

Abstract
Objectives:
To estimate a regional progression pattern of amyloid deposition from cross-sectional amyloid-sensitive PET data and evaluate its potential for in vivo staging of an individual's amyloid pathology.
Methods:
Multiregional analysis of florbetapir (18F-AV45)–PET data was used to determine individual amyloid distribution profiles in a sample of 667 participants from the Alzheimer's Disease Neuroimaging Initiative cohort, including cognitively normal older individuals (CN) as well as patients with mild cognitive impairment and Alzheimer disease (AD) dementia. The frequency of regional amyloid positivity across CN individuals was used to construct a 4-stage model of progressing amyloid pathology, and individual distribution profiles were used to evaluate the consistency of this hierarchical stage model across the full cohort.
Results:
According to a 4-stage model, amyloid deposition begins in temporobasal and frontomedial areas, and successively affects the remaining associative neocortex, primary sensory-motor areas and the medial temporal lobe, and finally the striatum. Amyloid deposition in these brain regions showed a highly consistent hierarchical nesting across participants, where only 2% exhibited distribution profiles that deviated from the staging scheme. The earliest in vivo amyloid stages were mostly missed by conventional dichotomous classification approaches based on global florbetapir-PET signal, but were associated with significantly reduced CSF Aβ42 levels. Advanced in vivo amyloid stages were most frequent in patients with AD and correlated with cognitive impairment in individuals without dementia.
Conclusions:
The highly consistent regional hierarchy of PET-evidenced amyloid deposition across participants resembles neuropathologic observations and suggests a predictable regional sequence that may be used to stage an individual's progress of amyloid pathology in vivo.
A distinct regional progression pattern of cerebral amyloid deposits has been estimated from case series of neuropathologic examinations and forms the basis of widely used staging schemes for the characterization of an individual's extent of amyloid pathology at autopsy.1,2 Interestingly, early phases of amyloid deposition are mainly found in individuals who were cognitively unimpaired at last clinical evaluation, arguing for a protracted preclinical phase of amyloid pathology.2–7
The advent of amyloid-sensitive PET imaging methods allows detecting cerebral amyloid deposits in living individuals with very high sensitivity and specificity.8–10 However, in contrast to the neuropathologic staging schemes, PET imaging data are mainly being used for the characterization of global amyloid burden into dichotomous positive/negative categories. Recent imaging–pathologic correlation studies found that amyloid positivity as conventionally defined usually corresponds to relatively advanced phases of amyloid pathology.11,12
We explored the feasibility of in vivo PET-based amyloid staging using a large sample of amyloid-sensitive florbetapir (18F-AV45)-PET data covering the full clinical spectrum of Alzheimer disease (AD), and adopting established neuropathologic approaches for determining regional staging models of amyloid pathology and other proteinopathies.1,2,13–16 Specifically, this includes estimation of a regional progression sequence based on the frequency of regional amyloid positivity across participants and evaluation of the regional hierarchy of amyloid deposition along this sequence across individual deposition profiles. The identified in vivo amyloid stages were characterized with respect to conventional dichotomous florbetapir-PET classifications, CSF Aβ42 levels, as well as clinical diagnosis and cognition.
METHODS
Participants.
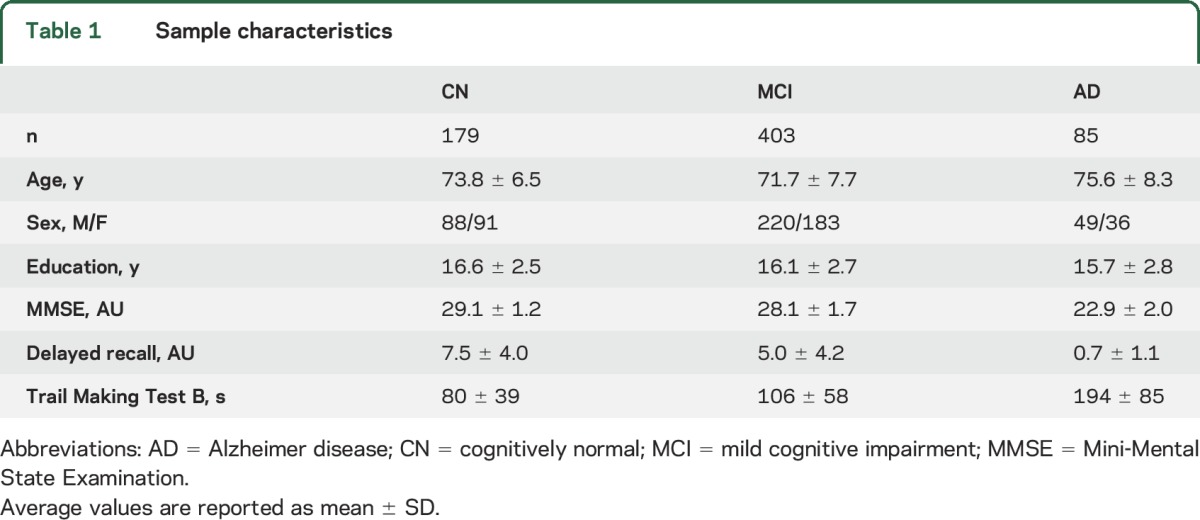
All data used in the present study were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) (adni-info.org). The present study sample consisted of a total of 667 participants enrolled in the ADNI-GO and ADNI-2 cohorts, who had concurrent 18F-AV45-PET and high-resolution structural MRI scans available. The sample included 179 cognitively normal (CN) older individuals, 403 patients with mild cognitive impairment (MCI), and 85 patients with AD dementia. Detailed diagnostic criteria have been reported previously17,18 and are published on the ADNI website (adni.loni.usc.edu/methods/). Sample characteristics are summarized in table 1.
