

Abstract
High resolution mass spectrometry is a key technology for in-depth protein characterization. High-field Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) enables high-level interrogation of intact proteins in the most detail to date. However, an appropriate complement of fragmentation technologies must be paired with FTMS to provide comprehensive sequence coverage, as well as characterization of sequence variants, and post-translational modifications. Here we describe the integration of front-end electron transfer dissociation (FETD) with a custom-built 21 tesla FT-ICR mass spectrometer, which yields unprecedented sequence coverage for proteins ranging from 2.8 to 29 kDa, without the need for extensive spectral averaging (e.g., ~60% sequence coverage for apo-myoglobin with four averaged acquisitions). The system is equipped with a multipole storage device separate from the ETD reaction device, which allows accumulation of multiple ETD fragment ion fills. Consequently, an optimally large product ion population is accumulated prior to transfer to the ICR cell for mass analysis, which improves mass spectral signal-to-noise ratio, dynamic range, and scan rate. We find a linear relationship between protein molecular weight and minimum number of ETD reaction fills to achieve optimum sequence coverage, thereby enabling more efficient use of instrument data acquisition time. Finally, real-time scaling of the number of ETD reactions fills during method-based acquisition is shown, and the implications for LC-MS/MS top-down analysis are discussed.
Keywords: FTMS, Ion-ion reaction, Fourier transform mass spectrometry, Electrospray ionization, MS/MS, MS2, Top-down, ETD
Graphical abstract
Introduction
Continued development of high-end technologies is needed to advance the field of top-down proteomics [1]. Full characterization of proteoforms, not just transcribed gene products (RNA), is central to unraveling the most complex cellular processes. Intact protein analysis can provide structural information that cannot be obtained through traditional, bottom-up proteomics [2]. For example, antibody-drug conjugates (ADCs), the most rapidly growing class of therapeutic compounds [3], require top-down analysis. Although these compounds are very effective in the treatment and tissue/cell specific targeting of various types of cancers [4], they can be highly heterogeneous [5] due to inherent problems in manufacturing. The heterogeneity arises from differences in antibody primary structure, the spatial and number distribution of drug conjugates [6], and the various linker species used for the antibody-drug tether. That heterogeneity cannot be captured appropriately with any combination of enzymatic approaches and necessitates that ADCs be analyzed intact to ensure that they are manufactured consistently and are safe for the public. Enzymatic approaches remove the inherent heterogeneity of the system and render extraction of the desired structural information problematic, even for the highest-performance commercial mass spectrometers. To achieve complete protein characterization, one must obtain primary sequence, site-localization of any post-translational modifications, and preferably higher-order structure characteristics. High-impact applications for top-down mass spectrometry, such as ADC characterization, should help drive the effort to close the analytical gap, in terms of proteins identified per experiment, quantification strategies, and diversity in protein vs. peptide chemistry, between bottom-up and top-down proteomics.
Closing the gap between top-down and bottom-up approaches will require advancement in sample preparation, front-end separations, and instrumentation. Thus, intact protein analysis continues to advance with the development of higher performance mass spectrometers [7] and new/versatile ion activation approaches [8–14]. With these advancements, routine implementation of top-down proteomics is an increasingly attainable goal. The inherently open-sourced technology and analytical merits of Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) makes it well suited for top-down protein characterization [15]. State of the art ion introduction methods [16], cell design [17], operation [18], magnetic field strength [7], and homogeneity [19] provide contemporary FT-ICR MS instruments with excellent sensitivity and dynamic range. Former FT-ICR MS issues with cryogen supply and consumption have been obviated by the availability of superconducting magnets operating without liquid nitrogen and with essentially zero liquid helium consumption. Finally, FT-ICR MS is very easily coupled with orthogonal technologies such as laser-based [13, 20] or electron-based dissociation techniques[10] as well as a host of other front-end hardware modifications.
Electron transfer dissociation (ETD) for top-down- or middle-down-size polypeptides has produced unprecedented sequence coverage [21–23]. Fragment ions produced by ETD retain labile post-translational modifications (PTMs) [24], and thus enable identification of their sequence location(s). However, fragmentation of intact proteins by ETD produces fragment ions spread over a relatively narrow m/z domain about the precursor. Product ion spectra exhibit very high spectral complexity surrounding the precursor m/z, often resulting in multiple overlapping product ion isotopic distributions. Consequently, these overlapping isotopic envelopes remain unresolved at even moderate resolving power. Ion–ion proton transfer (IIPT) reactions provide a route to simplify and better use the analytical m/z range; however, direct improvement in resolving power provides a similar effect. Also, the ETD process inherently produces a fairly uniform distribution of ions for each product ion channel; therefore, the precursor ion signal is distributed over dozens (or even hundreds in the case of proteins) of product ions. These aspects demand that top-down mass spectrometry instrumentation possess ultra-high mass resolution, broad dynamic range, and heightened sensitivity to take full advantage of data produced by ETD.
