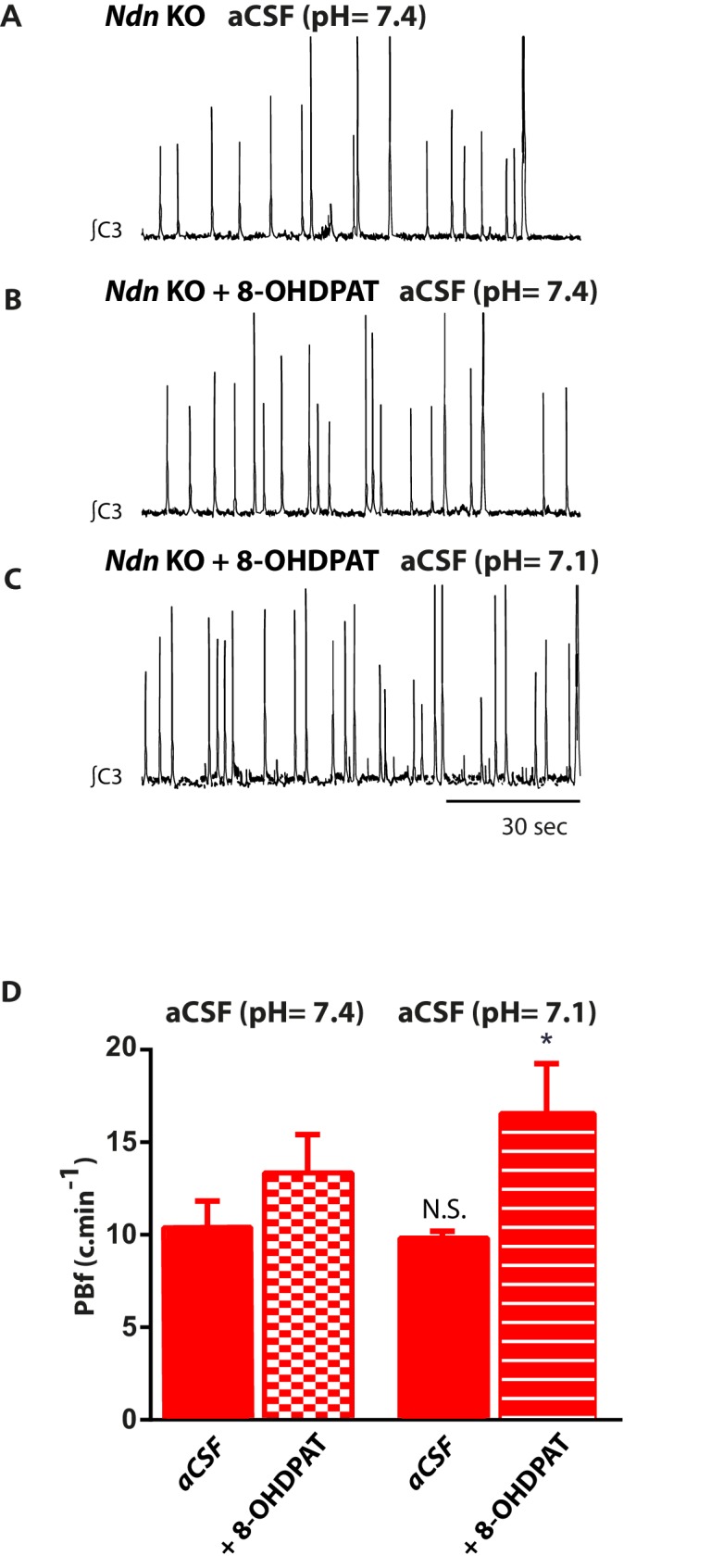
Figure 4—figure supplement 2. Effects of Fluoxetine and of the 5-HT1A-R agonist 8OHDPAT on the resting PBf and the PBf response to acidosis in wild-type medulla preparations.


We then assessed whether increasing extracellular 5-HT could rescue chemoreflex sensitivity in this preparation. Bath application of Fluoxetine (20 µM) prior to acidosis did not affect baseline PBf of Ndn-KO preparations (Figure 4K,L), but instead significantly increased PBf responses to acidosis to levels indistinguishable from WT controls (Figure 4K,L). Qualitatively similar responses were observed in experiments in which a 5-HT1A receptor agonist (8OHDPAT) was substituted for Fluoxetine (Figure 4—figure supplement 1A–D). We therefore conclude that the central chemoreceptor hyposensitivity characteristic of the Ndn-KO model can be restored by pharmacological manipulations that increase extracellular 5-HT and/or stimulate 5-HT1A-R activity.
Early life Fluoxetine-treatment has deleterious long-term respiratory consequences in WT mice
Although Fluoxetine had beneficial but transient effects on apnea incidence in Ndn-KO mice, we observed deleterious and long-lasting effects on respiratory function in WT controls. Early life Fluoxetine-treatment induced a significant increase in the number of apneic mice, the frequency of apneas, and the cumulative distribution of apneas at all timepoints measured (0, 15 and 45 DAT, Figure 3—figure supplement 2A–E), such that measurements at 45 DAT in WT mice (Figure 3—figure supplement 2) were similar to those obtained in Ndn-KO mice (Figure 3—figure supplement 1). The sensitivity of WT brainstem-spinal cord preparations, treated with Fluoxetine or with 8OHDPAT, to acute acidosis was similarly affected (Figure 4—figure supplement 2A–D). In neutral aCSF, neither Fluoxetine (Figure 4—figure supplement 2C) or 8OHDPAT (Figure 4—figure supplement 2D) affected resting PBf of WT en bloc preparations but instead abolished the normal increases in PBf responses to acidosis. Thus, we confirm that Fluoxetine treatment abolishes the capacity of WT mice to respond to acidosis (Voituron et al., 2010), and we propose a role for 5-HT1A-R activity in this response. We show here, for the first time, adverse effects of Fluoxetine on breathing outcomes.
