

Abstract
Type I interferons (IFN-I), such as IFN-α and IFN-β are important messengers in the host response against bacterial infections. Knowledge about the role of IFN-I in infections by nontuberculous mycobacteria (NTM) is limited. Here we show that macrophages infected with pathogens of the Mycobacterium avium complex produced significantly lower amounts of IFN-β than macrophages infected with the opportunistic pathogen M. smegmatis. To dissect the molecular mechanisms of this phenomenon, we focused on the obligate pathogen Mycobacterium avium ssp paratuberculosis (MAP) and the opportunistic M. smegmatis. Viability of both bacteria was required for induction of IFN-β in macrophages. Both bacteria induced IFN-β via the cGAS-STING-TBK1-IRF3/7-pathway of IFN-β activation. Stronger phosphorylation of TBK1 and higher amounts of extracellular bacterial DNA in the macrophage cytosol were found in M. smegmatis infected macrophages than in MAP infected macrophages. After intraperitoneal infection of mice, a strong Ifnb induction by M. smegmatis correlated with clearance of the bacteria. In contrast, MAP only induced weak Ifnb expression which correlated with bacterial persistence and increased number of granulomas in the liver. In mice lacking the type I interferon receptor we observed improved survival of M. smegmatis while survival of MAP was similar to that in wildtype mice. On the other hand, treatment of MAP infected wildtype mice with the IFN-I inducer poly(I:C) or recombinant IFN-β impaired the survival of MAP. This indicates an essential role of IFN-I in clearing infections by MAP and M. smegmatis. The expression level of IFN-I is decisive for transient versus persistent NTM infection.
KEYWORDS: cGAS, extracellular DNA, mycobacterium smegmatis, mycobacterium paratuberculosis, non tuberculous mycobacteria, STING, Type I interferon
Introduction
Type I interferons (IFN-I) such as IFN-α and IFN-β are pleiotropic cytokines involved in various infections and inflammatory reactions as well as in cancer surveillance.1 The protective role of IFN-I against many viral infections is well established.2 IFN-I are also induced during bacterial infection. Here they have been shown to be either protective or detrimental, depending on the type of bacteria and the route of infection.3
IFN-I are produced by many cell types.2 However, under conditions of bacterial infection or inflammation IFN-I are produced in high amounts by cells of the monocyte/macrophage lineage.4,5 A hierarchy exists for the induction of IFN-I. Usually, IFN-β is induced first which then via binding to the unique type I IFN receptor (IFNAR) activates the IFN cascade in an autocrine and paracrine fashion.2 IFN-I expression is activated by the interaction of pathogen associated molecular pattern (PAMP) with membrane-bound pattern recognition receptors (PRRs) of the Toll-like receptor (TLR) family. Alternatively, PAMPs that reach the cytosol induce IFN-I responses via cytosolic PRRs including cyclic GMP-AMP synthase (cGAS). The critical role of cGAS for IFN-β induction in bacteria infected macrophages was recently emphasized. DNA released from bacteria activates cytosolic cGAS to synthesize cyclic GMP-AMP (cGAMP). This triggers the STING-TBK-1-IRF3 signaling pathway to initiate transcription of Ifnb.6-9
Most of our knowledge of IFN-I in mycobacterial infection is derived from studies on M. tuberculosis. The bacteria have been shown to induce IFN-I response in human and murine macrophages. Even though the particular role of IFN-I during the host response against M. tuberculosis infection is not completely understood, most of such studies suggest that IFN-I promote rather than control M. tuberculosis infection.9-11 At the molecular level, eDNA of M. tuberculosis represents the critical ligand for IFN-I induction. The ESX-1 type VII secretion system of M. tuberculosis appears to be essentially for higher permeability of the phagosomal membrane thus allowing eDNA to access the cytosol of host cells. By this mechanism, M. tuberculosis is able to induce IFN-I expression via the cGAS-STING-TBK1-IRF3 pathway.7,9,12,13
Besides M. tuberculosis, the knowledge on IFN-I during infection with other mycobacterial species is limited. Some evidence supporting a beneficial role of IFN-I comes from experiments demonstrating that treatment of mice with recombinant murine IFN-β enhanced their resistance to systemic M. avium infection.14 In addition, infection of mice deficient for the IFN-I receptor (IFNAR−/−) revealed that IFN-I is involved in the early control of M. bovis BCG infection.15 Although these experiments imply that the M. bovis BCG might induce host IFN-I, infection of murine macrophages and dendritic cells with BCG resulted in lower expression of IFN-β compared with M. tuberculosis.16 This shows that in vitro studies reflect only to a limited extent the sum of effects induced in vivo.
The group of nontuberculous mycobacteria (NTM) comprises saprophytic, non-pathogenic, opportunistic and obligate pathogenic mycobacterial species some of which cause disease in humans and animals.17,18 M. smegmatis is an environmental organism and considered as a saprophytic NTM which is not able to survive in macrophages.19 Nevertheless, M. smegmatis is not “non-virulent” as it was isolated from skin or soft-tissue lesions of humans.18,20 In addition, it was recently described that intravenous infection of mice with a high dose of M. smegmatis was fatal for C57BL/6 mice.21
In contrast, Mycobacterium avium ssp paratuberculosis (MAP) is an obligate pathogenic NTM of the Mycobacterium avium complex. It is the causative agent of paratuberculosis (Johne's disease), a chronic transmural inflammation of the small intestine in ruminants. MAP has also been discussed as a possible etiological agent in the development of human diseases such as Crohn's disease, a chronic inflammatory enteritis in humans,22 type I diabetes,23,24 and multiple sclerosis.25 One of the key factors that enable MAP to survive inside host cells is its ability to silence the host immune response. This includes inhibition of phagosomal acidification and maturation,19,26 reduction of TNF-α expression,27 reduction of caspase activity,28 and inhibition of CD4+ T cells activation.29
In the present study, we aimed to study IFN-β induction upon infection with NTM by focusing on MAP or M. smegmatis. Using in vitro cell culture systems and in vivo mouse models, we found that both NTM induced Ifnb via the cGAS-STING-TBK1-IRF3/7 axis. In both cases viable bacteria were needed for Ifnb triggering. Higher phosphorylation of TBK1 and higher amounts of eDNA detected in macrophages infected with M. smegmatis indicated that this mycobacterium activated the signaling cascade more efficiently. Upon mouse infection, M. smegmatis but not MAP was largely cleared. However, upon infection of IFNAR−/− mice M. smegmatis could be re-isolated in high numbers after 21 days, indicating that IFN-I contributes to clearing of mycobacterial infection. Similarly, administration of recombinant IFN-β reduced the number of MAP in vivo. These data suggest that the attenuation of IFN-β activation allows MAP to evade the host immune defense. Overall, our results support the hypothesis that transient vs. persistent NTM infection is determined by the extent of IFN-β induction.
