

Abstract
Native mass spectrometry (MS) with electrospray ionization (ESI) has evolved as an invaluable tool for the characterization of intact native proteins and non-covalently bound protein complexes. Here we report the structural characterization by high resolution native top-down MS of human thrombin and its complex with the Bock thrombin binding aptamer (TBA), a 15-nucleotide DNA with high specificity and affinity for thrombin. Accurate mass measurements revealed that the predominant form of native human α-thrombin contains a glycosylation mass of 2205 Da, corresponding to a sialylated symmetric biantennary oligosaccharide structure without fucosylation. Native MS showed that thrombin and TBA predominantly form a 1:1 complex under near physiological conditions (pH 6.8, 200 mM NH4OAc), but the binding stoichiometry is influenced by the solution ionic strength. In 20 mM ammonium acetate solution, up to two TBAs were bound to thrombin, whereas increasing the solution ionic strength destabilized the thrombin-TBA complex and 1 M NH4OAc nearly completely dissociated the complex. This observation is consistent with the mediation of thrombin-aptamer binding through electrostatic interactions and it is further consistent with the human thrombin structure that contains two anion binding sites on the surface. Electron capture dissociation (ECD) top-down MS of the thrombin-TBA complex performed with a high resolution 15 Tesla Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer showed the primary binding site to be at exosite I located near the N-terminal sequence of the heavy chain, consistent with crystallographic data. High resolution native top-down MS is complementary to traditional structural biology methods for structurally characterizing native proteins and protein-DNA drug complexes.
Keywords: Native mass spectrometry, electron capture dissociation, protein-DNA complex, gas phase structure, binding interface
Graphical abstract
INTRODUCTION
Thrombin is a multifunctional protein with key functions in hemostasis and thrombosis [1]. It is proteolytically generated from the zymogen prothrombin upon membrane surface activation in response to vascular injury. The enzymatically active α-thrombin cleaves the fibrinogen to fibrin, which polymerizes to form a matrix for the insoluble nascent clot. It also activates a number of other procoagulant protein factors, including factor V, VIII and XI, via limited proteolysis and induces platelet aggregation. These coagulant factors covalently crosslink fibrin and further amplify the prothrombotic cascade. In addition to its procoagulant activities, thrombin also performs a number of anticoagulant functions. Upon binding to thrombomodulin, thrombin activates protein C, which in turn cleaves and inactivates factors Va and VIIIa, two essential coagulation cofactors, and thereby inhibits the amplification and progression of the coagulation cascade [2]. The procoagulant and anticoagulant activities of thrombin are tightly regulated by the allosteric control mechanism of thrombin through an extensive interaction network to maintain the fine balance of hemostasis and thrombosis under physiological and pathological conditions [3]. Because of its central role in hemostasis and thrombosis, thrombin has captured intense research interest and has been a prominent protein target for anticoagulant and cardiovascular disease therapy.
Recently, a novel class of thrombin binding drugs has received heightened interest in the inhibitory treatment of thrombosis. Thrombin binding aptamers (TBAs) are short strands of DNA/RNA oligomers with high specificity and affinity for thrombin. The first thrombin binding aptamer, a 15-mer oligonucleotide, 5′-GGTTGGTGTGGTTGG-3′, was discovered through an in vitro combinatorial selection and polymerase chain reaction process by Bock et al [4]. An increasing number of modified TBAs have since been developed and used for diagnostic and therapeutic applications [5–9]. Unlike other thrombin-binding biomolecules and proteins, TBAs are smaller mass DNA molecules with high chemical and physiological stabilities, easier to synthesize and have high specificity and affinity for thrombin. TBA has a G-nucleotide rich sequence and forms a uniquely ordered G-quadruplex structure, which has high shape complementarity with the thrombin surface anion binding site and is postulated to be the molecular basis for its high thrombin specificity. The structure of TBA, initially reported using nuclear magnetic resonance (NMR) spectroscopy, revealed that TBA in solution adopts a unimolecular chair-like quadruplex conformation consisting of two stacked G-quartets connected by two TT loops and a TGT loop [10–12]. This structure was subsequently confirmed by the X-ray crystallographic structure of TBA in complex with thrombin, despite the fine structural differences in the connectivity of the TBA central bases between these two models [13, 14].
Gross et al. studied the interaction between TBA and several metal ions with similar ionic radii by ESI-MS, and showed enhanced adduct formation with TBA compared with control oligonucleotides, supporting the G-quadruplex structure of TBA in eight-coordinate bonding with metal ions [15]. They further showed that metal binding provided additional protection to the TBA structure and less hydrogen/deuterium (H/D) exchange was observed compared with the free TBA. Balthasart et al. studied the interactions between ammonium ions and five DNA G-quadruplexes including TBA by ESI-MS and demonstrated that the G-quadruplex-(NH4)n complexes were observed under the softest instrumental conditions and the ammonium ions stabilized the G-quadruplex structures [16]. Hong et al. also reported that TBA−K+ and TBA−NH4+ both showed the characteristic feature of the antiparallel G4 structure by circular dichroism (CD) and ESI-MS [17]. However, regardless of the presence or absence of alkali metal ions, the charge state distributions of TBA were almost identical, suggesting that metal binding to TBA hardly altered the structure of TBA in ESI in solution. Recently, high-resolution crystallographic structures of the human thrombin-TBA complex have been reported that provide atomic level details of the interactions between the human thrombin and TBA [18, 19]. Thus far, no mass spectrometric structural characterization of the human thrombin-TBA complex has been reported.
