

ABSTRACT
Introduction: Although known as cytolytic viruses, group B coxackieviruses (CVB) are able to establish a persistent infection in vitro and in vivo. Viral persistence has been reported as a key mechanism in the pathogenesis of CVB-associated chronic diseases such as type 1 diabetes (T1D). The impact of CVB4 persistence on human pancreas ductal-like cells was investigated. Methods: A persistent CVB4 infection was established in ductal-like cells. PDX-1 expression, resistance to CVB4-induced lysis and CAR expression were evaluated. The profile of cellular microRNAs (miRNAs) was investigated through miRNA-sequencing. Viral phenotypic changes were examined, and genomic modifications were assessed by sequencing of the viral genome. Results: The CVB4 persistence in ductal-like cells was productive, with continuous release of infectious particles. Persistently infected cells displayed a resistance to CVB4-induced lysis upon superinfection and expression of PDX-1 and CAR was decreased. These changes were maintained even after virus clearance. The patterns of cellular miRNA expression in mock-infected and in CVB4-persistently infected ductal-like cells were clearly different. The persistent infection-derived virus (PIDV) was still able to induce cytopathic effect but its plaques were smaller than the parental virus. Several mutations appeared in various PIDV genome regions, but amino acid substitutions did not affect the predicted site of interaction with CAR. Conclusion: Cellular and viral changes occur during persistent infection of human pancreas ductal-like cells with CVB4. The persistence of cellular changes even after virus clearance supports the hypothesis of a long-lasting impact of persistent CVB infection on the cells.
KEYWORDS: CAR, Coxsackievirus B4, miRNA, pancreatic cells, PDX-1, persistence
Introduction
type B coxsackieviruses (CVB) are small non-envelopped RNA viruses that belong to the Picornaviridae family and the Enterovirus genus. This highly diverse genus currently includes 7 species that are involved in human diseases, and CVB (1–6) are classified in the Enterovirus-B species.1,2 CVB can cause severe acute clinical features such as meningitis, encephalitis, myocarditis, pancreatitis, hepatitis or fulminant sepsis in newborns 2,3 but these viruses are also reported to play a role in the development of chronic diseases like dilated cardiomyopathy or type 1 diabetes (T1D).4,5
In T1D patients, the infection of pancreatic cells with enteroviruses, especially CVB, has been reported.6-9 The pancreas is well known as one of the major targets of CVB. This tropism is supported by the expression of CAR, the specific receptor of CVB in the pancreas.10,11 In humans, pancreatic islets and especially β cells,6,7,12 and rarely exocrine cells13,14 were found to be susceptible to enteroviral infection.
CVB can effectively induce lytic infection in pancreatic cells.15-17 However this scenario of acute massive infection is not compatible with T1D which is an autoimmune disease with a progressive evolution, and the implication of the virus is believed to rely on the immune response.5 Therefore, the most likely situation would be a persistent CVB infection probably with low grade viral replication. Indeed, CVB are able to establish persistent infection in vitro as well as in vivo.18 Viral persistence has been suggested as a major mechanism in the enteroviral pathogenesis of CVB-related chronic diseases such as T1D.19,20
Previous in vitro studies confirmed that CVB can persist in pancreatic islets. The persistent infection of β cells resulted in the production of significant levels of IFNα,21 and in a disturbance in the function of these cells.22 The persistence of CVB4 in human pancreatic ductal cells resulted in an impaired formation and viability of islet-like cell aggregates.23
Two main types of persistent viral infections have been described in vitro: steady-state infections and carrier-state infections. In steady-state infections, almost all the cells are infected with no lytic replication cycle, whereas in carrier-state culture systems, only a small proportion of cells are involved with a significant virus replication.18,24 Data available on CVB persistence in vitro are compatible with a carrier-state persistent infection.18,25,26
Virus persistence is thought to result from a coevolution between the host cell and the virus, and this supposes changes in some of their characteristics 18
Viral factors during CVB persistence have been studied in several reports. These factors include genomic changes such as mutations with amino acid substitutions,27 a deletion in the 5′ non coding region,28-31 or the persistence as a stable and atypical double-stranded RNA genomic form.32 Among host factors, the role of a reduced expression of receptors 18,33,34 and the cellular activation status have been reported.35-37 Other cellular factors such as miRNAs could also play a role in viral persistence.38 MicroRNAs are a recently discovered class of small non coding RNAs that act via endogenous RNA interference.39,40 An association between miRNAs and non-lytic herpesvirus and hepatitis C virus infection has been reported.41,42 Recent reports support the role of miRNAs in enterovirus pathogenesis.43
An improved understanding of the impact of viral persistence on the host cell and the virus could provide new insights into the enteroviral pathogenesis of T1D.
In this study, we investigated the changes in host cell and virus during persistent CVB4 infection in a model of human pancreatic ductal-like cells.
Results
Persistent CVB4 infection in ductal-like cells
CVB4 infection in Panc-1 cells usually results in acute lysis with cytopathic effect (CPE) observable within a few days, depending of the infection dose (Fig. 1a). When the acute infection step was controlled namely by removing excess virus, cell proliferation gradually resumed and was maintained in the presence of the virus. After a few weeks of subculture (an average of 5 weeks), the morphology of infected cells was quite similar to that of uninfected cells (See Fig. 1a). The persistent infection was productive, and infectious particles were continuously released in the supernatants with a mean viral titer between 3 and 7 logs TCID50/mL (Fig. 1b). The mean amount of intracellular viral RNA was constant between 5 and 6 logs copies/ng of total RNA throughout the follow-up, as shown in Fig. 1c.
Figure 1.
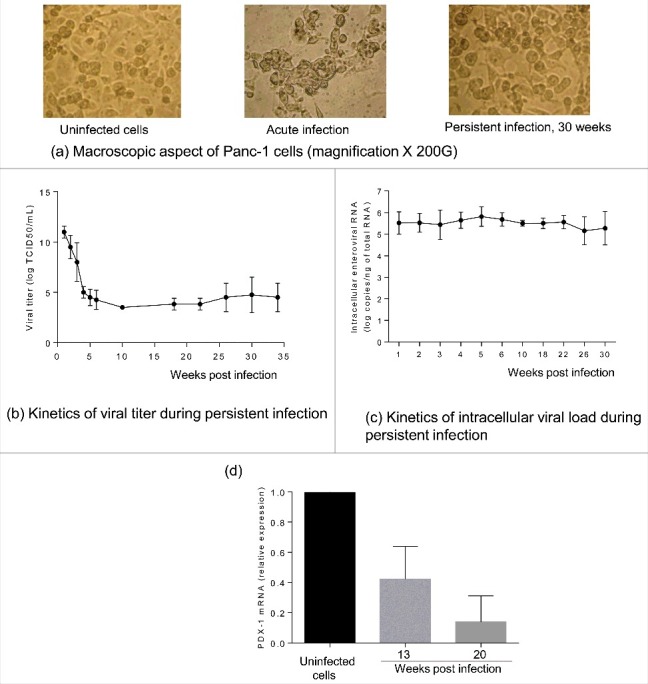
Characterization of persistent CVB4 infection in Panc-1 cells. A persistent CVB4 infection was established in Panc-1 cells. Cells were observed under an inverted microscope (initial magnification X 200) (a). Viral titer in supernatants was determined using end point dilution assay (b). Intracellular viral RNA was quantified using a real-time RT-qPCR (c). PDX-1 mRNA was assessed during persistent infection by using real-time RT-qPCR and expressed as fold-change as compared with uninfected cells (d). Results presented (b-e) are mean +/− SD of 3 independent persistent infections.
In the model of ductal-like cells persistently infected with CVB4 the impact of the infection on the cells was investigated. The expression of PDX-1 mRNA was reported to decline in CVB4 persistently infected cells.23 This observation was confirmed in the current study by using real-time RT-qPCR. The expression of PDX-1 at 20 weeks of chronic infection was around 14%, as compared with uninfected cells (Fig. 1d).