Results
M. smegmatis induces higher Ifnb expression in macrophages than M. avium subspecies
To investigate the role of IFN-I in NTM infections, we infected murine RAW 264.7 macrophages (RAW) with the MAP strains MAP 6783 and MAP K10, M. avium ssp hominis suis strain 04A/1287 (MAH), M. avium ssp avium (MAA) strains 44156 and 44158, and M. smegmatis mc2 as well as M. bovis BCG Pasteur for 5h. All tested mycobacterial strains induced Ifnb expression in RAW cells (Fig. 1A). However, compared to all other tested mycobacterial species M. smegmatis induced significantly higher Ifnb levels (Fig. 1A).
Figure 1.
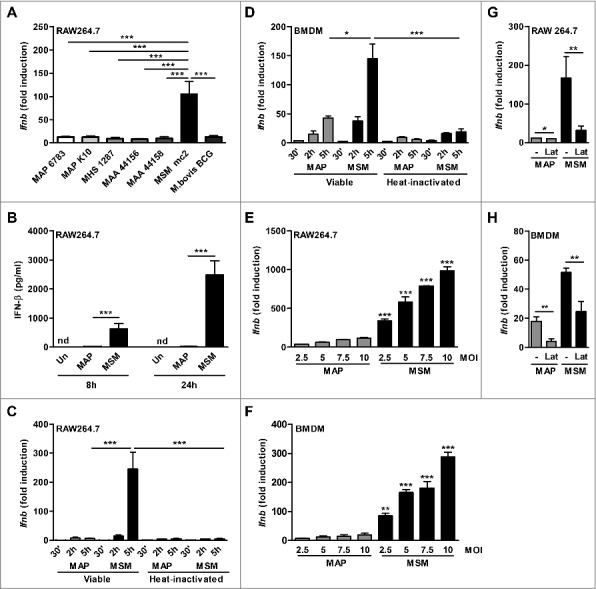
Ifnb induction by NTM mycobacteria in macrophages. (A) qPCR for Ifnb in RAW cells infected with viable MAP 6783, MAP K10, MAH 1287, MAA 44156, MAA 44158, M. bovis BCG Pasteur or M. smegmatis mc2 (MSM) (MOI 5) for 5 h. (B) IFN-β ELISA of supernatant from RAW cells infected with MAP or MSM. (C) qPCR for Ifnb in RAW cells and (D) BMDM infected with viable (V) or heat inactivated (HI) MAP or MSM (MOI 5) for 30 min (’), 2 h and 5 h. (E) RAW cells and (F) BMDM were infected with MAP or MSM (MOIs 2.5, 5.0, 7.5, 10) for 5 h. *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA with Tukey post test, means ± SEM. (G) RAW cells and (H) BMDM were pre-treated with 5 µM latrunculin B (Lat) for 1 h and subsequently infected with MAP or MSM for 5 h. Data are representative of 3 independent experiments, *p<0.05, **p<0.01 by 2 tailed student's t-test means ± SEM.
To dissect the molecular mechanism of Ifnb induction by the different mycobacterial strains, we focused on MAP strain MAP 6783 (MAP) and M. smegmatis. While abundant levels of IFN-β were secreted after M. smegmatis infection of RAW cells, MAP induced much lower amounts (Fig. 1B). Similar results were observed at mRNA level. Ifnb expression in M. smegmatis infected macrophages significantly increased within 5 h after infection, whereas the levels in MAP infected macrophages remained similarly low over time (Fig. 1C). Likewise, in BMDM, Ifnb strongly increased over time upon M. smegmatis and only moderately upon MAP infection (Fig. 1D). Interestingly, for both MAP and M. smegmatis Ifnb induction was dependent on the viability of the bacteria (Fig. 1C and D) and increased in a dose dependent manner (Fig. 1E and F). However, at equivalent MOIs the Ifnb induction by MAP was always significantly lower than by M. smegmatis (Fig. 1E and F). This difference in Ifnb triggering was not due to divergent numbers of intracellular MAP or M. smegmatis as observed by confocal microscopy (Fig. S1).
Endocytosis or phagocytosis was required for Ifnb induction. RAW cells or BMDM pre-treated with latrunculin B before both MAP and M. smegmatis infection exhibited significantly reduced Ifnb expression (Fig. 1G and H). This suggested that Ifnb induction by NTM is mediated via intracellular receptors or via receptors that need to be internalized.
MAP and M. smegmatis induce Ifnb expression via the cGAS-STING-TBK1-IRF3/7 signaling pathway
Induction of Ifnb expression by M. tuberculosis has been shown to require cytosolic signaling via cGAS and the STING-TBK1-IRF3 axis.7 Hence, we were interested to determine whether the same pathway is involved in Ifnb induction in macrophages infected with MAP or M. smegmatis. First, we wanted to exclude a potential contribution of TLR's. Induction of Ifnb was not significantly influenced in BMDM from mice lacking UNC93B, a trafficking protein required for the signaling of TLR3, 7, and 9. This indicated that intracellular TLRs did not contribute to Ifnb expression (Fig. 2A). Furthermore, Ifnb expression was independent of MyD88 and TRIF thus excluding any other TLR mediated signaling. In contrast, Ifnb expression was significantly reduced in IRF3−/−, IFNAR−/- and IRF7−/- macrophages infected with both mycobacterial species (Fig. 2B), although all cells, except IRF3−/−, were fully responsive to LPS stimulation (Fig. S2). IRF3 is known to be essential for induction of IFNβ by LPS.30 Immunoblot analysis revealed that phosphorylation of TBK1 was markedly induced in macrophages infected with M. smegmatis, but not with MAP (Fig. 2C).
Figure 2.
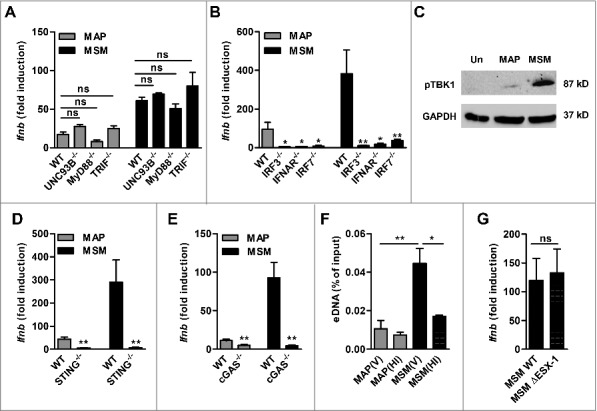
MAP and M. smegmatis (MSM) induce Ifnb expression via the cGAS-STING-TBK1-IRF3/7 signaling pathway. (A) qPCR for Ifnb in BMDM from WT, UNC93B−/−, MyD88−/−, TRIF−/−, (B) IRF3−/−, IFNAR/−, IRF7−/- mice infected with MAP or MSM. *p<0.05, **p<0.01, ns (non-significant) by one-way ANOVA with Tukey post test, means ± SEM. (C) Immunoblot of phosphoTBK1 in RAW cells infected with MAP or MSM for 5 h. (D) qPCR for Ifnb in WT, STING−/− and (E) cGAS−/− mice infected with MAP or MSM for 5 h. (F) qPCR analysis of eDNA in RAW cells infected with viable (V) or heat inactivated (HI) MAP or MSM for 5 h. eDNA was extracted and shown as percentage of the input control. (G) qPCR for Ifnb in RAW cells infected with MSM WT or MSM ΔESX-1 for 5 h. Data are representative of 3 independent experiments, *p<0.05, **p<0.01 by 2 tailed student's t-test means ± SEM.