Conclusion
Previously, a pleiotropic function of Necdin has been reported in different neuronal populations and at different developmental stages. Concerning the 5-HT system, an expression of Necdin was observed in virtually all 5-HT neurons (Zanella et al., 2008) and an alteration of the 5-HT system in embryonic and postnatal development was partially described in both Ndn-KO (Ndntm1-Stwand Ndntm1-Mus) mouse models, with alterations in 5-HT axonal bundle projections (Lee et al., 2005; Pagliardini et al., 2005) and 5-HT fibers containing swollen 5-HT ‘varicosities’ (Pagliardini et al., 2005; Zanella et al., 2008). Furthermore, an alteration of 5-HT metabolism (Zanella et al., 2008) was observed in mutant neonates suggesting that it might alter 5-HT modulation of the Respiratory Rhythm Generator. Finally, an in vitro exogenous application of 5-HT on brainstem-spinal cord preparations of Ndn mutant mice alleviates the incidence of apneas (Pagliardini et al., 2005; Zanella et al., 2008). Despite those observations, the pathological mechanism responsible for the serotonopathy in Ndn-KO mice and the causal link between this serotonopathy and the breathing alterations were not investigated. Here, we aimed to answer those questions.
Noticeably, all previous studies have been performed on heterozygous Ndn-deficient mice, with a deletion of the Ndn paternal allele only (Ndn+m/-p), the maternal allele being normally silent. However, we have shown that, due to a faint and variable expression of the Ndn maternal allele (+m), Ndn+m/-p mice present a variability in the severity of respiratory phenotype compared with the Ndn-/- mice (here named Ndn-KO) (Rieusset et al., 2013). For instance, reduction of 5-HT neurons was not previously found significant in the Ndn + m/-p mice (Zanella et al., 2008) but has been found significantly reduced in the Ndn-/- mice. In order to avoid such variability and to get consistent results, we chose here to study Ndn-/- mice.
Here, we have shown that Necdin plays a pleiotropic role in the development of 5-HT neuronal precursors that guides the development of central serotonergic circuits and the physiological activity of mature 5-HT neurons. Our results suggest that Necdin controls the level of SERT expression in 5-HT neurons and that lack of Necdin increases the quantity and activity of SERT leading to an increased reuptake and intra-cellular accumulation of 5-HT, as visualized by 5-HT LPAs, leading to a reduction in available extracellular 5-HT. Importantly, in vivo inhibition of SERT activity, genetically or pharmacologically (Fluoxetine treatment), is sufficient to prevent the formation of those 5-HT LPAs and suppresses the apnea observed in Ndn-KO mice. We also demonstrate, using an ex vivo approach, that the altered chemosensitivity to CO2/acidosis is caused by a central 5-HT deficit and is rescued by Fluoxetine-treatment. We conclude that an increase of 5-HT reuptake is the main cause of breathing deficits (central apnea and hypercapnia response) in Ndn-KO mice.
Unexpectedly, we reveal an adverse and long-term effect of early life administration of Fluoxetine on the breathing (apneas, chemosensitivity to CO2/acidosis) of healthy mice. Previous adverse effects have been observed on anxiety and depression (Glover and Clinton, 2016; Millard et al., 2017) after an early postnatal administration of Fluoxetine but the respiratory deficits are reported here for the first time and should be further investigated in another study.
Respiratory failure in patients with PWS constitute a challenging issue since it is the most common cause of death for 73% of infants and 49% of children, (Butler et al., 2017). Death is often linked to respiratory infection or respiratory disorder and may be sudden, with some reported cases of sudden death occurring at night (Gillett and Perez, 2016). In PWS patients, any environmental acute respiratory challenge caused by, for instance, a respiratory tract infection, high altitude or intense physical activity further exacerbates their inherent disability (blunted response to hypoxima/hypercapnia) to adapt an respiratory response. Until now, the underlying pathology for respiratory failure remained elusive and did not appear to be impacted by recent advancements in treatment modalities (Butler et al., 2017). Although oxygen treatment is efficient in preventing the hypoxemia induced by central apneas (Urquhart et al., 2013), such treatment is physically constraining. Within the context of PWS, the current study points towards a critical link between Necdin, serotonopathy, and chemosensing, a function in which brainstem serotonergic circuits play a critical role. Since our study shows that Fluoxetine can suppress apnea and restore chemosensitivity, we propose that Fluoxetine might be an appropriate ‘acute’ treatment that could be considered for Prader-Willi infants/children when they present the first signs of any breathing difficulties.
Materials and methods
Animals
Mice were handled and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (N.R.C., 1996) and the European Communities Council Directive of September 22th 2010 (2010/63/EU, 74). Experimental protocols were approved by the institutional Ethical Committee guidelines for animal research with the accreditation no. B13-055-19 from the French Ministry of Agriculture. All efforts were made to minimize the number of animals used. Necdin is an imprinted gene, paternally expressed only (Figure 2—figure supplement 3 and Figure 4—figure supplement 3). In order to avoid a variability in our results due to a stochastic and faint expression of the maternal allele (Rieusset et al., 2013), we worked with the Ndntm1-Mus strain and decided to study Ndn-/- mice (named here Ndn-KO), instead of Ndn+m/-p mice as it has been done previously.
