

Abstract
Oligonucleotides (ONs) modified with 2′-O-(pyren-1-yl)methylribonucleotides have been explored for a range of applications in molecular biology, nucleic acid diagnostics, and materials science for more than 25 years. The first part of this review provides an overview of synthetic strategies toward 2′-O-(pyren-1-yl)methylribonucleotides and is followed by a summary of biophysical properties of nucleic acid duplexes modified with these building blocks. Insights from structural studies are then presented to rationalize the reported properties. In the second part, applications of ONs modified with 2′-O-(pyren-1-yl)methyl-RNA monomers are reviewed, which include detection of RNA targets, discrimination of single nucleotide polymorphisms, formation of self-assembled pyrene arrays on nucleic acid scaffolds, the study of charge transfer phenomena in nucleic acid duplexes, and sequence-unrestricted recognition of double-stranded DNA. The predictable binding mode of the pyrene moiety, coupled with the microenvironment-dependent properties and synthetic feasibility, render 2′-O-(pyren-1-yl)methyl-RNA monomers as a promising class of pyrene-functionalized nucleotide building blocks for new applications in molecular biology, nucleic acid diagnostics, and materials science.
Graphical Abstract
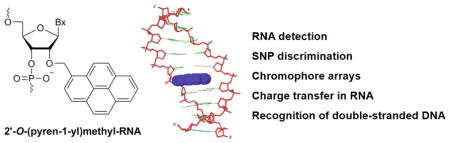
This review highlights the synthesis, biophysical properties, and wide range of applications of oligonucleotides modified with 2′-O-(pyren-1-yl)methyl-RNA monomers reported over the past 25 years.
1. Introduction
The past decades have witnessed the development of a great variety of pyrene-functionalized nucleotides.1 These efforts have been fueled by an aspiration for building blocks that can modulate the stability of - and bestow new function to - nucleic acid structures and enable new applications in nucleic acid based nanotechnology, diagnostics, and therapeutics. Pyrene moieties are of particular interest due to their microenvironment-sensitive fluorescence properties,2–6 π-stacking ability,7,8 and differential binding modes that depend on the nature of the nucleic acid targets. For example, localization of pyrene moieties in the groove of nucleic acid duplexes typically results in strong fluorescence emission in the 370–420 nm ‘monomer’ region as quenching interactions between pyrene and nucleobase moieties are minimized. At the same time, reduced duplex thermostability is frequently observed as the normally stabilizing hydration spine is perturbed by the bulky and hydrophobic pyrene moiety. In contrast, intercalating pyrene moieties typically result in weak fluorescence emission and increased duplex thermostability because pyrene and nucleobase moieties engage in strong stacking interactions (the aromatic surface area of pyrene (~220 Å) is similar to that of an A:T Watson-Crick base pair (~269 Å)).8 Pyrene chromophores may also display excimer fluorescence if a pyrene in an electronically excited state forms a π-stacking dimer with a pyrene in its ground state and the interplanar distance is less than 4 Å.7
Pyrene moieties can be linked to nucleotides in numerous ways, e.g., by replacing the nucleobase moiety, through attachment to the nucleobase moiety, or through attachment to the sugar moiety. The attachment point and linker length have a significant impact on the binding and photophysical properties of the resulting pyrene-functionalized oligonucleotides. For a review of pyrene-functionalized nucleotide monomers and their applications, please consult reference 1. 2′-O-(Pyren-1-yl)methyl-RNA monomers, in which the pyrene moiety is linked via a methylene group to the O2′-position of a ribonucleotide, have enjoyed particularly prominent attention (Figure 1). Applications of oligonucleotides (ONs) modified with 2′-O-(pyren-1-yl)methyl-RNA monomers (also abbreviated O2′-PyMe-ONs in the following) include (a) detection of single-stranded RNA targets using pyrene’s monomer or excimer fluorescence emission, (b) development of probes for discrimination of single nucleotide polymorphisms (SNPs), (c) formation of self-assembled pyrene arrays on nucleic acid scaffolds, (d) the study of charge transfer phenomena in RNA duplexes, and (e) sequence-unrestricted recognition of double-stranded DNA. This focused review will discuss these applications after providing an overview of i) synthetic strategies towards 2′-O-(pyren-1-yl)methyl-RNA monomers, ii) thermal denaturation and fluorescence properties of nucleic acid duplexes modified with these building blocks, and iii) insights from structural studies.
Figure 1.

Structure of 2′-O-(pyren-1-yl)methyl-RNA monomers A, C, G, and U.
2. Synthesis of 2′-O-(pyren-1-yl)methyl RNA building blocks
Despite their structural simplicity, 2′-O-alkylated ribonucleotides are challenging to synthesize due to the presence of multiple nucleophilic groups, which either necessitate regioselective O2′-functionalization or the use of protecting groups prior to O2′-functionalization and subsequent removal of the protecting groups.9
In 1991, Yamana and coworkers reported the first synthesis of 2′-O-(pyren-1-yl)methyluridine phosphoramidite 5 (Scheme 1).10 Uridine 1 is O3′,O5′-ditritylated with modest regioselectivity and yield,11 followed by O2′-arylmethylation of 2 using the Williamson ether synthesis approach and detritylation. Key intermediate 3 is then O5′-dimethoxytritylated and O3′-phosphitylated to afford target phosphoramidite 5 in ~10% yield over five steps from uridine.
Scheme 1.

Original route to 2′-O-(pyren-1-yl)methyluridine phosphoramidite 5:10 (a) TrCl, pyridine (27%)11; (b) (i) KOH, 1-chloromethylpyrene, benzene/dioxane, (ii) 80% aq. AcOH (65% from 2)10; (c) DMTr-Cl, DMAP, pyridine (72%);12 (d) NC(CH2)2OP(Cl)N(i-Pr)2, (i-Pr)2NEt, CH2Cl2 (76%).12
Our group found the initial O3′,O5′-tritylation of uridine to be cumbersome and therefore devised an alternative route12 to key intermediate 3 that is based on trialkylborate13 mediated ring opening of O2,O2′-anhydrouridine 6 using 1-pyrenemethanol in anhydrous DMSO at high temperatures (Scheme 2). Key intermediate 3 is obtained in slightly higher overall yield (~18% vs ~23%) and this route is, in our opinion, more practical across a wider range of reaction scales.
Scheme 2.

Alternative route to key intermediate 3:12 (a) (PhO)2CO, NaHCO3, DMF (90%)14; (b) 1-pyrenemethanol, BH3·THF, DMSO (25%).12
Wada and coworkers recently reported a route to a potential precursor of 3 in which uridine 1 is converted to 3-N-benzoyl-2′-O-trimethylsilyl-3′,5′-O-(di-tert-butylsilanediyl)uridine 7 in 96% yield via a telescoped three step procedure (Scheme 3). This intermediate is then O2′-pyrenylmethylated in ~53% yield under reductive conditions using 1-pyrenecarbaldehyde, triethylsilane, and trimethylsilyl trifluoromethanesulfonate as a Lewis acid.15 Although the global deprotection of the resulting precursor 8 to give 3 has not been reported yet, this route is a promising alternative pathway to key intermediate 3.
Scheme 3.