Overall, these data suggested that cytosolic signaling might be required for Ifnb activation in MAP and M. smegmatis infected macrophages. Indeed, Ifnb expression was significantly reduced in STING−/− (Fig. 2D) and cGAS−/− macrophages (Fig. 2E) as well as in cGAS and STING knock down RAW cells (Fig. S3). The knock out macrophages remained fully responsive to LPS stimulation (Fig. S2).
The above data showed a differential magnitude of activation of the cGAS-STING-TBK1 pathway. This might be due to distinct release of eDNA from the mycobacteria bearing phagosomes. To test this, we extracted DNA from the cytosol of infected macrophages and probed for the presence of mycobacterial eDNA by qPCR. Enhanced amounts of eDNA were detected in the cytosol of macrophages infected with viable M. smegmatis when compared with MAP infected macrophages. Furthermore, the amount of eDNA in the cytosol of macrophages was significantly lower when the macrophages were treated with heat-inactivated M. smegmatis or MAP (Fig. 2F). These data indicate that the differential release of mycobacterial eDNA into the cytosol of infected macrophages is responsible for a diverse level of activation of the cGAS-STING-TBK1 pathway by the different mycobacteria species.
It was shown before that M. tuberculosis activates Ifnb expression by releasing eDNA into the cytosol of host cells in an ESX-1 dependent manner.7,9 M. smegmatis bears an ESX-1 secretion system. Therefore, by using a M. smegmatis ESX-1 deletion mutant, we tested whether the release of eDNA by M. smegmatis was dependent of ESX-1. The mutant induced Ifnb levels comparable to the wildtype (WT) bacteria (Fig. 2G). Moreover, no difference in bacterial burden was observed in mice infected either with M. smegmatis WT or the ESX-1 mutant (Fig. S4).
These results clearly demonstrate that MAP and M. smegmatis release DNA into the cytosol of the host cell to stimulate Ifnb induction via cGAS, STING, TBK1, and IRF3 which includes IFNAR and IRF7 dependent feedback signaling. Therefore, both mycobacteria appear to stimulate the cytosolic sensors cGAS and STING, but to different degrees.
M. smegmatis is not able to persist in infected mice
It is well known that MAP is able to persist in intraperitoneally (i.p.) infected mice over long periods of time.31-35 However, little is known for M. smegmatis in that respect. To better understand the fate of M. smegmatis after infection of mice, C57BL/6 mice were infected i.p. with M. smegmatis or MAP and CFUs were determined from liver, spleen, and mesentery at 21 d post infection (dpi). MAP could be isolated from these organs at high numbers. In contrast, M. smegmatis was barely detectable in such tissues, indicating that the mice were able to clear M. smegmatis, but not MAP (Fig. 3A to C).
Figure 3.
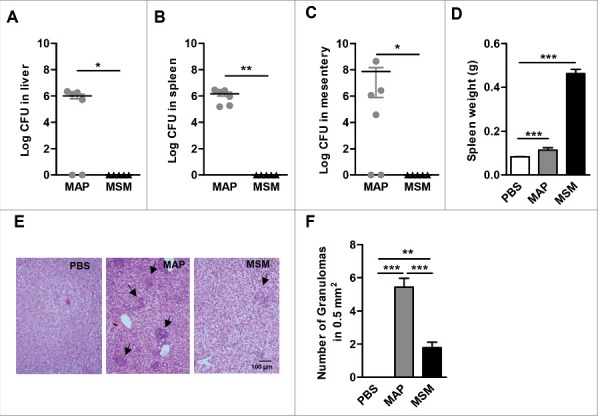
Pathology of MAP and M. smegmatis (MSM) infected mice. C57BL/6 mice (n = 3–6/group) were infected MAP or MSM. At day 21 post infection, (A) livers (B) spleens and (C) mesentery were plated. *p<0.05, ** p<0.01 by Mann-Whitney U test, means ± SEM. (D) spleen weight was determined. (E) Representative HE stained liver sections. Arrows heads point at granulomas. (F) Numbers of granulomas in livers. Data are representative of 2 independent experiments. *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA with Tukey post test, means ± SEM.
MAP infected mice displayed moderately enhanced spleen weight. In contrast, mice infected with M. smegmatis exhibited severe splenomegaly suggesting a strong inflammatory response (Fig. 3D). Granulomas were found in the liver of mice infected with both mycobacteria. However, the number and the size of granuloma in the liver of MAP infected mice were significantly larger (Fig. 3E and F). These results indicate that unlike MAP, M. smegmatis is efficiently cleared by the murine host.
High or low induction of Ifnb upon M. smegmatis or MAP infection, respectively, correlates with lower or higher bacterial burden in mice
We wanted to understand whether the IFN-β response in mice during MAP or M. smegmatis infection correlates with differential persistence of the bacteria. To this end, we used the firefly luciferase-based heterozygous IFN-β+/Δβ-luc reportermouse that allows Ifnb reporter expression concomitant with IFN-β production.36 IFN-β+/Δβ-luc mice were infected i.p. with MAP and M. smegmatis (108 CFUs/mouse) and luciferase expression was analyzed for up to 14 d post infection by non-invasive in vivo imaging. Ifnb promoter activity was weakly induced by MAP (Fig. 4A). In contrast, M. smegmatis infected mice displayed considerably higher levels of luciferase expression. In addition, in M. smegmatis infected mice the luciferase signal could be detected after 3 d of infection and increased activity was detectable until 14 d post infection (Fig. 4A).
Figure 4.
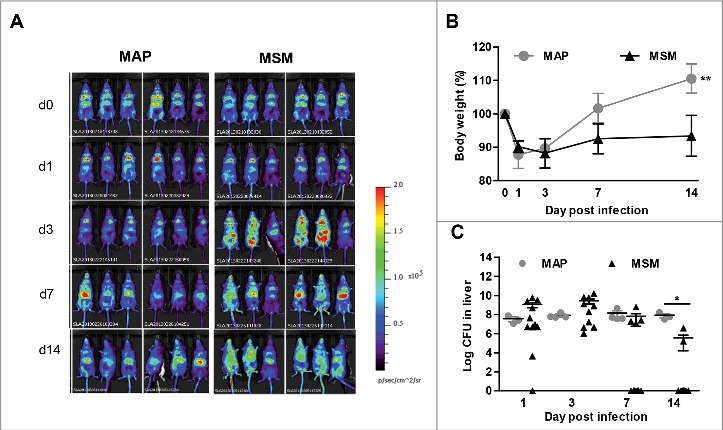
Induction of IFN-β after MAP or M. smegmatis (MSM) infection in vivo. (A) IFN-β+/Δβ-luc (n = 6/group) were infected with MAP or MSM. At day 1, 3, 7, and 14 post infection, mice were injected with D-luciferin and visualized for luciferase activity. (B) Body weights of mice infected with MAP or MSM. **p<0.01 by Two way ANOVA with Bonferroni post test, means ± SEM. (C) C57BL/6 mice (n = 3–10/group) were infected with MAP or MSM. Numbers of CFU in liver were determined. Data are representative of 2 independent experiments. *p<0.05 by Mann-Whitney U test, means ± SEM.