Table 1.
Sample characteristics
Standard protocol approvals, registrations, and patient consents.
Data collection and sharing in ADNI was approved by the institutional review board of each participating institution and written informed consent was obtained from all participants.
Imaging data.
Acquisition and standardized preprocessing steps of the multicentric MRI and PET imaging data in ADNI have been reported previously19,20 and are described in detail on the ADNI website (adni.loni.usc.edu/methods/). Briefly, structural MRI data were acquired on 3T scanning platforms using T1-weighted sagittal 3D magnetization-prepared rapid gradient echo sequences. Florbetapir-PET scans were acquired during a 50- to 70-minute interval following a 370 MBq bolus injection of florbetapir (18F). All ADNI imaging data undergo standardized preprocessing steps aimed at increasing data uniformity across the multicenter scanner platforms.
Procedures for preprocessing of the imaging data have been described in detail previously.19,20 Briefly, florbetapir-PET scans were rigidly coregistered to the corresponding structural MRI scan, corrected for partial volume effects (PVE),21,22 and spatially normalized to an aging/AD-specific reference template using the registration measures from the corresponding MRI scans.
Regional florbetapir-PET uptake values were sampled from 52 brain regions defined in the Harvard–Oxford structural atlas,23 covering the whole cerebral cortex, as well as hippocampus, amygdala, striatum, and thalamus. The atlas labels were propagated to the customized template space using nonlinear image registration, and multiplied with a binary gray matter (GM) mask of the reference template thresholded at 50% GM probability.20 Regional florbetapir-PET uptake means were converted to standard uptake value ratios by scaling to the mean uptake of the whole cerebellum (SUVRCer) in non-PVE-corrected data.22,24
Individual regional amyloid deposition profiles, i.e., the presence or absence of amyloid pathology in each of the 52 brain regions, were determined based on a cutoff of regional SUVRCer = 0.92. This cutoff lies in between the 2 most commonly used cutoffs of SUVRCer = 1.10 and SUVRCer = 1.17 for establishing amyloid positivity based on the global cortical signal10,25–27 in non-PVE-corrected florbetapir-PET data and was extrapolated to the PVE-corrected florbetapir-PET data of this study using linear regression.26 More detailed information on cutoff selection is provided in appendix e-1 at Neurology.org.
Data analysis.
Determination of a model of regionally progressing amyloid pathology.
In analogy to the approach used in previous neuropathologic staging studies,1,2,13,14 a model of regionally progressing amyloid pathology was estimated based on the frequency of regional amyloid positivity across participants, where the proportion of participants who exhibit amyloid pathology in a given brain region is used as an indicator of this region's temporal involvement in the course of spatially progressing amyloid pathology. This approach critically depends on a high regional variance in amyloid deposition across the examined participants. Thus, due to our focus on the characterization of early phases of amyloidosis, and given that amyloid load is believed to reach global levels before the emergence of cognitive impairments,28 we restricted the determination of the progression model to the sample of cognitively normal older individuals.
In order to operationalize the regionally detailed progression model into a manageable staging system, the 52 ranked anatomical regions were merged into 4 larger anatomical divisions (1–4) based on equally sized proportions of the observed range of involvement frequencies (illustrated in figures 1 and e-1). Additional analyses also explored regionally less or more detailed 3- or 5-stage models (appendix e-1).
Figure 1. Model of regional amyloid progression and staging scheme.

Brain renderings on the left illustrate the frequency of regional amyloid positivity across individuals on a color scale from black/blue (lowest) to yellow/red (highest). The 52 brain regions are merged into 4 larger anatomic divisions based on equal partitions of the frequency range (1–4). In the resulting 4-stage model of regional amyloid progression (I–IV), incremental stages are defined by involvement of higher numbered anatomic divisions (in red), in addition to the affected areas of the previous stage (blue).
In vivo staging of individual amyloid pathology.
Regardless of diagnosis, individual amyloid deposition profiles were used to stage each individual's florbetapir-PET scan according to the estimated 4-stage model of regional amyloid progression. For this, an anatomical division was defined positive for amyloid pathology if at least 50% of the regions included in this division exhibited suprathreshold signal in the respective participant. Participants who only exhibited amyloid deposition in the first division were classified as being in stage I, and successive stages II-IV were characterized by additional involvement of anatomical divisions 2, 3, and 4, respectively. Participants who exhibited amyloid positivity in any division >1 without concurrent amyloid positivity in the previous divisions were classified as being nonstageable. A high proportion of stageable vs nonstageable participants is an indicator of the general validity of the hierarchical staging scheme.15,16 Given that this proportion may be biased in the CN group, because it is also used for determining the staging model in this study, we further conducted sensitivity analyses across independent training and test samples for this group (appendix e-1).