Over the past decades, native mass spectrometry (MS) [20] has evolved as a powerful tool for direct probing of native state proteins and various non-covalent protein complexes under near physiological conditions [21–33]. Combined with top-down MS, the information derived from such native MS strategies is more comprehensive that that derived from the traditional bottom-up MS approach. This is because the physiologically protein complexes and their higher order assemblies are the subject of the analysis, rather than the denatured proteins. Such analyses have been made possible by the development of advanced MS instrumentation. Rich structural biology information can be obtained from native MS experiments regarding protein complex stoichiometry [34, 35], protein-protein interaction interfaces [26], and protein-lipid interactions [24]. The native MS approach has significant advantages in specificity, sensitivity and analytical speed compared with traditional structural biology methods such as X-ray crystallography and NMR. In previous studies, we have demonstrated that native top-down MS with electron capture dissociation (ECD) can yield protein complex structural information regarding the location of subunit interaction interfaces [26], sites of ligand binding [23], and possibly surface topology [25], and the MS-derived information is consistent with published studies using solution and solid state techniques. Here we applied high resolution native MS to the characterization of native human thrombin and its complex with thrombin binding aptamer to provide structural elucidation and insights complementary to those obtained from conventional structural biology methods.
EXPERIMENTAL SECTION
Materials and Sample Preparation
The thrombin binding aptamer (5′-GGTTGGTGTGGTTGG-3′) was purchased from Integrated DNA Technologies, Inc. (Coralville, IA). Human α-thrombin was purchased from Haematologic Technologies, Inc. (Esses Junction, VT). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. All solutions were prepared in Milli-Q water (Millipore Corp., Billerica, MA). The thrombin samples were exchanged into aqueous ammonium acetate solutions using 10 kDa MWCO Amicon Ultra centrifugal columns (Millipore Corp., Billerica, MA). The thrombin sample was then treated with TBA solutions in the ammonium acetate solution with the appropriate ionic strength for one hour at room temperature prior to the MS analysis.’
Native Top-Down Mass Spectrometry
Top-down MS of native human thrombin and thrombin-TBA complex was performed on a ultrahigh resolution 15-Tesla SolariX hybrid Qh-FTICR mass spectrometer (Bruker) and a Synapt G1 HDMS QTOF mass spectrometer (Waters). Solutions containing human α-thrombin (10 μM) were prepared with 20 mM ammonium acetate (pH 6.8). For the thrombin-TBA complex, the thrombin solution was incubated with excess TBA (1 mM) in serial diluted aqueous solutions adjusted to different ionic strengths by ammonium acetate. The protein solutions were electrosprayed via nanoESI glass emitters (Proxeon/Thermo Scientific, Inc.) at flow rates of 20–50 nL/min.
For FTICR MS measurements, the electrospray source temperature was controlled by a source heater and countercurrent nitrogen drying gas flow of 2.5 L/min to provide optimal desolvation. The temperature was set between 180–200°C to allow desolvation of the native protein samples while maintaining the integrity of the non-covalent complex. MS experiments were performed in broadband mode (m/z 600–10000) with the following settings: 1 M data size, capillary voltage 1000–1200 V, 0.5 sec source accumulation time, 1 sec ion accumulation time, 0.05 sec ion cooling time, 1 ms time of flight. The skimmer potential was tuned between 35–80 V to provide source activation of the thrombin and thrombin-TBA complex for sufficient desolvation. ECD-MS experiments of thrombin and the thrombin-TBA complex were performed on all charge states of the precursor to maximize product ion yield, with the following settings: 0.01 sec ECD pulse length, 1 V bias, 15 V ECD lens. ECD mass spectra were collected from data averaging of up to 800 scans.
For the native MS experiments using the Synapt G1 HDMS mass spectrometer, the full mass spectral scan was acquired over the range of m/z 200–10000, with the following instrument settings: capillary voltage 1000–1500 kV, sample cone voltage 150 V, extraction cone voltage 5.5 V, source temperature 100°C, source backing pressure 5–6 Torr.
Data Analysis
MS/MS data were processed with DataAnalyis and Biotools (Bruker Daltonics, Inc.). Briefly, monoisotopic masses ((M+H)+) were extracted by DataAnalysis software using a modified Thrash algorithm (SNAP ver 2.0, Bruker) with the following settings: quality factor threshold 0.5; S/N threshold 2; maximum charge state, ≤ protein precursor charge state. Data were calibrated by internal product ions and further assigned using Biotools based on protein sequences determined by accurate mass measurements. The assigned ions were manually confirmed to ensure the quality of the assignments.