To correlate the dynamics of MAP and M. smegmatis infections with the IFN-β response, we infected C57BL/6 mice with MAP or M. smegmatis and analyzed them at day 1, 3, 7, and 14 post infection. While MAP infected mice were able to recover quickly from the initial body weight loss, M. smegmatis infected mice did not recover during the observation period (Fig. 4B). Bacterial plating revealed that MAP was able to persist in the liver. Similar numbers of MAP could be recovered from the liver at each time point. In contrast, the bacterial load with M. smegmatis in the liver continuously declined (Fig. 4C). These results suggested that the increased levels of IFN-β in M. smegmatis infected mice might promote mycobacterial clearance.
IFNAR is necessary to clear mycobacterial infection
To demonstrate the involvement of IFN-I in the host response to mycobacterial infection, C57BL/6 WT and IFNAR−/− mice which are non-responsive to IFN-I were infected with MAP or M. smegmatis. Mice infected with MAP initially lost weight and recovered after 2 d of infection independent of whether they were WT or IFNAR−/−. In contrast, M. smegmatis infected mice recovered their body weights upon day 21. Remarkably, upon M. smegmatis infection weight loss of IFNAR−/- mice was more dramatic than of WT mice and the animals did not recover during the observation period (Fig. 5A and B). At necropsy, increases in spleen weight were similar when WT to IFNAR−/- mice were compared (Fig. 5C).
Figure 5.
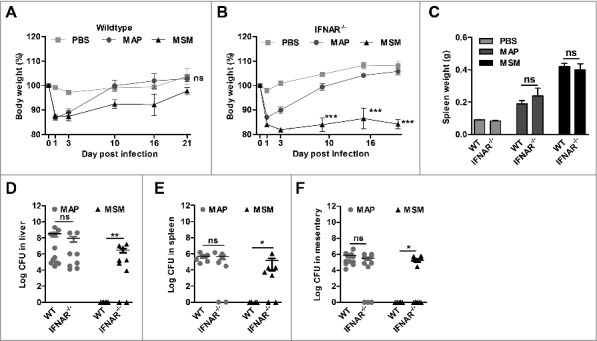
Clearance of M. smegmatis (MSM) depends on IFN-I signaling. C57BL/6 mice and IFNAR−/− mice (n = 5–6/group) were injected with PBS, MAP and MSM. (A and B) body weight was monitored. ***p<0.001 by Two way ANOVA with Bonferroni post test, means ± SEM. (C) spleen weights at 21 dpi. *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA with Tukey post test, means ± SEM. (D) Numbers of CFU in liver, (E) spleen and (F) mesentery were determined by plating 21 dpi. Data are representative of 2 independent experiments, *p<0.05, ** p<0.01, ns (non-significant), by Mann-Whitney U test, means ± SEM.
The analysis of tissue lysates revealed that C57BL/6 WT mice were able to clear M. smegmatis infection within 3 weeks. In contrast, high numbers of M. smegmatis could be recovered from the organs of the majority of infected IFNAR−/- mice (Fig. 5D to F). These data show that functional IFN-I signaling is necessary for the host to control M. smegmatis infection, whereas in case of MAP infection IFN-I signaling did not play a key role.
IFN-β promotes control of MAP infections
IFN-I is obviously involved in the control of M. smegmatis infection. On the other hand, the low amounts of IFN-β induced by MAP might not be sufficient to elicit a significant in vivo effect against this bacterium. Thus, we hypothesized that exogenous administration of IFN-β might help to reduce the number of bacteria. To test this, MAP infected mice were treated with either recombinant IFN-β (rIFN-β) or poly(I:C) (Fig. 6A). The spleen weights were similar in MAP infected and in MAP infected IFN-β treated mice, while the addition of poly(I:C) increased the spleen weight (Fig. 6B). Interestingly, determination of the bacterial numbers in liver, spleen and mesentery revealed that the treatment of MAP infected mice with poly(I:C) or rIFN-β led to a significant effect on bacterial persistence. In contrast to the liver, CFU in spleen and mesentery were significantly reduced compared with untreated MAP infected mice (Fig. 6C to E).
Figure 6.
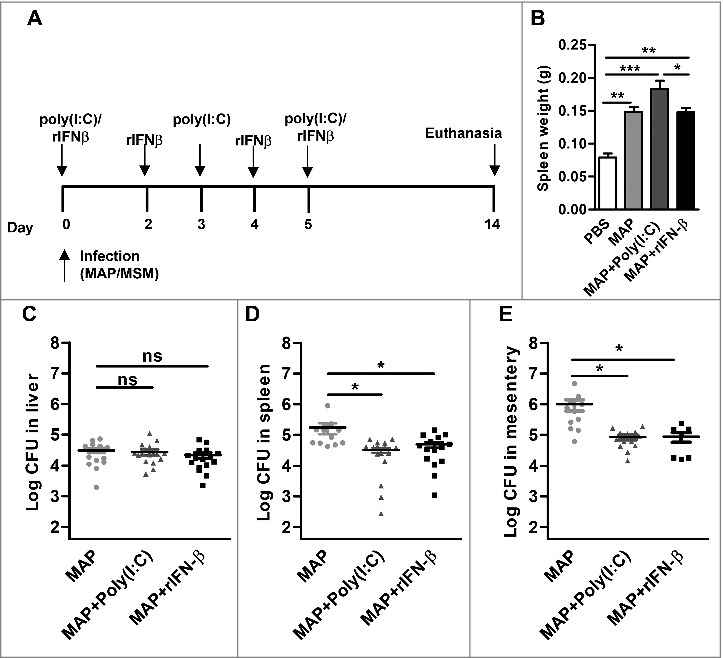
IFN-β supports MAP clearance. C57BL/6 (n = 3–10/group) were infected with PBS, MAP, MAP + 100 µg poly(I:C)/mouse, or MAP + 1000 units rIFN-β. Mice were killed at 14 dpi. (A) Schematic representation of the experiment. (B) spleen weight was determined. (C) Numbers of CFU in liver, (D) spleen and (E) mesentery were determined by plating. Data are representative of 2 independent experiments, *p<0.05, **p<0.01, ***p<0.001, ns (non-significant), by one-way ANOVA with Tukey post test, means ± SEM.
Taken together, these results clearly indicate that in case of MAP and M. smegmatis IFN-I contribute to mycobacterial clearance. Therefore, subversion of IFN-β induction in MAP infected macrophages and also in mice might be an immune evasion strategy of MAP and most likely of other pathogenic NTM that adds to their persistence.