Fluoxetine was obtained from Sigma (Saint-Quentin Fallavier, France) for cell culture and en bloc medullary experiments and from Mylan pharma for in vivo experiments.
Transgenic mice
We bred ePet-EYFP-expressing (Scott et al., 2005a; Scott et al., 2005b) or Slc6a4-Cre Knock-in (Zhuang et al., 2005) mice with Ndn-KO (Muscatelli et al., 2000) mice, all on C57BL/6 background. Protocols of genotyping mice have been previously described for Pet-EYFP (Hawthorne et al., 2010), Ndn-KO (Rieusset et al., 2013) and Sert-Cre Knock-in mice (Zhuang et al., 2005), in which the Slc6a4 gene was replaced by Cre was referred to in the text as Slc6a4-KO. Breeding of Slc6a4-KO with Ndn-KO mice was referred to in the text as Ndn-Slc6a4-DKO.
Immunohistochemistry and quantification
Tissue preparation and IHC were performed as previously described (Rieusset et al., 2013). Antibodies used were: rabbit polyclonal anti-Necdin (07–565; Millipore, Bedford, MA, USA; 1:500), mouse monoclonal anti-GFP (Interchim, NB600-597; 1:500), goat polyclonal anti-5HT (Immunostar, 20079; 1:300). Sections were examined on a Zeiss Axioplan two microscope with an Apotome module.
Brainstem structures were sampled by selecting the raphe obscurus area and counting was performed on three sagittal sections/animal of 100 µm which represent the entire PET1-YFP positive cell population of the raphe obscurus (ROb/B2) and pallidus (RPa/B1), both nuclei being difficult to separate. For each section, a Z-stack composed of 10 confocal images (8 µm focal spacing) was acquired. For quantification, stereological method has been applied on each Z-stack image using the eCELLence software developed by Glance Vision Technologies (Italy). The total cell number/per animal was obtained by summing the sub-total of cells counted for the 3 Z-stacks.
Images of 5-HT LPAs were acquired using a confocal microscope (Olympus). Between 4 and 8 fibers/brain region for each animal (3WT and 3 KO) were analyzed for the presence of 5-HT LPAs (>1.8 µm2) on 100 µm long fiber. The size of 5-HT LPAs was quantified using Image J. 5-HT LPA diameter has been defined ad arbitrium as the size of the largest 5-HT punctiform labelling found in the WT fibers.
Organotypic slice cultures and time lapse experiments
Slice cultures from E11.5 embryonic mouse brainstems were prepared from Pet-EYFP and Ndn KO/Pet-EYFP mice. Thick coronal sections (250 µm) brainstem were cut using a tissue chopper and cultured in Neurobasal medium (Thermofisher) containing 2% B27 (Thermofisher), 4% horse serum, 10 µg/ml insulin, 200 mM HEPES, 1% Antibiotic Antimycotic (Thermofisher). For time lapse experiments, the dishes were mounted in a CO2 incubation chamber (5% CO2 at 37°C) fitted onto an inverted confocal microscope (LSM510, Zeiss). Acquisitions of the region containing raphe Pet-EYFP +neurons were performed every 10 min for up to 15 hr. Cell coordinates, velocity, and tortuosity (total length of the track/direct distance from the first to the last point) were calculated using MtrackJ plugin of Image J.
Electrophysiology patch-clamp
Sagittal slices that included the raphe (400 μm thick) were cut from brainstems of 2 week old Pet-EYFP and Ndn-KO/Pet-EYFP mice. Whole-cell recordings were made from YFP+ cells in the region of the B4 raphe nucleus. During recordings, slices were continuously perfused with artificial cerebrospinal-fluid (aCSF) at 37°C. Patch pipettes (4–5 MΩ) were filled with an internal solution with the following composition (in mM): 120 KGlu, 10 KCl, 10 Na2-phosphocreatine, 10 HEPES, 1 MgCl2, 1 EGTA, 2 ATP Na2, 0.25 GTP Na; pH = 7.3 adjusted with KOH. Current clamp at i = 0 were recorded with a HEKA amplifier and acquired using PatchMaster software (HEKA). Offline analysis was performed with Clamfit 10.3.
In vitro recordings from en bloc brainstem-spinal cord preparations
As previously reported (Berner et al., 2012), the medulla and cervical cord of P0-P1 neonatal mice were dissected, placed in a 2 ml in vitro recording chamber, bubbled with carbogen, maintained at 27°C and superfused (3.5–4.5 ml per min) with aCSFcomposed with (mM): 129.0 NaCl, 3.35 KCl, 21 NaHCO3, 1.26 CaCl2, 1.15 MgCl2, 0.58 NaH2PO4, and 30.0 D-glucose (‘Normal aCSF’: pH 7.4) or using the same components except with 10 mM NaHCO3 (‘Acidified aCSF’: pH 7.1). Inspiratory discharges of respiratory motoneurons were monitored by extracellular recording with glass suction electrodes applied to the proximal cut end of C4 and C3 spinal nerves roots. Axoscope software and Digidata 1320A interface (Axon Instruments, Foster, CA, USA) were used to collect electrophysiological data. Offline analysis was performed with Spike 2 (Cambridge Electronic Design, UK) and Origin 6.0 (Microcal Software, Northampton, MA, USA) software for PC. Burst frequency was analyzed and calculated as the number of C4 bursts per minute. The values of inspiratory burst frequency were calculated as the mean of the last 3 min of any condition: ACSF (7.4) and ACSF (7.1). Standardized experiments in WT and Ndn-KO preparations were repeated on different preparations from different litters. For a given preparation, only one drug was applied and only one trial was performed.