Technological advances in fragmentation efficiency and scan rate appropriate for LC time-scale have firmly cemented ETD as an indispensable tool for biomolecule analysis [25–30]. A particularly valuable recent advance is the adaptation of ETD reagent ion source inside the front-end ionization source (FETD) [31]. This manuscript details a multi-laboratory/industry collaboration to pair the complementary technology behind the high field FT-ICR MS instruments at the National High Magnetic Field Laboratory (NHMFL) with FETD. We are able to generate ETD product ion spectra with high sequence coverage for proteins up to 29 kDa with very little need for signal averaging. Further, we explore the relationship between acheivable sequence coverage and number of ETD fragment ion fills to an external ion accumulation device. Prediction of the minimum number of ETD fragment ion fills to achieve maximal sequence coverage for a given protein MW was optimized for on-line top-down LC-MS/MS performance.
Methods
Instrumentation
All data were acquired with a 21 T FT-ICR mass spectrometer described in detail elsewhere [7]. In brief, this hybrid system consists of a linear rf ion trap (Velos Pro; ThermoFisher Scientific) coupled with a custom-built FT-ICR mass analyzer. The Velos Pro is equipped with a commercial Orbitrap Fusion API inlet/FETD reagent [31] source (Thermo FisherScientific). The instrument control software is modified to control the ETD electronics. Q00 of the Velos is shortened to accommodate the reagent ion source (RIS) inside the existing source housing. The stacked ring ion guide (SRIG) DC offset voltage is used to repel ions produced by electrospray during reagent anion production within the RIS. A DC voltage was appropriated as the second of two DC potentials used to create a differential offset between adjacent rods of Q0 within the Velos to provide low-pass mass filtering of ions produced by the RIS. Owing to the flat-pole geometry of Q0, the mass resolution of this device is fairly poor and represents only half of the circuit required to constitute a mass-resolving quadrupole. Nevertheless, it provided adequate filtering of the higher m/z ions (>216 m/z) generated from the RIS to produce 90%–95% pure fluoranthene anions at the cost of about 50% drop in transmission efficiency (from ~1.5 ms to ~3.0 ms increase in reagent ion injection period). Alternatively, a broadband waveform can be applied to the linear ion trap prior to reaction, to remove the most common contaminant, the fluoranthene-nitrogen adduct species (m/z 216). In this implementation, we are able to use one or both approaches simultaneously to ensure gas-phase reagent ion purity. ETD reaction periods were normalized to roughly the time required for two half-lives of precursor decay (e.g., 45 ms for melittin and 8 ms for carbonic anhydrase).
The schematic representation in Figure 1a shows the multi-pole storage device (MSD) that accumulates multiple ETD fragment ion fills. All ETD reactions were carried out in the high pressure cell of a Velos Pro dual cell rf ion trap assembly. ETD reactions were conducted as described by Syka et al. [9] by use of an axial rf pseudopotential for charge sign-independent trapping. Automatic gain control (AGC) targets were 1E5 charges for the analyte ion and 2E5 charges for fluoranthene reagent anions, with charge state-dependent reaction periods. For carbonic anhydrase analysis, these AGC target values were increased to 2E5 analyte ions and 4E5 for reagent ions. The MSD has an axial DC potential applied to enable ion manipulation along the z-axis of the device [32]. In addition, this MSD is equipped with an auxiliary rf supply for m/z-dependent axial ejection to the ICR cell [16] that greatly reduces time-of-flight mass discrimination associated with gated ion trapping in the ICR cell. The relationship between signal-to-noise ratio (S/N) and multiple fills was explored with and without ETD reaction (see Supplemental Information). We found that S/N increases linearly over a wide range of fills, illustrating that neither MSD ion storage capacity nor ICR cell ion capacity are exceeded in these experiments.
Figure 1.