Route to potential intermediate 8:15 (a) (i) (t-Bu)2Si(OTf)2, DMF; (ii) Me3SiCl, imidazole; (iii) BzCl, (i-Pr)2NEt, pyridine (96% from 1); (b) PyCHO, Et3SiH, TMSOTf, 1,4-dioxane (53%).
The other canonical 2′-O-(pyren-1-yl)methylribonucleotides have been obtained via a different strategy (Scheme 4).16,17 Thus, the free nucleosides are first O2′-pyrenylmethylated via Williamson’s approach in modest regioselectivity and yield. This is followed by N-acylation of the exocyclic amines via transient protection protocols, O5′-dimethoxytritylation, and O3′-phosphitylation. This reaction sequence affords the protected target nucleotides in 9–12% overall yield over four steps from the free nucleosides.
Scheme 4.

Synthetic routes to 2′-O-(pyren-1-yl)methyl-adenosine, -cytidine and -guanosine phosphoramidites:16,17 (a) PyCH2Cl, NaH, and DMF (9a and 9c) or DMSO (9g); (b) (i) TMSCl, pyridine, (ii) BzCl (10a and 10c) or isobutyrylchloride (10g), pyridine, (iii) aq. NH3; (c) DMTCl, pyridine; (d) (i-Pr2N)2PO(CH2)2CN, tetrazole, CH2Cl2 (12a and 12c) or NC(CH2)2OP(Cl)N(i-Pr)2, (i-Pr)2NEt, CH2Cl2 (12g). Pg = protecting groups.
3. Thermal denaturation properties of O2′-PyMe-modified ONs
Several research teams have used the abovementioned phosphoramidites to incorporate 2′-O-(pyren-1-yl)methyl-RNA monomers A, C, G, and U into oligonucleotides via solid-phase synthesis and study the hybridization properties of the resulting ONs. For example, the teams of Murakami and Yamana,18,19 and later our group,12,17 have shown that DNA strands modified with 2′-O-(pyren-1-yl)methyl-RNA monomers generally form greatly stabilized duplexes with complementary DNA (cDNA), with increases in thermal denaturation temperatures (Tm’s) of up to 13 °C per incorporation relative to corresponding unmodified DNA duplexes. Studies in which the nature of the 2′-O-(pyren-1-yl)methyl-RNA monomers and the 3′-flanking nucleotide were systematically varied, have shown that the duplex stabilizing effect is strongly sequence context dependent.17 Thus, ONs in which the 2′-O-(pyren-1-yl)methyl-RNA monomer is flanked by a 3′-purine consistently display significantly greater cDNA affinity (approximate order: 3′-A>3′-G≫3′-T>3′-C). This is indicative of pyrene intercalation in DNA duplexes, as 3′-flanking purines result in stronger stacking interactions. The four canonical 2′-O-(pyren-1-yl)methyl-RNA monomers have differential effects on duplex stability.17 Thus, the G monomer has a considerably lower stabilizing effect than the other 2′-O-(pyren-1-yl)methyl-RNA monomers and is even destabilizing in some sequence contexts.
DNA strands that are modified with 2′-O-(pyren-1-yl)methyl-RNA monomers display significantly lower affinity towards complementary RNA (cRNA) and, in fact, often result in destabilized heteroduplexes relative to unmodified control duplexes.12,17,19 In other words, a pronounced preference for DNA binding is observed with DNA-selectivity values – defined as ΔΔTm (DNA-RNA) = ΔTm (vs DNA) - ΔTm (vs RNA) – typically in the range of 7–14 °C for short, singly modified ONs.12,18 The observed DNA-selectivity is consistent with the known B-type duplex preference for intercalators (vide infra).
2′-O-(Pyren-1-yl)methylribonucleotides have also been incorporated into RNA and 2′-O-methyl-RNA strands. Singly modified RNA strands display similar or slightly higher affinity towards cDNA relative to unmodified RNA strands, but decreased cRNA affinity.19,20 The effect upon incorporation of a single 2′-O-(pyren-1-yl)methylribonucleotide into 2′-O-methyl-RNA strands varies from moderately stabilizing with A-rich RNA targets to moderately destabilizing with mixed-sequence RNA targets (ΔTm between -3.9 °C to +3.9 °C),16,21 whereas the affinity against 2′-O-methyl-RNA targets with repeating pyrimidine-purine steps generally is lower than with unmodified 2′-O-methyl-RNA strands. In summary, these observations suggest that 2′-O-(pyren-1-yl)methylribonucleotides are affinity-enhancing modifications in DNA-like duplexes, whereas they are less stabilizing, and at times even destabilizing, in RNA-like duplexes.
4. Other biophysical and structural studies of O2′-CH2Py-modified nucleic acid duplexes
Biophysical and structural studies have been performed on duplexes that are modified with 2′-O-(pyren-1-yl)methylribonucleotides to determine the binding mode of the pyrene moiety and thereby rationalize the observed thermal denaturation and fluorescence trends.
The circular dichroism (CD) profiles in the region below 300 nm of duplexes between U-modified DNA strands and cDNA or cRNA, are similar to those of the corresponding unmodified duplexes, suggesting that the global helix geometries are largely unperturbed. However, induced CD peaks were observed in the 335–350 nm region, indicating a coupling between the transition moments of the planar and achiral pyrene moiety and the chiral base-pairs. Shifts to longer wavelength are observed upon hybridization with cDNA targets, whereas only very weak induced CD peaks were observed upon cRNA binding.18,19
Pyrene absorption maxima of O2′-CH2Py-modified DNA strands are shifted prominently to the red upon cDNA binding (Δγ up to 5 nm), whereas the shifts are less pronounced with cRNA. 12,17,19 On the other hand, O2′-CH2Py-modified RNA strands display pronounced hypsochromic shifts upon hybridization with cRNA (Δγ down to -5 nm).19 These observations suggest that the pyrene moieties are located in very different local environments in DNA:DNA, DNA:RNA and RNA:RNA duplexes. Given that hybridization-induced bathochromic shifts of pyrene absorption maxima are indicative of ground-state electronic interactions between intercalating pyrene moieties and flanking nucleobases,22 it can be inferred that the pyrene moiety of 2′-O-(pyren-1-yl)methylribonucleotides is intercalating in DNA:DNA duplexes and - to a lesser degree - in DNA:RNA duplexes, whereas it is located in extrahelical positions in RNA:RNA duplexes. This is consistent with the observation that intercalators prefer to intercalate into B-type DNA duplexes, which are more elongated and with more efficient nucleobase overlap, rather than A-type RNA duplexes, which are more compressed with less efficient nucleobase overlap.23
Further support for this binding hypothesis is provided from fluorescence emission spectra of O2′-CH2Py-modified duplexes. In general, low emission levels - consistent with efficient nucleobase-mediated quenching of pyrene fluorescence4,6 - are observed for duplexes between O2′-CH2Py-modified DNA strands and cDNA or cRNA, whereas duplexes between O2′-CH2Py-modified RNA strands and cRNA exhibit much more prominent levels of emission.19 Furthermore, fluorescence anisotropy measurements suggest that pyrene moieties in O2′-CH2Py-modified RNA duplexes rotate more freely consistent with an extrahelical location, whereas they are more confined in O2′-CH2Py-modified DNA duplexes consistent with stacking and intercalation.19 The fluorescence properties of O2′-CH2Py-modified ONs and their duplexes with cDNA/cRNA will be discussed in subsequent sections.
Additional support for the proposed binding models has been obtained from molecular dynamics (MD) simulations of O2′-CH2Py-modified duplexes.19 Thus, the final structure of a 10-mer U-modified RNA duplex suggests that the duplex retains its canonical A-type helix geometry, with the pyrene moiety pointing away from the helix where it is free from π-π stacking interactions with flanking nucleobases (Figure 2a). In contrast, the corresponding U-modified DNA duplex adopts a typical B-type geometry in which the pyrene moiety is intercalating into the duplex, engaging in π-π stacking interactions with flanking base-pairs (Figure 2b). Our group observed similar outcomes in MD simulations of U-modified DNA duplexes.24 Strong structural support for an intercalating binding mode of the pyrene moiety in DNA duplexes was obtained from a 2D NOESY spectrum of a U-modified DNA duplex.19 NOE crosspeaks between the hydrogens from the pyrene moiety and hydrogens from the flanking base-pairs were observed; the absence of NOE crosspeaks between U’s H1′ proton and protons from the 3′-flanking nucleobase – which are normally observed in DNA duplexes – was also noted (Figure 2c). These observations are consistent with a 3′-intercalative binding mode of the pyrene (see Figure 2d).
Figure 2.