RT-qPCR
For RT-qPCR, mice were sacrificed at P1, the brainstem dissected, and tissues were rapidly collected and frozen in liquid nitrogen prior to RNA isolation using standard conditions. RNA, reverse transcription and real time PCR were conducted as previously described (Rieusset et al., 2013). Sequences of the various primer pairs used for qPCR, as well as the slope of the calibration curve established from 10 to 1 × 109 copies and qPCR efficiency E, were as follow: Tph2: F: 5’-GAGCTTGATGCCGACCAT-3’; R: 5’-TGGCCACATCCACAAAATAC-3’; Slc6a4: F:5’-CATATGCTACCAGAATGGTGG-3’; R:5’-AAGATGGCCATGATGGTGTAA-3’. For each sample, the number of cDNA copies was normalized according to relative efficiency of RT determined by the standard cDNA quantification. Finally, gene expression was expressed as the cDNA copy number quantified in 5 µL aliquots of RT product.
Western blot
Newborn mice were sacrificed and brainstems were immediately dissected and snap-frozen in liquid nitrogen and stored at −80°C until protein extraction. Protein extraction was conducted as previously described (Felix et al., 2012). Membranes were blocked with PBS containing 5% BSA for 1 hr, followed by an overnight incubation at 4°C with the following primary antibodies: guinea pig anti-SERT (1/2000, Frontier Institute), mouse anti-B3 tubulin (1/2000, ThermoFisher Scientific). Membranes were then washed and incubated 2 hr with either anti-guinea pig (1/1000, ThermoFisher Scientific), or anti-mouse (1/2000; DAKO) horseradish peroxidase-conjugated secondary antibodies. Visualisation was performed using the Super signal West-pico chemolumniscent substrate (Pierce, Thermo Scientific, France). Quantification was performed using ImageJ.
Biochemical analysis of the medullary serotonergic system
Pregnant mice were killed by cervical dislocation at gestational day E18.5 and fetuses were removed, decapitated, and the medulla dissected and stored at −80°C until measurements. Medullary 5-HT, its precursor L-tryptophan (L-Trp), and its main metabolite, 5-hydroxy-indol acid acetic (5-HIAA), were measured with high-pressure liquid chromatography separation and electrochemical detection (Waters System: pump P510, electrochemical detector EC2465; Atlantis column DC18; mobile phase: citric acid, 50 mM; orthophosphoric acid, 50 mM; sodium octane sulfonic acid, 0.112 mM; EDTA, 0.06 mM; methanol, 5%; NaCl, 2 mM; pH 2.95). Contents are expressed in nanograms per medulla.
Raphe primary neuronal culture and live cell uptake assay
Raphe primary cell culture
Newborn mice (n = 6 per culture) were decapitated, brainstems extracted, the meninges removed and the medial part of the brainstem dissected. Tissues were enzymatically digested at 37°C for 30 min with HBSS containing 2 mg/mL of filter-sterilized papain. Cells were resuspended in Neurobasal medium (Thermofisher) containing 2% B27 (Thermofisher), 0.5 mM L-glutamine, glucose (50 mM), 50 ng/ml NGF, 10 ng/ml bFGF, 10 µg/ml insulin. 2 × 105 cells were plated on round 14 mm glass coverslip pre-coated with Polyethyleneimine (20 µg/ml). Cells were cultured during 8 days in presence of 5% of NU serum (Becton Dickinson) during the first 2 days. Immunocytochemistry was performed to verify presence of 5-HT+ neurons in the culture.
Live cell imaging of (4-(4-(dimethylamino)styryl)-N-methylpyridinium (ASP+) uptake
Cells were placed in a bath chamber on the stage of an inverted microscope (Nikon eclipse TE300) and perfused (2 ml/min) with Krebs medium (mM): 150 NaCl; 2.5 KCl; 2 CaCl2; 2 MgCl2; 2.5 Hepes acide; 2.5 Hepes-Na; pH 7,4. Time-lapse cell acquisition was started when ASP+ (1, 2, 5, 10, 15 or 20 µM) was added to the perfusion. ASP+ was excited at 488 nm and fluorescence was captured at 607 nm every 10 s for 5 min using Metamorph software (MolecularDevices). Each ASP+ concentration was tested on three different cultures for WT and Ndn-KO and one for Ndn/Slc6a4-DKO. Cells placed on the coverslip were replaced for each concentration tested. For each ASP+ cells, an ROI of the same surface was delineated on the soma in order to measure pixel intensity in arbitrary fluorescence units. 6 ROI were determined at each measurement. Data were background subtracted and ASP+ fluorescence intensity was expressed as a function of initial fluorescence intensity.