(a) Conceptual layout for the 21 T FT-ICR mass spectrometer with which all experiments were conducted. This system features a dual cell linear ion trap, a multipole storage device, a dynamically harmonized cell, and a 21 tesla actively shielded superconducting magnet. (b) Experimental event sequence, showing how multiple fill experiments were conducted
Data Acquisition and Processing
Data were acquired with a dynamically harmonized ICR cell [17] by use of the standard LTQ-FT Ultra FTMS acquisition electronics (ThermoFisher Scientific), except for replacement of the preamplifier (Stahl Electronics, Mettenheim, Germany). Data acquisition for all direct infusion experiments were acquired with four microscans unless otherwise noted. Time-domain ICR transient duration was 0.76 s. Data was stored as the standard Thermo file .raw format, and transient data were stored as .dat files and processed with Predator [33] (8 Mword time-domain data, one zero-fill, magnitude mode). Externally calibrated FT-ICR mass spectra were deconvolved by use of Thrash or Xtract with a S/N threshold of 6. Neutral masses were submitted as input to Prosight Lite [34]. Search tolerance for precursor mass and product ions was set to ±10 ppm. Fractional sequence coverage was determined directly from search results for each protein.
Sample Preparation
A set of standard proteins was used to test the relationship between number of fills and fractional sequence coverage: melittin, ubiquitin, cytochrome c, hemoglobin α subunit, apomyoglobin, histone H1, and carbonic anhydrase (molecular weight 3–30 kDa). All proteins were used as received (Sigma Aldrich, St. Louis, MO, USA) without further purification. Stock solutions were prepared for each protein in ultra-pure 18 MΩ water at a concentration of ~1 mM. Each stock solution was diluted directly into an electrospray solution (49:49:2; H2O:MeOH:HOAc) to a final protein concentration of 0.5 μM. We also prepared an equimolar mixture of three proteins at 0.5 μM each into electrospray solution (melittin, ubiquitin, and cytochrome c). Samples were directly infused (50 μm i.d., 360 μm o.d. polyimide-coated fused silica capillary, 500 nL/min flow rate) into the mass spectrometer by use of a custom nano-ESI source (University of Washington Proteomics Resource NSI source) at an ESI voltage of 1.8–2.5 kV. Capillary tips were produced by means of either an in-house developed grinding process or laser pulling (Sutter Instruments, Novato, CA, USA).
Results and Discussion
Front-End ETD Interfaced to a 21 T FT-ICR Mass Analyzer
The Velos Pro linear ion trap is equipped with electronics from a first generation ETD unit, such that the available DC outputs could be reassigned to the new FETD related devices, including a SRIG DC bias, a Q0 x-rod differential DC for low-pass mass filtering, and command current for the glow discharge. A DC bias applied to the SRIG allowed us to gate ions from the ESI source, such that ion injection from the RIS and the ESI source were decoupled. Application of a −50 V DC bias to the SRIG was capable of the rejection of all ions from the ESI source, whereas ion transmission from the RIS remained unaffected. During normal transmission from the ESI source, the SRIG bias was set to 0 V DC. Reagent ion purity remains a potential issue for implementing FETD with a Velos Pro system due to incomplete reagent ion isolation prior to ion–ion reaction. The main contributing contaminant species, the fluoranthene:nitrogen adduct (m/z 216), undergoes ion–ion reaction primarily through proton transfer. Although most good ETD reagents react with protein cations to a small degree via ion–ion proton transfer (or produces hydrogen loss), the mixing of two or more reagents with uncharacterized propensities for ETD/ion–ion proton transfer reaction pathways complicates product ion interpretation and lowers the yield of sequence-informative product ions. Therefore, two measures were taken to remove contaminant species from the reagent ion beam to achieve high quality ETD spectra. The first measure is to operate Q0 as a simple low-pass mass filter by application of a differential DC bias between adjacent rods. The planar Q0 electrode geometry limits performance but still achieves removal of 95%–99% of ions at or above the m/z of the fluoranthene:nitrogen adduct (m/z 216). Because the quadrupole geometry is not ideal for isolation, transmission efficiency was sacrificed for specificity (~50% loss in transmission efficiency). The second measure to ensure reagent ion purity is through the application of a broadband “clean-up” waveform. This waveform is constructed such that ions within a 20 Da window centered at the fluoranthene:nitrogen adduct are ejected from the linear ion trap prior to ion–ion reaction. ETD is a versatile technology that in principle may be coupled to any mass spectrometry platform, but benefits from hardware that places a true quadrupole mass filter [35] downstream from the RIS.
