

Abstract
IL-22 is known to induce intestinal epithelial cells (IECs) to produce the chemokine CXCL8. However, IECs exist in a cytokine network during mucosal inflammation, such that IL-22 must act in concert with potent pro-inflammatory cytokines like TNF-α and IL-1. Our studies show that IL-22 alone increased CXCL8 secretion from HT-29 cells, but the levels were minimal compared to that of the cells treated with TNF-α or IL-1 only. More significantly, costimulation with IL-22 and TNF-α enhanced both CXCL8 secretion and mRNA levels well over that of TNF-α stimulation alone. A similar enhancing effect was seen with IL-22 and IL-1-stimulated CXCL8 secretion. The enhancing effect of IL-22 on TNF-α-induced CXCL8 secretion was then determined to require the p38 MAPK, but not STAT1/3, PI3K, Akt, JNK, ERK or IкBα. These experiments indicate that the more significant effect of IL-22 on IECs responses may not be in inducing CXCL8 by itself, but of enhancing TNF-α- and IL-1-induced CXCL8 secretion to augment the contribution of IECs to local inflammatory responses.
Keywords: CXCL8, intestinal epithelial, IL-22, IL-1, p38 MAPK, TNF
INTRODUCTION
The epithelial cells that line the intestinal tract are constantly bombarded by a variety of bacteria and potential pathogens. Intestinal epithelial cells (IECs) respond to various microbial stimuli in the environment, but also respond to cytokines produced by T cells and other cell types [1, 2]. In response to microbial or cytokine stimuli, IECs produce a variety of cytokines, including important chemokines like CXCL8 [3]. Due to their abundance, IECs are able to amplify inflammatory signals and recruit immune cells to the site of injury.
Inflammatory bowel disease (IBD) is characterized by a dramatic inflammation of the intestinal tissue with neutrophil accumulation on the intestinal crypts [2, 4]. Several studies have shown an increase in tissue levels of TNF-α in IBD [5, 6] such that current treatments for IBD have targeted TNF through monoclonal antibodies [1]. IECs respond to TNF-α by releasing several important cytokines and chemokines, suggesting that IECs play an important role in mucosal inflammation, such as in IBD [2, 4].
IL-22 is a recently identified member of the IL-10-related cytokine family and is produced by activated Th17, Th1, Th22 and other cell types [7, 8]. Interestingly, a major target for IL-22 appears to be epithelial cells and keratinocytes, which express the IL-22 receptor [7]. Important to this study, the expression of the IL-22 receptor by both the Caco-2 and HT-29 cell lines has been confirmed [9], making these cell lines excellent models to study the effect of IL-22 on IECs. IL-22 is highly expressed in many different chronic inflammatory diseases, including IBD [8–11]. Furthermore, a risk locus for the ulcerative colitis form of IBD has been localized near the IL-22 gene on chromosome 12q15 [11]. This suggests an important role for IL-22 in the pathology of IBD. IL-22 stimulation of IECs has been shown to induce CXCL8 production [9] suggesting a pro-inflammatory function for this cytokine. Other studies support IL-22 as a pro-inflammatory cytokine [8, 10] or as protective cytokine with anti-inflammatory effects [8, 12].
In IECs, IL-22 strongly signals through the JAK/STAT pathways, activating mainly STAT3 and STAT1 [9]. However, IL-22 can activate MAPK signaling pathways, activating Akt/PKB, JNK, and ERK1/2 pathways in IECs [9] and also p38 MAPK in a variety of other cell types [8, 13, 14].