For comparison, florbetapir-PET scans were also conventionally classified as amyloid-positive or amyloid-negative based on the commonly used cutoffs of SUVRCer = 1.10 and SUVRCer = 1.17, applied to the global composite SUVRCer values that are made available in the ADNI database (Jagust Lab, UC Berkley; adni.loni.usc.edu/methods/pet-analysis).
In vivo amyloid stages in relation to a CSF biomarker of amyloid pathology.
Amyloid stages were studied in relation to CSF Aβ42 levels, which are well-known to decrease as cortical amyloid deposition increases.29,30 CSF measures of Aβ42 from the same study time point were available for 90% (n = 603) of the study sample. Methods for CSF biomarker quantification in the ADNI cohort are based on the xMAP Luminex platform and Innogenetics/Fujirebio AlzBio3 immunoassay kits and are described in detail elsewhere31 (adni.loni.usc.edu/methods/). Differences in CSF Aβ42 levels across increasing amyloid stages were assessed using Spearman correlation and Mann-Whitney U tests between the first 2 amyloid stages and the group without any evidence of regional amyloid deposition (stage 0).
In vivo amyloid stages in relation to clinical diagnosis and cognition.
Differences in the distribution of amyloid stages between clinical diagnoses were assessed using χ2 test. In addition, for each diagnostic group separately, we assessed associations between progressing amyloid stage and cognitive performance in tests of episodic memory (30-minute delayed recall [DR] of the Rey Auditory Verbal Learning Test) and executive function (Trail Making Test B [TMT-B]) using Spearman correlations. For comparison, associations between increasing amyloid load and cognitive performance were also assessed using continuous global composite SUVRCer values and the conventional lower cutoff for defining presence of detectable amyloid deposits (SUVRCer > 1.10).
RESULTS
Model of regionally progressing amyloid pathology.
The examined brain regions differed markedly with respect to the presence of amyloid pathology across CN individuals (figures 1 and e-1). Amyloid deposition was most frequently observed in the basal part of the temporal lobe (inferior temporal and fusiform gyrus), the anterior cingulate gyrus, and the parietal operculum (∼55%–45% of individuals), followed by wide parts of the temporal, frontal, and parietal associative cortex (∼40%–30%). Amyloid deposits in primary sensory-motor cortices (precentral and postcentral, occipital pole, Heschl gyrus) and anterior medial temporal lobe structures were considerably less frequent (∼25%–15%), and only a small minority showed amyloid positivity in the posterior medial temporal lobe and the striatum (∼10%–5%). None of the participants exhibited amyloid positivity in the thalamus. This pattern was very robust against variations in the cutoff used for defining regional amyloid positivity (appendix e-1), and was used to define a 4-stage model of regional amyloid progression by grouping the brain regions from highest to lowest frequency into 4 anatomic divisions (figures 1 and e-1).
In vivo staging of individual amyloid pathology.
Assessing regional amyloid deposition across the 4 anatomic divisions in individual florbetapir-PET scans yielded a highly consistent hierarchical deposition pattern across participants, allowing us to classify 98% (410/418) of participants with detectable regional amyloid deposition into any of the 4 successive amyloid stages (figures 2 and 3; table e-1). Sensitivity analyses across independent training and test samples indicated negligible bias in the proportion of stageable vs nonstageable participants in the CN group (appendix e-1). In vivo amyloid stages were positively associated with age in the CN (p = 0.006) and MCI groups (p = 0.002), but not in the AD group (p = 0.139) (appendix e-1).
Figure 2. Staging of individual amyloid burden.