RESULTS AND DISCUSSION
High Resolution Native MS of Human Thrombin
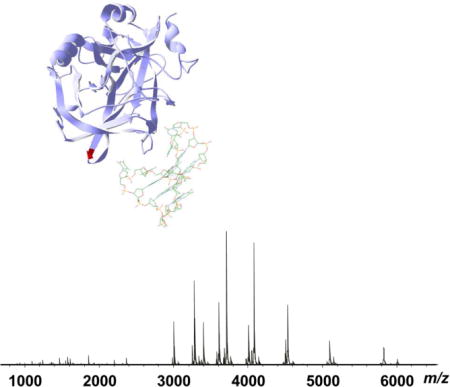
Native MS of human thrombin was performed using both Synapt G1 QTOF and Solarix 15 T hybrid Qh-FTICR mass spectrometers. The intact mass of the human thrombin sample was measured to be 36029 Da (average mass, QTOF data) and 36006.3 Da (monoisotopic mass, FTMS data) (Figure 1). The accurate mass corresponds to the sum of the light and heavy chains of human α-thrombin (UniProt ID P00734_THRB_Human, light chain residues 328–363, heavy chain residues 364–622) with an additional modification of 2207.37 Da (monoisotopic mass). This modification is consistent with a common glycan with a sialylated symmetric biantennary structure. The MS results showed that the human α-thrombin sample was highly homogeneous and the predominant form contained a single glycosylation at the conserved N-glycosylation site Asn53 on the heavy chain, as confirmed by ECD fragmentation (Supplemental Figure 1). Both the light chain and heavy chain do not carry other modifications besides four disulfide bonds. This is consistent with the proteolytic processing of the α-thrombin from its zymogen prothrombin, and therefore no modifications were found on the N-termini of both light and heavy chains. Our results agree with the mass of the human α-thrombin previously reported by Sall et al., where a homogeneous glycosylation pattern was also observed [36]. A minor molecular species with the monoisotopic mass of 36154.16 Da, corresponding to the fucosylated form of the biantennary glycan, was also observed in our FTICR MS experiments, but the relative abundance is less than 10% of the overall thrombin abundance. The carbohydrate structure of human thrombin has been previously characterized by Nilsson et al. through sequential exoglycosidase digestion of intact thrombin and sugar and methylation analysis of the oligosaccharides by GC and LC/MS [37]. Despite the difference in the extent of fucosylation, our study confirmed the sialylated biantennary core glycan structure as the predominant glycosylation in the human α-thrombin. Fucosylation is one of the most common modifications of the glycan structure of glycoproteins or glycolipids, and it comprises the attachment of a fucose residue to N- or O-glycans and glycolipids. Fucosylation is regulated by fucosyltransferase enzyme activities; increased levels of fucosylation have been implicated in a number of pathological conditions, including inflammation and cancer [38]. The microheterogeneity in the observed fucosylation levels in different human thrombin samples may be a reflection of the specific sample and physiological conditions.
Figure 1.
High resolution native MS of human α-thrombin. (a) Primary sequence of human α-thrombin (UnitProt P00734, chain 328–363 and 364–622); (b) Native FT-ICR mass spectrum of human α-thrombin (10 μM in 20 mM NH4OAc), pH 6.8. The accurate mass measurement of the native human α-thrombin is shown for the 11+ precursor (inset). The annotated mass values are monoisotopic masses for the corresponding molecular species.
Native MS and Top-Down MS of Human Thrombin-TBA complex
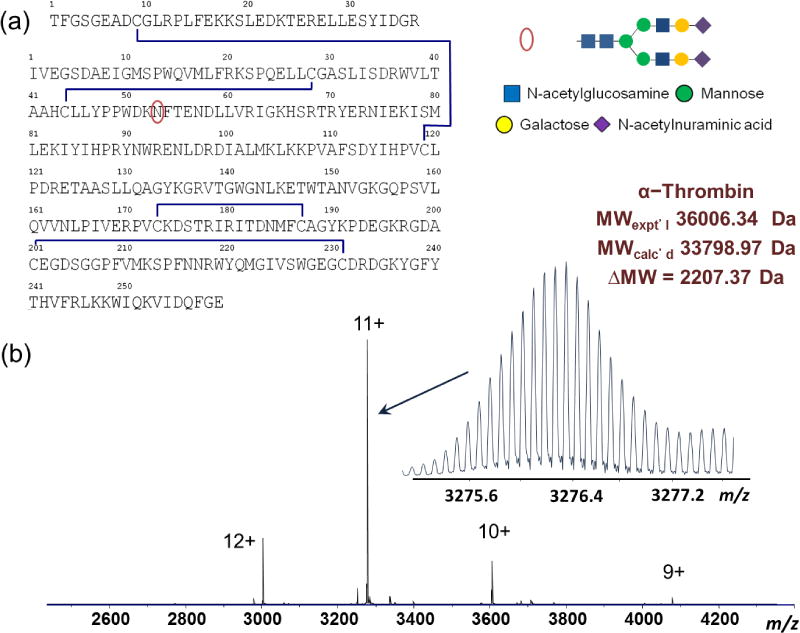
Native MS of the human α-thrombin-TBA complex was performed on both FTICR and QTOF MS instruments. High resolution MS of the TBA in the positive ion mode measured its accurate monoisotopic mass to be 4723.82 Da, consistent with the 15-mer oligonucleotide structure (Figure 2a). TBA has high propensity to form adducts with metal ions (Supplemental Figure 2a) and the multiple adduct ions add significant complexity to the interpretation of the thrombin-TBA complex MS data; therefore we chose the desalted form of TBA (Supplemental Figure 2b) for the ESI-MS study of the thrombin-TBA complex. Metal cations, especially monovalent alkali metal cations K+ and Na+, help stabilize the G-quadruplex structure of TBA, which is essential for the thrombin-TBA interaction. However, the presence or absence of the metal adducts did not change the overall charge state distributions of TBA, suggesting that metal cations did not change the structure of TBA significantly. This is consistent with the report by Hong et al. [17]. Furthermore, in our experiments, we did not observe significant difference in the MS and MS/MS between thrombin-TBA and thrombin-TBA-K+/Na+; the presence of metal cations did not enhance the thrombin-aptamer complex binding affinity, nor did it change the complex dissociation behaviors under CAD or ECD conditions. This observation is in line with previous reports that despite the role of metal cations in stabilizing the G-quadruplex structure of TBA, the presence of metal cations is not the prerequisite for the formation of the three dimensional structure of TBA. TBA has high propensity to fold into the distinct G4 structure autonomously or is facilitated by other factors such as temperature [39], molecular crowding [40], and most notably, by the presence of thrombin [41]. Nagatoishi et al. reported formation of the TBA G-quadruplex under cation deficient conditions and showed that the presence of thrombin alone induced the formation of the TBA G-quadruplex structure [41]. Other groups also reported that thrombin is able to act as molecular chaperon and induces formation of the quadruplex structure of TBA [42]. Together, these findings support the concept that the thrombin-TBA complex observed in our study represents or closely approximates its native state in the solution and gas phase.