In vivo recordings of breathing parameters by plethysmography
Breathing of unrestrained, non-anesthetized mice was recorded using constant air flow whole-body plethysmography filled with air or 4% CO2 in air (EMKA Technologies, Paris, France). Neonatal mice (P0-P1) were recorded in 25 ml chambers (calibrated by injecting 50 µl of air) maintained at neonatal thermoneutral ambient temperature (32 ± 0.5°C). For adolescent and adult mice (P15-P30-P60), four plethysmography 200 ml chambers containing air or (calibrated by injecting 1 ml of air) maintained at 25 ± 0.5°C were used to allow simultaneous measurements. Analog signals were obtained using an usbAMP device equipped with four inputs and processed using EMKA technologies IOX software (EMKA Technologies, Paris, France). For neonatal mice, we measured mean respiratory frequency (Rf, expressed in cycles per minute) during quiet periods when mice breathed air or 5 min after breathing hypercapnic air. For adolescent and adult mice respiratory parameters (frequency, tidal volume, minute ventilation) were recorded over 30 min after an initial 30 min period of stabilization in the apparatus.,Apnea was defined as a prolonged expiratory time (four times eupneic expiratory time), which corresponds to a threshold of 1 s.
Statistical analysis
Analyses were performed using two-tailed non-parametric statistical tools due to the size of the samples (GraphPad, Prism software). Values are indicated as following: (Q2 (Q1, Q3), n; statistical test, p-value) where Q2 is the median, Q1 is the first quartile and Q3 is the third quartile and scatter dot plots report Q2 (Q1, Q3). Histograms report the mean ±SEM. The level of significance was set at a p-value less than 0.05. Appropriate tests were conducted depending on the experiment and are indicated in the figure legends. Mann-Whitney (MW) test was performed to compare two unmatched groups: differences between WT and Ndn-KO (Figure 1 and Figure 2—figure supplement 1). Kolmogorov-Smirnov test was performed to compare the cumulative distribution of two unmatched groups: differences between WT and Ndn-KO in apnea accumulation over time (Figure 3E; Figure 3—figure supplement 1E; Figure 3—figure supplement 2E). Chi-square test was performed to compare two groups of animal (WT and Ndn-KO) with categorical outcome variable (apnea or no apnea) (Figure 3C; Figure 3—figure supplement 2C). Kruskal-Wallis (KW) followed by a post hoc test Dunn test was performed to compare three or more independent groups (Figure 2G,H; Figure 3D,F–H); Friedman test followed by a post hoc test Dunn test was performed to compare matched groups (Figure 4—figure supplement 2C,D). Two-way ANOVA followed by Bonferroni post-hoc test was performed to compare two factors (Figure 2B). Two-way repeated-measure (RM) ANOVA was performed to compare two factors (genotype compared either to time, drug treatment or respiratory challenge) with repeated measure matched by time or respiratory challenge (Figure 3—figure supplement 1D; Figure 3—figure supplement 2D;Figure 4D–F,L and Figure 4—figure supplement 1D); genotype and respiratory challenge. ANCOVA was performed to compare slopes of two regression lines (WT versus Ndn-KO: Figure 2E). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Acknowledgements
We thank Camille Dumon and Magdalena Assael for their technical help and the members of the animal facility, genotyping and imaging platforms of INMED laboratory. We thank Pr Keith Dudley for comments and careful reading of the manuscript. Opinions expressed are those of the authors exclusively. This study has been supported by INSERM, CNRS and ANR (Prageder N° ANR-14-CE13-0025-01) grants. YS was supported by Stiftelsen Frimurare Barnhuset i Stockholm grants and N.K.H. Kronprinsessan Lovisas Forening for barnasjukvard; AB was supported by FPWR fellowship grant.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Valéry Matarazzo, Email: valery.matarazzo@inserm.fr.
Françoise Muscatelli, Email: francoise.muscatelli@inserm.fr.
Joseph G Gleeson, Howard Hughes Medical Institute, The Rockefeller University, United States.
Funding Information
This paper was supported by the following grants:
Institut National de la Santé et de la Recherche Médicale to Valéry Matarazzo, Laura Caccialupi, Fabienne Schaller, Nazim Kourdougli, Alessandra Bertoni, Clément Menuet, Patricia Gaspar, Françoise Muscatelli.
Centre National de la Recherche Scientifique to Laurent Bezin, Pascale Durbec, Gérard Hilaire, Françoise Muscatelli.
Agence Nationale de la Recherche PRAGEDER ANR14-CE13-0025-01 to Valéry Matarazzo, Fabienne Schaller, Yuri Shvarev, Clément Menuet, Nicolas Voituron, Gérard Hilaire, Françoise Muscatelli.
Stiftelsen Frimurare Barnhuset i Stockholm to Yuri Shvarev.
Kronprinsessan Lovisas Forening for Barnasjukvard to Yuri Shvarev.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Supervision, Investigation, Writing—original draft, Project administration, Writing—review and editing.
Conceptualization, Formal analysis, Supervision, Validation, Investigation, Visualization, Methodology, Writing—original draft, Writing—review and editing.
Formal analysis, Validation, Investigation, Visualization, Methodology.
Formal analysis, Validation, Investigation, Visualization, Methodology.
Formal analysis, Investigation, Visualization, Methodology.
Formal analysis, Investigation, Visualization, Methodology.
Investigation, Visualization, Methodology, Writing—review and editing.
Investigation, Visualization, Methodology, Writing—review and editing.
Resources, Investigation, Visualization, Methodology.
Resources, Methodology, Writing—review and editing.
Methodology, Writing—review and editing.
Visualization, Methodology.
Conceptualization, Validation, Visualization.
Conceptualization, Data curation, Formal analysis, Supervision, Funding acquisition, Validation, Investigation, Visualization, Writing—original draft, Project administration, Writing—review and editing.
Ethics
Animal experimentation: Mice were handled and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (N.R.C., 1996) and the European Communities Council Directive of September 22th 2010 (2010/63/EU, 74). Experimental protocols were approved by the Institutional Ethical Committee guidelines for animal research with the accreditation no. B13-055-19 from the French Ministry of Agriculture.