Discussion
We observed that the opportunistic pathogenic M. smegmatis induced abundant Ifnb expression during macrophage infection. This observation is in agreement with Shah et al., see ref.37 who showed that IFN-β production induced by M. smegmatis and 2 other opportunistic pathogenic NTM, M. fortuitum and M. kansasii, was more pronounced than induction by M. tuberculosis.37 Interestingly, Ifnb induction by M. smegmatis was significantly higher than that induced by all the slow growing pathogenic NTM or M. bovis BCG tested here (Fig. 1A). Remarkably, the ESX-1 secretion system is absent in the Mycobacterium species tested in our study besides M. smegmatis. The membrane-interacting activity of M. tuberculosis ESAT-6 secreted by ESX-1 type VII secretion system is known to be essential for Ifnb induction. M. smegmatis expresses an ESX-1 secretion system. However, it was shown recently that the membrane-interacting activity of M. tuberculosis ESAT-6 is not present in its ortholog in M. smegmatis.38 Thus, it was possible that the molecular pathway of IFN-β induction and the relevance of IFN-β in the pathogenesis of infections between NTM and M. tuberculosis are different. This prompted us to dissect the pathways of IFN-β induction and the role of IFN-β during the host response to such infections in more detail. We focused on M. smegmatis and MAP.
M. smegmatis was recently found to cause fatal disease after intravenous infection of C57BL/6 mice.21 Similarly, we showed that even i.p. infection of mice with M. smegmatis caused severe disease, as indicated by the severe and sustained body weight loss during the first 2 weeks of infection (Fig. 4B, Fig. 5A and B). However, the animals were finally able to clear the infection by day 21 (Fig. 5D to F) and recover body weight. The clearance of the bacterial load was associated with enhanced expression of Ifnb. IFN-β production and signaling were essential to control the infection as IFNAR−/− mice lacking a functional IFN-I system were no longer able to restrict bacterial growth.
Similar to M. tuberculosis, the induction of Ifnb expression was dependent on the cGAS-STING-TBK1-IRF3 pathway of IFN-β induction (Fig. 2D and E) which appears to be activated by eDNA of M. smegmatis. Accordingly, high amounts of DNA of M. smegmatis could be detected in the cytosol of host macrophages (Fig. 2F). How this DNA is released from the phagosome into the cytosol is still unclear. We could exclude any involvement of the ESX-1 secretion system since deletion of the ESX-1 system in M. smegmatis neither influenced Ifnb expression by macrophages nor the outcome of infection in vivo (Fig. 2G and Fig. S4). This is in striking contrast to M. tuberculosis infections where the ESX-1 secretion system is required.9,10,12 Indeed, this situation is reminiscent of infections by extracellular pathogens such as pathogenic streptococci. In such cases, IFN-β is also produced by macrophages and is necessary for bacterial clearance.6,39 Streptococci induce IFN-β production in macrophages also via the cytosolic cGAS-STING-TBK1-IRF3 pathway and similar to our experiments with M. smegmatis this pathway requires bacterial viability, phagocytic uptake, and the release of bacterial DNA into the cytosol.6,39 The mechanism by which DNA breaches the vesicular membrane is also not fully resolved. It seems that streptococcal toxins partially regulate DNA access to the cytosol, although other factors are likely to play a role.40
Pathogenicity of MAP is mostly unresolved. Similar to many other pathogenic mycobacteria, the survival and persistence of MAP is one key feature of MAP pathogenicity. In the present study, we found that similar to MAA, MAH and M. bovis BCG, MAP induced very weak expression of Ifnb in murine macrophages in vitro (Fig. 1A). Similarly, in vivo MAP induced only weak IFN-β (Fig. 4A). The weak induction correlated with MAP survival for more than 21 d and with increased granuloma formation in the liver. MAP survival was not influenced in mice lacking IFNAR (Fig. 5D and F). This demonstrates that the low level IFN-β response induced by MAP does not substantially contribute to the infection outcome. Nevertheless, clearance of MAP can be significantly enhanced by repeated administration of rIFN-β or poly(I:C) (Fig. 6D and E). Bacterial loads of spleen and mesentery were significantly lower in this case while no difference could be found in the liver. Since the induction of IFN-β can potentially vary among different tissues,41 it is possible that the amount of Ifnb in the liver of mice stimulated with MAP + rIFN-β or MAP + poly(I:C) did not reach the threshold needed for effective clearance (Fig. 6C). This indicates that IFN-β, when present in sufficient amounts, contributes to clearance also of MAP. Overall these data show that IFN-β contributes to controlling NTM infection in mice. How NTM clearance is promoted by IFN-β needs further studies since IFN-I can influence a multitude of immune cells.42 One possible mechanism linking NTM clearance and IFN-I production might be the differential production of nitric oxide (NO) as IFNAR signaling is known to trigger NO production.43 Treatment of macrophages with recombinant IFNαβ results in the restriction of M. tuberculosis growth by NO.44
Our in vitro experiments revealed that Ifnb induction by M. smegmatis and even the low response induced by MAP takes place via the cytosolic cGAS-STING-TBK1-IRF3/7 pathway. Activation required viability and cellular uptake. This indicates that mycobacterial components which are produced in the phagosome contribute to Ifnb induction by both mycobacterial species. The cytosolic cGAS-dependent sensing of bacterial DNA has been described for infections by M. tuberculosis and other intracellular pathogens, including Listeria and Chlamydia.7,8,45 We were able to detect mycobacterial eDNA in the cytosol of macrophages infected with MAP and M. smegmatis (Fig. 2F). The higher amounts in the cytosol of macrophages infected by viable M. smegmatis suggest that in cells infected by such bacteria the eDNA release from the phagosome is more efficient than in MAP infected cells. This might explain the stronger cGAS-STING-TBK1 activation. MAP inhibits the phagosomal maturation process and survives in macrophages. In contrast, M. smegmatis is found at 1 h post infection in acidified and maturing phagosomes which leads to the killing of M. smegmatis.19 Bacterial viability is necessary for Ifnb induction by M. smegmatis. Most probably bacterial autolysis and degradation might liberate amounts of DNA necessary to efficiently activate the signaling pathway. However, understanding the detailed molecular mechanism requires further studies.
In conclusion, our data demonstrate that IFN-I are necessary to control M. smegmatis and MAP infections. The persistence of each bacterium is determined by the amount of bacterial eDNA released into the cytosol of the host cell and the ability to activate IFN-I via the cGAS-STING-TBK1-IRF3/7 pathway. It seems that NTM that are unable to survive in macrophages release high amounts of eDNA by a yet unknown mechanism. This leads to the induction of high levels of IFN-I which helps to control infection. As MAP and other pathogenic NTM seem to restrict the release of eDNA, the incomplete induction of the host IFN-I response appears to be a specific immune evasion mechanism of these NTM that contributes to their persistence.