Structures obtained from MD simulations of a U-modified (a) RNA duplex (5′-r-CCUCAUGAGG-3′ and cRNA) and (b) DNA duplex (5′-d-CCUCATGAGG-3′ and cDNA). (c) Observed NOE contacts between nucleobase and pyrene protons in a U-modified DNA duplex (5′-C1C2U3A4G5C6T7A8G9G10 : 3′-G10G9A8T7C6G5A4U3C2C1). (d) Illustration of a 3′-intercalative binding mode. Panels a–c are reproduced from reference 19 with permission from Oxford University Press.
The ability to precisely control the pyrene moiety’s position in different O2’-CH2Py-modified duplexes has been utilized in a variety of applications, which will be highlighted next.
5. Applications of O2′-CH2Py-modified ONs in nucleic acid based nanotechnology, diagnostics, and therapeutics
Oligonucleotides modified with 2′-O-(pyren-1-yl)methyl-RNA monomers have been utilized for several explorative applications including detection of single-stranded RNA targets using the pyrene moiety’s monomer or excimer fluorescence emission; development of probes for SNP discrimination; formation of self-assembled pyrene arrays on nucleic acid scaffolds; the study of charge transfer phenomena in RNA duplexes: and sequence-unrestricted recognition of double-stranded DNA.
5.1 Detection of single-stranded RNA targets based on pyrene monomer fluorescence
Research over the past decade has revealed many new roles for RNA species in gene expression and regulation. To clarify these roles, there is an increased demand for probes that enable detection of RNA species in homogenous assays and cells. Pyrene-modified oligonucleotides have been extensively studied as model probes toward this end due to the high quantum yield and position-dependent emission characteristics of pyrene.1
In 1999, Murakami and coworkers reported that singly U-modified DNA probes of the sequence 5′-CATGUBTAC (B = A/C/G/T) only are weakly fluorescent, presumably due to stacking interactions between pyrene and nucleobase moieties resulting in efficient quenching of pyrene fluorescence, whereas the corresponding duplexes with cRNA are more emissive.18 The emission increases are highly sequence-context dependent, ranging from 1.3-fold increases when a 2′-deoxyadenosine is flanking the U monomer on the 3′-side over to 250-fold increases for probes with 3′-flanking 2′-deoxycytidines. These variations are a result of different quenching efficiencies of the 3′-flanking nucleobases,4,6 and sequence-context dependent binding preferences of the pyrene moieties (e.g., more efficient stacking and intercalation with 3′-flanking purines). Binding to cDNA, on the other hand, does not result in major emission changes, as the fluorescence of the intercalating pyrene moiety is efficiently quenched by electron transfer from the excited pyrene to the flanking nucleobases.18 It should be noted that no attempts were made to remove dissolved oxygen, a potential quencher of pyrene fluorescence. Our group has studied U-modified DNA probes in AT-rich sequences and consistently observed decreased emission upon cDNA/cRNA binding, highlighting that U-modified DNA probes are too sequence-context dependent to be useful for diagnostic applications.12 DNA strands modified with one of the other 2′-O-(pyren-1-yl)methylribonucleotides, i.e., A, C or G, also generally result in decreased emission upon cDNA binding.17
In 2001, Yamana and coworkers demonstrated that U-modified RNA probes result in prominent fluorescence increases upon duplex formation with cRNA, irrespective of the nature of the 3′-flanking nucleotide (13–33 fold increases, Figure 3a).20 The resulting duplexes are strongly fluorescent with quantum yields of up to ~24%. In contrast, the emission of duplexes with singly mismatched RNA targets is 4- to 30-fold less intense, provided the mismatched base-pair is close to the 2′-O-(pyren-1-yl)methyluridine monomer, suggesting that these probes can discriminate SNPs in RNA. Importantly, SNP discrimination can be realized under non-stringent conditions, i.e., conditions where mismatched duplexes are formed, which avoids the need for rigorous washing protocols to remove mismatched duplexes. Duplex formation between U-modified RNA probes and cDNA, on the other hand, does not result in substantial emission changes and quantum yields are <1% (Figure 3b). These observations are consistent with the proposed binding modes, i.e., the pyrene moieties are projected into the non-quenching minor groove in matched RNA duplexes, whereas they are more likely to intercalate in mismatched RNA duplexes or in duplexes with cDNA (Figure 3c).
Figure 3.