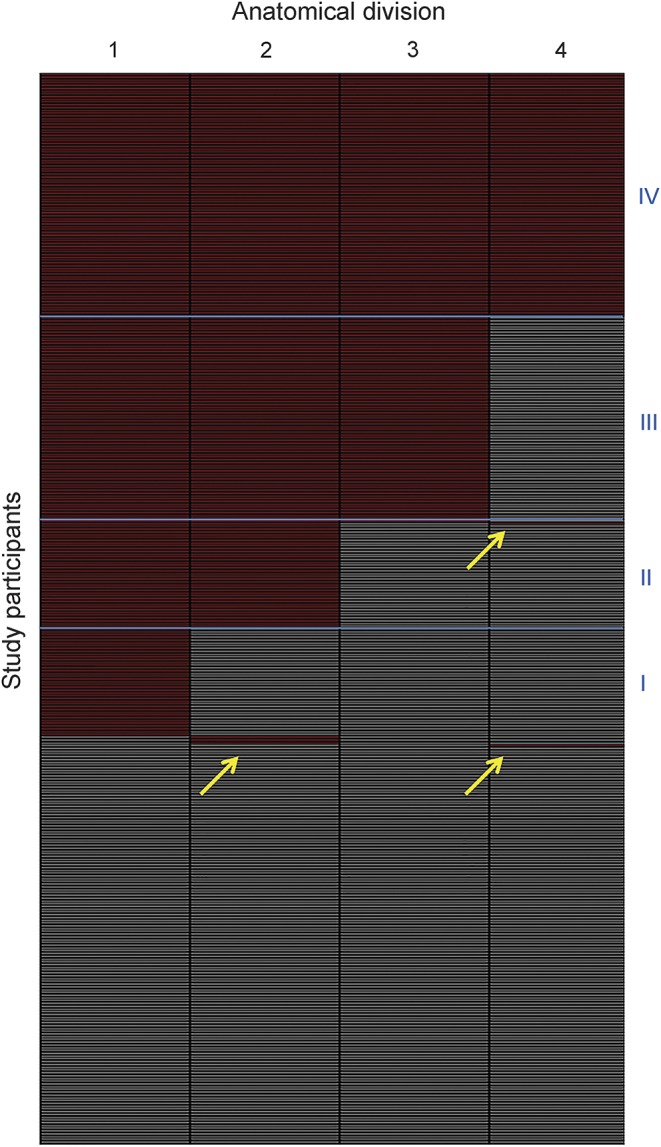
Each row of the matrix corresponds to a study participant and each column to one of the 4 anatomic divisions (1–4). Absence or presence of amyloid is denoted by gray and red, respectively. Amyloid deposition in the 4 anatomic divisions shows a consistent hierarchical nesting across participants, allowing us to stage 98% of the participants with detectable regional amyloid deposits into one of the 4 amyloid stages predicted by the regional progression model (blue boxes, I–IV). Only 8 participants showed distribution profiles that did not conform to the model (yellow arrows; 2 individuals [1 cognitively normal (CN), 1 Alzheimer disease] with deposition profiles of 1-1-0-1; 5 individuals [2 CN, 3 mild cognitive impairment (MCI)] with 0-1-0-0; and 1 individual [MCI] with 0-0-0-1).
Figure 3. Proportions of in vivo amyloid stages by clinical diagnosis.

The relative proportions of in vivo amyloid stages were plotted for each diagnostic group separately and across all participants. Among participants exhibiting stageable amyloid deposition, there is a shift from lower to higher in vivo amyloid stages across the cognitively normal (CN) over the mild cognitive impairment (MCI) to the Alzheimer disease (AD) group. Unstageable cases were relatively rare across all diagnostic categories.
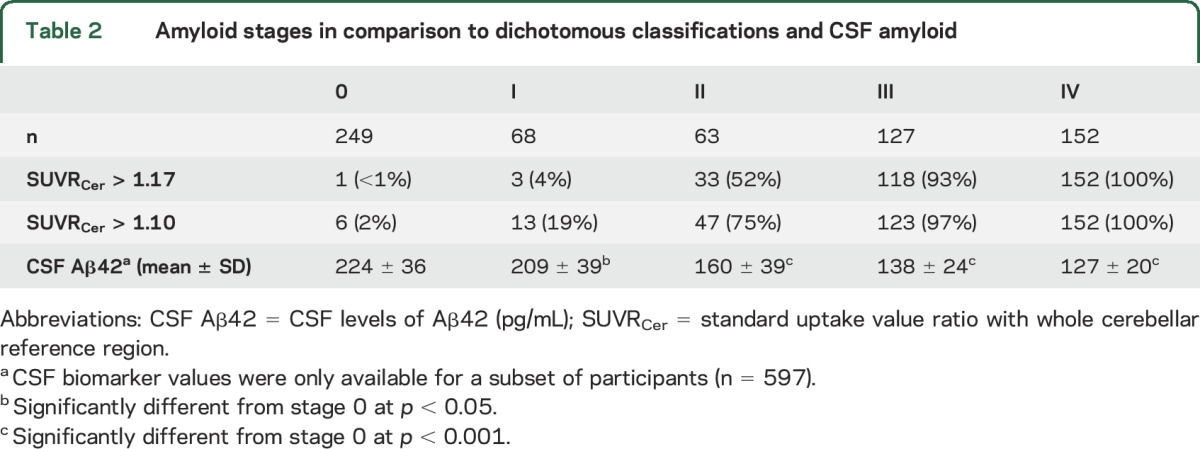
Table 2 compares the in vivo amyloid staging scheme to conventional dichotomous classifications. Based on a global cortical cutoff of SUVRCer = 1.17, all stage IV participants and the vast majority of stage III participants were identified as being amyloid-positive, whereas half of stage II and the vast majority of stage I participants were classified as being amyloid-negative. Even at a much lower cutoff of SUVRCer = 1.10, 25% of stage II participants and the large majority of stage I participants (81%) were still classified as amyloid-negative. However, characterizing the different amyloid stages in terms of an external CSF-based amyloid biomarker demonstrated a continuous decline in Aβ42 levels across progressing amyloid stages (table 2; ρ595 = −0.80, p < 0.001), which was already noticeable at the earliest amyloid stages I (−7%, p = 0.01) and II (−29%, p < 0.001) when compared to Aβ42 levels of stage 0.
Table 2.