Resistance of CVB4 persistently infected cells to lysis upon superinfection
The impact of superinfection on persistently infected cells was investigated. Uninfected and CVB4 persistently infected cells were infected with CVB4 (from the same stock that was used to establish persistent infection) at a MOI of 10. On day 2 post infection, cell viability remained unchanged in persistently infected cells, while a dramatic decrease was observed in previously uninfected cells (Fig. 2a). The level of infectious particles in supernatants collected at 24h and 72h post infection was determined. The virus replication was highly effective in uninfected cells with a viral titer reaching 1010 TCID50/mL at 72h post infection. In contrast, viral titers before and after superinfection were similar in CVB4 persistently-infected cells, as shown in Fig. 2b. At 72h post infection, cells were harvested, washed and the level of intracellular viral RNA was evaluated by real-time RT-qPCR. Upon infection, intracellular viral RNA in uninfected cells was around 6 log copies/ng of total RNA, compatible with an acute infection. The intracellular viral load remained unchanged after superinfection in CVB4 persistently infected cells (Fig. 2c). These observations suggest that persistent infection protected cells from superinfection. We reasoned that this might be due to reduced expression of CAR, the main receptor used by CVB4. The level of CAR mRNA in persistently-infected cells gradually declined and reached less than 3% at 22 weeks post-infection, compared with uninfected cells (Fig. 3).
Figure 2.
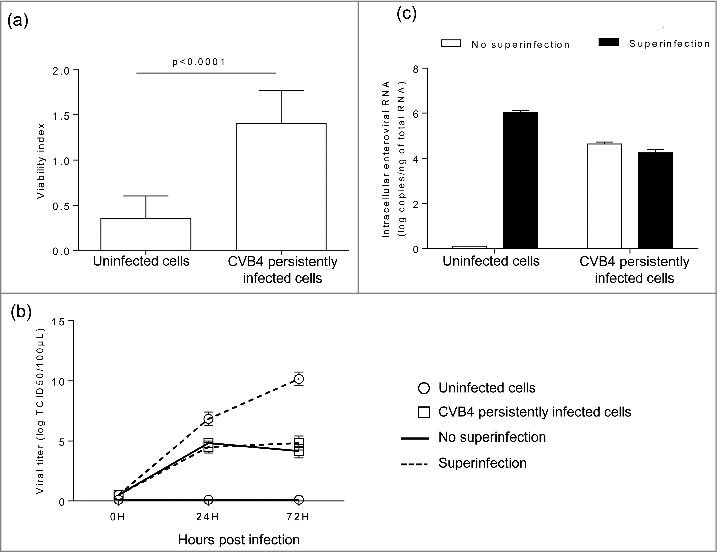
Acute CVB4 superinfection of persistently infected cells did not induce cell lysis. CVB4 and uninfected cells were infected with CVB4 at a MOI of 10. Cell viability was assessed at 48h post infection by using the cristal violet assay (a). Supernatants were collected and viral progeny was determined (b), Cells were harvested, washed, and intracellular viral RNA was quantified by using RT-qPCR (c). Results are mean+/−SD of 3 independent experiments.
Figure 3.
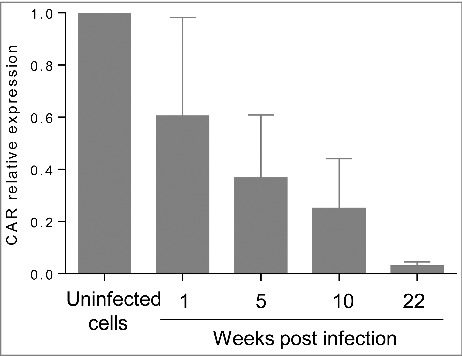
CAR expression is significantly decreased during persistent infection. CAR mRNA was quantified by real-time RT-qPCR during persistent infection. Results are mean+/−SD of 3 independent experiments.
Persistent reduced expression of PDX-1 and CAR in CVB4 persistently infected ductal-like cells after viral clearance
We hypothesized that changes induced in persistently-infected cells could persist after viral clearance. To test this hypothesis, persistent infection was completely cured by fluoxetine, as previously reported.44 One year after the end of treatment we found that PDX-1 expression was still significantly reduced in persistently infected cells that had been cured by fluoxetine (18.2% and 17.8% respectively in untreated and cured cells, compared with uninfected cells) (Fig. 4a). In addition, CVB4 superinfection studies were performed in CVB4 persistently infected cells that were cured by fluoxetine, one year after the end of treatment. There was no cell lysis in cured cells (Fig. 4b); nevertheless the cells were readily infected, as supported by quantity of viral progeny in supernatants (Fig. 4c), and of intracellular viral RNA (Fig. 4d). CAR expression was evaluated in cell cultures cured with fluoxetine. CAR mRNA levels increased significantly in cured cells compared with persistently infected cells (65% versus 3.8% of the expression in uninfected cells, p < 0.0001) (Fig. 4e). However, the membrane expression of CAR on infected and cured cells remained similar (Fig. 4f).
Figure 4.
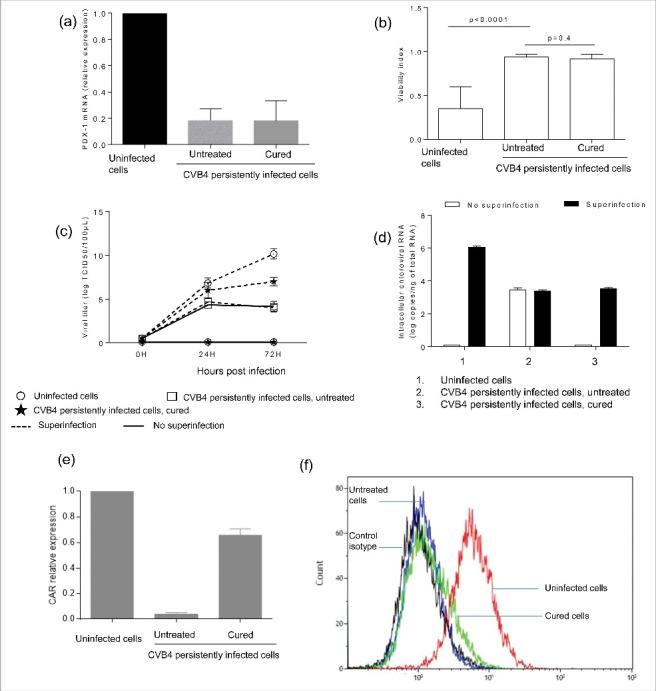
Persistence of changes induced in CVB4 persistently infected cells after virus clearance. CVB4 persistent infection was cured using fluoxetine, and then PDX-1 mRNA expression was evaluated in CVB4 peristently infected cells that were cured (a). Cured cells and untreated cells were infected with CVB4 at a MOI of 10. Cell viability (b), viral progeny (c), and intracellular viral RNA (d) were investigated. CAR mRNA was quantified by real-time RT-qPCR (e). The membrane expression of CAR was evaluated by flow cytometry (f). Results are mean+/−SD of 3 independent experiments, and one representative experiment is shown for flow cytometry.
Pattern of cellular miRNAs in ductal-like cells persistently infected with CV-B4
The expression of miRNAs in CVB4 persistently infected and uninfected cells was evaluated through miRNA sequencing. The miRNA profile was clearly different in persistently infected cells, as compared with uninfected cells (Fig. 5a). A total of 81 miRNAs displayed a significant differential expression (fold change ≥ 3, p< 0.05) including 65 upregulated and 16 downregulated miRNAs (Fig. 5b). The expression pattern determined by miRNA sequencing was confirmed by taqman RT-qPCR for 2 miRNAs that were described in the literature to be related to enteroviral infection: miR-146a-5p (upregulated) and miR-23b (downregulated) 45-48 (Fig. 5c–d). The levels of miR-146a-5p and miR-23b in ductal-like cells acutely infected with CV-B4 were also determined. These miRNAs were upregulated and downregulated respectively in ductal-like cells persistently infected with CVB4 whereas their levels in cells acutely infected harvested on day 2 p.i were unchanged compared with uninfected cells (see Fig. 5d).