Steady-state fluorescence emission spectra of (a) 5′-r-ACAUGCAGUGUUGAU (---) and the corresponding duplex with RNA (___), and (b) 5′-d-ACATGCAGUGTTGAT (---) and the corresponding duplex with RNA (___) (λεξ = 338 nm). Note the vastly different Y-axis scales. (c) U-modified RNA probes result in large emission increases upon RNA binding, but less pronounced increases upon DNA binding. Panels a and b are reproduced from reference 20 with permission from Wiley.
Murakami and coworkers later extended this design principle to U-modified 2′-O-methyl-RNA probes, with the goal of developing more enzymatically stable probes for diagnostic in vivo applications.25 The fluorescence of a model probe – featuring a 3′-flanking cytosine – was found to increase 334-fold upon cRNA binding, whereas significantly less prominent increases were observed with cDNA. A set of seven U-modified 2′-O-methyl-RNA probes were evaluated for their ability to detect potential acceptor sites for antisense ONs on folded 16S-rRNA from E. coli. Three of the selected target regions were predicted to be single-stranded in nature whereas the remaining four targets were predicted to be at least partially embedded in intramolecular double-stranded structures. At 37 °C, five of the U-modified 2′-O-methyl-RNA probes – the three probes targeting the single-stranded regions and two probes targeting stem regions – resulted in major emission increases at 375 nm upon incubation with the folded 16S-rRNA under physiologically relevant conditions, suggesting that these regions are accessible to antisense ONs.
In 2013, our group expanded the concept of U-modified ONs with RNA-like backbones for RNA detection, demonstrating that DNA probes with central segments of alternating locked nucleic acid (LNA)26 and 2′-O-(pyren-1-yl)methyluridine monomers display very pronounced and highly mismatch-sensitive increases in pyrene emission upon RNA binding (Figure 4).27 Thus, cRNA binding resulted in 21-to 83-fold increases in emission intensities, whilst the emission level of mismatched duplexes was 2.1- to 19-fold lower. This approach relies on the ability of LNA monomers to increase the North-type character of flanking 2′-deoxyribonucleotides and thereby tune the central region toward a more RNA-like geometry.28,29 Presumably, LNA-induced conformational tuning of flanking 2′-O-(pyren-1-yl)methyluridine monomers places the pyrene moiety in the minor groove upon RNA binding, whereby pyrene-nucleobase interactions normally leading to quenching of fluorescence are minimized. The presence of mismatched base-pairs, on the other hand, increases the likelihood of intercalation and quenching of the pyrene moieties. Support for the proposed binding modes was obtained from absorption spectroscopy. Thus, U-modified DNA/LNA mixmer probes result in significant hypsochromic shifts of pyrene absorption maxima upon cRNA binding (Δλ ~ -6 nm), whereas less pronounced shifts were observed upon binding with mismatched RNA targets. Quantum yields of the brightest duplexes with cRNA were estimated to approach 40%. A major advantage of U-modified DNA/LNA-mixmer probes over the corresponding RNA probes is that they are prepared under mild DNA-synthesis conditions and more chemically and enzymatically stable.
Figure 4.

(a) DNA probes with central segments of alternating LNA and U-monomers result in large emission enhancements upon binding with cRNA, whereas duplexes with mismatched RNA targets only are scarcely emissive. (b) Fluorescence intensity at λem = 376 nm of single-stranded probes (SSP) and duplexes with complementary or centrally mismatched RNA targets (mmRNA, nature of mismatched nucleotide is specified) (λex = 350 nm). Lower case a, c, g and t denote LNA monomers. Hybridization-induced increases (i.e., the intensity ratio between a matched duplex and single-stranded probe) and discrimination factors (i.e., the intensity ratio between a matched and mismatched duplex) are listed above the corresponding histograms. Adapted from reference 27 with permission from the Royal Society of Chemistry.
In a related subsequent study, Tang and co-workers reported on the emission characteristics of DNA probes with deoxyribonucleotides, ribonucleotides, 2′-O-methylribonucleotides or 2′-deoxy-2′-fluororibonucleotides flanking a central 2′-O-(pyren-1-yl)methyluridine monomer.30 The proximal nucleotides were found to have a distinctive effect on the emission levels of the pyrene moiety upon RNA binding, with the most prominent emission increases being reported for probes with flanking 2′-deoxy-2′-fluororibonucleotides nucleotides (2′-F > 2′-OMe > 2′-OH ≫ 2′-H). The brightest duplexes have fluorescence quantum yields approaching 30%. Increasing the number of flanking RNA-like nucleotides beyond one on either side of the U monomer had only limited additional impact on emission characteristics. This suggests that the nucleotides that directly flank the U monomer have the greatest influence on the binding mode of the pyrene moieties. Molecular dynamic simulations suggest that the pyrene moiety intercalates in duplexes with RNA when the central 2′-O-(pyren-1-yl)methyluridine is flanked by 2′-deoxyribonucleotides, whereas the pyrene is positioned in the minor grove when 2′-deoxy-2′-fluororibonucleotides, 2′-O-methylribonucleotides, or ribonucleotides flank the U monomer. DNA probes in which U monomers were flanked by 2′-deoxy-2′-fluororibonucleotides were used to detect in vitro T7 RNA transcripts with excellent mismatch specificity, underscoring the diagnostic potential of these class of probes.
5.2 Detection of single-stranded RNA targets based on pyrene excimer fluorescence
The large Stoke’s shift (>130 nm) of pyrene–pyrene excimers renders excimer-forming probes attractive for nucleic acid diagnostics as this may reduce background fluorescence from unbound probes. For a comprehensive review of excimer-forming probes, see reference 1. In 2002, Murakami and coworkers reported the first example of RNA detection probes that rely on pyrene excimer signals from consecutively arranged 2′-O-(pyren-1-yl)methylribonucleotides.31 Thus, duplex formation between 2′-O-methyl-RNA probes with two sequential U monomers and cRNA was shown to result in 12- to 97-fold increases in excimer emission at 480 nm, presumably because the pyrene moieties stack in the minor groove of the formed duplex (Figure 5a). In contrast, hybridization with RNA targets which places mismatched nucleotides near the two U monomers only results in negligible excimer emission, suggesting that these probes can be used to discriminate RNA SNPs under non-stringent conditions. Duplex formation with cDNA also only results in low levels of excimer emission, presumably because the pyrene moieties intercalate into the formed duplex. A set of three 2′-O-methyl-RNA probes with two sequential U monomers were evaluated for their ability to detect potential acceptor sites for antisense ONs on natively folded 5S-rRNA from E. coli. As expected, only the probe targeting a single-stranded region resulted in prominent excimer emission on incubation with the 5S-rRNA target, whereas probes targeting sequences that are partially or fully embedded within double-stranded stem regions did not result in significant excimer signal.
Figure 5.

(a) Fluorescence emission spectra of a 2′-O-methyl-RNA probe with two sequential U monomers in absence (----) or presence of cRNA (___) (λex = 342 nm). (b) Illustration of a microarray-based RNA detection platform based on such probes. (c) Fluorescence microscopic images of probe-functionalized slides incubated with RNA that is complementary to the CF3 probe (340/15 nm excitation and 480/30 nm emission band-pass filters used). Sequences of immobilized probes are indicated on the right top corner of each image. CF3: 5′-thiol-C6-PEG-d(T)12-2′-OMe-RNA-GAUGUGUUUCUCCUC, JN2: 5′-thiol-C6-PEG-d(T)12-2′-OMe-RNA-GAGGGCUUCGUGCGC, CM3: 5′-thiol-C6-d(T)12-2′-OMe-RNA-UUUUCAUUGUUUUCC and cRNA-CF3: 5′-r-GAGGAGAAACACAUC. Adapted from reference 32 with permission from Elsevier.
In a subsequent study, Murakami and coworkers immobilized 2′-O-methyl-RNA probes with two sequential U monomers onto glass slides, allowing for solid-phase label-free detection of RNA targets (Figure 5b).32 The signal-to-background ratios upon cRNA binding were found to be highly linker-dependent, with the largest ratios (~22) being observed when the probes were immobilized on the glass slides using PEG-(dT)12-linkers (Figure 5c – upper left). Incubation with scrambled RNA targets did not result in substantially elevated signal-to-background ratios (Figure 5c – upper right and lower left), nor did incubation with cDNA targets (not shown), suggesting that the RNA-selective recognition character observed for these probes in homogeneous solutions, is maintained on surfaces. This microarray, therefore, presents itself as an interesting tool for RNA expression analysis.
Murakami and coworkers later utilized this class of probes to detect Krüppel mRNA (Kr-mRNA) in Drosophila.33 Kr-mRNA is highly abundant during early stages of differentiation and has very specific spatiotemporal expression patterns, rendering it an interesting biological model system. As expected, significant excimer emission was observed when U-modified 2′-O-methyl-RNA probes were incubated with total embryonic Drosophila mRNA that was collected 3–5 hours post-fertilization, i.e., a time point where Kr-mRNA is highly abundant. In contrast, excimer emission levels were 4-fold lower when using total embryonic mRNA extracts that were collected 0–2 hours post-fertilization, i.e., a time point when Kr-mRNA is scarcely expressed. Detection of Kr-mRNA was also explored in fixed Drosophila embryos. Prominent excimer signals were observed when 2′-O-methyl-RNA probes with two consecutive U monomers were incubated with stage-5 embryos in which Kr-mRNA expression is prominent, whereas only very low levels of excimer emission were observed when using stage-1 embryos (Figure 6 – left and right image, respectively). The specific localization of the excimer signal in the central region of the stage-5 embryo, agrees with reported Kr-mRNA expression patterns in early-differentiated embryos. As expected, a scrambled U-modified 2′-O-methyl-RNA control probe did not result in excimer emission (Figure 6 – middle image). These results demonstrate that 2′-O-methyl-RNA probes with consecutive U monomers can be used to study mRNA expression patterns with high spatiotemporal resolution.
Figure 6.