Figure 2.
High resolution native MS of human α-thrombin-TBA complex. (a) Positive ion mass spectrum of TBA; (b) mass spectra of thrombin-TBA complex and thrombin, acquired from solution containing 200 mM NH4OAc. The mass difference shows the incorporation of one TBA in the complex. The mass values are annotated as monoisotopic mass. The accurate mass measurements are shown for the 11+ ions (inset). The filled blue circles indicate the number of TBAs in each complex.
Native MS of the human thrombin-TBA complex was measured with different ammonium acetate concentrations. ESI-MS of the thrombin-TBA complex in 200 mM NH4OAc showed the primary complex to be 40730 Da, a mass increase corresponding to a 1:1 binding stoichiometry between thrombin and TBA (Figure 2b). A small amount of the thrombin-TBA2 complex (45481 Da, average mass) was also observed under this condition. Native MS performed on the QTOF MS instrument showed consistent results with the FTICR MS data (Figure 3). The complex stability can be affected by increasing the solution ionic strength. Under diluted NH4OAc (20 mM), the thrombin-TBA2 formed the predominant complex, whereas under increased NH4OAc condition (1 M), the thrombin-TBA complex was significantly destabilized and dissociated into the free thrombin and TBA (Figures 3b and 3c). The influence of ionic strength on binding affinity suggests that electrostatic interactions are a predominant factor in the binding interactions between thrombin and TBA, and that the electrostatic shielding effect from increased ionic concentration is significant to counteract other contributions including shape recognition and hydrophobic interactions. This has been confirmed by the recently reported structures of the thrombin-TBA complexes (Protein Data Bank, PDB 4DII and PDB 4DIH) and studies by Lin et al. [43]. Hianik et al. also showed that increased concentrations of NaCl resulted in the weakening of interactions between thrombin and different aptamers as measured by electrochemical sensors [44].
Figure 3.

Native MS of human α-thrombin-TBA complex under different ionic concentrations. (a) 200 mM NH4OAc; (b) 20 mM NH4OAc; (c) 1 M NH4OAc. Blue circles indicate the number of TBAs in each complex. The charge states of major ions in each spectrum are labeled.
The presence of the thrombin-TBA2 complex under low ionic concentrations suggests that there are at least two TBA binding sites on thrombin with possibly different specificities and affinities. Under near physiological conditions, the binding site with higher specificity binds TBA to form the predominant 1:1 thrombin-TBA structure, whereas under lower ionic strength or high TBA concentration, the low specificity binding site binds an additional TBA to form the thrombin-TBA2 complex. This is consistent with the thrombin structure that contains two anion binding sites with different specificities on the surface.
Our results are corroborated by a number of studies using other analytical and structural approaches. Bornhop and coworkers studied the binding interactions between human α-thrombin and two different DNA aptamers using backscattering interferometry [45]. It was reported that binding of the first aptamer molecule resulted in a significant change in the binding affinity of the second aptamer molecule. Specifically, the binding affinity of the “Bock” aptamer to preformed thrombin-“Tasset” aptamer complexes was found to be 7-fold higher compared to binding to thrombin alone. This result suggests that at least two binding sites are present on the thrombin surface, consistent with our results by native MS. However, it should be noted that increased binding affinity for the second aptamer was not observed in our results, as one would expect that the predominant complex would be the thrombin-TBA2 complex instead of the thrombin-TBA complex, given the excess ratio of the TBA relative to thrombin. This suggests that the aptamer used in our study, the Bock aptamer, preferentially binds to a specific site on the thrombin surface, whereas the Tasset aptamer binds to a distinctly different binding site on the thrombin surface.
ECD-MS was performed on the thrombin-TBA complex (at 200 mM NH4OAc) (Figure 4). The ECD fragmentation was applied to all precursor charge states to increase the fragmentation efficiency and product ion yield. Adding dilute DTT solutions (20 μM) to the protein sample also helped improve the top-down MS efficiency over the time course of ECD data collection while maintaining the thrombin-TBA complex integrity. ECD fragmentation yielded a total of 13 c ions and 19 z’ ions, among which 11 c ions showed a mass increase of 4723 Da, corresponding to the mass of the intact TBA (Figure 4). No fragmentation of the TBA molecule was detected. The dissociation coverage map for the thrombin-TBA complex is similar to that obtained for the thrombin protein alone (Supplemental Figure 1). ECD-MS/MS of protein-ligand complexes yields product ions of the protein chain, some of which are still bound to the ligand (i.e., the TBA ligand in this case). The sequence of these ligand-bound fragments was used to analyze the ligand binding regions. The ECD fragmentation map suggests that the region around Lys21 is the primary TBA binding site (Figure 4a).
Figure 4.