Amyloid stages in comparison to dichotomous classifications and CSF amyloid
Among participants exhibiting stageable amyloid deposition, distribution of in vivo amyloid stages differed significantly between diagnostic categories (χ2 = 72.98, p < 0.001; figure 3 and table e-1), with higher in vivo amyloid stages being more frequent in patients with MCI or AD compared to CN individuals. Amyloid stages III or IV (indicating extraneocortical involvement) were observed in 96% of patients with AD, 66% of patients with MCI, and 42% of CN individuals with detectable amyloid deposition. Within diagnostic groups separately, higher amyloid stages were associated with worse DR performance in CN (ρ79 = −0.40, p < 0.001), and with worse DR (ρ248 = −0.30, p < 0.001) and TMT-B performance (ρ245 = 0.33, p < 0.001) in MCI, but did not associate with cognition in AD (p > 0.86) (table e-2). By contrast, among participants identified as being amyloid-positive by a conventional global cutoff of 1.10, SUVRCer values correlated with cognition in MCI (DR: ρ213 = −0.29, p < 0.001, TMT-B: ρ212 = 0.35, p < 0.001) and AD (DR: ρ72 = −0.25, p = 0.035), but not in CN (p > 0.17).
DISCUSSION
In analogy to neuropathologic staging studies, we extrapolated a regional pattern of amyloid progression on the basis of florbetapir-PET amyloid deposition profiles across participants, and assessed its utility for in vivo staging of an individual's amyloid burden. The approach suggested a progression pattern from temporobasal and frontomedial areas, over the remaining associative neocortex, to primary sensory-motor cortex, the medial temporal lobe, and finally the striatum. Individual deposition profiles showed a highly consistent hierarchical nesting across these brain regions, arguing for the general validity of the progression model and enabling an in vivo staging of an individual's progress of amyloid pathology on the basis of cross-sectional imaging data.
The estimated pattern of regional amyloid progression in the florbetapir-PET data shows a remarkable resemblance with previous neuropathologic estimates. Particularly the progression from the associative neocortex over primary sensory-motor areas and the medial temporal lobe to the striatum is well-described in the neuropathologic literature and corresponds to phases 1 to 3 of the widely used Thal scheme of 5 progressive phases of cerebral amyloidosis.2,4 While the first Thal phase encompasses the whole associative neocortex without further regional distinction, previous neuropathologic studies specifically implicated basal portions of the temporal and frontomedial cortex in the initial phase of amyloid accumulation (corresponding to the first stage of the Braak A-B-C scheme of amyloid progression).1,3,5,6 However, other cross-sectional 11C-PiB-PET estimates based on regional correlates of increasing global SUVR values pointed to medial frontal, medial parietal, and lateral temporoparietal areas as the sites of initial amyloid accumulation, without noticeable involvement of the inferior temporal cortex.12,32 Besides possible radiotracer-related differences, these discrepancies are likely to be attributable to the different analytic approaches. A recent 18F-florbetaben-PET study used a similar approach to the one employed in our study and reported a very similar pattern of most frequently observed amyloid deposits in basal temporal and frontomedial cortices, and least frequent deposits in the medial temporal lobe (the striatum was not investigated).33 However, differences in involvement frequencies across neocortical regions were rather small, which probably relates to a high proportion of participants with globally progressed amyloid pathology in the clinical sample used for model determination. Accordingly, no attempts were made in this previous study to use the estimated progression pattern in the context of an in vivo staging scheme.
In our individual staging analyses, only 2% of participants with detectable amyloid deposition violated the proposed hierarchy of regional involvement (figures 2 and 3 and table e-1). The consistent regional hierarchy of amyloid deposition in this large series of florbetapir-PET scans across the full clinical spectrum of AD strongly indicates that PET-measured amyloid deposition follows a predictable regional sequence that can be used analogously to established neuropathologic approaches for staging an individual's pathologic state along this sequence. However, in this cross-sectional data it remains unknown whether individuals inevitably progress from earlier to later stages, as well as under which circumstances and at which rates this progression occurs. A distinct advantage of the presented in vivo staging approach is that it will allow studying these questions through longitudinal amyloid-PET assessments and analysis of such data is warranted.
The earliest in vivo amyloid stages identify participants with regionally restricted amyloid deposits in specific neocortical association areas that are probably entirely missed in clinical routine binary visual assessment,11 and mostly missed by semiquantitative classification approaches based on suprathreshold global cortical signal, even at a relatively lenient cutoff (table 2). Decreased CSF Aβ42 levels compared to the stage 0 group corroborate the neurobiologic relevance of the regionally increased florbetapir-PET signal in these participants. Of course, the consistency of hierarchical amyloid deposition across participants has its limitation with respect to regional detail. Thus, while regionally more fine-grained staging schemes may potentially further increase the sensitivity to detect earliest focal amyloid deposits, they will also generally result in higher numbers of participants who show deviating deposition patterns (i.e., are nonstageable; appendix e-1). Moreover, the distinct amyloid progression pattern as estimated here and in neuropathologic case series1,2,34 may be different in genetically determined forms of amyloid accumulation, particularly with respect to the occurrence of striatal vs cortical amyloid.35–38 Different staging schemes may have to be developed for these populations.