Molecular Weight and ETD Fragment Ion Fills
As discussed in depth by Riley et. al. [36], achieving maximum S/N is especially critical for top-down product ion mass spectra because the original precursor ion signal is distributed among many available fragmentation channels and fragment ion charge distributions. The two primary means for increasing S/N are spectral averaging and/or increasing the trapped ion population. Spectral averaging produces a gain in S/N that is proportional to n1/2, in which n is the number of spectra averaged. However, for FTMS analyzers, spectral averaging is very costly in terms of time and may not always be appropriate, especially for on-line LC-MS/MS experiments. For this reason, it is preferable to improve S/N by increasing the ion population, which is possible with our hardware configuration. This can be done by means of a multiple ETD reaction fill scheme [22, 37] or by increasing ETD reaction capacity [36, 38]. Here, we demonstrate a multiple ETD reaction fill scheme (see Figure 1b). We repeat ion–ion reactions at relatively low precursor target ion number (typically 1E5 precursor and 2E5 reagent anion targets) and accumulate the reaction products in the MSD until maximum sequence coverage has been achieved. This device is estimated to have a storage capacity in excess of 50 million charges (see Supplemental Figure 1, 2) and has been designed to match or exceed the analytical charge capacity of the ICR cell such that there is no bottleneck in the number of ions/charges that can be supplied for mass analysis. Use of the MSD allows the ion trap assembly to be optimized as an ETD reaction vessel operated under conditions in which the pseudo-first order kinetic approximation holds true. Therefore, optimal and reproducible ETD fragmentation performance is attained. In this regard, the MSD effectively bridges the ion capacity gap between a single ETD reaction event in the linear rf ion trap and the ICR cell. It should be noted that the linear rf ion trap also has a very high charge capacity if operated as a storage-only device (tens of millions of charges), however, at those target values ETD performance is diminished.
The multiple fill scheme described above was used to explore the relationship between number of fills (or cumulative AGC ion target value) and fractional sequence coverage across a series of proteins of different molecular weight. Because the number of sequence-informative dissociation channels grows linearly with molecular weight, so too should the number of ETD fragment ion fills to reach maximal protein sequence coverage. It should be noted that this is not the case for internal fragments, which increase faster than linearly with increasing MW (or N amino acid residues). Figure 2a–g show the appropriate number of fills required to reach maximal sequence coverage for each of the seven protein standards. For example, melittin (MW = ~2.8 kDa) reaches maximal sequence coverage within only 3E5–5E5 cumulative ion target (~3–5 ETD fragment ion fills; Figure 2a). However, for the much larger histone H1 (21 kDa) sequence coverage reaches a plateau by 1.5E6–2.0E6 cumulative ion target (~15–20 fills; Figure 2f). These data illustrate a classic case of diminishing returns with regard to sequence coverage observed at higher numbers of fragment ion fills (>20), which deleteriously increase analysis time. The presently reported sequence coverage values redefine the current state-of-the-art for intact protein mass spectrometry (4 μ scans per spectrum). Although comparable sequence coverage levels have been observed previously, they were obtained by more extensive spectral averaging and by combining results from multiple fragmentation techniques [39, 40]. Extensive spectral averaging places a tremendous performance bottleneck on top-down LC-MS/MS. When viewed in terms of information per unit time, the benefit of this approach becomes much more apparent (see below).
Figure 2.

Fractional sequence coverage versus cumulative ion target (1 ETD fragment ion fill = 1E5 analyte charges) melittin (a), ubiquitin (b), cytochrome c (c), hemoglobin subunit (d), apo-myoglobin (e), histone H1 (f), and carbonic anhydrase (g). Low MW proteins, with fewer possible dissociation channels, require fewer fills to achieve reasonable coverage than do their larger counterparts
From Figure 2a–g, we extracted the lowest number of fills at which sequence coverage is maximized (90–95 the maximum value observed) via curve-fit and plotted optimal fill number versus analyte MW, to reveal a linear relationship. Although this linear fit model represents the data well, we expect nonlinearities based on protein tertiary structure, intact cysteine disulfide bonds, gas-phase intramolecular interactions, etc. Therefore, the linear model is a generalization that we shall apply to all proteins during LC-MS acquisition. It is worth noting that as we build up information from large-scale intact protein analysis, our model could be adapted to accommodate any observed non-linearity in the MW versus cumulative target based on sample type or other characteristics. Within this linear fit model, each data point represents the highest scan duty cycle without sacrificing sequence coverage for interrogated intact proteins. This model serves as a guide for more advanced data acquisition rules applied to LC-MS/MS ETD acquisition for top-down proteomics to more appropriately utilize instrument time as a function of the observed precursor MW.