Materials and Methods
Bacterial strains and growth conditions
M. smegmatis mc2155 (ATCC 19420),19 M. bovis BCG Pasteur, M. avium ssp avium DSM44156, M. avium ssp avium DSM44158, M. avium ssp and hominissuis strain 04A/128746 were grown in Middlebrook 7H9 (MB) medium or on solid MB 7H10 agar (Beckton Dickinson) supplemented with 0.2–0.5% glycerol, 10% OADC (0.06% oleic acid/ 5% albumin/ 2% dextrose/ 0.003% catalase enrichment (Carl Roth). M. avium ssp paratuberculosis strain 6783 (DSM44135) and K10 strain (ATCC BCA-968) were grown in MB medium with mycobactin J (2 mg/l, IDVET) supplementation. The M. smegmatis ΔESX-1 mutant (provided by William R Jacobs Jr, Albert Einstein College of Medicine, New York, USA) was cultured in MB medium supplemented with 50 μg/ml hygromycin B (Roche).
Mouse strains
C57BL/6 mice were purchased from the Janvier. IRF3−/−,47 IRF7−/−,48 IFNAR−/−,49 STING−/−,45 and transgenic IFN-β-luciferase reporter mice (IFN-β+/Δβ-luc),36 cGAS−/-,50 MyD88−/- and TRIF−/− (the Jackson Laboratory), UNC93B−/-51 were bred at the Helmholtz Center for Infection Research (HZI), Braunschweig, Germany. Female mice aged between 8–12 weeks were used for all mouse infection experiments. Mouse infection experiments were approved by the Lower Saxony Federal State Office for Consumer Protection and Food Safety, Germany (LAVES: reference number 08/1504).
Macrophage infection
The mouse macrophage cell line RAW264.7 (ATCC® TIB-71™) was maintained in DMEM medium (Thermo Fisher) supplemented with 10% FCS (Biochrom), 1% glutamine (Thermo Fisher), 100 units/ml penicillin, 100µg/ml streptomycin (Thermo Fisher). Bone marrow derived macrophages were cultured in DMEM medium in the presence of 20% L929-cell conditioned medium for 10 d. For infection experiments, macrophages were cultured in antibiotic free medium overnight and infected with a suspension containing prevalently single mycobacteria at MOI of 5 or as indicated for 5h. For some experiments, macrophages were pre-treated with 5 µM latrunculin (Calbiochem) for 1 h prior infection.
Extraction of DNA from the cytosol of macrophages and detection of eDNA
To obtain DNA from the cytosol 1 × 107 RAW cells were seeded on 10 cm cell culture dishes and infected with mycobacteria at MOIs of 5. After 5 h the cells were scraped in 1 ml sucrose containing homogenization buffer (20 mM HEPES (Thermo Fisher)/250 mM sucrose/ 0.5 mM EDTA (Carl Roth)).52 Cell membranes were disrupted with a tissue grinder and 50 µl of lysates were taken as input controls. Each remainder lysate was centrifuged for 15 min at 15,000 x g. Post-phagosomal supernatants were incubated with 5 μg/ml DNase-free RNase (Roche) for 1 h followed by incubation with 20 mg/ml proteinase K (Carl Roth) overnight at 37°C. DNA was prepared by phenol:chloroform:isoamlyalcohol (25:24:1) extraction and isopropanol precipitation in the presence of 20 μg glycogen. DNA extraction of the input controls was performed by beating 4 times for 10 sec. in the presence of 0.1 mm silica beads and subjected to the extraction procedure as described above. Mycobacterial eDNA was detected by quantitative real-time PCR (qPCR) using primers to detect mycobacterial 16s rDNA (5’-for-TCCGAACTGAGACCGGCTTT-3’, 5’-rev-TCCAGGGCTTCACACATGCT-3’). Amounts of eDNA were calculated according to the Eq. (2)−(Ct[cytosol] -Ct[Input x DF])x100, where DF is dilution factor of respective input DNA.
qPCR
RNA was reverse transcribed using M-MLV reverse transcriptase (Promega) as described by the manufacturer and qPCR was performed using a Mx3005P qPCR system (Agilent Technologies). Ct values were normalized to the housekeeping gene Rps9 (Δct) and expressed as fold change to the untreated control (∆∆ct). Primer sequences: Ifnb-for_5’-ACCACAGCCCTCTCCATCAACTA-3’; Ifnb-rev_5’-CTCTTCTGCATCTTCCTCCGTCAT-3’; Rps9-for_5′-CTGGACGAGGGCAAGATGAAG-3′; Rps9-rev_5′-TGACGTTGGCG GATGAGCACA-3′.
Immunoblot analysis
Cells were lysed in cell extraction buffer (Thermo Fisher) supplemented with 1x protease inhibitor cocktail P8340 (Thermo Fisher), 0.5 mM AEBSF (CalBiochem) and 1x Halt phosphatase inhibitor cocktail (Thermo Fisher). Cells lysates were separated by 12.5% SDS-PAGE and transferred onto nitrocellulose membranes. Blots were incubated with anti-phospho-TBK1/NAK (Ser172)(D52C2) and anti-GAPDH (Cell Signaling).
ELISA
Cell culture supernatants were collected from infected RAW cells at 8 h and 24 h. IFN-β was analyzed following the manufacturer's instruction (IFN-β ELISA Biomedical Laboratories).
In vivo infections
C57BL/6 mice were infected i.p. with 107–108 bacteria re-suspended in 200 μl PBS (Thermo Fisher). For some experiments, mice were injected i.p. with 100 μg/ml poly(I:C) (GE Healthcare) or i.v. with 1000 units of recombinant IFN-β (Biolegend) before and after infection with MAP. Treatment with poly(I:C) and recombinant IFN-β was repeated every 3–4 and 2–3 d for 1 week, respectively.
Organ plating and histopathology
Mice were killed at 1–3 weeks. Livers, spleens and mesentery were homogenized and plated on MB agar. Number of bacteria (CFU/gram) was expressed in a logarithmic scale. Histology, hematoxylin eosin (HE), was performed in the mouse pathology platform at HZI, Braunschweig.
In vivo imaging
IFN-β+/Δβ-luc mice were injected i.p. with MAP or M. smegmatis. Mice were anesthetized with isoflurane using the gas anesthesia system (Caliper Life Sciences). Prior to image acquisition, 3 mg of luciferin (Caliper Life Sciences) dissolved in 100 µl PBS was injected i.p. Images were obtained at the consecutive time points thereafter using the IVIS-200 system (Caliper Life Sciences). The software living image (Caliper Life Sciences) was used for image and quantification of emission intensities.
Statistical analysis
Data are expressed as means ± SEM by using GraphPad Prism 5.03 (GraphPad, San Diego, CA, USA). Depending on the experiment, 2 tailed student's t-test, Mann-Whitney U-test, one-way ANOVA with Tukey post test and 2 way ANOVA with Bonferroni post test were used. The difference between samples and controls was considered statistically significant at a level of *p<0.05, **p<0.01 and ***p<0.001.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to A. Beineke (Univ. Veterinary Medicine Hannover) for expert advice in tissue pathology. We thank T. Basler (Univ. Veterinary Medicine Hannover) for the help in mycobacterial cultures. We also thank M. Hornef (Univ. Hospital Aachen) for providing the MyD88−/− and TRIF−/− mice, C. Rice (Rockefeller Univ.) for providing cGAS−/− mice, and M. Brinkmann (Helmholtz Center for Infection Research) for providing the UNC93B−/− mice. We are grateful to W. R. Jacobs Jr. (Albert Einstein College of Medicine) for providing the M. smegmatis ΔESX-1 mutant strain.