Additional files
References
- Abdala AP, Dutschmann M, Bissonnette JM, Paton JF. Correction of respiratory disorders in a mouse model of Rett syndrome. PNAS. 2010;107:18208–18213. doi: 10.1073/pnas.1012104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebischer J, Sturny R, Andrieu D, Rieusset A, Schaller F, Geib S, Raoul C, Muscatelli F. Necdin protects embryonic motoneurons from programmed cell death. PLoS One. 2011;6:e23764. doi: 10.1371/journal.pone.0023764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieu D, Meziane H, Marly F, Angelats C, Fernandez PA, Muscatelli F. Sensory defects in Necdin deficient mice result from a loss of sensory neurons correlated within an increase of developmental programmed cell death. BMC Developmental Biology. 2006;6:56. doi: 10.1186/1471-213X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieu D, Watrin F, Niinobe M, Yoshikawa K, Muscatelli F, Fernandez PA. Expression of the Prader-Willi gene Necdin during mouse nervous system development correlates with neuronal differentiation and p75NTR expression. Gene Expression Patterns. 2003;3:761–765. doi: 10.1016/S1567-133X(03)00138-8. [DOI] [PubMed] [Google Scholar]
- Arens R, Gozal D, Burrell BC, Bailey SL, Bautista DB, Keens TG, Ward SL. Arousal and cardiorespiratory responses to hypoxia in Prader-Willi syndrome. American Journal of Respiratory and Critical Care Medicine. 1996;153:283–287. doi: 10.1164/ajrccm.153.1.8542130. [DOI] [PubMed] [Google Scholar]
- Barrett KT, Dosumu-Johnson RT, Daubenspeck JA, Brust RD, Kreouzis V, Kim JC, Li A, Dymecki SM, Nattie EE. Partial raphe dysfunction in neurotransmission is sufficient to increase mortality after anoxic exposures in mice at a critical period in postnatal development. Journal of Neuroscience. 2016;36:3943–3953. doi: 10.1523/JNEUROSCI.1796-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner J, Shvarev Y, Zimmer A, Wickstrom R. Hypoxic ventilatory response in Tac1-/- neonatal mice following exposure to opioids. Journal of Applied Physiology. 2012;113:1718–1726. doi: 10.1152/japplphysiol.00188.2012. [DOI] [PubMed] [Google Scholar]
- Bush JR, Wevrick R. Loss of Necdin impairs myosin activation and delays cell polarization. Genesis. 2010;48:540–553. doi: 10.1002/dvg.20658. [DOI] [PubMed] [Google Scholar]
- Butler MG, Manzardo AM, Heinemann J, Loker C, Loker J. Causes of death in Prader-Willi syndrome: Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genetics in Medicine : Official Journal of the American College of Medical Genetics. 2017;19:635–642. doi: 10.1038/gim.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Hamilton J, Narang I. Clinically important age-related differences in sleep related disordered breathing in infants and children with Prader-Willi Syndrome. PLoS One. 2014;9:e101012. doi: 10.1371/journal.pone.0101012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Paterson DS, Hoffman JM, Mokler DJ, Borenstein NS, Belliveau RA, Krous HF, Haas EA, Stanley C, Nattie EE, Trachtenberg FL, Kinney HC. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303:430–437. doi: 10.1001/jama.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MS, Popa N, Djelloul M, Boucraut J, Gauthier P, Bauer S, Matarazzo VA. Alteration of forebrain neurogenesis after cervical spinal cord injury in the adult rat. Frontiers in Neuroscience. 2012;6 doi: 10.3389/fnins.2012.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festen DA, de Weerd AW, van den Bossche RA, Joosten K, Hoeve H, Hokken-Koelega AC. Sleep-related breathing disorders in prepubertal children with Prader-Willi syndrome and effects of growth hormone treatment. The Journal of Clinical Endocrinology & Metabolism. 2006;91:4911–4915. doi: 10.1210/jc.2006-0765. [DOI] [PubMed] [Google Scholar]
- Gillett E, Perez I. Disorders of sleep and ventilatory control in Prader-Willi syndrome. Diseases. 2016;4:23. doi: 10.3390/diseases4030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover ME, Clinton SM. Of rodents and humans: A comparative review of the neurobehavioral effects of early life SSRI exposure in preclinical and clinical research. International Journal of Developmental Neuroscience. 2016;51:50–72. doi: 10.1016/j.ijdevneu.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Arens R, Omlin KJ, Ward SL, Keens TG. Absent peripheral chemosensitivity in Prader-Willi syndrome. Journal of Applied Physiology. 1994;77:2231–2236. doi: 10.1152/jappl.1994.77.5.2231. [DOI] [PubMed] [Google Scholar]
- Gérard M, Hernandez L, Wevrick R, Stewart CL. Disruption of the mouse necdin gene results in early post-natal lethality. Nature Genetics. 1999;23:199–202. doi: 10.1038/13828. [DOI] [PubMed] [Google Scholar]