Instrument-Controlled ETD Fragment Ion Fills
Incorporation of the empirical relationship determined in Figure 3 directly into the mass spectrometry acquisition software enables method-based acquisitions in which the number of ETD fragment ion fills scales with MW, as shown in Figure 4. This approach is based on real-time charge state determination to obtain MW. Owing to software limitations, charge states above 25+ are not assigned in real time. The sample is a mixture of three standard proteins (melittin, ubiquitin, and cytochrome c). The top panels in Figure 4 show ETD mass spectra of melittin [M + 5H]5+ (MW = 2.8 kDa) with 3 ETD fragment ion fills (left) resulting in a total acquisition time of 4.14 s, ubiquitin [M + 10H]10+ (MW = 8.6 kDa) with 8 ETD fragment ion fills (right) resulting in a total acquisition time of 4.54 s, and cytochrome c [M + 16H]16+ (MW = 12 kDa) with 11 ETD fragment ion fills (middle) resulting in a total acquisition time of 7.38 s. The number of fills for each of these MS/MS acquisitions was assigned on-the-fly by means of a short processing step immediately after acquisition of the FTMS survey acquisition data. First, the FTMS survey spectrum was converted to neutral masses, and the number of fills was then interpolated from the empirical relationship outlined in Figure 3 for all targets in the data-dependent acquisition list from the mass spectrometry software. This simple approach is superior to conventional use of a single ion target value, which leads to spending too much acquisition time for smaller proteins and too little acquisition time (and thus reduced S/N ratio) for larger proteins. Overall sequence coverage and identification rates for larger proteins are thereby greatly improved in an optimized amount of time, as shown in Figure 2a–g. If this experiment had been conducted with a fixed number of fills (11 for optimized cytochrome c sequence coverage) for each of the three target species, an additional 10.42 s would have been required for the scan cycle.
Figure 3.
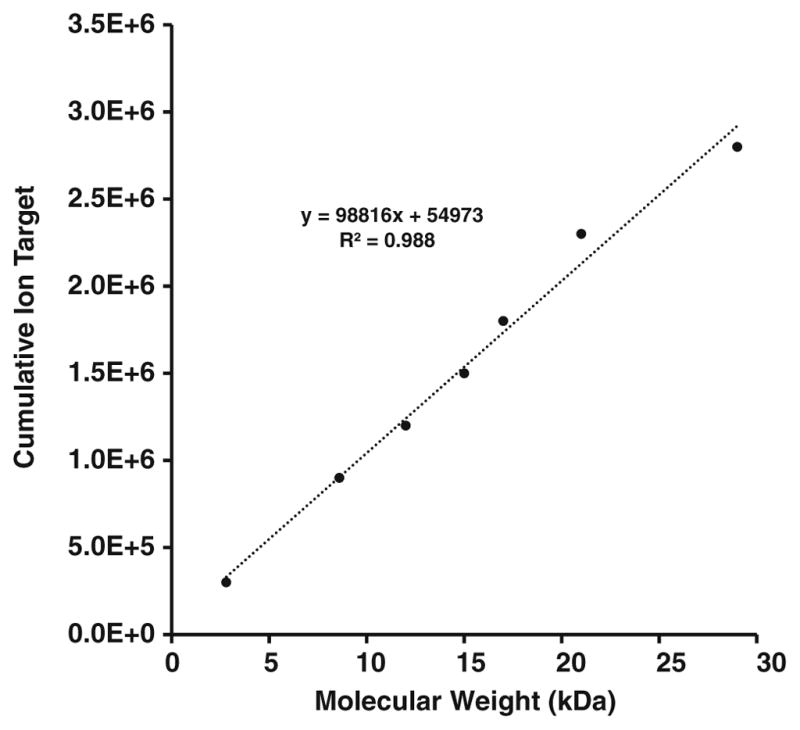
Minimum precursor AGC cumulative ion target required for optimized sequence coverage. The highly linear relationship (R2 = 0.988) evidently results from a linear increase in number of dissociation channels with increasing MW
Figure 4.