Funding
This work was supported by grants from the German Research Foundation (DFG) to RG (Ge522/6–1 and Go983/4–1). RG and SW were additionally supported by the German Federal Ministry of Education and Research (BMBF, ZooMAPII: 01KI1003A and 01KI1003C). NR was supported by Hannover Biomedical Research School (HBRS) and the Center for Infection Biology (ZIB) at Hannover Medical School. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author contributions
Designed experiments: NR SW RG. Performed the experiments: NR AN KA SL AS NJ KL JS. Analyzed the data: NR UK SW RG. Wrote the paper: NR SW RG.
References
- [1].Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 2007; 6:975-90; https://doi.org/ 10.1038/nrd2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Trinchieri G. Type I interferon: friend or foe? J Exp Med 2010; 207:2053-63; PMID:20837696; https://doi.org/ 10.1084/jem.20101664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe 2016; 19:760-9; PMID:27281568; https://doi.org/ 10.1016/j.chom.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Solodova E, Jablonska J, Weiss S, Lienenklaus S. Production of IFN-beta during listeria monocytogenes infection is restricted to monocyte/macrophage lineage. Plos One 2011; 6:e18543; https://doi.org/ 10.1371/journal.pone.0018543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stockinger S, Kastner R, Kernbauer E, Pilz A, Westermayer S, Reutterer B, Soulat D, Stengl G, Vogl C, Frenz T, et al. Characterization of the Interferon-Producing Cell in Mice Infected with Listeria monocytogenes. Plos Pathogens 2009; 5:e1000355; PMID:19325882; https://doi.org/ 10.1371/journal.ppat.1000355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, Trieu-Cuot P, Golenbock DT, Kaminski PA. Group B streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe 2016; 20:49-59; PMID:27414497; https://doi.org/ 10.1016/j.chom.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The cytosolic sensor cGAS detects mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 2015; 17:811-9; PMID:26048136; https://doi.org/ 10.1016/j.chom.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, et al. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J 2014; 33:1654-66; PMID:24970844; https://doi.org/ 10.15252/embj.201488029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 2012; 11:469-80; PMID:22607800; https://doi.org/ 10.1016/j.chom.2012.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 2007; 178:3143-52; PMID:17312162 [DOI] [PubMed] [Google Scholar]
- [11].Dorhoi A, Yeremeev V, Nouailles G, Weiner J, 3rd Jorg S, Heinemann E, Oberbeck-Muller D, Knaul JK, Vogelzang A, Reece ST, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol 2014; 44:2380-93; PMID:24782112; https://doi.org/ 10.1002/eji.201344219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, et al. Mycobacterium tuberculosis differentially Activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 2015; 17:799-810; PMID:26048138; https://doi.org/ 10.1016/j.chom.2015.05.003 [DOI] [PubMed] [Google Scholar]
- [13].Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for mycobacterium tuberculosis. Cell Host Microbe 2015; 17:820-8; PMID:26048137; https://doi.org/ 10.1016/j.chom.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Denis M. Recombinant murine beta interferon enhances resistance of mice to systemic Mycobacterium avium infection. Infect Immun 1991; 59:1857-9; PMID:2019446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kuchtey J, Fulton SA, Reba SM, Harding CV, Boom WH. Interferon-alphabeta mediates partial control of early pulmonary Mycobacterium bovis bacillus Calmette-Guerin infection. Immunology 2006; 118:39-49; PMID:16630021; https://doi.org/ 10.1111/j.1365-2567.2006.02337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, Bishai WR. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med 2015; 21:401-6; PMID:25730264; https://doi.org/ 10.1038/nm.3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Biet F, Boschiroli ML. Non-tuberculous mycobacterial infections of veterinary relevance. Res Vet Sci 2014; 97 Suppl:S69-77; PMID:25256964; https://doi.org/ 10.1016/j.rvsc.2014.08.007 [DOI] [PubMed] [Google Scholar]
- [18].Pierre-Audigier C, Jouanguy E, Lamhamedi S, Altare F, Rauzier J, Vincent V, Canioni D, Emile JF, Fischer A, Blanche S, et al. Fatal disseminated Mycobacterium smegmatis infection in a child with inherited interferon gamma receptor deficiency. Clin Infect Dis 1997; 24:982-4; PMID:9142806 [DOI] [PubMed] [Google Scholar]
- [19].Kuehnel MP, Goethe R, Habermann A, Mueller E, Rohde M, Griffiths G, Valentin-Weigand P. Characterization of the intracellular survival of Mycobacterium avium ssp. paratuberculosis: phagosomal pH and fusogenicity in J774 macrophages compared with other mycobacteria. Cell Microbiol 2001; 3:551-66; PMID:11488816 [DOI] [PubMed] [Google Scholar]
- [20].Wallace RJ, Jr, Nash DR, Tsukamura M, Blacklock ZM, Silcox VA. Human disease due to Mycobacterium smegmatis. J Infect Dis 1988; 158:52-9; PMID:3392420 [DOI] [PubMed] [Google Scholar]
- [21].Sweeney KA, Dao DN, Goldberg MF, Hsu T, Venkataswamy MM, Henao-Tamayo M, Ordway D, Sellers RS, Jain P, Chen B, et al. A recombinant Mycobacterium smegmatis induces potent bactericidal immunity against Mycobacterium tuberculosis. Nat Med 2011; 17:1261-8; PMID:21892180; https://doi.org/ 10.1038/nm.2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Naser SA, Sagramsingh SR, Naser AS, Thanigachalam S. Mycobacterium avium subspecies paratuberculosis causes Crohn's disease in some inflammatory bowel disease patients. World J Gastroenterol 2014; 20:7403-15; PMID:24966610; https://doi.org/ 10.3748/wjg.v20.i23.7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Paccagnini D, Sieswerda L, Rosu V, Masala S, Pacifico A, Gazouli M, Ikonomopoulos J, Ahmed N, Zanetti S, Sechi LA. Linking chronic infection and autoimmune diseases: Mycobacterium avium subspecies paratuberculosis, SLC11A1 polymorphisms and type-1 diabetes mellitus. PLoS One 2009; 4:e7109; PMID:19768110; https://doi.org/ 10.1371/journal.pone.0007109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Naser SA, Thanigachalam S, Dow CT, Collins MT. Exploring the role of Mycobacterium avium subspecies paratuberculosis in the pathogenesis of type 1 diabetes mellitus: a pilot study. Gut Pathog 2013; 5:14; PMID:23759115; https://doi.org/ 10.1186/1757-4749-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Frau J, Cossu D, Coghe G, Lorefice L, Fenu G, Porcu G, Sardu C, Murru MR, Tranquilli S, Marrosu MG, et al. Role of interferon-beta in Mycobacterium avium subspecies paratuberculosis antibody response in Sardinian MS patients. J Neurol Sci 2015; 349:249-50; PMID:25598492; https://doi.org/ 10.1016/j.jns.2015.01.004 [DOI] [PubMed] [Google Scholar]