Differential interference contrast (DIC) and fluorescence images of fixed Drosophila embryos. (i–iii): DIC images, (iv–vi): fluorescence images, (vii–ix): merged images. Kr-F mRNA probe: 5′-[2′-OMe-RNA]-UCC UAA UUU UGU GCU; scrambled probe: 5′-[2′-OMe-RNA]-AGU CAC UUU UUC GUU. 340/15 nm excitation and 480/30 nm emission band-pass filters used. Adapted from reference 33 with permission from Elsevier.
5.3 Formation of self-assembled pyrene arrays on nucleic acid scaffolds
The use of nucleic acids as supramolecular scaffolds for arrangement of chromophores has received considerable interest due to the prospect of developing DNA-based light-harvesting complexes.34–37 Pyrene arrays utilizing 2′-O-(pyren-1-yl)methylribonucleotides as the key components are highlighted in this section.
The observation that duplexes between 2′-O-methyl-RNA probes with two consecutive U monomers and cRNA emit pyrene-pyrene excimer emission (see section 5.2), suggests that formation of π-stacking pyrene arrays in the minor groove is possible. In 2005, Nakamura, Yamana and co-workers demonstrated that the excimer signal in duplexes with RNA grows progressively more intense as the number of sequential U monomers in 2′-O-methyl-RNA probes increases.38 Similar results were later observed when using 2′-O-methyl-RNA probes with consecutive A, C and U monomers.16 The CD spectra of duplexes between 2′-O-methyl-RNA probes with 2–4 consecutive incorporations of 2′-O-(pyren-1-yl)methylribonucleotides and cRNA exhibit positive Cotton effects in the 300–360 nm region, which is consistent with exciton coupling between pyrene moieties.16 MD simulations provided additional support for the formation of stable pyrene arrays in the minor groove (Figure 7). The minimized structure was used to derive a simulated CD spectrum for the modified duplex, which was found to closely resemble the experimental CD profile, thus validating pyrene array formation in the minor groove.
Figure 7.

Molecular dynamics minimized structure of duplex between 5′-[2′-OMe-RNA]-U8-U4-U8 and cRNA. Reproduced from reference 16 with permission from the Royal Society of Chemistry.
In 2008, Nakamura, Yamana, and coworkers described an alternative strategy for formation of pyrene arrays, which is based on RNA duplexes with interstrand zipper arrangements of 2′-O-(pyren-1-yl)methylribonucleotides.39 Two complementary 15-mer RNA strands - each containing a central stretch of five separated 2′-O-(pyren-1-yl)methylribonucleotides - were hybridized in a manner that positions A and U monomers opposite of each other (Figure 8). This architecture allows the pyrene moieties to stack with each other as evidenced by positive Cotton effects in the pyrene region and prominent excimer emission (quantum yield of 22%). Interestingly, the formed duplexes are much more stable than duplexes between individual probe strands and cRNA, suggesting that array formation counteracts the destabilizing effect of 2′-O-(pyren-1-yl)methylribonucleotides in RNA duplexes.
Figure 8.

Formation of pyrene arrays in RNA duplexes with interstrand zipper arrangements of 2′-O-(pyren-1-yl)methylribonucleotides. Adapted from reference 39 with permission from the American Chemical Society.
In 2012, Nakamura, Yamana, and coworkers extended this concept to 2′-O-methyl RNA duplexes.21 The duplexes are less thermostable and fluorescent, suggesting that the O2′-methyl group interferes with excimer formation.
5.4 Study of charge transfer phenomena in RNA duplexes
While charge transfer (CT) phenomena have been extensively studied in DNA due to their potential applications in nanoscale devices,40,41 less attention has been devoted to the study of CT in RNA, even though the base stacking and structural dynamics of the duplex – two major factors influencing the CT efficiency – are substantially different.
In 2009, Yamana and coworkers studied CT in RNA duplexes in which the photoexcitable electron donor U and the electron acceptor 2′-O-(p-nitrobenzyl)uridine (UNB) were separated by a bridging (rA)n-(U)n sequence (Figure 9– upper panel).42 The free energy for electron transfer from excited-state pyrene (Pyr*) to the uracil base and from Pyr* to UNB was estimated to be approximately -0.5 and -1.09 eV, respectively. Electron injection and the subsequent electron transfer processes were, accordingly, expected to be energetically favorable. Indeed, long-range CT was observed as evidenced by the quenching of pyrene fluorescence, the magnitude of which was found to be strongly dependent on the donor-acceptor distance. Thus, the emission quantum yields increased steadily from ~0.8% to ~12.3% as the NB monomer was moved further away from the U monomer (~15% in absence of UNB), demonstrating that excess electrons can move through RNA π-stacks (Figure 9 – lower panel).
Figure 9.