Scaling of the number of fills as a function of molecular weight during method-based operation of the mass spectrometer. Bottom: Positive ESI mass spectrum of a mixture of three proteins (melittin, ubiquitin, cytochrome c). The ETD product ion spectra of melittin [M + 5H]5+ was acquired with three fills (left), ubiquitin [M + 10H]10+ with eight fills (right), and cytochrome c [M + 16H]16+ with 11 fills (top). All values were determined on-the-fly during method-based acquisition from the linear expression shown in Figure 3. That equation was incorporated directly into the ITCL programming of the system
FETD Top-Down Analysis for Larger Proteins
ETD analysis of carbonic anhydrase (M + 34H)34+ validates the utility of high mass resolution combined with the multiple fill approach. We acquired ETD product ion mass spectra for different number of averaged transients at resolving power settings ranging from 37,500 at m/z 400 (0.095 s acquisition period) to 600,000 at m/z 400 (1.52 s acquisition period). The results are shown in Figure 5. Sequence coverage increased monotonically with increased acquisition period for two reasons: (1) more overlapping isotopic distributions are resolved; and (2) signal-to-noise increases with the square root of acquisition period for an undamped signal. However, due primarily to ion-neutral collisions and to a lesser degree ion–ion interactions, the transient does damp somewhat, resulting in a net decrease in sequence coverage. This results in a net decrease in performance for acquisition periods that span durations longer than that for which a stable cyclotron orbit is observed for these large fragments. Angular velocities of ions excited to large cyclotron orbit at 21 T result in dramatically shortened mean-free-path lengths than at lower magnetic field strength (e.g., 9.4 T and 14.5 T). All evidence that we have gathered points toward collisional damping as the primary performance limitation, and as a result we are actively working to reduce the base pressure in the UHV region of the system (UHV chamber pressure <1E-11 Torr at the time of these measurements).
Figure 5.
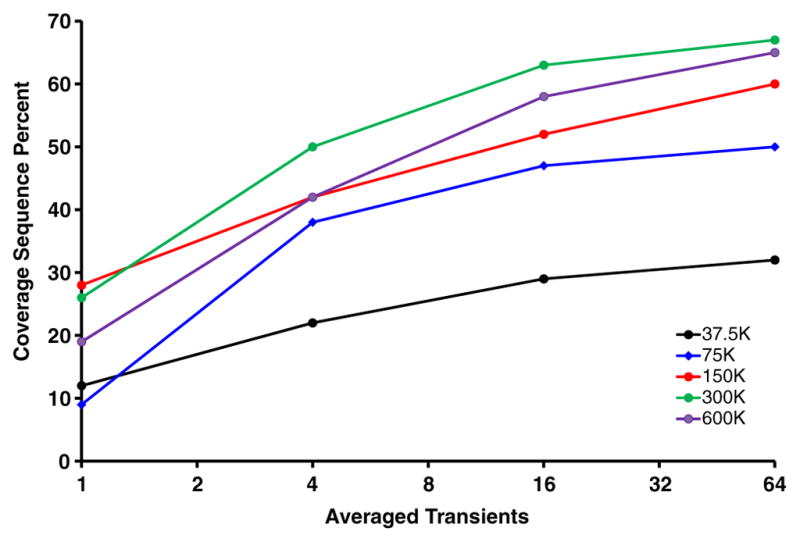
Fractional sequence coverage for carbonic anhydrase as a function of the number of averaged spectra. These data were acquired over a range of resolving power (m/z 400) settings from 37.5 to 600 k
In Figure 6a and b, we demonstrate the maximum achievable sequence coverage for carbonic anhydrase by use of ETD with the 21 T instrument (87%; 94% if bonds with adjacent proline residues are ignored since they cannot be cleaved via ETD). This experiment was conducted via direct infusion and we were not sample-limited. In this case, we were able to perform extensive spectral averaging (1500 transients acquired) to improve the data quality. Although these acquisition conditions are clearly incompatible with LC-MS/MS acquisitions, these data provide strong evidence that ETD-based fragmentation provides extensive fragment ion coverage across the entire backbone of the protein of interest, not just coverage near the N- and C-termini (see Supplemental Figures 3 and 4 for mass accuracy information on fragment ion assignments). Previous observations of fragment ion coverage only in the termini are likely a consequence of ETD reaction periods that are too long and allow for secondary electron transfer to larger fragment ions or due to limitations of the instrumentation being utilized (e.g., insufficient resolving power). In fact, these data strongly support the idea that ETD provides relatively unbiased fragmentation throughout the entire protein. Future effort will be focused on the ability to produce these large fragment ions in high numbers very quickly, as may be possible via ion parking and IIPT.
Figure 6.

(a) Fragment ion map generated from carbonic anhydrase [M + 34H]34+ illustrating 87% sequence coverage from ETD fragmentation alone. The signal was averaged over 1500 acquisitions by use of 16 fills with a reaction period of 6.0 ms (RP at m/z 400 = 600,000) (b) Mass spectrum of the underlying data. Insets include 5, 50, and 100 Da ranges expanded to illustrate the spectral complexity and dynamic range within the spectrum. Large fragment ions are observed at m/z > 1800
Supplementary Material
Acknowledgments
This work was supported by NSF Division of Materials Research through DMR-11-57490, the State of Florida, and National Institutes of Health grants AI 033993 (DFH) and GM 037537 (DFH).