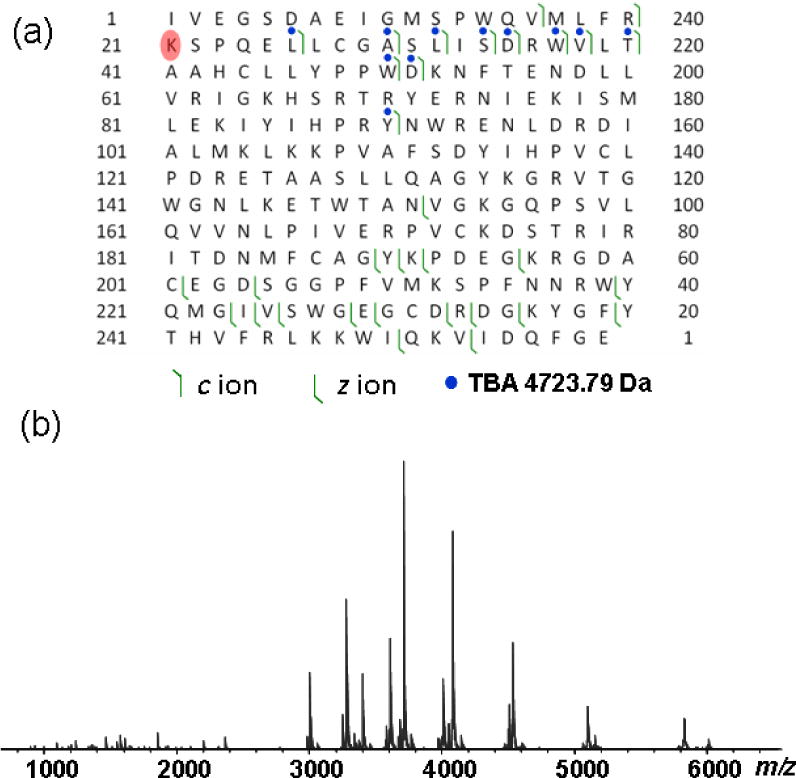
ECD fragmentation of the human α-thrombin-TBA complex. (a) Product ion map of the thrombin-TBA complex. Only the heavy chain is shown. Blue circles indicate the mass increase of 4723.8 Da, corresponding to the mass of intact TBA. The highlighted residue (Lys21) in red indicates the interaction site; (b) ECD mass spectrum of the thrombin-TBA complex.
Correlation of Native MS with Thrombin Structure
Human α-thrombin has been structurally characterized extensively over the past decades, given its central role in hemostasis and thrombosis and its clinical importance. The primary sequence of human α-thrombin contains a 36 residue light chain and a 259 residue heavy chain, linked by an interchain disulfide bond. The light chain is not enzymatically active, whereas the heavy chain contains the active catalytic site residues Ser205, His43 and Asp99, and two allosteric binding sites on the surface. A distinct feature of the thrombin structure is its uneven surface charge distribution, resulting in high electrostatic field strengths on the thrombin surface. Most notably, two contiguous surface patches with high populations of positively charged residues extend along the opposite side of the thrombin surface, known as the anion binding exosite I and exosite II (Figure 5a). These two exosites have different substrate binding specificities; exosite I is the fibrinogen recognition site and exosite II is the heparin binding site.
Figure 5.
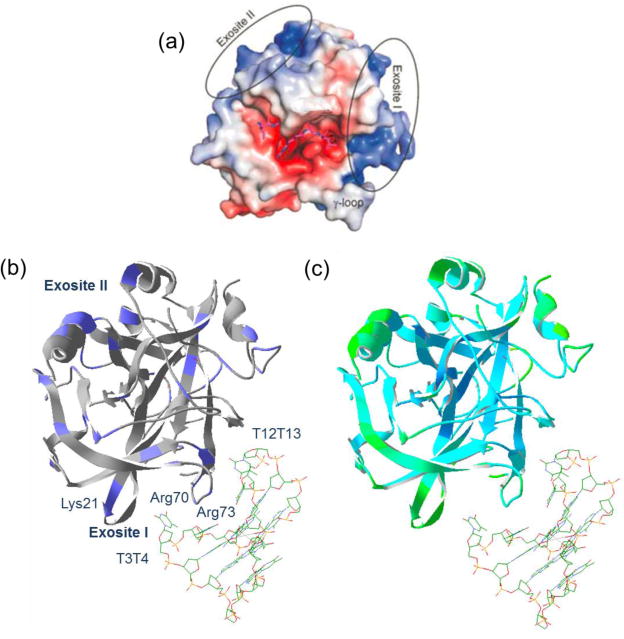
Structures of human thrombin and thrombin-TBA complex. (a) The surface structure representation of the electrostatic properties of human α-thrombin (PDB 1TB6). The blue color shows the positively charged surface areas and the red color indicates the negatively charged surface areas. The positions of the two anion binding exosites are highlighted; (b) the structure of the thrombin-TBA complex (PDB 4DII). The thrombin structure is shown in ribbons, and the TBA molecule is shown in the stick structure. Positively charged residues are colored blue on the structure. The identified and other potential binding sites are labeled; (c) the structure of thrombin-TBA complex (PDB 4DII) with the B-factor highlighted. The green color represents regions with high flexibility; the cyan color represents regions with low flexibility.