During an inflammatory response, cytokines are not isolated from one another, but rather they work in concert within a signaling network. Although the role of IL-22 in inducing proinflammatory chemokine responses alone has been explored [9, 10], the effect of IL-22 along with the potent pro-inflammatory cytokines TNF-α and IL-1 has not yet been explored for responses by IECs. As TNF-α, IL-1, and IL-22 can all induce the production of CXCL8, we hypothesized that IL-22, specifically in the presence of TNF-α or IL-1, may greatly modulate the production of CXCL8 production by IECs. Therefore, using the HT-29 and Caco-2 colonic carcinoma cell lines as models for IECs, we explored the effect of IL-22 along with TNF-α or IL-1 on the production of CXCL8. Moreover, we investigated the intracellular signaling pathways activated by IL-22 and TNF-α together to determine specifically how the effect occurred. The exploration of any IL-22 and TNF-α synergy could be very important to our understanding of the role of IL-22 within IBD-relevant signaling pathways and immune responses in the context of IECs, particularly given the known importance of TNF-α in chronic inflammatory diseases such as IBD.
MATERIALS AND METHODS
Antibodies
Recombinant human TNF-α and human IL-1β were obtained from R&D Systems (Minneapolis, MN) and recombinant human Interleukin-22 (IL-22) from Cell Signaling Technologies (Danvers, MA). Mouse monoclonal antibodies against human total IкBα and STAT3, rabbit monoclonal antibodies against human phosphorylated IкBα and Tyr705 phosphorylated STAT3, rabbit antibodies to β-actin, as well as HRP-conjugated anti-rabbit and HRP-conjugated anti-mouse detection antibodies were obtained from Cell Signaling Technologies.
Cell Culture for Cytokine Secretion
The human adenocarcinoma cell lines, HT-29 (ATCC HTB-38) and Caco-2 (ATCC HTB-37; American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Scientific Hyclone Laboratories, Logan, UT) with 10% fetal bovine serum (FBS; Hyclone), 3.7g/l sodium bicarbonate, 2mM L-glutamine (Hyclone), non-essential amino acids (Cellgro, Manassas, VA), penicillin (25 IU) and streptomycin (25μg/ml). The cells were cultured at 37°C in a 90% air-10% CO2 humid environment. For chemokine secretion experiments, the cells were cultured at 2 × 105 cells/well (Caco-2) or 4 × 105 cells/well (HT-29) in 24-well culture plates for 24 h. The medium was then changed to serum-free DMEM containing insulin, transferrin, and selenium (ITS-DMEM; BD Biosciences, Bedford, MA) and the cells were treated with recombinant human TNF-α or IL-1β and/or recombinant human IL-22 as indicated. In some experiments, the cells were pre-treated with specific inhibitors before cytokine stimulation. These included the PI3K inhibitor, Wortmannin (Sigma Aldrich) at 10nM for 30 minutes; the Akt inhibitor V, Triciribine (Calbiochem/EMD Chemicals, Gibbstown, NJ) at 10μM for 1 hour; the STAT3 inhibitor VI S31-201 (EMD Millipore) at 100μM; the JAK2/STAT3 inhibitor, WP1066 (Calbiochem) at 5μM for 1 hour; the STAT1 inhibitor, Fludarabine (Selleckchem, Houston, TX) at 25μM; the Tyk2 inhibitor Bayer-18 (Symansis, New Zealand) at 40nM or 60nM; the JNK Inhibitor SP600125 (EMD Millipore, Billerica, MA) at 25μM for 1 hour; the ERK1/2 inhibitor PD98059 (Calbiochem) at 25μM; or the p38 MAPK inhibitor SB203580 (Promega, Madison, WI) at 1μM. After incubating the cultures for 24 hours, the culture supernatants were collected and stored at −80°C and adherent cells were trypsin-EDTA treated and counted using a hemocytometer.
Secreted human CXCL8 was quantified by using the specific DuoSet ELISA kit (R&D Systems). The absorbances of the samples were measured using a Bio-Tek ELx808 microplate reader (Bio-Tek Instruments Inc., Winooski, VT) and the resulting chemokine concentration values were normalized to 105 cells.