Footnotes
Electronic supplementary material The online version of this article (doi:10. 1007/s13361-017-1702-3) contains supplementary material, which is available to authorized users.
References
- 1.Kelleher NL, Thomas PM, Ntai I, Compton PD, LeDuc RD. Deep and quantitative top-down proteomics in clinical and translational research. Expert Rev Proteomics. 2014;11:649–651. doi: 10.1586/14789450.2014.976559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catherman AD, Skinner OS, Kelleher NL. Top-down proteomics: facts and perspectives. Biochem Biophys Res Commun. 2014;445:683–693. doi: 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal P, Bertozzi CR. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconj Chem. 2015;26:176–192. doi: 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feld J, Barta SK, Schinke C, Braunschweig I, Zhou Y, Verma AK. Linked-in: design and efficacy of antibody drug conjugates in oncology. Oncotarget. 2013;4:397–412. doi: 10.18632/oncotarget.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005;14:2436–2446. doi: 10.1110/ps.051478705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behrens CR, Ha EH, Chinn LL, Bowers S, Probst G, Fitch-Bruhns M, Monteon J, Valdiosera A, Bermudez A, Liao-Chan S, Wong T, Melnick J, Theunissen JW, Flory MR, Houser D, Venstrom K, Levashova Z, Sauer P, Migone TS, van der Horst EH, Halcomb RL, Jackson DY. Antibody-drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharm. 2015;12:3986–3998. doi: 10.1021/acs.molpharmaceut.5b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendrickson CL, Quinn JP, Kaiser NK, Smith DF, Blakney GT, Chen T, Marshall AG, Weisbrod CR, Beu SC. 21 Tesla Fourier transform ion cyclotron resonance mass spectrometer: a national resource for ultrahigh resolution mass analysis. J Am Soc Mass Spectrom. 2015;26:1626–1632. doi: 10.1007/s13361-015-1182-2. [DOI] [PubMed] [Google Scholar]
- 8.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Infrared multiphoton dissociation of large multiply charged ions for biomolecule sequencing. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 9.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 11.Cannon JR, Holden DD, Brodbelt JS. Hybridizing ultraviolet photodissociation with electron transfer dissociation for intact protein characterization. Anal Chem. 2014;86:10970–10977. doi: 10.1021/ac5036082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cannon JR, Cammarata MB, Robotham SA, Cotham VC, Shaw JB, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal Chem. 2014;86:2185–2192. doi: 10.1021/ac403859a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riley NM, Rush MJ, Rose CM, Richards AL, Kwiecien NW, Bailey DJ, Hebert AS, Westphall MS, Coon JJ. The negative mode proteome with activated ion negative electron transfer dissociation (AI-NETD) Mol Cell Proteomics. 2015;14:2644–2660. doi: 10.1074/mcp.M115.049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 15.Garcia BA. What does the future hold for top-down mass spectrometry? J Am Soc Mass Spectrom. 2010;21:193–202. doi: 10.1016/j.jasms.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 16.Kaiser NK, Savory JJ, Hendrickson CL. Controlled ion ejection from an external trap for extended m/z range in FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 2014;25:943–949. doi: 10.1007/s13361-014-0871-6. [DOI] [PubMed] [Google Scholar]
- 17.Nikolaev EN, Boldin IA, Jertz R, Baykut G. Initial experimental characterization of a new ultra-high resolution FTICR cell with dynamic harmonization. J Am Soc Mass Spectrom. 2011;22:1125–1133. doi: 10.1007/s13361-011-0125-9. [DOI] [PubMed] [Google Scholar]
- 18.Kaiser NK, McKenna AM, Savory JJ, Hendrickson CL, Marshall AG. Tailored ion radius distribution for increased dynamic range in FT-ICR mass analysis of complex mixtures. Anal Chem. 2013;85:265–272. doi: 10.1021/ac302678v. [DOI] [PubMed] [Google Scholar]
- 19.Vladimirov G, Kostyukevich Y, Hendrickson CL, Blakney GT, Nikolaev E. Effect of magnetic field inhomogeneity on ion cyclotron motion coherence at high magnetic field. Eur J Mass Spectrom (Chichester, Eng) 2015;21:443–449. doi: 10.1255/ejms.1375. [DOI] [PubMed] [Google Scholar]