Exosite I consists of two main N-terminal sequence stretches, Arg35–Leu41 and Lys70–Lys81, including a cluster of basic residues Arg35, Lys36, Lys70, Arg73, Arg75, and Arg77 (chymotrypsin numbering, in which Lys36 corresponds to Lys21 in the heavy chain). These basic residues collectively make important contributions to the overall positive electrostatic surface potential at exosite I, which interacts with the negative charge of the complementary TBA domain. Exosite II is located in the C-terminal region and includes key residues Lys191, Lys196, and Arg197. The crystallographic structure of the thrombin-TBA complex initially reported by Tulinsky et al. showed that the TBA molecule to be sandwiched between exosites I and II on two thrombin molecules, suggesting both exosites contribute to TBA binding [13]. High resolution structures of the human thrombin-TBA complex at 2.05 Å and 1.80 Å were recently reported and it was shown that the exosite I is the primary binding site for TBA, whereas the secondary interface is the result of crystal packing optimization rather than specific recognition [18, 19]. TBA binding is through interactions between the two TT loops (T3T4 and T12T13) and multiple residues on the exosite I including Arg70, Arg73 and Lys21 (Figure 5b). The T3T4 loop is sandwiched between the exosite I sequence stretches Arg20–Leu26 (Arg35–Leu41 in chymotrypsin numbering) and Arg70–Arg73 (Arg75–Arg77a in chymotrypsin numbering). ECD produced fragment ions flanking Lys21, therefore supporting the interaction between Lys21 and TBA; however, no fragment ions were observed near Arg70 and Arg73. The low fragmentation efficiency around the Arg70–Arg73 region is likely related to the lower structural flexibility near these residues, as reflected by their lower crystallographic B-factor (Figure 5c). This has been previously reported by Gross et al. [46]. Considering the practical limit in the native protein ECD experiment, our result does not exclude the possibility of the TBA interaction involving these residues. Despite the existing challenges, the native top-down MS approach showed evidence of the thrombin-TBA interaction site that is consistent with the high resolution crystallographic structural results.
CONCLUSIONS
Native ESI-MS and MS/MS were performed to characterize a protein-nucleic acid complex, the human α-thrombin/Bock aptamer. The glycosylation mass of α-thrombin was determined through high resolution, accurate mass measurement of the intact thrombin. Native MS of thrombin-TBA complex under different solution ionic strength conditions revealed the binding stoichiometry change that is reflective of the electrostatic nature of thrombin-DNA aptamer interactions. Native top-down MS allows the inference of the putative binding sites between thrombin and DNA aptamers, which is corroborated by previous solution and solid phase structural data. The measurement of protein-nucleic acid complexes by native ESI-MS dates back to the 1990s (e.g., see [47, 48]). Tandem MS of protein-nucleic acid complexes has recently showed progress to address complexes of larger DNA/RNA molecules, beyond the earlier work with single nucleotides [23, 49]. The sites of peptide binding to a 31-nucleotide sequence of TAR RNA were recently elucidated by native top-down MS [50]. As larger protein-nucleic acid complexes are probed by native top-down MS, better models will be needed to help us better understand the dissociation process of complexes composed of large, highly negatively charged molecules bound to proteins [51]. Overall, our work demonstrated that high resolution native top down MS is a powerful tool for the structural characterization of intact proteins and their non-covalent complexes with proteins, DNA/RNA, drugs, and other binding partners and ligands, and it may provide structural insights complementary to that obtained from traditional structural methods.
Supplementary Material
Supplemental Figure 1. ECD fragmentation product ion map of human α-thrombin heavy chain. N-linked glycosylation at Asn53 is indicated from the product ions labeled with the green dots with the corresponding mass increase of the carbohydrate group.
Supplemental Figure 2. FT-ICR mass spectrum of the TBA before (a) and after (b) desalting by ultracentrifugation. The charge state 4+ ions are shown.
Acknowledgments
Support from the US National Institutes of Health (R01GM103479 and S10RR028893 to J.A.L) and the US Department of Energy (UCLA Institute of Genomics and Proteomics; DE-FC03-02ER63421) are acknowledged.
References
- 1.Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stassen JM, Arnout J, Deckmyn H. The hemostatic system. Curr Med Chem. 2004;11:2245–2260. doi: 10.2174/0929867043364603. [DOI] [PubMed] [Google Scholar]
- 3.Licari LG, Kovacic JP. Thrombin physiology and pathophysiology. J Vet Emerg Crit Care. 2009;19:11–22. doi: 10.1111/j.1476-4431.2009.00383.x. [DOI] [PubMed] [Google Scholar]
- 4.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 5.Cai B, Yang X, Sun L, Fan X, Li L, Jin H, Wu Y, Guan Z, Zhang L, Zhang L, Yang Z. Stability and bioactivity of thrombin binding aptamers modified with d-/l-isothymidine in the loop regions. Org Biomol Chem. 2014;12:8866–8876. doi: 10.1039/c4ob01525h. [DOI] [PubMed] [Google Scholar]
- 6.Avino A, Fabrega C, Tintore M, Eritja R. Thrombin binding aptamer, more than a simple aptamer: Chemically modified derivatives and biomedical applications. Curr Pharm Des. 2012;18:2036–2047. doi: 10.2174/138161212799958387. [DOI] [PubMed] [Google Scholar]