Figure 5.
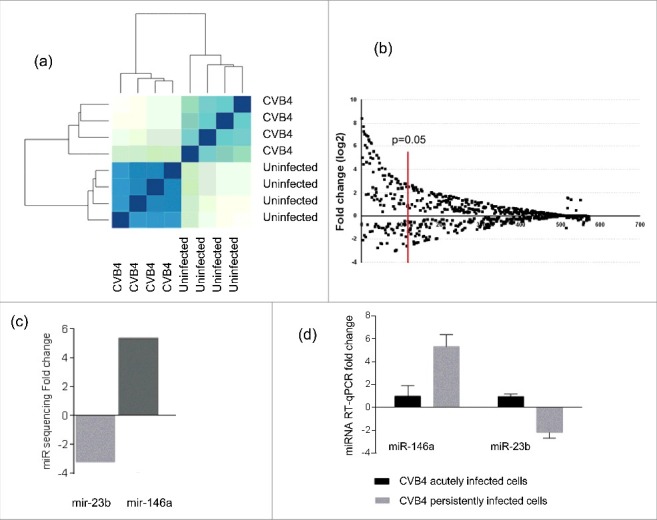
Cellular microRNA profile during CVB4 persistent infection. MiRNA sequencing was performed on CVB4 and uninfected Panc-1 cells. The profile is compared between CVB4 and uninfected cells (a). MiRNAs with a fold change ≥ 3 and p<0.05 were considered as differentially expressed (b). The fold-change of miR-146a and miR-23b expression in persistently infected cells determined by miR-sequencing is shown (c). Taqman RT-qPCR was used to to quantify miR-146a and miR-23b in CVB4 acutely and persistently infected cells. RT-qPCR results are mean +/− SD of 3 independent experiments (d).
Three miRNAs were selected for further study, including the 2 previous and miR-138–5p that was predicted to target the 3′ non coding region of CVB4-E2 using RegRNA 2.0 software (with default settings). The transfection of mimics of these 3 miRNAs (Fig. 6a–c) had no impact on viral replication (Fig. 6d–e).
Figure 6.
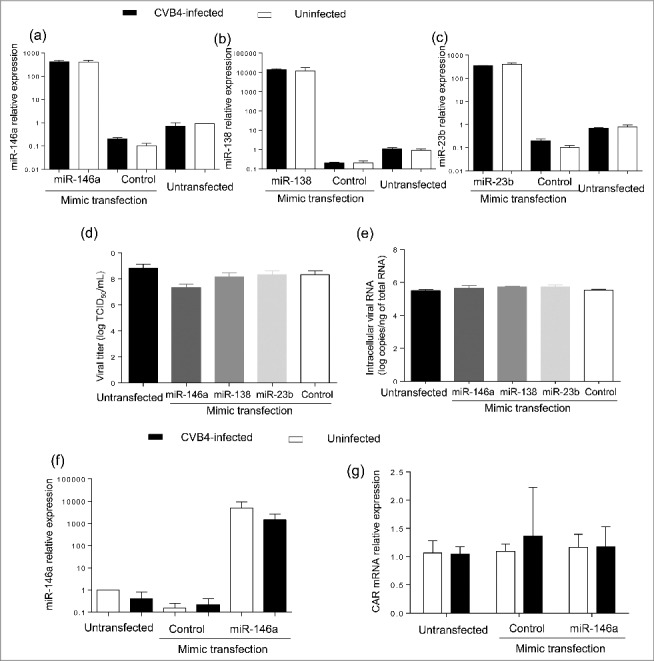
Impact of miRNA mimic transfection on virus replication and CAR mRNA expression in Panc-1 cells. Panc-1 cells were transfected with miR-146a, miR-138 and miR-23b mimics at 20nM, and then infected with with CVB4 at MOI of 1, 24h after transfection. Levels of MiRNAs were quantified in cells (a-c). Viral titers in supernatants (d) and intracellular viral RNA levels (e) were determined at 48h post infection. MiR-146a mimic was transfected in Panc-1 cells at 50 nM, which were subsequently inoculated with CVB4 at MOI of 0.01. The expression of miR-146a (f) and CAR mRNA (g) was assessed in cells. Results are mean+/−SD of 3 independent experiments.
MiR-146a-5p was predicted to target CAR gene using miRWalk 2.0 software (with default settings). The transfection of miR-146a mimic (Fig. 6f) did not impact CAR mRNA expression (Fig. 6g).
Characterization of persistent infection-derived CVB4
The persistent infection of ductal-like cells cultures resulted in cellular modifications. We wondered whether changes affecting the virus also occurred during the persistent infection. Similarly to the parental virus, the persistent infection-derived virus (PIDV) can induce cytopathic effect (CPE) in several cell lines such as HEp-2, Panc-1 or RD cells (data not shown). The aspect of plaques induced on HEp-2 cells by the PIDV was compared with the parental virus. The virus emerging from persistent infection induced only tiny plaques on HEp-2 cells, while the parental virus was able to produce larger size plaques (Fig. 7a). The levels of infectious particles and viral RNA of PIDV and parental virus suspension were determined. The proportion of infectious particles, as evaluated by the ratio between viral titer and the viral RNA load, was lower in PIDV compared with parental virus (p = 0.01) (Fig. 7b).
Figure 7.
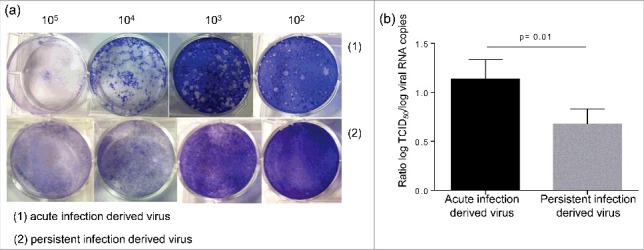
Characterization of persistent infection derived virus. The aspect of the plaque induced by the persistent infection derived virus and the parental virus were compared (a). The ratio between the viral titer and the viral RNA load was evaluated for both viruses (b).
Thereafter, whole viral genome deep sequencing was performed on both parental virus and PIDV, using 2 independent acute and persistent infections. A total of 105 cumulated mutations were observed in various regions of PIDV (as compared with parental virus), with 40 amino-acid substitutions.VP1 and 2A proteins were more commonly affected, with 9 and 11 amino-acid substitutions respectively. Only one silent mutation (A1854G) was shared by viruses derived from 2 independent cultures of ductal-like cells persistently infected with CVB4 E2.
Recently it was reported that amino acid residues highly conserved among type B coxsackieviruses located in the north rim of the canyon (VP1) and in the puff region (VP2), represent the footprints of CVB3 on CAR.49
To infestigate whether changes had occurred in these regions, mutations in PIDV with a frequency ≥ 20% were integrated in silico in the nucleotide sequence of the CVB4 E2 reference strain. Amino acid sequences were obtained by using blastx (NCBI). VP1 and VP2 sequences were aligned with sequences corresponding to CVB3/28 and CVB3/Nancy strains, using Uniprot software. Fig. 8 shows that the footprints of CVB4 E2 on CAR were conserved in PIDV. In addition, a modeling of interaction between CAR and VP1, based on residues described for CVB3 showed similar patterns for both the initial stock virus and PIDV (see Fig. 9).
Figure 8.

CVB4 footprints on CAR during persistent infection in Panc-1 cells. Mutations with a frequency ≥ 20% obtained by deep sequencing of 2 CVB4 independent acute infections and 2 independent persistent infections in Panc-1 cells, were integrated in the sequence of the CVB4E2 published strain (Accession number: AF311939.1). Translated amino acid sequences were obtained using blastx (NCBI). VP1(a) and VP2(b) sequences were aligned along with sequences corresponding to CVB3/28 and CVB3/Nancy strains, using Uniprot software. Footprints of the virus on CAR are shown in blue boxes.
Figure 9.