- 20.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc. 2013;135:12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, English AM, Bai DL, Ugrin SA, Shabanowitz J, Ross MM, Hunt DF, Wang WH. Analysis of monoclonal antibody sequence and post-translational modifications by time-controlled proteolysis and tandem mass spectrometry. Mol Cell Proteom. 2015;15:1479–1488. doi: 10.1074/mcp.O115.056721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson LC, English AM, Wang WH, Bai DL, Shabanowitz J, Hunt DF. Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry. Int J Mass Spectrom. 2015;377:617–624. doi: 10.1016/j.ijms.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Fornelli L, Compton PD, SharmaSCanterbury J, Mullen C, Zabrouskov V, Fellers RT, Thomas PM, Licht JD, Senko MW, Kelleher NL. Unabridged analysis of human histone H3 by differential top-down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression. Mol Cell Proteom. 2015;15:776–790. doi: 10.1074/mcp.M115.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol Cell Proteom. 2007;6:1942–19451. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of electron-transfer dissociation on a hybrid linear ion trap-Orbitrap mass spectrometer. Anal Chem. 2007;79:3525–3534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Udeshi ND, Compton PD, Shabanowitz J, Hunt DF, Rose KL. Methods for analyzing peptides and proteins on a chromatographic time-scale by electron-transfer dissociation mass spectrometry. Nat Protoc. 2008;3:1709–1717. doi: 10.1038/nprot.2008.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Compton PD, Strukl JV, Bai DL, Shabanowitz J, Hunt DF. Optimization of electron transfer dissociation via informed selection of reagents and operating parameters. Anal Chem. 2012;84:1781–1785. doi: 10.1021/ac202807h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Good DM, Wenger CD, McAlister GC, Bai DL, Hunt DF, Coon JJ. Post-acquisition ETD spectral processing for increased peptide identifications. J Am Soc Mass Spectrom. 2009;20:1435–1440. doi: 10.1016/j.jasms.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadygov RG, Good DM, Swaney DL, Coon JJ. A new probabilistic database search algorithm for ETD spectra. J Proteome Res. 2009;8:3198–3205. doi: 10.1021/pr900153b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Earley L, Anderson LC, Bai DL, Mullen C, Syka JE, English AM, Dunyach JJ, Stafford GC, Jr, Shabanowitz J, Hunt DF, Compton PD. Front-end electron transfer dissociation: a new ionization source. Anal Chem. 2013;85:8385–8390. doi: 10.1021/ac401783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilcox BE, Hendrickson CL, Marshall AG. Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. J Am Soc Mass Spectrom. 2002;13:1304–1312. doi: 10.1016/S1044-0305(02)00622-0. [DOI] [PubMed] [Google Scholar]
- 33.Blakney GT, Hendrickson CL, Marshall AG. Predator data station: a fast data acquisition system for advanced FT-ICR MS experiments. Int J Mass Spectrom. 2011;306:246–252. [Google Scholar]
- 34.Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, Thomas PM. ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics. 2015;15:1235–1238. doi: 10.1002/pmic.201570050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senko MW, Remes PM, Canterbury JD, Mathur R, Song Q, Eliuk SM, Mullen C, Earley L, Hardman M, Blethrow JD, Bui H, Specht A, Lange O, Denisov E, Makarov A, Horning S, Zabrouskov V. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem. 2013;85:11710–11714. doi: 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- 36.Riley NM, Mullen C, Weisbrod CR, Sharma S, Senko MW, Zabrouskov V, Westphall MS, Syka JE, Coon JJ. Enhanced dissociation of intact proteins with high capacity electron transfer dissociation. J Am Soc Mass Spectrom. 2015;3:520–531. doi: 10.1007/s13361-015-1306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW, Marshall AG. High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 Tesla. Anal Chem. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- 38.Rose CM, Russell JD, Ledvina AR, McAlister GC, Westphall MS, Griep-Raming J, Schwartz JC, Coon JJ, Syka JE. Multipurpose dissociation cell for enhanced ETD of intact protein species. J Am Soc Mass Spectrom. 2013;24:816–8127. doi: 10.1007/s13361-013-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E, Denisov E, Lange O, Remes P, Taylor D, Splendore M, Wouters ER, Senko M, Makarov A, Mann M, Horning S. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteom. 2009;8:2759–2769. doi: 10.1074/mcp.M900375-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Riley NM, Sun L, Hebert AS, Yan X, Westphall MS, Rush MJ, Zhu G, Champion MM, Mba Medie F, Champion PA, Coon JJ, Dovichi NJ. Coupling capillary zone electrophoresis with electron transfer dissociation and activated ion electron transfer dissociation for top-down proteomics. Anal Chem. 2015;87:5422–5429. doi: 10.1021/acs.analchem.5b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.