Based on clinicopathologic correlations between striatal amyloid deposition (corresponding to Thal phase 3) and the occurrence of dementia,2,4,34 it has been suggested to incorporate an a priori distinction between cortical and striatal amyloid deposition in the analysis of amyloid-sensitive PET data, representing a pathologic 2-stage model that would possibly be more predictive of clinical status.39,40 In our data-driven estimation of an in vivo staging scheme, we provide evidence that (1) PET-measured striatal amyloid deposition (anatomical division 4 in our model) is extremely unlikely to occur without concomitant cortical amyloid (figure 2) and that (2) advanced in vivo amyloid stages are most frequently observed in patients with AD and associate with cognitive impairments in individuals without dementia. Comparison with conventional continuous measures of global amyloid load indicate that these show a higher dynamic range in clinically overt disease stages, whereas the presented in vivo staging approach may be particularly useful for stratification of early, preclinical stages of AD.
A limitation of our in vivo imaging approach to amyloid staging is that the presence of regional amyloid necessarily needs to be defined by a signal cutoff that aims to separate noise from specific tracer binding. While we oriented this cutoff on most commonly used cutoffs for defining amyloid positivity on the basis of global cortical florbetapir-PET SUVRCer, cutoff definition is an arbitrary undertaking per se, and the transferability of global signal cutoffs to regional signals remains largely unexplored. Furthermore, in this study we used a constant regional cutoff, whereas it may be argued that differential cutoffs should be used due to differing noise levels between brain regions.33,35,40 Although the invariance of the regional amyloid pattern under varying cutoffs (appendix e-1) indicates little interregional variability in noise levels in our PVE-corrected data, the choice of cutoff definition might well affect the regional staging scheme and future research should investigate possible advantages of region-specific cutoffs for the presented in vivo amyloid staging approach. Moreover, as is the case for cutoffs applied to the global signal,12 the height of the regional cutoff affects the absolute numbers of individuals with detectable amyloid deposits. Thus, cutoff selection as well as other methodical choices, such as the regional detail of the staging scheme, should weigh off priorities for sensitivity or specificity depending on the actual research context or clinical application of the staging approach.
Pending replication of our findings in independent cohorts and using different radiotracers, the presented in vivo amyloid staging approach may become a valuable tool for characterizing an individual's amyloid burden for diagnostic purposes or for sample stratification in clinical trials.
GLOSSARY
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CN
cognitively normal
- DR
delayed recall
- GM
gray matter
- MCI
mild cognitive impairment
- PVE
partial volume effects
- SUVRCer
standard uptake value ratios scaled to the mean uptake of the whole cerebellum
- TMT-B
Trail Making Test B
Footnotes
Supplemental data at Neurology.org
Editorial, page 2029
Contributor Information
Collaborators: Alzheimer's Disease Neuroimaging Initiative, Michael Weiner, Paul Aisen, Michael Weiner, Paul Aisen, Ronald Petersen, Clifford R. Jack, Jr, William Jagust, John Q. Trojanowki, Arthur W. Toga, Laurel Beckett, Robert C. Green, Andrew J. Saykin, John Morris, Enchi Liu, Robert C. Green, Tom Montine, Ronald Petersen, Paul Aisen, Anthony Gamst, Ronald G. Thomas, Michael Donohue, Sarah Walter, Devon Gessert, Tamie Sather, Laurel Beckett, Danielle Harvey, Anthony Gamst, Michael Donohue, John Kornak, Clifford R. Jack, Jr, Anders Dale, Matthew Bernstein, Joel Felmlee, Nick Fox, Paul Thompson, Norbert Schuff, Gene Alexander, Charles DeCarli, William Jagust, Dan Bandy, Robert A. Koeppe, Norm Foster, Eric M. Reiman, Kewei Chen, Chet Mathis, John Morris, Nigel J. Cairns, Lisa Taylor-Reinwald, J.Q. Trojanowki, Les Shaw, Virginia M.Y. Lee, Magdalena Korecka, Arthur W. Toga, Karen Crawford, Scott Neu, Andrew J. Saykin, Tatiana M. Foroud, Steven Potkin, Li Shen, Zaven Kachaturian, Richard Frank, Peter J. Snyder, Susan Molchan, Jeffrey Kaye, Joseph