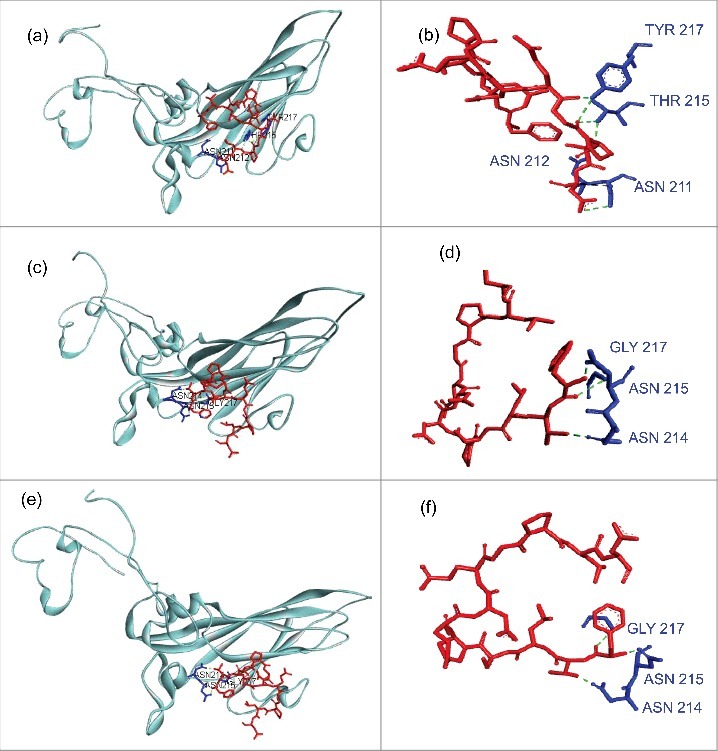
Modeling of interaction between CAR and Coxsackievirus B VP1 north canyon rim. (a-b) Interaction between CAR and Coxsackievirus B3. VP1 model (PDB:1COV) was used. CAR residues that represent the virus footprint (as described by Organtini et al., 2014) have been modeled using CAR D1 domain (PDB:1F5W) as template and are shown in red. VP1 north canyon rim residues are shown in blue. Only Hydrogen bonds that link CAR to a VP1 amino-acid involved in CAR recognition (Organtini et al., 2014) are displayed. Bounds were observed with ASN 211, ASN 212, THR 215 and TYR 217. (c-d) Interaction between CAR and Coxsackievirus B4 E2. The CVB4 E2 strain used in our laboratory (stock virus) was sequenced on a Ion PGM™ deep sequencing platform, and mutations with a prevalence higher that 20% were introduced in the CVB4 E2 published full genome sequence (AF311939.1). The VP1 model was built using PDB:1COV as template. Bounds were observed with 3 VP1 amino-acids (ASN 214, ASN 215, GLY 217) including 2 residues observed in CVB3 model (ASN 214, ASN 215 that correspond to ASN 211 and ASN 212 in CVB3 reading frame). (e-f) Interaction between CAR and Coxsackievirus B4 derived from persistent infection in Panc-1 cells (PIDV). PIDV sequence was determined as described above. The VP1 model was built using PDB:1COV as template. Bounds were observed with the same residues (ASN 214, ASN 215, GLY 217) as compared with the initial stock virus. Nucleotides sequences were translated in proteins using BlastX (NCBI). Models were created using Swiss model (https://swissmodel.expasy.org/). Docking data were obtained from Zdock server (zdock.umassmed.edu/) and interactions investigated using Biova/Discovery Studio 2016 software (Accelry Inc.). Conventional hydrogen bounds are shown in green dotted and carbon hydrogen bound in black dotted.
Discussion
The persistence of cytolytic viruses such as enteroviruses may be viewed as the result of host immune pressure, or as a pathogenesis mechanism. Group B coxsackieviruses associated chronic diseases such as T1D have been linked at least partially to virus persistence. In T1D, primary target cells are β cells within the pancreatic islets, but evidence of virus detection in others cell populations such as pancreatic ductal cells, has also been reported.9 The role of pancreatic ductal cells in the homeostasis of pancreas is important since they are thought to contribute to the replenishment of β cells through transdifferentiation.50
Interestingly, CVB4 persistence in pancreatic ductal cells was previously reported to induce a decrease of PDX1, also known as insulin promoter factor 1, which is a transcription factor necessary for pancreatic development, including β cells maturation, and duodenal differentiation.23 In this study we confirmed this finding using real-time RT-qPCR. Moreover we have observed several changes in ductal-like cells persistently infected with CVB4.
The persistently infected cells displayed a resistance to lysis when exposed to a superinfection with CVB4. In addition, viral progeny and intracellular RNA remained unchanged after superinfection, suggesting a lack of entry or a limited entry of the challenging virus in these cells. Reduced virus entry could be explained by a decrease of cell susceptibility, as supported by the significant decrease of the CAR molecule in cells during CVB4 persistent infection. Our results are in agreement with previous reports. Indeed, resistance to lysis associated with a decrease of CAR expression was found during CVB3 persistence in a murine cardiac myocyte cell line.18
The persistently infected cells were completely cured by using fluoxetine, and then subcultured for one year. The expression of PDX-1 was still reduced in cured cells at similar levels than in untreated cells. In addition, fluoxetine cured cells conserved the resistance to CVB4-induced lysis observed in persistently infected cell cultures. A similar observation was reported in CVB3 chronically-infected murine myocytes that were cured using soluble CAR.18
It is noteworthy that the level of CAR mRNA increased significantly in cured cells whereas membrane expression did not change. Indeed CAR was barely detectable at the membrane of cured or untreated CVB4 persistently infected cells. This discrepancy between mRNA and membrane expression may be explained by the fact that flow cytometry evaluated CAR membrane isoforms only while mRNA levels reflect the expression of both membrane and solubles isoforms.51,52 It cannot be excluded that the expression of CAR soluble isoforms, but not of membrane isoforms, increased in cured cells in our experiments.
Altogether, observations in cured and untreated persistently infected cells suggest that the resistant phenotype was associated with a decrease in membrane CAR expression; however cured cells remained susceptible to CVB4 infection. This resistance phenotype probably involves many other factors such as reprogramming or epigenetic changes, and deserves further investigation.
To the best of our knowledge, this is the first observation of an impact of enteroviral persistence onto the cellular miRNAs.
In this study, the patterns of cellular miRNAs in mock-infected and in CVB4- persistently infected ductal-like cells were clearly different. The technical approach is noteworthy. Indeed miRNA sequencing was performed instead of analyzing a predefined list of miRNAs. This method yielded a high number of miRNAs with significant differential expression (65 upregulated and 16 downregulated). In addition, our results suggest that miRNA expression pattern is different between CVB4 acute and persistent infection. Recently Kim et al. investigated the expression of cellular miRNAs in human pancreatic islets acutely infected with CVB5. The authors reported on day 7 post infection, a modified expression for 33 miRNAs (including 6 upregulated and 27 downregulated) out of 754 analyzed miRNAs.53 MiR-663b was the only one of our list that was reported in their study.
We have investigated the impact on CVB4 replication of 3 miRNAs (miR-138, miR-23b and miR-146a-5p) potentially playing a role in enterovirus infection. The transfection of miRNAs mimics in ductal-like cells did not induce any change in viral titers determined in supernatants and in intracellular enteroviral RNA levels. Furthermore, miR-146a was predicted to potentially target CAR, and its impact on CAR expression was evaluated. However, in cells transfected with miR-146a mimic (infected with CVB4 or not) the expression of CAR was not different compared with non-transfected cells. It cannot be excluded that the selected miRNAs have other effects than those evaluated in this study, e.g. they can promote antiviral responses or at the opposite participate in the escape to the immune response.43
The decrease of CAR expression during chronic infection may result from the combined effect of several miRNAs or from other epigenetic changes.