- 7.Pagano B, Martino L, Randazzo A, Giancola C. Stability and binding properties of a modified thrombin binding aptamer. Biophys J. 2008;94:562–569. doi: 10.1529/biophysj.107.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martino L, Virno A, Randazzo A, Virgilio A, Esposito V, Giancola C, Bucci M, Cirino G, Mayol L. A new modified thrombin binding aptamer containing a 5′–5′ inversion of polarity site. Nucl Acid Res. 2006;34:6653–6662. doi: 10.1093/nar/gkl915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tasset DM, Kubik MF, Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes1. J Mol Biol. 1997;272:688–698. doi: 10.1006/jmbi.1997.1275. [DOI] [PubMed] [Google Scholar]
- 10.Schultze P, Macaya RF, Feigon J. Three-dimensional solution structure of the thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG) J Mol Biol. 1994;235:1532–1547. doi: 10.1006/jmbi.1994.1105. [DOI] [PubMed] [Google Scholar]
- 11.Wang KY, McCurdy S, Shea RG, Swaminathan S, Bolton PH. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry. 1993;32:1899–1904. doi: 10.1021/bi00059a003. [DOI] [PubMed] [Google Scholar]
- 12.Macaya RF, Schultze P, Smith FW, Roe JA, Feigon J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc Natl Acad Sci USA. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padmanabhan K, Padmanabhan KP, Ferrara JD, Sadler JE, Tulinsky A. The structure of α-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J Biol Chem. 1993;268:17651–17654. doi: 10.2210/pdb1hut/pdb. [DOI] [PubMed] [Google Scholar]
- 14.Kelly JA, Feigon J, Yeates TO. Reconciliation of the x-ray and NMR structures of the thrombin-binding aptamer d(GGTTGGTGTGGTTGG) J Mol Biol. 1996;256:417–422. doi: 10.1006/jmbi.1996.0097. [DOI] [PubMed] [Google Scholar]
- 15.Vairamani M, Gross ML. G-quadruplex formation of thrombin-binding aptamer detected by electrospray ionization mass spectrometry. J Am Chem Soc. 2003;125:42–43. doi: 10.1021/ja0284299. [DOI] [PubMed] [Google Scholar]
- 16.Balthasart F, Plavec J, Gabelica V. Ammonium ion binding to DNA G-quadruplexes: Do electrospray mass spectra faithfully reflect the solution-phase species? J Am Soc Mass Spectrom. 2013;24:1–8. doi: 10.1007/s13361-013-0649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong ES, Yoon HJ, Kim B, Yim YH, So HY, Shin SK. Mass spectrometric studies of alkali metal ion binding on thrombin-binding aptamer DNA. J Am Soc Mass Spectrom. 2010;21:1245–1255. doi: 10.1016/j.jasms.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 18.Russo Krauss I, Merlino A, Giancola C, Randazzo A, Mazzarella L, Sica F. Thrombin-aptamer recognition: A revealed ambiguity. Nucl Acid Res. 2011;39:7858–7867. doi: 10.1093/nar/gkr522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo Krauss I, Merlino A, Randazzo A, Novellino E, Mazzarella L, Sica F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucl Acids Res. 2012;40:8119–8128. doi: 10.1093/nar/gks512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leney AC, Heck AJR. Native mass spectrometry: What is in the name? J Am Soc Mass Spectrom. 2017;28:5–13. doi: 10.1021/jasms.8b05378. [DOI] [PubMed] [Google Scholar]
- 21.Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 22.Loo JA, Berhane B, Kaddis CS, Wooding KM, Xie Y, Kaufman SL, Chernushevich IV. Electrospray ionization mass spectrometry and ion mobility analysis of the 20S proteasome complex. J Am Soc Mass Spectrom. 2005;16:998–1008. doi: 10.1016/j.jasms.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Yin S, Loo JA. Elucidating the site of protein-ATP binding by top-down mass spectrometry. J Am Soc Mass Spectrom. 2010;21:899–907. doi: 10.1016/j.jasms.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Ah Young AP, Jiang J, Zhang J, Khoi Dang X, Loo JA, Zhou ZH, Egea PF. Conserved SMP domains of the ermes complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci U S A. 2015;112:E3179–3188. doi: 10.1073/pnas.1422363112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Wolff JJ, Van Orden SL, Loo JA. Native top-down electrospray ionization-mass spectrometry of 158 kDa protein complex by high-resolution Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2014;86:317–320. doi: 10.1021/ac4033214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Reza Malmirchegini G, Clubb RT, Loo JA. Native top-down mass spectrometry for the structural characterization of human hemoglobin. Eur J Mass Spectrom. 2015;21:221–231. doi: 10.1255/ejms.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrera NP, Robinson CV. Advances in the mass spectrometry of membrane proteins: From individual proteins to intact complexes. Ann Rev Biochem. 2011;80:247–271. doi: 10.1146/annurev-biochem-062309-093307. [DOI] [PubMed] [Google Scholar]
- 28.Laganowsky A, Reading E, Hopper JT, Robinson CV. Mass spectrometry of intact membrane protein complexes. Nature Protoc. 2013;8:639–651. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marty MT, Hoi KK, Gault J, Robinson CV. Probing the lipid annular belt by gas-phase dissociation of membrane proteins in nanodiscs. Angew Chem Int Ed Engl. 2016;55:550–554. doi: 10.1002/anie.201508289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Politis A, Park AY, Hall Z, Ruotolo BT, Robinson CV. Integrative modelling coupled with ion mobility mass spectrometry reveals structural features of the clamp loader in complex with single-stranded DNA binding protein. J Mol Biol. 2013;425:4790–4801. doi: 10.1016/j.jmb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Rosati S, Rose RJ, Thompson NJ, van Duijn E, Damoc E, Denisov E, Makarov A, Heck AJ. Exploring an orbitrap analyzer for the characterization of intact antibodies by native mass spectrometry. Angew Chem Int Ed Engl. 2012;51:12992–12996. doi: 10.1002/anie.201206745. [DOI] [PubMed] [Google Scholar]