Quinn, Betty Lind, Sara Dolen, Lon S. Schneider, Sonia Pawluczyk, Bryan M. Spann, James Brewer, Helen Vanderswag, Judith L. Heidebrink, Joanne L. Lord, Ronald Petersen, Kris Johnson, Rachelle S. Doody, Javier Villanueva-Meyer, Munir Chowdhury, Yaakov Stern, Lawrence S. Honig, Karen L. Bell, John C. Morris, Beau Ances, Maria Carroll, Sue Leon, Mark A. Mintun, Stacy Schneider, Daniel Marson, Randall Griffith, David Clark, Hillel Grossman, Effie Mitsis, Aliza Romirowsky, Leyla deToledo-Morrell, Raj C. Shah, Ranjan Duara, Daniel Varon, Peggy Roberts, Marilyn Albert, Chiadi Onyike, Stephanie Kielb, Henry Rusinek, Mony J de Leon, Lidia Glodzik, Susan De Santi, P. Murali Doraiswamy, Jeffrey R. Petrella, R. Edward Coleman, Steven E. Arnold, Jason H. Karlawish, David Wolk, Charles D. Smith, Greg Jicha, Peter Hardy, Oscar L. Lopez, MaryAnn Oakley, Donna M. Simpson, Anton P. Porsteinsson, Bonnie S. Goldstein, Kim Martin, Kelly M. Makino, M. Saleem Ismail, Connie Brand, Ruth A. Mulnard, Gaby Thai, Catherine Mc-Adams-Ortiz, Kyle Womack, Dana Mathews, Mary Quiceno, Ramon Diaz-Arrastia, Richard King, Myron Weiner, Kristen Martin-Cook, Michael DeVous, Allan I. Levey, James J. Lah, Janet S. Cellar, Jeffrey M. Burns, Heather S. Anderson, Russell H. Swerdlow, Liana Apostolova, Po H. Lu, George Bartzokis, Daniel H.S. Silverman, Francine Parfitt, Heather Johnson, Martin R. Farlow, Ann Marie Hake, Brandy R. Matthews, Scott Herring, Christopher H. van Dyck, Richard E. Carson, Martha G. MacAvoy, Howard Chertkow, Howard Bergman, Chris Hosein, Sandra Black, Bojana Stefanovic, Curtis Caldwell, Ging-Yuek Robin Hsiung, Howard Feldman, Benita Mudge, Michele Assaly, Andrew Kertesz, John Rogers, Dick Trost, Charles Bernick, Donna Munic, Diana Kerwin, Marek-Marsel Mesulam, Kristina Lipowski, Chuang-Kuo Wu, Nancy Johnson, Carl Sadowsky, Walter Martinez, Teresa Villena, Raymond Scott Turner, Kathleen Johnson, Brigid Reynolds, Reisa A. Sperling, Keith A. Johnson, Gad Marshall, Meghan Frey, Jerome Yesavage, Joy L. Taylor, Barton Lane, Allyson Rosen, Jared Tinklenberg, Marwan Sabbagh, Christine Belden, Sandra Jacobson, Neil Kowall, Ronald Killiany, Andrew E. Budson, Alexander Norbash, Patricia Lynn Johnson, Thomas O. Obisesan, Saba Wolday, Salome K. Bwayo, Alan Lerner, Leon Hudson, Paula Ogrocki, Evan Fletcher, Owen Carmichael, John Olichney, Charles DeCarli, Smita Kittur, Michael Borrie, T-Y Lee, Rob Bartha, Sterling Johnson, Sanjay Asthana, Cynthia M. Carlsson, Steven G. Potkin, Adrian Preda, Dana Nguyen, Pierre Tariot, Adam Fleisher, Stephanie Reeder, Vernice Bates, Horacio Capote, Michelle Rainka, Douglas W. Scharre, Maria Kataki, Earl A. Zimmerman, Dzintra Celmins, Alice D. Brown, Godfrey D. Pearlson, Karen Blank, Karen Anderson, Andrew J. Saykin, Robert B. Santulli, Eben S. Schwartz, Kaycee M. Sink, Jeff D. Williamson, Pradeep Garg, Franklin Watkins, Brian R. Ott, Henry Querfurth, Geoffrey Tremont, Stephen Salloway, Paul Malloy, Stephen Correia, Howard J. Rosen, Bruce L. Miller, Jacobo Mintzer, Crystal Flynn Longmire, Kenneth Spicer, Elizabether Finger, Irina Rachinsky, John Rogers, Andrew Kertesz, Dick Drost, Nunzio Pomara, Raymundo Hernando, Antero Sarrael, Susan K. Schultz, Laura L. Boles Ponto, Hyungsub Shim, Karen Elizabeth Smith, Norman Relkin, Gloria Chaing, Lisa Raudin, Amanda Smith, Kristin Fargher, and Balebail Ashok Raj
AUTHOR CONTRIBUTIONS
Michel J. Grothe: study concept, analysis and interpretation of data, drafting/revising the manuscript for content. Henryk Barthel: study concept, interpretation of data, drafting/revising the manuscript for content. Jorge Sepulcre: study concept, contribution of analytic tools, drafting/revising the manuscript for content. Martin Dyrba: analysis of data, contribution of analytic tools. Osama Sabri: interpretation of data, drafting/revising the manuscript for content. Stefan Teipel: interpretation of data, drafting/revising the manuscript for content.
STUDY FUNDING
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904 and DOD ADNI [Department of Defense award number W81XWH-12-2-0012]). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration, private pharmaceutical companies and nonprofit organizations, as a $60 million, 5-year public–private partnership. ADNI is funded by the NIA, the NIBIB, and through contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California.