- Hawthorne AL, Wylie CJ, Landmesser LT, Deneris ES, Silver J. Serotonergic neurons migrate radially through the neuroepithelium by dynamin-mediated somal translocation. Journal of Neuroscience. 2010;30:420–430. doi: 10.1523/JNEUROSCI.2333-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilaire G, Voituron N, Menuet C, Ichiyama RM, Subramanian HH, Dutschmann M. The role of serotonin in respiratory function and dysfunction. Respiratory Physiology & Neurobiology. 2010;174:76–88. doi: 10.1016/j.resp.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Richerson GB. Contributions of 5-HT neurons to respiratory control: neuromodulatory and trophic effects. Respiratory Physiology & Neurobiology. 2008;164:222–232. doi: 10.1016/j.resp.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman JR, Speck DF. Substance P immunoreactive projections to the ventral respiratory group in the rat. Peptides. 1994;15:803–808. doi: 10.1016/0196-9781(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Kachidian P, Poulat P, Marlier L, Privat A. Immunohistochemical evidence for the coexistence of substance P, thyrotropin-releasing hormone, GABA, methionine-enkephalin, and leucin-enkephalin in the serotonergic neurons of the caudal raphe nuclei: a dual labeling in the rat. Journal of Neuroscience Research. 1991;30:521–530. doi: 10.1002/jnr.490300309. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Broadbelt KG, Haynes RL, Rognum IJ, Paterson DS. The serotonergic anatomy of the developing human medulla oblongata: implications for pediatric disorders of homeostasis. Journal of Chemical Neuroanatomy. 2011;41:182–199. doi: 10.1016/j.jchemneu.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima T, Hasegawa K, Yoshikawa K. Necdin promotes tangential migration of neocortical interneurons from basal forebrain. Journal of Neuroscience. 2010;30:3709–3714. doi: 10.1523/JNEUROSCI.5797-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwako K, Hosokawa A, Nishimura I, Uetsuki T, Yamada M, Nada S, Okada M, Yoshikawa K. Disruption of the paternal necdin gene diminishes TrkA signaling for sensory neuron survival. Journal of Neuroscience. 2005;25:7090–7099. doi: 10.1523/JNEUROSCI.2083-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau T, Proissl V, Ziegler J, Schloss P. Visualization of neurotransmitter uptake and release in serotonergic neurons. Journal of Neuroscience Methods. 2015;241:10–17. doi: 10.1016/j.jneumeth.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Lee S, Walker CL, Karten B, Kuny SL, Tennese AA, O'Neill MA, Wevrick R. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Human Molecular Genetics. 2005;14:627–637. doi: 10.1093/hmg/ddi059. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang Y, Zhang Y, Zhu W, Xu X, Niinobe M, Yoshikawa K, Lu C, He C. Nogo-A inhibits necdin-accelerated neurite outgrowth by retaining necdin in the cytoplasm. Molecular and Cellular Neuroscience. 2009;41:51–61. doi: 10.1016/j.mcn.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Maejima T, Masseck OA, Mark MD, Herlitze S. Modulation of firing and synaptic transmission of serotonergic neurons by intrinsic G protein-coupled receptors and ion channels. Frontiers in Integrative Neuroscience. 2013;7:40. doi: 10.3389/fnint.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard SJ, Weston-Green K, Newell KA. The effects of maternal antidepressant use on offspring behaviour and brain development: Implications for risk of neurodevelopmental disorders. Neuroscience & Biobehavioral Reviews. 2017;80:743–765. doi: 10.1016/j.neubiorev.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Miller J, Wagner M. Prader-Willi syndrome and sleep-disordered breathing. Pediatric Annals. 2013;42:e210–e214. doi: 10.3928/00904481-20130924-10. [DOI] [PubMed] [Google Scholar]
- Miller NL, Wevrick R, Mellon PL. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Human Molecular Genetics. 2009;18:248–260. doi: 10.1093/hmg/ddn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouri A, Sasaki A, Watanabe K, Sogawa C, Kitayama S, Mamiya T, Miyamoto Y, Yamada K, Noda Y, Nabeshima T. MAGE-D1 regulates expression of depression-like behavior through serotonin transporter ubiquitylation. Journal of Neuroscience. 2012;32:4562–4580. doi: 10.1523/JNEUROSCI.6458-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P, Cremer H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Human Molecular Genetics. 2000;9:3101–3110. doi: 10.1093/hmg/9.20.3101. [DOI] [PubMed] [Google Scholar]
- Nixon GM, Brouillette RT. Sleep and breathing in Prader-Willi syndrome. Pediatric Pulmonology. 2002;34:209–217. doi: 10.1002/ppul.10152. [DOI] [PubMed] [Google Scholar]
- Oz M, Libby T, Kivell B, Jaligam V, Ramamoorthy S, Shippenberg TS. Real-time, spatially resolved analysis of serotonin transporter activity and regulation using the fluorescent substrate, ASP+ Journal of Neurochemistry. 2010;114:no–1029. doi: 10.1111/j.1471-4159.2010.06828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Ren J, Wevrick R, Greer JJ. Developmental abnormalities of neuronal structure and function in prenatal mice lacking the prader-willi syndrome gene necdin. The American Journal of Pathology. 2005;167:175–191. doi: 10.1016/S0002-9440(10)62964-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Rent J, Wevrick R, Greer JJ. Neurodevelopmental abnormalities in the brainstem of prenatal mice lacking the Prader-Willi syndrome gene Necdin. Advances in Experimental Medicine and Biology. 2008;605:139–143. doi: 10.1007/978-0-387-73693-8_24. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Hilaire G, Weese-Mayer DE. Medullary serotonin defects and respiratory dysfunction in sudden infant death syndrome. Respiratory Physiology & Neurobiology. 2009;168:133–143. doi: 10.1016/j.resp.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavone M, Caldarelli V, Khirani S, Colella M, Ramirez A, Aubertin G, Crinò A, Brioude F, Gastaud F, Beydon N, Boulé M, Giovannini-Chami L, Cutrera R, Fauroux B. Sleep disordered breathing in patients with Prader-Willi syndrome: A multicenter study. Pediatric Pulmonology. 2015;50:1354–1359. doi: 10.1002/ppul.23177. [DOI] [PubMed] [Google Scholar]