- 32.Thompson NJ, Hendriks LJ, de Kruif J, Throsby M, Heck AJ. Complex mixtures of antibodies generated from a single production qualitatively and quantitatively evaluated by native orbitrap mass spectrometry. MAbs. 2014;6:197–203. doi: 10.4161/mabs.27126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson NJ, Rosati S, Heck AJ. Performing native mass spectrometry analysis on therapeutic antibodies. Methods. 2014;65:11–17. doi: 10.1016/j.ymeth.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Spirig T, Malmirchegini GR, Zhang J, Robson S, Sjodt M, Liu M, Kumar KK, Dickson CF, Gell DA, Lei B, Loo JA, Clubb RT. Staphylococcus aureus uses a novel multidomain receptor to break apart human hemoglobin and steal its heme. J Biol Chem. 2013;288:1065–1078. doi: 10.1074/jbc.M112.419119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen J, Zhang H, Gross ML, Blankenship RE. Native electrospray mass spectrometry reveals the nature and stoichiometry of pigments in the FMO photosynthetic antenna protein. Biochemistry. 2011;50:3502–3511. doi: 10.1021/bi200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sall DJ, Kaiser RE. Characterization of the interaction between human. Alpha.-thrombin and methyl 3-(2-methyl-1-oxopropoxy) [1]benzothieno[3,2-b]furan-2-carboxylate (ly806303) using electrospray mass spectrometry and tandem mass spectrometry. J Med Chem. 1993;36:2350–2355. doi: 10.1021/jm00068a012. [DOI] [PubMed] [Google Scholar]
- 37.Nilsson B, Horne MK, 3rd, Gralnick HR. The carbohydrate of human thrombin: Structural analysis of glycoprotein oligosaccharides by mass spectrometry. Arch Biochem Biophys. 1983;224:127–133. doi: 10.1016/0003-9861(83)90196-0. [DOI] [PubMed] [Google Scholar]
- 38.Ma B, Simala-Grant JL, Taylor DE. Fucosylation in prokaryotes and eukaryotes. Glycobiology. 2006;16:158R–184R. doi: 10.1093/glycob/cwl040. [DOI] [PubMed] [Google Scholar]
- 39.Baldrich E, Restrepo A, O’Sullivan CK. Aptasensor development: Elucidation of critical parameters for optimal aptamer performance. Anal Chem. 2004;76:7053–7063. doi: 10.1021/ac049258o. [DOI] [PubMed] [Google Scholar]
- 40.Miyoshi D, Karimata H, Sugimoto N. Hydration regulates thermodynamics of G-quadruplex formation under molecular crowding conditions. J Am Chem Soc. 2006;128:7957–7963. doi: 10.1021/ja061267m. [DOI] [PubMed] [Google Scholar]
- 41.Nagatoishi S, Tanaka Y, Tsumoto K. Circular dichroism spectra demonstrate formation of the thrombin-binding DNA aptamer G-quadruplex under stabilizing-cation-deficient conditions. Biochem Biophys Res Commun. 2007;352:812–817. doi: 10.1016/j.bbrc.2006.11.088. [DOI] [PubMed] [Google Scholar]
- 42.Baldrich E, O’Sullivan CK. Ability of thrombin to act as molecular chaperone, inducing formation of quadruplex structure of thrombin-binding aptamer. Anal Biochem. 2005;341:194–197. doi: 10.1016/j.ab.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 43.Lin PH, Chen RH, Lee CH, Chang Y, Chen CS, Chen WY. Studies of the binding mechanism between aptamers and thrombin by circular dichroism, surface plasmon resonance and isothermal titration calorimetry. Colloids and Surfaces B: Biointerfaces. 2011;88:552–558. doi: 10.1016/j.colsurfb.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 44.Hianik T, Ostatna V, Sonlajtnerova M, Grman I. Influence of ionic strength, pH and aptamer configuration for binding affinity to thrombin. Bioelectrochemistry. 2007;70:127–133. doi: 10.1016/j.bioelechem.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Olmsted IR, Xiao Y, Cho M, Csordas AT, Sheehan JH, Meiler J, Soh HT, Bornhop DJ. Measurement of aptamer-protein interactions with back-scattering interferometry. Anal Chem. 2011;83:8867–8870. doi: 10.1021/ac202823m. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Native electrospray and electron-capture dissociation FTICR mass spectrometry for top-down studies of protein assemblies. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sannes-Lowery KA, Hu P, Mack DP, Mei HY, Loo JA. HIV-1 tat peptide binding to TAR RNA by electrospray ionization mass spectrometry. Anal Chem. 1997;69:5130–5135. doi: 10.1021/ac970745w. [DOI] [PubMed] [Google Scholar]
- 48.Ganguly AK, Pramanik BN, Tsarbopoulos A, Covey TR, Huang E, Fuhrman SA. Mass spectrometric detection of the noncovalent GDP-bound conformational state of the human H-ras protein. J Am Chem Soc. 1992;114:6559–6560. [Google Scholar]
- 49.Yin S, Xie YM, Loo JA. Mass spectrometry of protein-ligand complexes: Enhanced gas-phase stability of ribonuclease-nucleotide complexes. J Am Soc Mass Spectrom. 2008;19:1199–1208. doi: 10.1016/j.jasms.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneeberger EM, Breuker K. Native top-down mass spectrometry of TAR RNA in complexes with a wild-type tat peptide for binding site mapping. Angew Chem Int Ed. 2017;56:1254–1258. doi: 10.1002/anie.201610836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogorzalek Loo RR, Loo JA. Salt bridge rearrangement (SaBRe) explains the dissociation behavior of noncovalent complexes. J Am Soc Mass Spectrom. 2016;27:975–990. doi: 10.1007/s13361-016-1375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. ECD fragmentation product ion map of human α-thrombin heavy chain. N-linked glycosylation at Asn53 is indicated from the product ions labeled with the green dots with the corresponding mass increase of the carbohydrate group.
Supplemental Figure 2. FT-ICR mass spectrum of the TBA before (a) and after (b) desalting by ultracentrifugation. The charge state 4+ ions are shown.