It is well admitted that persistence results from adaptations of both host cell and virus.24 We therefore analyzed some features of the virus emerging from persistent infection, compared with the parental strain. Both viruses were able to induce a cytopathic effect on several cell lines. However the plaques induced on HEp-2 cells by the persistent infection derived virus (PIDV) were smaller than those induced by the parental strain. This phenotypic change was previously observed during CVB3 persistence in murine cardiac myocyte cell line.18
In addition, the proportion of infectious particles compared with the levels of viral RNA was lower in PIDV, than in parental strain, which may be explained by genomic changes resulting in an impaired replication of the virus.
We report in this study for the first time, a whole genome deep sequencing analysis of an enterovirus emerging from persistent infection. As compared with the parental strain, several mutations appeared throughout the viral genome during the persistence of CVB4 in ductal-like cells. The mutations with amino-acid substitutions affected mainly VP1, VP2, 3D, 2A and 2C regions. Whether these mutations play a role in viral adaptation or phenotypic changes requires further investigations.
In previous studies various regions of enterovirus genome were analyzed, but no consistent mutation has been reported to be associated with enterovirus persistence.27,54-57 However, another team focused on the 5′ non coding region and observed deletions with size ranging from 7 to 49 nucleotides. These terminally-deleted persistent variants were not able to induce CPE, and have been observed in vitro as well as in vivo.28-31 Genome sequencing did not reveal such variants in our system, in agreement with the fact that PIDV was able to induce a CPE in our experiments.
Furthermore, we demonstrated that the amino-acid changes occurring during persistence in VP1 and VP2 viral capsid proteins did not alter CAR footprints on the virus. This finding suggests that CVB4 can replicate in a CAR-restricted environment, but still maintains its interaction properties with CAR. However the efficiency of PIDV binding to CAR requires additional studies. It cannot be excluded that alternative receptors are used by PIDV. In a previous report, Schmidtke et al. sequenced the genome of a CVB3 strain emerging from persistent infection in a low CAR – expressing fibroblast cell line. This strain exerted higher lytic properties than the parental virus, although it exhibited reduced CAR-binding properties. As a matter of fact the virus emerging from persistent infection acquired the ability to recognize additional receptors, and 4 VP1 amino-acid substitutions were identified to be associated with this phenotype.27
In conclusion, cellular and viral changes occurring during CVB4 persistence in pancreatic ductal-like cells have been investigated. Persistently-infected cells gained a sustained resistance to CVB4 induced cell lysis. CAR expression was reduced and the miRNA profile was modified. Phenotypic and genotypic changes of the virus were also observed reflecting virus adaptation during the persistence. The persistence of CVB in pancreatic cells induced changes that were maintained even after virus clearance. Further investigations are needed to elucidate the mechanisms of the persistence of CVB4 in pancreatic cells and of the sustained impact of the virus on these cells.
Material and methods
Cells and virus
The human ductal cell line Panc-1 (ATCC) was cultured in Dulbecco's modified Eagle's medium supplemented with 10% of heat inactivated fetal calf serum (FCS), 1% of L-glutamine and 1% of penicillin and streptomycin. HEp-2 cells (BioWhittaker) were grown in minimum essential medium supplemented with 10% of FCS, 1% of L-glutamine, 1% of non-essential amino acids and 1% of penicillin and streptomycin. HEp-2 cells were used for the production and titration of the virus.
The diabetogenic CVB4 E2 strain kindly provided by Ji-Won Yoon (Julia McFarlane Diabetes Research Center, Calgary, Alberta, Canada) was propagated in HEp-2 cells. Briefly, after 3 freeze-thaw cycles, the suspension was collected and clarified at 2000 g for 10 min at 4°C. Aliquots of virus preparations were stored at −80°C. The CVB4 E2 strain was isolated by Yoon et al. from the pancreas of a child with recently diagnosed type 1 diabetes and is diabetogenic in mice.58
Persistent CVB4 infection in Panc-1 cells
The persistent infection in Panc-1 cells was established as described previously 23 with slight modifications. Briefly, a 25 cm2 Nunc® cell culture flask (Thermofisher Scientific, Villebon, France) containing an average of 106 cells was inoculated with CVB4E2 at a multiple of infection (MOI) of 0.01. During the acute lytic infection, the culture medium was regularly changed, and finally a stable equilibrium was found between viral replication and cell proliferation. The cells were scraped and subcultured once a week. Supernatants and cells were periodically collected for analysis. CVB4 and mock-infected cells were processed identically. Persistently infected cells can be frozen and stored in liquid nitrogen, with maintenance of infection after thawing.
Viral progeny in supernatants
The viral titer in supernatants of infected cells was assessed using the end-point dilution assay, and the Spearman-Karber statistical method was used to determine the tissue culture 50% infectious dose (TCID50).
Virus plaque assay
HEp-2 cells were cultured in a 6-well culture plate as confluent monolayers at a density of 106 cells/well. After 24 h, medium was removed and cells were washed once with PBS and overlaid with 3 mL of dilutions of the virus suspension, and then incubated at 37°C for 2 h. After removal of the supernatant, cells were overlaid with 3 ml of agar containing Eagle's minimal essential medium (MEM).Three days later, the cells were fixed with 10% PFA for 2 h. After removal of PFA and agar, cells were stained with 1% cristal violet for 30 minutes. After washing and drying the plaques were observed and counted.
Enterovirus RNA quantification
Total RNA was extracted from Panc-1 cells using TriReagent® RNA isolation reagent/ Chloroform procedure (Sigma). Extracted RNA was then dissolved in 50 µL of nuclease free water, quantified with a Nanodrop® spectrophotometer (Thermofisher Scientific).
The Affinityscript® QPCR cDNA Synthesis kit (Agilent technologies, Les Ulis, France) was used for the ARN retrotranscription step on a Perkin Elmer 2400 thermocycler (Villebon-sur-Yvette, France). Quantitative real-time PCR for cDNA amplification was performed with the Brilliant® II QPCR kit (Agilent technologies) on the Mx3000p® instrument (Agilent technologies). Enterovirus 71 RNA (Vircell, Granada, Spain) was used as standard for quantification. The oligonucleotides and the reaction conditions were described previously.59
Cell viability assay
The cell viability was assessed using the crystal violet assay. Cells seeded in a 96-well plate were washed once with PBS and incubated with crystal violet at 37°C for 20 minutes. Then, crystal violet was removed and cells layers were extensively washed in tap water by immersion in a large beaker. The dissolution of crystal violet remaining inside cells was performed by incubation with 1% SDS at 37°C for 15 minutes. Upon solubilization, the absorbance in each well was read at 595 nm.
Fluoxetine treatment
The treatment of persistently infected cells was performed as described previously.44 Briefly Panc-1 cells persistently infected with CVB4 were treated twice a week with fluoxetine at 5.5µM, and virus was completely cleared within 3 weeks.
PDX-1 and CAR gene expression
RNA extracts were treated with DNAse I (Qiagen, Les Ulis, France). The quantification of PDX-1 mRNA and GAPDH mRNA (house-keeping gene) was performed by using the Taqman gene expression assays (hs00236830 and hs03929097, respectively) (Thermofisher Scientific). The cDNA synthesis was performed with the High-capacity cDNA reverse transcription kit (Thermofisher Scientific), and the Taqman Universal Master Mix II, no UNG (Thermofisher Scientific) was used for real-time qPCR according to the manufacturer's instructions. CAR mRNA was quantified by real-time RT-qPCR by using the SuperScript III Platinum One-Step Quantitative RT-PCR System (Thermofisher Scientific) and oligonucleotides described by Lam and colleagues.60 The quantification was performed on the Mx3000p® instrument (Agilent technologies) with the following program: 50°C for 15 minutes, 95°C for 2 minutes, and 40 cycles of amplification consisting of 15 seconds at 95°C and 30 seconds at 60°C. The expression of the β-actin mRNA was used for normalization.