- [26].Rumsey J, Valentine JF, Naser SA. Inhibition of phagosome maturation and survival of Mycobacterium avium subspecies paratuberculosis in polymorphonuclear leukocytes from Crohn's disease patients. Med Sci Monit 2006; 12:BR130-9; PMID:16572045 [PubMed] [Google Scholar]
- [27].Basler T, Holtmann H, Abel J, Eckstein T, Baumer W, Valentin-Weigand P, Goethe R. Reduced transcript stabilization restricts TNF-alpha expression in RAW264.7 macrophages infected with pathogenic mycobacteria: evidence for an involvement of lipomannan. J Leukoc Biol 2010; 87:173-83; PMID:19850884; https://doi.org/ 10.1189/jlb.0309207 [DOI] [PubMed] [Google Scholar]
- [28].Kabara E, Coussens PM. Infection of Primary Bovine Macrophages with Mycobacterium avium Subspecies paratuberculosis Suppresses Host Cell Apoptosis. Front Microbiol 2012; 3:215; PMID:22833736; https://doi.org/ 10.3389/fmicb.2012.00215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zur Lage S, Goethe R, Darji A, Valentin-Weigand P, Weiss S. Activation of macrophages and interference with CD4+ T-cell stimulation by Mycobacterium avium subspecies paratuberculosis and Mycobacterium avium subspecies avium. Immunology 2003; 108:62-9; PMID:12519304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun 2003; 306:860-6; PMID:12821121 [DOI] [PubMed] [Google Scholar]
- [31].Cooney MA, Steele JL, Steinberg H, Talaat AM. A murine oral model for Mycobacterium avium subsp. paratuberculosis infection and immunomodulation with Lactobacillus casei ATCC 334. Front Cell Infect Microbiol 2014; 4:11; PMID:24551602; https://doi.org/ 10.3389/fcimb.2014.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Koc A, Bargen I, Suwandi A, Roderfeld M, Tschuschner A, Rath T, Gerlach GF, Hornef M, Goethe R, Weiss S, et al. Systemic and mucosal immune reactivity upon Mycobacterium avium ssp. paratuberculosis infection in mice. PLoS One 2014; 9:e94624; PMID:24728142; https://doi.org/ 10.1371/journal.pone.0094624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suwandi A, Bargen I, Roy B, Pils MC, Krey M, Zur Lage S, Basler T, Rohde M, Falk CS, Hornef MW, et al. Experimental colitis is exacerbated by concomitant infection with Mycobacterium avium ssp. paratuberculosis. Inflamm Bowel Dis 2014; 20:1962-71; PMID:25144571; https://doi.org/ 10.1097/MIB.0000000000000157 [DOI] [PubMed] [Google Scholar]
- [34].Meissner T, Eckelt E, Basler T, Meens J, Heinzmann J, Suwandi A, Oelemann WM, Trenkamp S, Holst O, Weiss S, et al. The Mycobacterium avium ssp. paratuberculosis specific mptD gene is required for maintenance of the metabolic homeostasis necessary for full virulence in mouse infections. Front Cell Infect Microbiol 2014; 4:110; PMID:25177550; https://doi.org/ 10.3389/fcimb.2014.00110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ghosh P, Steinberg H, Talaat AM. Virulence and immunity orchestrated by the global gene regulator sigL in Mycobacterium avium subsp. paratuberculosis. Infect Immun 2014; 82:3066-75; PMID:24799632; https://doi.org/ 10.1128/iai.00001-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol 2009; 183:3229-36; PMID:19667093; https://doi.org/ 10.4049/jimmunol.0804277 [DOI] [PubMed] [Google Scholar]
- [37].Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VA, Vogel SN, Fitzgerald KA, Sutterwala FS, Briken V. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-beta and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J Immunol 2013; 191:3514-8; PMID:23997220; https://doi.org/ 10.4049/jimmunol.1301331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].De Leon J, Jiang G, Ma Y, Rubin E, Fortune S, Sun J. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J Biol Chem 2012; 287:44184-91; PMID:23150662; https://doi.org/ 10.1074/jbc.M112.420869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S, Drobits B, Li XD, Knapp S, Kovarik P. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog 2011; 7:e1001345; PMID:21625574; https://doi.org/ 10.1371/journal.ppat.1001345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, Golenbock DT. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe 2008; 4:543-54; PMID:19064255; https://doi.org/ 10.1016/j.chom.2008.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Prakash A, Smith E, Lee CK, Levy DE. Tissue-specific positive feedback requirements for production of type I interferon following virus infection. J Biol Chem 2005; 280:18651-7; PMID:15767254; https://doi.org/ 10.1074/jbc.M501289200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol 2015; 15:87-103; PMID:25614319; https://doi.org/ 10.1038/nri3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, Castro F, Cardoso V, Gaifem J, Wu X, et al. Type I IFN Inhibits Alternative Macrophage Activation during Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-gamma Signaling. J Immunol 2016; 197:4714-26; PMID:27849167; https://doi.org/ 10.4049/jimmunol.1600584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pahari S, Khan N, Aqdas M, Negi S, Kaur J, Agrewala JN. Infergen stimulated macrophages restrict mycobacterium tuberculosis growth by autophagy and release of nitric oxide. Sci Rep 2016; 6:39492; PMID:28000752; https://doi.org/ 10.1038/srep39492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol 2014; 193:2394-404; PMID:25070851; https://doi.org/ 10.4049/jimmunol.1302718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Basler T, Geffers R, Weiss S, Valentin-Weigand P, Goethe R. Mycobacterium avium subspecies induce differential expression of pro-inflammatory mediators in a murine macrophage model: evidence for enhanced pathogenicity of Mycobacterium avium subspecies paratuberculosis. Immunobiology 2008; 213:879-88; PMID:18926302; https://doi.org/ 10.1016/j.imbio.2008.07.009 [DOI] [PubMed] [Google Scholar]
- [47].Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000; 13:539-48; PMID:11070172 [DOI] [PubMed] [Google Scholar]
- [48].Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005; 434:772-7; PMID:15800576; https://doi.org/ 10.1038/nature03464 [DOI] [PubMed] [Google Scholar]
- [49].Spanier J, Lienenklaus S, Paijo J, Kessler A, Borst K, Heindorf S, Baker DP, Kroger A, Weiss S, Detje CN, et al. Concomitant TLR/RLH signaling of radioresistant and radiosensitive cells is essential for protection against vesicular stomatitis virus infection. J Immunol 2014; 193:3045-54; PMID:25127863; https://doi.org/ 10.4049/jimmunol.1400959 [DOI] [PubMed] [Google Scholar]
- [50].Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014; 505:691-5; PMID:24284630; https://doi.org/ 10.1038/nature12862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 2006; 7:156-64; PMID:16415873; https://doi.org/ 10.1038/ni1297 [DOI] [PubMed] [Google Scholar]
- [52].Luhrmann A, Haas A. A method to purify bacteria-containing phagosomes from infected macrophages. Methods Cell Sci 2000; 22:329-41; PMID:11549946 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.