Upper: illustration of electron transfer process in RNA duplexes modified with U and 2′-O-(p-nitrobenzyl)uridine (NB). Lower: probe sequences and fluorescence quantum yields of duplexes with rA20 (λex = 345 nm). Adapted from reference 42 with permission from the American Chemical Society.
CT phenomena in RNA duplexes have also been studied using a chemical assay in which 2′-O-(pyren-1-yl)methyluridine and 5-bromouridine were used as a photoexcitable electron donor and kinetic electron trap, respectively.43 Reductive decomposition of 5-bromouridine - a consequence of long-distance electron transfer that proceeds through up to ten A-U base pairs - was observed. No decomposition was observed in equivalent DNA duplexes when the donor and acceptor were separated by more than four A-T base pairs. In DNA duplexes, the direct stacking between the intercalating pyrene and flanking pyrimidines prevents efficient escape of the excess electron from the contact ion-pair due to rapid charge recombination. In RNA duplexes, charge recombination is slow due to the spatial separation of the positive and negative charges, as the pyrene occupies an extrahelical position.
Charge transfer was also demonstrated to occur in RNA duplexes containing base pairs between A monomers and 5-(p-nitrophenyl)uridine. Evidently, photo-induced charge transfer occurs from an excited pyrene moiety located in the minor groove, through the central base pair, and out to the NB moiety that is located in the major groove.44
In summary, these results demonstrate the value of 2′-O-(pyren-1-yl)methylribonucleotides in RNA CT studies since the position of the pyrene moiety is predictable.
5.5 Sequence-unrestricted recognition of double-stranded DNA
Substantial efforts have been devoted to the development of probes that can recognize double-stranded DNA (dsDNA) due to the considerable promise for gene regulation and modification, and detection of biologically interesting sequences. Notable examples include triplex-forming oligonucleotides (TFOs), peptide nucleic acids (PNAs), polyamides, optimized TFOs and PNAs, and CRISPR/Cas.45–51 Despite these advances, there remains a need for hybridization-based probes that enable efficient sequence-unrestricted recognition of dsDNA with high binding specificity, ease-of-use, and straightforward cellular delivery.
Our group is pursuing the development of double-stranded oligodeoxyribonucleotides probes that are energetically activated for dsDNA-recognition through modification with +1 interstrand zipper motifs52 of intercalator-functionalized nucleotides such as 2′-O-(pyren-1-yl)methylribonucleotides (Figure 10 top).53,54 This particular structural motif forces the two intercalating pyrene moieties of the 2′-O-(pyren-1-yl)methyl-RNA monomers to compete for the same inter-base-pair region within the probe duplex. This results in a violation of the nearest neighbor exclusion principle,55 which asserts that intercalation is anti-cooperative at adjacent sites due to limitations in local helix expandability (every intercalation event unwinds the duplex by ~3.4 Å) and to prevent disruption of stabilizing stacking interactions between nucleobases and the first intercalator.56,57 Consequentially, a partially unwound and destabilized region is formed, which manifests itself as low probe Tm’s.x,y Support for this structure has been obtained from 2D NOESY NMR experiments and MD simulations (Figure 10 - bottom left).24,56 Other interstrand zipper arrangements of 2′-O-(pyren-1-yl)methyl-RNA monomers do not result in probe destabilization, since the pyrene moieties intercalate in separate base pair steps and therefore do not cause a violation of the nearest neighbor exclusion principle.58 The two strands that comprise these so-called “Invader probes”, display very high affinity toward complementary DNA regions, since duplex formation – as discussed in Section 3 – results in strongly stabilizing stacking interactions between the pyrene and flanking nucleobase moieties (Figure 10 - bottom right). The higher stability of the products (i.e., the two probe-target duplexes formed as part of the recognition complex) relative to the reactants (i.e., the double-stranded Invader probe and DNA target region) provides the driving force for dsDNA-recognition. As an example, the Invader probe 5′-GTGAUATGC:3′-CACTAUACG exhibits a ΔTm of -2 °C relative to the unmodified DNA duplex ([Na+] = 110 mM, pH 7), whereas the corresponding duplexes between the individual probe strands and cDNA have ΔTm’s of +12.5 and +11.5 °C, respectively.53
Figure 10.

Principle of dsDNA-recognition by Invader probes (top). Lowest energy structure from MD simulations of 5′-GTGAUATGC:3′-CACTAUACG (bottom left) and 5′-GTGAUATGC:3′-CACTATACG (bottom right) – clearly, the Invader probe is partially unwound and distorted. Lower panel adapted from 24 with permission from the Royal Society of Chemistry.
We have shown that all four natural 2′-O-(pyren-1-yl)methyl-RNA monomers can be used as activating components of Invader probes. However, probes with +1 interstrand zippers of the pyrimidine monomers are especially activated for dsDNA-recognition since particularly stable probe-target duplexes are formed when the pyrene-functionalized monomers are flanked by large 3′-purines.17 Furthermore, we have shown that incorporation of additional +1 interstrand zippers of 2′-O-(pyren-1-yl)methyl-RNA monomers results in further thermodynamic activation of the probes for recognition of dsDNA.54
Invader probes with +1 interstrand zippers of 2′-O-(pyren-1-yl)methyl-RNA monomers have been shown to recognize DNA hairpin structures comprised of a double-stranded stem region that is isosequential to the probe, and a single-stranded T10 loop. Thus, incubation of Invader probes with a sequence-matched DNA hairpin results in the formation of ternary recognition complexes that move more slowly on non-denaturing PAGE gels (Figure 11). Conversely, incubation with DNA hairpins with stem regions that differ in nucleotide sequence at one or more positions relative to the probes, does not result in hairpin recognition, highlighting the excellent mismatch discrimination of Invader probes.54
Figure 11.

3′-Digoxigenin (DIG) labeled DNA hairpins (DH) (34.4 nM) were incubated with a 1000- or 500-fold excess of pre-annealed Invader probes X1:X2 or X9:X10 in HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.2, 10% sucrose, 1.44 mM spermine tetrahydrochloride) for 17 h at room temperature. RC = recognition complex. Sequence differences at the 6- or 8-position are highlighted in red. Sequences for Invader probes are X1: 5′-GGUATATATAGGC, X2: 3′-CCAUATATATCCG, X9: 5′-GGUAUATATAGGC, and X10: 3′-CCAUAUATATCCG. Adapted from reference 54 with permission from the Royal Society of Chemistry.
Invader probes based on 2′-O-(pyren-1-yl)methyl-RNA monomers have been used in colorimetric 96-well plate sandwich assays for recognition of 28-mer mixed-sequence dsDNA fragments specific to different food pathogens.59 Thus, amine-terminated Invader capture probes were attached to plates and co-incubated with biotinylated Invader signaling probes and dsDNA targets (Figure 12). Two ternary complexes, i.e., one in solution and another one tethered to the well surface, are formed upon successful recognition of dsDNA targets. Incubation with streptavidin-horseradish peroxidase conjugates (SA-HRP) and subsequent removal of unbound species results in the formation of a fluorescent product upon addition of a HRP substrate. The dsDNA fragments were detected without pre-amplification at concentrations down to 20 pM with excellent binding specificity.
Figure 12.

Principle of Invader-based sandwich assay for detection of mixed-sequence dsDNA. Invader capture probe (black); Invader signaling probe (blue); dsDNA target (red); biotin (B); streptavidin-horseradish peroxide conjugates (HRP); QuantaBlu (QB). Reproduced from reference 59 with permission from the Royal Society of Chemistry.
Perhaps the most exciting demonstration of mixed-sequence dsDNA-recognition using Invader probes was presented in a recent study in which 14-mer Cy3-labeled Invader probes with three +1 interstrand zippers of 2′-O-(pyren-1-yl)methyl-RNA monomers were shown to signal the presence of a high copy-number gender-specific chromosomal DNA region in fixed interphase and metaphase nuclei from bovine kidney cells under non-denaturing conditions (Figure 13 – top and middle panels).54 No signals were observed when a fully base-paired Invader probe differing in nucleotide sequence at three positions relative to the DYZ-1 satellite dsDNA target was used, or when Y-chromosome-specific probes were incubated with nuclei from female bovine fibroblast cells (Figure 13 – bottom panel). These observations underscore that very specific recognition of mixed-sequence chromosomal DNA targets is possible with Invader probes.
Figure 13.