- Ptak K, Yamanishi T, Aungst J, Milescu LS, Zhang R, Richerson GB, Smith JC. Raphé neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. Journal of Neuroscience. 2009;29:3720–3737. doi: 10.1523/JNEUROSCI.5271-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Lee S, Pagliardini S, Gérard M, Stewart CL, Greer JJ, Wevrick R. Absence of Ndn, encoding the Prader-Willi syndrome-deleted gene necdin, results in congenital deficiency of central respiratory drive in neonatal mice. Journal of Neuroscience. 2003;23:1569–1573. doi: 10.1523/JNEUROSCI.23-05-01569.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieusset A, Schaller F, Unmehopa U, Matarazzo V, Watrin F, Linke M, Georges B, Bischof J, Dijkstra F, Bloemsma M, Corby S, Michel FJ, Wevrick R, Zechner U, Swaab D, Dudley K, Bezin L, Muscatelli F. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genetics. 2013;9:e1003752. doi: 10.1371/journal.pgen.1003752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rood BD, Calizo LH, Piel D, Spangler ZP, Campbell K, Beck SG. Dorsal raphe serotonin neurons in mice: immature hyperexcitability transitions to adult state during first three postnatal weeks suggesting sensitive period for environmental perturbation. Journal of Neuroscience. 2014;34:4809–4821. doi: 10.1523/JNEUROSCI.1498-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter B, Buschatz D, Trowitzsch E, Aksu F, Andler W. Respiratory control in children with Prader-Willi syndrome. European Journal of Pediatrics. 1997;156:65–68. doi: 10.1007/s004310050555. [DOI] [PubMed] [Google Scholar]
- Scott MM, Krueger KC, Deneris ES. A differentially autoregulated Pet-1 enhancer region is a critical target of the transcriptional cascade that governs serotonin neuron development. Journal of Neuroscience. 2005a;25:2628–2636. doi: 10.1523/JNEUROSCI.4979-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MM, Wylie CJ, Lerch JK, Murphy R, Lobur K, Herlitze S, Jiang W, Conlon RA, Strowbridge BW, Deneris ES. A genetic approach to access serotonin neurons for in vivo and in vitro studies. PNAS. 2005b;102:16472–16477. doi: 10.1073/pnas.0504510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedky K, Bennett DS, Pumariega A. Prader Willi syndrome and obstructive sleep apnea: co-occurrence in the pediatric population. Journal of Clinical Sleep Medicine. 2014;10:403–409. doi: 10.5664/jcsm.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazaki R, Nishimura I, Yoshikawa K. Necdin is required for terminal differentiation and survival of primary dorsal root ganglion neurons. Experimental Cell Research. 2002;277:220–232. doi: 10.1006/excr.2002.5558. [DOI] [PubMed] [Google Scholar]
- Tan HL, Urquhart DS. Respiratory complications in children with Prader Willi syndrome. Paediatric Respiratory Reviews. 2017;22:52–59. doi: 10.1016/j.prrv.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Tennese AA, Gee CB, Wevrick R. Loss of the Prader-Willi syndrome protein necdin causes defective migration, axonal outgrowth, and survival of embryonic sympathetic neurons. Developmental Dynamics. 2008;237:1935–1943. doi: 10.1002/dvdy.21615. [DOI] [PubMed] [Google Scholar]
- Teran FA, Massey CA, Richerson GB. Serotonin neurons and central respiratory chemoreception: where are we now? Progress in Brain Research. 2014;209:207–233. doi: 10.1016/B978-0-444-63274-6.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toward MA, Abdala AP, Knopp SJ, Paton JF, Bissonnette JM. Increasing brain serotonin corrects CO2 chemosensitivity in methyl-CpG-binding protein 2 (Mecp2)-deficient mice. Experimental Physiology. 2013;98:842–849. doi: 10.1113/expphysiol.2012.069872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge S, Narboux-Nême N, Gaspar P. Genetic models of serotonin (5-HT) depletion: what do they tell us about the developmental role of 5-HT? The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology. 2011;294:1615–1623. doi: 10.1002/ar.21248. [DOI] [PubMed] [Google Scholar]
- Urquhart DS, Gulliver T, Williams G, Harris MA, Nyunt O, Suresh S. Central sleep-disordered breathing and the effects of oxygen therapy in infants with Prader-Willi syndrome. Archives of Disease in Childhood. 2013;98:592–595. doi: 10.1136/archdischild-2012-303441. [DOI] [PubMed] [Google Scholar]
- Voituron N, Shvarev Y, Menuet C, Bevengut M, Fasano C, Vigneault E, El Mestikawy S, Hilaire G. Fluoxetine treatment abolishes the in vitro respiratory response to acidosis in neonatal mice. PLoS One. 2010;5:e13644. doi: 10.1371/journal.pone.0013644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella S, Watrin F, Mebarek S, Marly F, Roussel M, Gire C, Diene G, Tauber M, Muscatelli F, Hilaire G. Necdin plays a role in the serotonergic modulation of the mouse respiratory network: implication for Prader-Willi syndrome. Journal of Neuroscience. 2008;28:1745–1755. doi: 10.1523/JNEUROSCI.4334-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. Journal of Neuroscience Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]