PDX-1 and CAR mRNA relative expression in infected vs. noninfected cells was determined with the 2−ΔΔCt formula.61
Flow cytometry
Anti-human CAR mouse IgG1 monoclonal antibody (clone RmcB) and negative control mouse IgG1 monoclonal antibody were purchased from Merck Millipore (Molsheim, France) and used at the supplier's recommended concentrations. Both antibodies were phycoerythrin (PE) – conjugated. Panc-1 cells were detached by trypsinization, washed once in PBS and suspended in FACS Buffer (PBS, 1% BSA). Cell suspensions were incubated at 4°C for 30 min with the appropriate antibodies, and washed 3 times with FACS Buffer. The cells were then fixed in 4% paraformaldehyde, washed once with PBS, and analyzed by flow cytometry on a Navios Flow cytometer (Beckman Coulter, Inc.).
Sequencing of microRNAs
Cells were collected from 4 replicates of CVB4 and mock persistent infection, and washed with PBS. RNA extraction was performed using the miRNeasy mini kit (Qiagen) with a step of on-column DNA digestion using DNase I (Qiagen). Extracted RNA was quantified with a Nanodrop® spectrophotometer (Thermofisher) and the quality was assessed using the 2100 Bioanalyzer (Agilent technologies).
Small RNA librairies were prepared from 1µg of total RNA using the Ion Total RNA-Seq Kit v2.0 (Life Technologies, Carlsbad, CA, USA). Barcoded libraries were quantified and assessed for quality using the Agilent 2100 BioAnalyzer (Agilent Technologies). Libraries were pooled in equimolar amounts and sequenced on a Ion PROTON™ Platform using a Ion P1™ Chip Kit v2 and the Ion P1™ Sequencing 200 kit v3 (Life Technologies).
Primary analysis transforming signal to DNA sequences was done with the default parameters on a Torrent Server 4.0.2 (Life Technologies). Demultiplexing was done with 0 errors allowed in barcodes. Raw reads were analyzed with ncProSeq 1.5.1.62 For each sample, reproducibility of counts within the 2 barcodes was investigated by Spearman correlation. Each pair of barcodes were then pooled and a new ncProSeq analysis was performed on all samples. The differential Expression of the raw counts obtained from ncProSeq was performed with DESeq2.63
Quantification of miRNAs by RT-PCR
The expression of miRNAs was quantified using the TaqMan® MicroRNA kits (Thermofisher Scientific). The Taqman MicroRNA Reverse Transcription Kit was used for cDNA synthesis, and the real-time qPCR was performed with the Taqman Small RNA assay (primers and probe) and the Taqman Universal PCR Master Mix II according to the manufacturer recommendations, on the Mx3000p® thermocycler (Agilent technologies). RNU6B was used for normalization and the relative expression was determined using the 2−ΔΔCt formula.61
Transfection of miRNA mimics/inhibitors
Panc-1 cells were seeded at 0.5×105 cells in 0.5 mL complete medium per well in a 24-well plate at 70% confluence. After 24 h, the cells were transfected with specific mirVana™ miRNA mimic, miRNA inhibitor or negative controls (Thermofisher Scientific), by using Lipofectamine® RNAiMAX Transfection Reagent (Thermofisher Scientific). After 24h, the transfected or non-transfected cells were then inoculated with CVB4 at MOI 0.01. The virus was removed after 2h by washing 3 times and fresh medium was added. The plate was incubated at 37°C, 5% CO2.
CVB4E2 genome sequencing
Full genome sequencing was performed on virus suspensions resulting from acute and persistent CVB4E2 infection. 2 mL of virus suspension was concentrated 10-fold by using a Vivaspin 6 device (Sartorius, Dourdan, France) with a membrane molecular weight cut-off at 1,000,000 Dalton. The resulting 200µL were RNAse treated and then used for automated RNA extraction on the Magtration System 12GC with the MagDEA Viral DNA/RNA 200 reagents (Precision System Science Co, Ltd, Japan), with a DNase treatment step. cDNA synthesis was performed with the SuperScript III first-strand synthesis system (Invitrogen) and random hexamer primers. The synthesized cDNA was amplified by Multiple Displacement Amplification technology (MDA) following the protocol of the Quantitect Whole transcriptome kit (Qiagen). The amplified cDNA was used to prepare barcoded librairies by using the Ion Xpress Plus Fragment library kit (Thermofisher). After quality assessement and quantification on the Agilent 2100 BioAnalyzer (Agilent), librairies were pooled in equimolar amounts and sequenced on a Ion PGM™ Platform using a Ion 318™ Chip Kit v2 and the Ion PGM™ HiQ sequencing kit (Life Technologies).
The raw sequences were obtained from the intensities on the Torrent-Server software version 5.0.4 (Life Technologies) with no trimming and default calibration. Demultiplexing was performed by allowing one error by index. Alignment on CVB4E2 was made with the tmap tool with default settings for Torrent Server 5.0.4. The detection of variants was performed with the TSVC tool from Torrent 5.0.4 Server with parameters “Somatic-Low-stringency.”
Statistical analysis
Data are presented as the mean ± the standard deviation. Comparisons were performed using Student t or Mann Whitney U tests, as appropriate. P < 0.05 was considered statistically significant. All analyses were performed with Graph Pad Prism 6.03 software (GraphPad Software, La Jolla, CA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Jacques Trauet for technical assistance.
Funding
This work was supported by Ministère de l'Education Nationale de l'Enseignement Supérieur et de la Recherche, Université Lille 2 (Equipe d'accueil 3610) and Center Hospitalier Régional et Universitaire de Lille, and by EU FP7 (GA-261441-PEVNET : Persistent virus infection as a cause of pathogenic inflammation in type 1 diabetes - an innovative research program of biobanks and expertise).
References
- [1].Knowles N, Hovi T, Hyypiä T. Picornaviridae. In: Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses San Diego: King, A.M.Q., Adams, M.J., Carstens, E.B. and Lefkowitz, E.J; 2012. page 855-80. [Google Scholar]
- [2].Tapparel C, Siegrist F, Petty TJ, Kaiser L. Picornavirus and enterovirus diversity with associated human diseases. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis 2013; 14:282-93; http://dx.doi.org/ 10.1016/j.meegid.2012.10.016 [DOI] [PubMed] [Google Scholar]
- [3].Romero JR. Pediatric group B coxsackievirus infections. Curr Top Microbiol Immunol 2008; 323:223-39; PMID:18357772 [DOI] [PubMed] [Google Scholar]
- [4].Hober D, Alidjinou EK. Enteroviral pathogenesis of type 1 diabetes: queries and answers. Curr Opin Infect Dis 2013; 26:263-9; PMID:23549392; http://dx.doi.org/ 10.1097/QCO.0b013e3283608300 [DOI] [PubMed] [Google Scholar]
- [5].Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol 2010; 6:279-89; PMID:20351698; http://dx.doi.org/ 10.1038/nrendo.2010.27 [DOI] [PubMed] [Google Scholar]
- [6].Dotta F, Censini S, van Halteren AGS, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Del Prato S, et al.. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A 2007; 104:5115-20; PMID:17360338; http://dx.doi.org/ 10.1073/pnas.0700442104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009; 52:1143-51; PMID:19266182; http://dx.doi.org/ 10.1007/s00125-009-1276-0 [DOI] [PubMed] [Google Scholar]
- [8].Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia 2011; 54:2417-20; PMID:21597997; http://dx.doi.org/ 10.1007/s00125-011-2192-7 [DOI] [PubMed] [Google Scholar]