Detection of chromosomal DNA using Invader probes. Fluorescence micrographs from fluorescence in situ hybridization experiments using Y-chromosome specific Invader probes under non-denaturing conditions. Invader probe 5′-Cy3-AGCCCUGTGCCCTG:3′-TCGGGACACGGGAC-Cy3 was incubated with nuclei from male bovine kidney cells in interphase (upper panel) or metaphase (middle panel), or with nuclei from female bovine fibroblast cells (lower panel). Images viewed using Cy3 (left column) or DAPI (middle column) filter settings; overlays are shown in the right column. Adapted from reference 54 with permission from the Royal Society of Chemistry.
6. Summary and perspectives
In the 25 years since their original synthesis, a wide range of applications for oligonucleotides modified with 2′-O-(pyren-1-yl)methyl-ribonucleotides have been proposed and continue to be discovered. As we have summarized in this Review, these applications include detection of RNA targets, discrimination of single nucleotide polymorphisms, formation of self-assembled pyrene arrays on nucleic acid scaffolds, the study of charge transfer phenomena in nucleic acid duplexes, and sequence-unrestricted recognition of double-stranded DNA. These applications are facilitated by predictable binding modes and biophysical properties of the pyrene moiety, which depend on the specific nucleic acid context. Thus, in B-type duplexes, such as DNA duplexes, the pyrene moiety of 2′-O-(pyren-1-yl)methyl-ribonucleotides is intercalating, leading to significant stabilization but low fluorescence emission levels. In A-type duplexes, such as RNA duplexes, the pyrene moiety is located in an extrahelical position, which typically results in prominent emission and duplex destabilization unless pyrene arrays are formed. Few, if any, pyrene-functionalized nucleotide monomers have been as thoroughly characterized and display such binding predictability and versatility in biophysical properties and applications. This renders the relatively easy-to-synthesize 2′-O-(pyren-1-yl)methyl-ribonucleotides as promising building blocks for new applications in molecular biology and materials science.
Acknowledgments
The Hrdlicka laboratory has enjoyed support from numerous sources over the past ten years including Award Number R01 GM088697 from the National Institute of General Medical Sciences, National Institutes of Health; Awards IF13-001 and IF14-012 from the Higher Education Research Council, Idaho State Board of Education; and The Office of Naval Research (N00014-10-1-0282).
Biographies
Patrick J. Hrdlicka received his PhD degree in 2006 from the University of Southern Denmark working with Prof. Jesper Wengel, and was subsequently appointed as an Assistant Professor at the University of Idaho. He was promoted to Associate Professor in 2011 and Professor in 2017. His research interests include development and characterization of chemically functionalized oligonucleotides and nanomaterials and their utilization in molecular biology, diagnostics, and materials science.
Saswata Karmakar received his PhD degree in Chemistry in 2013 from the University of Idaho, for his work with Prof. Hrdlicka on chemically modified oligonucleotides for mixed-sequence recognition of double-stranded DNA. Dr. Karmakar conducted his postdoctoral research with Prof. Eric T. Kool at Stanford University and joined Translate Bio (former RaNA Therapeutics) in 2015 where he is a Senior Scientist. His current research focus includes the design and synthesis of chemically modified oligonucleotides for recognition of long non-coding RNAs for therapeutic applications.
References
- 1.Østergaard ME, Hrdlicka P. Chem Soc Rev. 2011;40:5771–5788. doi: 10.1039/c1cs15014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dougherty G, Pilbrow JR. Int J Biochem. 1984;16:1179–1192. doi: 10.1016/0020-711x(84)90215-5. [DOI] [PubMed] [Google Scholar]
- 3.Kalyanasundaram K, Thomas JK. J Am Chem Soc. 1977;99:2039–2044. [Google Scholar]
- 4.Manoharan M, Tivel KL, Zhao M, Nafisi K, Netzel TL. J Phys Chem. 1995;99:17461–17472. [Google Scholar]
- 5.Yao CX, Kraatz HB, Steer RP. Photochem Photobiol Sci. 2005;4:191–199. doi: 10.1039/b414577c. [DOI] [PubMed] [Google Scholar]
- 6.Wilson JN, Younjin C, Samuel T, Cuppoletti A, Kool ET. ChemBioChem. 2008;9:279–285. doi: 10.1002/cbic.200700381. [DOI] [PubMed] [Google Scholar]
- 7.Winnik FM. Chem Rev. 1993;93:587–614. [Google Scholar]
- 8.Matray TJ, Kool ET. J Am Chem Soc. 1998;120:6191–6192. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zatsepin TS, Romanova EA, Oretskaya TS. Russ Chem Rev. 2002;71:513–534. [Google Scholar]
- 10.Yamana K, Ohashi Y, Nunota K, Kitamura M, Nakano H, Sangen O, Shimidzu T. Tetrahedron Lett. 1991;32:6347–6350. [Google Scholar]
- 11.Zemlicka J. Collect Czech Chem Commun. 1964;29:1734–1735. [Google Scholar]
- 12.Karmakar S, Anderson BA, Rathje RL, Andersen S, Jensen TB, Nielsen PE, Hrdlicka PJ, Org J. Chem. 2011;76:7119–7131. doi: 10.1021/jo201095p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross BS, Springer RH, Tortorici Z, Dimock S. Nucleosides Nucleotides. 1997;16:1641–1643. [Google Scholar]
- 14.Roy SK, Tang JY. Org Proc Res Dev. 2000;4:170–171. [Google Scholar]
- 15.Uchiyama N, Ogata T, Oka N, Wada T. Nucleosides Nucleotides Nucleic Acids. 2011;30:446–456. doi: 10.1080/15257770.2011.592171. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura M, Shimomura Y, Ohtoshi Y, Sasa K, Hayashi H, Nakano H, Yamana K. Org Biomol Chem. 2007;5:1945–1951. doi: 10.1039/b705933g. [DOI] [PubMed] [Google Scholar]
- 17.Karmakar S, Guenther DC, Hrdlicka PJ. J Org Chem. 2013;78:12040–12048. doi: 10.1021/jo402085v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamana K, Iwase R, Furutani S, Tsuchida H, Zako H, Yamaoka T, Murakami A. Nucleic Acids Res. 1999;27:2387–2392. doi: 10.1093/nar/27.11.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura M, Fukunaga Y, Sasa K, Ohtoshi Y, Kanaori K, Hayashi H, Nakano H, Yamana K. Nucleic Acids Res. 2005;33:5887–5895. doi: 10.1093/nar/gki889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamana K, Zako H, Asazuma K, Iwase R, Nakano H, Murakami A. Angew Chem Int Ed. 2001;40:1104–1106. doi: 10.1002/1521-3773(20010316)40:6<1104::aid-anie11040>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura M, Fukuda M, Takada T, Yamana K. Org Biomol Chem. 2012;10:9620–9626. doi: 10.1039/c2ob26773j. [DOI] [PubMed] [Google Scholar]