DISCLOSURE
M. Grothe reports no disclosures relevant to the manuscript. H. Barthel received consultant and speaker honoraria as well as travel expenses from Piramal Imaging. J. Sepulcre is supported by the NIH (1K23EB019023). M. Dyrba reports no disclosures relevant to the manuscript. O. Sabri received consultant and speaker honoraria as well as travel expenses from Piramal Imaging. S. Teipel reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 2.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 3.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 1999;45:358–368. [DOI] [PubMed] [Google Scholar]
- 4.Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain 2015;138:1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging 1991;12:295–312. [DOI] [PubMed] [Google Scholar]
- 6.Giannakopoulos P, Hof PR, Vallet PG, Giannakopoulos AS, Charnay Y, Bouras C. Quantitative analysis of neuropathologic changes in the cerebral cortex of centenarians. Prog Neuropsychopharmacol Biol Psychiatry 1995;19:577–592. [DOI] [PubMed] [Google Scholar]
- 7.Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA. Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 2012;72:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teipel S, Drzezga A, Grothe MJ, et al. Multimodal imaging in Alzheimer's disease: validity and usefulness for early detection. Lancet Neurol 2015;14:1037–1053. [DOI] [PubMed] [Google Scholar]
- 9.Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer disease: phase 3 study. Alzheimers Dement 2015;11:964–974. [DOI] [PubMed] [Google Scholar]
- 10.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thal DR, Beach TG, Zanette M, et al. [(18)F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer's disease: specific detection of advanced phases of amyloid-beta pathology. Alzheimers Dement 2015;11:975–985. [DOI] [PubMed] [Google Scholar]
- 12.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 2015;138:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 14.Josephs KA, Murray ME, Whitwell JL, et al. Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol 2016;131:571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gertz HJ, Xuereb J, Huppert F, et al. Examination of the validity of the hierarchical model of neuropathological staging in normal aging and Alzheimer's disease. Acta Neuropathol 1998;95:154–158. [DOI] [PubMed] [Google Scholar]
- 16.Jellinger KA. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol 2008;116:1–16. [DOI] [PubMed] [Google Scholar]
- 17.Grothe MJ, Teipel SJ. Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer's disease correspond to dissociable functional brain networks. Hum Brain Mapp 2016;37:35–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grothe MJ, Heinsen H, Amaro E Jr, Grinberg LT, Teipel SJ. Cognitive correlates of basal forebrain atrophy and associated cortical hypometabolism in mild cognitive impairment. Cereb Cortex 2016;26:2411–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teipel S, Grothe MJ. Does posterior cingulate hypometabolism result from disconnection or local pathology across preclinical and clinical stages of Alzheimer's disease? Eur J Nucl Med Mol Imaging 2016;43:526–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teipel SJ, Kurth J, Krause B, Grothe MJ. The relative importance of imaging markers for the prediction of Alzheimer's disease dementia in mild cognitive impairment: beyond classical regression. Neuroimage Clin 2015;8:583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller-Gartner HW, Links JM, Prince JL, et al. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. J Cereb Blood Flow Metab 1992;12:571–583. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez-Escamilla G, Lange C, Teipel S, Buchert R, Grothe MJ. PETPVE12: an SPM toolbox for partial volume effects correction in brain PET: application to amyloid imaging with AV45-PET. Neuroimage 2017;147:669–677. [DOI] [PubMed] [Google Scholar]
- 23.Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 24.Catafau AM, Bullich S, Seibyl JP, et al. Cerebellar amyloid-beta plaques: how frequent are they, and do they influence 18F-florbetaben SUV ratios? J Nucl Med 2016;57:1740–1745. [DOI] [PubMed] [Google Scholar]
- 25.Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer’s disease and cognitively normal subjects. J Nucl Med 2012;53:378–384. [DOI] [PubMed] [Google Scholar]
- 26.Landau SM, Breault C, Joshi AD, et al. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013;54:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol 2011;68:1404–1411. [DOI] [PubMed] [Google Scholar]
- 28.Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landau SM, Lu M, Joshi AD, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol 2013;74:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimmer T, Riemenschneider M, Forstl H, et al. Beta amyloid in Alzheimer's disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry 2009;65:927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vlassenko AG, Benzinger TL, Morris JC. PET amyloid-beta imaging in preclinical Alzheimer's disease. Biochim Biophys Acta 2012;1822:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 2016;80:247–258. [DOI] [PubMed] [Google Scholar]
- 34.Beach TG, Sue LI, Walker DG, et al. Striatal amyloid plaque density predicts Braak neurofibrillary stage and clinicopathological Alzheimer's disease: implications for amyloid imaging. J Alzheimers Dis 2012;28:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Annus T, Wilson LR, Hong YT, et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement 2016;12:538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 2007;27:6174–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koivunen J, Verkkoniemi A, Aalto S, et al. PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer's disease. Brain 2008;131:1845–1853. [DOI] [PubMed] [Google Scholar]
- 38.Villemagne VL, Ataka S, Mizuno T, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol 2009;66:1537–1544. [DOI] [PubMed] [Google Scholar]
- 39.Beach TG, Thal DR, Zanette M, Smith A, Buckley C. Detection of striatal amyloid plaques with [18F]flutemetamol: validation with postmortem histopathology. J Alzheimers Dis 2016;52:863–873. [DOI] [PubMed] [Google Scholar]
- 40.Shah N, Frey KA, Muller ML, et al. Striatal and cortical beta-amyloidopathy and cognition in Parkinson's disease. Mov Disord 2016;31:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]