- [9].Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, Roivainen M. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 2004; 47:225-39; PMID:14727023; http://dx.doi.org/ 10.1007/s00125-003-1297-z [DOI] [PubMed] [Google Scholar]
- [10].Oikarinen M, Tauriainen S, Honkanen T, Vuori K, Karhunen P, Vasama-Nolvi C, Oikarinen S, Verbeke C, Blair GE, Rantala I, et al.. Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia 2008; 51:1796-802; PMID:18696046; http://dx.doi.org/ 10.1007/s00125-008-1107-8 [DOI] [PubMed] [Google Scholar]
- [11].Spagnuolo I, Patti A, Sebastiani G, Nigi L, Dotta F. The case for virus-induced type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 2013; 20:292-8; PMID:23743646; http://dx.doi.org/ 10.1097/MED.0b013e328362a7d7 [DOI] [PubMed] [Google Scholar]
- [12].Richardson SJ, Leete P, Bone AJ, Foulis AK, Morgan NG. Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl-1. Diabetologia 2013; 56:185-93; PMID:23064357; http://dx.doi.org/ 10.1007/s00125-012-2745-4 [DOI] [PubMed] [Google Scholar]
- [13].Foulis AK, Farquharson MA, Cameron SO, McGill M, Schönke H, Kandolf R. A search for the presence of the enteroviral capsid protein VP1 in pancreases of patients with type 1 (insulin-dependent) diabetes and pancreases and hearts of infants who died of coxsackieviral myocarditis. Diabetologia 1990; 33:290-8; PMID:2376300; http://dx.doi.org/ 10.1007/BF00403323 [DOI] [PubMed] [Google Scholar]
- [14].Foulis AK, McGill M, Farquharson MA, Hilton DA. A search for evidence of viral infection in pancreases of newly diagnosed patients with IDDM. Diabetologia 1997; 40:53-61; PMID:9028718; http://dx.doi.org/ 10.1007/s001250050642 [DOI] [PubMed] [Google Scholar]
- [15].Anagandula M, Richardson SJ, Oberste MS, Sioofy-Khojine A-B, Hyöty H, Morgan NG, Korsgren O, Frisk G. Infection of human islets of langerhans with two strains of Coxsackie B virus serotype 1: assessment of virus replication, degree of cell death and induction of genes involved in the innate immunity pathway. J Med Virol 2014; 86:1402-11; PMID:24249667; http://dx.doi.org/ 10.1002/jmv.23835 [DOI] [PubMed] [Google Scholar]
- [16].Elshebani A, Olsson A, Westman J, Tuvemo T, Korsgren O, Frisk G. Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res 2007; 124:193-203; PMID:17169456; http://dx.doi.org/ 10.1016/j.virusres.2006.11.004 [DOI] [PubMed] [Google Scholar]
- [17].Hodik M, Lukinius A, Korsgren O, Frisk G. Tropism analysis of two coxsackie B5 strains reveals virus growth in human primary pancreatic islets but not in exocrine cell clusters In Vitro. Open Virol J 2013; 7:49-56; PMID:23723955; http://dx.doi.org/ 10.2174/1874357901307010049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pinkert S, Klingel K, Lindig V, Dörner A, Zeichhardt H, Spiller OB, Fechner H. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J Virol 2011; 85:13409-19; PMID:21976640; http://dx.doi.org/ 10.1128/JVI.00621-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jaïdane H, Hober D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab 2008; 34:537-48; PMID:18951821; http://dx.doi.org/ 10.1016/j.diabet.2008.05.008 [DOI] [PubMed] [Google Scholar]
- [20].Jaïdane H, Sauter P, Sane F, Goffard A, Gharbi J, Hober D. Enteroviruses and type 1 diabetes: towards a better understanding of the relationship. Rev Med Virol 2010; 20:265-80; PMID:20629044; http://dx.doi.org/ 10.1002/rmv.647 [DOI] [PubMed] [Google Scholar]
- [21].Chehadeh W, Kerr-Conte J, Pattou F, Alm G, Lefebvre J, Wattré P, Hober D. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J Virol 2000; 74:10153-64; PMID:11024144; http://dx.doi.org/ 10.1128/JVI.74.21.10153-10164.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yin H, Berg A-K, Westman J, Hellerström C, Frisk G. Complete nucleotide sequence of a Coxsackievirus B-4 strain capable of establishing persistent infection in human pancreatic islet cells: effects on insulin release, proinsulin synthesis, and cell morphology. J Med Virol 2002; 68:544-57; PMID:12376963; http://dx.doi.org/ 10.1002/jmv.10236 [DOI] [PubMed] [Google Scholar]
- [23].Sane F, Caloone D, Gmyr V, Engelmann I, Belaich S, Kerr-Conte J, Pattou F, Desailloud R, Hober D. Coxsackievirus B4 can infect human pancreas ductal cells and persist in ductal-like cell cultures which results in inhibition of Pdx1 expression and disturbed formation of islet-like cell aggregates. Cell Mol Life Sci CMLS 2013; 70:4169-80; PMID:23775130; http://dx.doi.org/ 10.1007/s00018-013-1383-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frisk G. Mechanisms of chronic enteroviral persistence in tissue. Curr Opin Infect Dis 2001; 14:251-6; PMID:11964840; http://dx.doi.org/ 10.1097/00001432-200106000-00002 [DOI] [PubMed] [Google Scholar]
- [25].Heim A, Canu A, Kirschner P, Simon T, Mall G, Hofschneider PH, Kandolf R. Synergistic interaction of interferon-beta and interferon-gamma in coxsackievirus B3-infected carrier cultures of human myocardial fibroblasts. J Infect Dis 1992; 166:958-65; PMID:1328409; http://dx.doi.org/ 10.1093/infdis/166.5.985 [DOI] [PubMed] [Google Scholar]
- [26].Heim A, Brehm C, Stille-Siegener M, Müller G, Hake S, Kandolf R, Figulla HR. Cultured human myocardial fibroblasts of pediatric origin: natural human interferon-alpha is more effective than recombinant interferon-alpha 2a in carrier-state coxsackievirus B3 replication. J Mol Cell Cardiol 1995; 27:2199-208; PMID:8576936; http://dx.doi.org/ 10.1016/S0022-2828(95)91515-X [DOI] [PubMed] [Google Scholar]
- [27].Schmidtke M, Selinka HC, Heim A, Jahn B, Tonew M, Kandolf R, Stelzner A, Zell R. Attachment of coxsackievirus B3 variants to various cell lines: mapping of phenotypic differences to capsid protein VP1. Virology 2000; 275:77-88; PMID:11017789; http://dx.doi.org/ 10.1006/viro.2000.0485 [DOI] [PubMed] [Google Scholar]
- [28].Chapman NM, Kim K-S, Drescher KM, Oka K, Tracy S. 5′ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 2008; 375:480-91; PMID:18378272; http://dx.doi.org/ 10.1016/j.virol.2008.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kim K-S, Tracy S, Tapprich W, Bailey J, Lee C-K, Kim K, Barry WH, Chapman NM. 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 2005; 79:7024-41; PMID:15890942; http://dx.doi.org/ 10.1128/JVI.79.11.7024-7041.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim K-S, Chapman NM, Tracy S. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol 2008; 82:2033-7; PMID:18057248; http://dx.doi.org/ 10.1128/JVI.01774-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tracy S, Smithee S, Alhazmi A, Chapman N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J Med Virol 2015; 87:240-7; PMID:25111164; http://dx.doi.org/ 10.1002/jmv.24039 [DOI] [PubMed] [Google Scholar]
- [32].Tam PE, Messner RP. Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J Virol 1999; 73:10113-21; PMID:10559326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fechner H, Pinkert S, Wang X, Sipo I, Suckau L, Kurreck J, Dörner A, Sollerbrant K, Zeichhardt H, Grunert H-P, et al.. Coxsackievirus B3 and adenovirus infections of cardiac cells are efficiently inhibited by vector-mediated RNA interference targeting their common receptor. Gene Ther 2007; 14:960-71; PMID:17377597; http://dx.doi.org/ 10.1038/sj.gt.3302948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Werk D, Schubert S, Lindig V, Grunert H-P, Zeichhardt H, Erdmann VA, Kurreck J. Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of coxsackievirus B3 and its cognate coxsackievirus-adenovirus receptor. Biol Chem 2005; 386:857-63; PMID:16164410; http://dx.doi.org/ 10.1515/BC.2005.100 [DOI] [PubMed] [Google Scholar]