- 22.Asanuma H, Fujii T, Kato T, Kashida H. J Photochem Photobiol C. 2012;13:124–135. [Google Scholar]
- 23.Marin V, Hansen HF, Koch TR, Armitage BA. J Biomol Struct Dyn. 2004;21:841–850. doi: 10.1080/07391102.2004.10506974. [DOI] [PubMed] [Google Scholar]
- 24.Karmakar S, Madsen AS, Guenther DC, Gibbons BC, Hrdlicka PJ. Org Biomol Chem. 2014;12:7758–7773. doi: 10.1039/c4ob01183j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahara A, Iwase R, Sakamoto T, Yamaoka T, Yamana K, Murakami A. Bioorg Med Chem. 2003;11:2783–2790. doi: 10.1016/s0968-0896(03)00227-x. [DOI] [PubMed] [Google Scholar]
- 26.Kaur H, Babu BR, Maiti S. Chem Rev. 2007;107:4672–4697. doi: 10.1021/cr050266u. [DOI] [PubMed] [Google Scholar]
- 27.Karmakar S, Hrdlicka PJ. Chem Sci. 2013;4:3447–3454. doi: 10.1039/C3SC50726B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen M, Nielsen CB, Nielsen KE, Jensen GA, Bondensgaard K, Singh SK, Rajwanshi VK, Koshkin AA, Dahl BM, Wengel J, Jacobsen JP. J Mol Recognit. 2000;13:44–53. doi: 10.1002/(SICI)1099-1352(200001/02)13:1<44::AID-JMR486>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Petersen M, Bondensgaard K, Wengel J, Jacobsen JP. J Am Chem Soc. 2002;124:5974–5982. doi: 10.1021/ja012288d. [DOI] [PubMed] [Google Scholar]
- 30.Imincan G, Pei F, Yu L, Jin H, Zhang L, Yang X, Zhang L, Tang X. Anal Chem. 2016;88:4448–4455. doi: 10.1021/acs.analchem.6b00227. [DOI] [PubMed] [Google Scholar]
- 31.Mahara A, Iwase R, Sakamoto T, Yamana K, Yamaoka T, Murakami A. Angew Chem Int Ed. 2002;41:3648–3650. doi: 10.1002/1521-3773(20021004)41:19<3648::AID-ANIE3648>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Sakamoto T, Kobori A, Murakami A. Bioorg Med Chem Lett. 2008;18:2590–2593. doi: 10.1016/j.bmcl.2008.03.040. [DOI] [PubMed] [Google Scholar]
- 33.Ueda T, Kobori A, Yamayoshi A, Yoshida H, Yamaguchi M, Murakami A. Bioorg Med Chem. 2012;20:6034–6039. doi: 10.1016/j.bmc.2012.08.028. [DOI] [PubMed] [Google Scholar]
- 34.Malinovskii VL, Wenger D, Haener R. Chem Soc Rev. 2010;39:410–422. doi: 10.1039/b910030j. [DOI] [PubMed] [Google Scholar]
- 35.Bandy TJ, Brewer A, Burns JR, Marth G, Nguyen TN, Stulz E. Chem Soc Rev. 2011;40:138–148. doi: 10.1039/b820255a. [DOI] [PubMed] [Google Scholar]
- 36.Kashida H, Asanuma H. Phys Chem Chem Phys. 2012;14:7196–7204. doi: 10.1039/c2cp40520b. [DOI] [PubMed] [Google Scholar]
- 37.Ensslen P, Wagenknecht HA. Acc Chem Res. 2015;48:2724–2733. doi: 10.1021/acs.accounts.5b00314. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura M, Ohtoshi Y, Yamana K. Chem Commun. 2005:5163–5165. doi: 10.1039/b507808c. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura M, Murakami Y, Sasa K, Hayashi H, Yamana K. J Am Chem Soc. 2008;130:6904–6905. doi: 10.1021/ja801054t. [DOI] [PubMed] [Google Scholar]
- 40.Arnold AR, Grodick MA, Barton JK. Cell Chem Biol. 2016;23:183–197. doi: 10.1016/j.chembiol.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatramani R, Keinan S, Balaeff A, Beratan DN. Coord Chem Rev. 2011;255:635–648. doi: 10.1016/j.ccr.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maie K, Miyagi K, Takada T, Nakamura M, Yamana K. J Am Chem Soc. 2009;131:13188–13189. doi: 10.1021/ja902647j. [DOI] [PubMed] [Google Scholar]
- 43.Takada T, Otsuka Y, Nakamura M, Yamana K. Bioorg Med Chem. 2011;19:6881–6884. doi: 10.1016/j.bmc.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 44.Fukuda M, Nakamura M, Takada T, Yamana K. Tet Lett. 2010;51:1732–1735. [Google Scholar]
- 45.Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. Nucleic Acids Res. 2008;36:5123–5138. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen PE. Chem Biodiv. 2010;7:786–804. doi: 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]
- 47.Blackledge MS, Melander C. Bioorg Med Chem. 2013;21:6101–6114. doi: 10.1016/j.bmc.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hari Y, Obika S, Imanishi T. Eur J Org Chem. 2012;15:2875–2887. [Google Scholar]
- 49.Aiba Y, Sumaoka J, Komiyama M. Chem Soc Rev. 2011;40:5657–5668. doi: 10.1039/c1cs15039a. [DOI] [PubMed] [Google Scholar]
- 50.Bahal R, Sahu B, Rapireddy S, Lee CM, Ly DH. ChemBioChem. 2012;13:56–60. doi: 10.1002/cbic.201100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sander JD, Joung JK. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The following nomenclature describes the relative arrangement between two monomers positioned on opposing strands in a duplex. The number n describes the distance measured in number of base pairs and has a positive value if a monomer is shifted toward the 5′-side of its strand relative to a second reference monomer on the other strand. Conversely, n has a negative value if a monomer is shifted toward the 3′-side of its strand relative to a second reference monomer on the other strand.
- 53.Sau SP, Madsen AS, Podbevsek P, Andersen NK, Kumar TS, Andersen S, Rathje RL, Anderson BA, Guenther DC, Karmakar S, Kumar P, Plavec J, Wengel J, Hrdlicka PJ. J Org Chem. 2013;78:9560–9570. doi: 10.1021/jo4015936. and reference cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guenther DC, Anderson GH, Anderson BA, Karmakar S, Hrdlicka PJ. Chem Sci. 2015;6:5006–5015. doi: 10.1039/c5sc01238d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crothers DM. Biopolymers. 1968;6:575–584. doi: 10.1002/bip.1968.360060411. [DOI] [PubMed] [Google Scholar]
- 56.Tsai C, Jain S, Sobell HM. J Mol Biol. 1977;114:301–315. doi: 10.1016/0022-2836(77)90252-2. [DOI] [PubMed] [Google Scholar]
- 57.Williams LD, Egli M, Gao Q, Rich A. In: Structure and Function: Nucleic Acids. Sarma RH, Sarma MH, editors. Vol. 1. Adenine press; 1992. pp. 107–125. [Google Scholar]
- 58.Double-stranded probes with “0-zippers” also result in a violation of the nearest neighboring exclusion principle, but the effects are less pronounced than in +1 zipper probes - see reference 24.
- 59.Denn B, Karmakar S, Guenther DC, Hrdlicka PJ. Chem Commun. 2013;49:9851–9853. doi: 10.1039/c3cc45705b. [DOI] [PMC free article] [PubMed] [Google Scholar]