- [35].Feuer R, Whitton JL. Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr Top Microbiol Immunol 2008; 323:149-73; PMID:18357769 [DOI] [PubMed] [Google Scholar]
- [36].Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 2002; 76:4430-40; PMID:11932410; http://dx.doi.org/ 10.1128/JVI.76.9.4430-4440.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Feuer R, Mena I, Pagarigan RR, Hassett DE, Whitton JL. Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med Microbiol Immunol (Berl) 2004; 193:83-90; ; http://dx.doi.org/ 10.1007/s00430-003-0192-z [DOI] [PubMed] [Google Scholar]
- [38].Grey F. Role of microRNAs in herpesvirus latency and persistence. J Gen Virol 2015; 96:739-51; PMID:25406174; http://dx.doi.org/ 10.1099/vir.0.070862-0 [DOI] [PubMed] [Google Scholar]
- [39].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/ 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev 2015; 87:3-14; PMID:25979468; http://dx.doi.org/ 10.1016/j.addr.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].El-Diwany R, Wasilewski LN, Witwer KW, Bailey JR, Page K, Ray SC, Cox AL, Thomas DL, Balagopal A. Acute hepatitis C virus infection induces consistent changes in circulating microRNAs that are associated with nonlytic hepatocyte release. J Virol 2015; 89:9454-64; PMID:26157120; http://dx.doi.org/ 10.1128/JVI.00955-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim H, Choi H, Lee SK. Epstein-Barr Virus MicroRNA miR-BART20-5p Suppresses Lytic Induction by Inhibiting BAD-Mediated caspase-3-Dependent Apoptosis. J Virol 2016; 90:1359-68; http://dx.doi.org/ 10.1128/JVI.02794-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ho B-C, Yang P-C, Yu S-L. MicroRNA and Pathogenesis of Enterovirus Infection. Viruses 2016; 8:E11; http://dx.doi.org/ 10.3390/v8010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Alidjinou EK, Sané F, Bertin A, Caloone D, Hober D. Persistent infection of human pancreatic cells with Coxsackievirus B4 is cured by fluoxetine. Antiviral Res 2015; 116:51-4; PMID:25655448; http://dx.doi.org/ 10.1016/j.antiviral.2015.01.010 [DOI] [PubMed] [Google Scholar]
- [45].Ho B-C, Yu S-L, Chen JJW, Chang S-Y, Yan B-S, Hong Q-S, Singh S, Kao C-L, Chen H-Y, Su K-Y, et al.. Enterovirus-induced miR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor eIF4E. Cell Host Microbe 2011; 9:58-69; PMID:21238947; http://dx.doi.org/ 10.1016/j.chom.2010.12.001 [DOI] [PubMed] [Google Scholar]
- [46].Tong L, Lin L, Wu S, Guo Z, Wang T, Qin Y, Wang R, Zhong X, Wu X, Wang Y, et al.. MiR-10a* up-regulates coxsackievirus B3 biosynthesis by targeting the 3D-coding sequence. Nucleic Acids Res 2013; 41:3760-71; PMID:23389951; http://dx.doi.org/ 10.1093/nar/gkt058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wen B, Dai H, Yang Y, Zhuang Y, Sheng R. MicroRNA-23b inhibits enterovirus 71 replication through downregulation of EV71 VPl protein. Intervirology 2013; 56:195-200; PMID:23594713; http://dx.doi.org/ 10.1159/000348504 [DOI] [PubMed] [Google Scholar]
- [48].Zhang Q, Xiao Z, He F, Zou J, Wu S, Liu Z. MicroRNAs regulate the pathogenesis of CVB3-induced viral myocarditis. Intervirology 2013; 56:104-13; PMID:23183417; http://dx.doi.org/ 10.1159/000343750 [DOI] [PubMed] [Google Scholar]
- [49].Organtini LJ, Makhov AM, Conway JF, Hafenstein S, Carson SD. Kinetic and structural analysis of coxsackievirus B3 receptor interactions and formation of the A-particle. J Virol 2014; 88:5755-65; PMID:24623425; http://dx.doi.org/ 10.1128/JVI.00299-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lysy PA, Weir GC, Bonner-Weir S. Making β cells from adult cells within the pancreas. Curr Diab Rep 2013; 13:695-703; PMID:23925431; http://dx.doi.org/ 10.1007/s11892-013-0400-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dörner A, Xiong D, Couch K, Yajima T, Knowlton KU. Alternatively spliced soluble coxsackie-adenovirus receptors inhibit coxsackievirus infection. J Biol Chem 2004; 279:18497-503; PMID:14978041; http://dx.doi.org/ 10.1074/jbc.M311754200 [DOI] [PubMed] [Google Scholar]
- [52].Thoelen I, Magnusson C, Tågerud S, Polacek C, Lindberg M, Van Ranst M. Identification of alternative splice products encoded by the human coxsackie-adenovirus receptor gene. Biochem Biophys Res Commun 2001; 287:216-22; PMID:11549277; http://dx.doi.org/ 10.1006/bbrc.2001.5535 [DOI] [PubMed] [Google Scholar]
- [53].Kim KW, Ho A, Alshabee-Akil A, Hardikar AA, Kay TWH, Rawlinson WD, Craig ME. Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 2016; 65:996-1003; PMID:26558682; http://dx.doi.org/ 10.2337/db15-0956 [DOI] [PubMed] [Google Scholar]
- [54].Beaulieux F, Zreik Y, Deleage C, Sauvinet V, Legay V, Giraudon P, Kean KM, Lina B. Cumulative mutations in the genome of Echovirus 6 during establishment of a chronic infection in precursors of glial cells. Virus Genes 2005; 30:103-12; PMID:15744568; http://dx.doi.org/ 10.1007/s11262-004-4587-8 [DOI] [PubMed] [Google Scholar]
- [55].Duncan G, Colbère-Garapin F. Two determinants in the capsid of a persistent type 3 poliovirus exert different effects on mutant virus uncoating. J Gen Virol 1999; 80 (Pt 10):2601-5; PMID:10573152; http://dx.doi.org/ 10.1099/0022-1317-80-10-2601 [DOI] [PubMed] [Google Scholar]
- [56].Duncan G, Pelletier I, Colbère-Garapin F. Two amino acid substitutions in the type 3 poliovirus capsid contribute to the establishment of persistent infection in HEp-2c cells by modifying virus-receptor interactions. Virology 1998; 241:14-29; PMID:9454713; http://dx.doi.org/ 10.1006/viro.1997.8955 [DOI] [PubMed] [Google Scholar]
- [57].Pelletier I, Duncan G, Pavio N, Colbère-Garapin F. Molecular mechanisms of poliovirus persistence: key role of capsid determinants during the establishment phase. Cell Mol Life Sci CMLS 1998; 54:1385-402; PMID:9893712; http://dx.doi.org/ 10.1007/s000180050261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med 1979; 300:1173-9; PMID:219345; http://dx.doi.org/ 10.1056/NEJM197905243002102 [DOI] [PubMed] [Google Scholar]
- [59].Alidjinou EK, Sané F, Engelmann I, Hober D. Serum-dependent enhancement of coxsackievirus B4-induced production of IFNα, IL-6 and TNFα by peripheral blood mononuclear cells. J Mol Biol 2013; 425:5020-31; PMID:24120940; http://dx.doi.org/ 10.1016/j.jmb.2013.10.008 [DOI] [PubMed] [Google Scholar]
- [60].Lam WY, Cheung ACY, Tung CKC, Yeung ACM, Ngai KLK, Lui VWY, Chan PKS, Tsui SKW. miR-466 is putative negative regulator of Coxsackie virus and Adenovirus Receptor. FEBS Lett 2015; 589:246-54; PMID:25497012; http://dx.doi.org/ 10.1016/j.febslet.2014.12.006 [DOI] [PubMed] [Google Scholar]
- [61].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods San Diego Calif 2001; 25:402-8; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- [62].Chen C-J, Servant N, Toedling J, Sarazin A, Marchais A, Duvernois-Berthet E, Cognat V, Colot V, Voinnet O, Heard E, et al.. ncPRO-seq: a tool for annotation and profiling of ncRNAs in sRNA-seq data. Bioinforma Oxf Engl 2012; 28:3147-9; http://dx.doi.org/ 10.1093/bioinformatics/bts587 [DOI] [PubMed] [Google Scholar]
- [63].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550; PMID:25516281; http://dx.doi.org/ 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]