RNA Isolation and Reverse-Transcription Real-Time PCR Analysis
HT-29 and Caco-2 cells were cultured in 10% FCS-DMEM at 1 × 106 cells/well in 6-well tissue culture plates for 24 hours. The medium was then changed to serum-free ITS-DMEM and the cells were treated with TNF-α (2Ong/ml) with or without IL-22 (100ng/ml) for 6 hours. The cells were lysed and RNA isolated using the Qiagen RNeasy mini RNA extraction kit (Qiagen Inc., Valencia, CA). The concentration of RNA was then measured using a Nanodrop Spectrophotometer ND-1000 (Thermo Scientific) and the samples were stored at −80°C.
An RNA sample of 1 μg was used to generate cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) at 25°C for 10 minutes, 37°C for 120 minutes, and then the enzyme was heat inactivated at 85°C for 5 minutes. Samples were stored at −20°C. For the real-time PCR reaction, 4.5μl of a 1:10 dilution of the cDNA reaction product was used in a total volume of 45μl for analysis using primers for human CXCL8 and GAPDH (Qiagen). cDNA levels were quantified using the iQ SYBR Green supermix (Bio Rad Laboratories, Hercules, CA) and the Mini Opticon/MJ Mini personal thermal cycler (Bio Rad). The amplification conditions were 3 minutes at 95°C followed by a two-step cycle of 95°C for 15 seconds and 60°C for 60 seconds for a total of 40 cycles. CXCL8 transcripts were then normalized to GAPDH transcripts for all corresponding conditions and fold-changes in CXCL8 were calculated using the threshold cycle (Ct) values. Data was analyzed using the Bio-Rad CFX Manager Software.
Western Blot Analysis
Cells were cultured at 1 × 106 cells/well in 6-well tissue culture plates for 24 hours. The medium was then changed to serum-free ITS-DMEM and the cells were treated with TNF-α (20ng/ml) with or without IL-22 (100ng/ml) for the times indicated. The adherent cells were washed once with ice cold PBS before the addition of a 1× RIPA Buffer solution (Cell Signaling Technology, Danvers, MA) containing 100μM phenylmethylsulfonylfluoride and 100μM 4-nitrophenyl phosphate (Sigma-Aldrich). The cells were kept on ice for 5 minutes followed by scraping with a cell scraper. The whole cell extracts were collected and incubated on ice for 15 minutes before centrifuging at 13,000 × g in a cold centrifuge for 15 minutes. The supernatants were collected and stored at −80°C.
The extracts were diluted to equal amounts of protein in Laemmli buffer and were separated on 10% or 12% SDS-polyacrylamide resolving gels with 4% polyacrylamide stacking gels and Western blotted as previously published [15]. The membranes were blocked in 5% nonfat dry milk in Tris-buffered saline (TBS) with 0.1% (v/v) Tween-20 (TBS-T) or 5% BSA in TBS-T, as appropriate, for 1 hour at room temperature before incubating with the appropriate primary antibody overnight at 4°C with mixing. The blots were then washed with TBS-T and incubated with either horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Cell Signaling Technology). After 1 hour, the blots were washed with TBS-T and the bands were visualized using the SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific, Rockford, IL) followed by exposure to X-ray film. For multiple reprobes of the same membrane, the blots were washed with TBS-T followed by incubation with a stripping buffer [15] at 50°C for 12 minutes. The membranes were again washed with TBS-T and blocked in 5% milk TBS-T or 5% BSA prior to probing with primary antibodies. Blots were scanned and band densities were quantified using the Quantity One software (Bio Rad).
Statistical Analysis
For comparison of three or more separate experiments and analysis of significant difference, ANOVA and Fisher’s protected least significant difference test with a level of significance set at p<0.05 were performed using the Statview program (SAS Institute Inc., Cary, NC). Shown in all figures are the means ± the standard error of the mean (SEM) for three or more experiments unless otherwise indicated.
RESULTS
IL-22 Enhances TNF-α- or IL-1-Induced CXCL8 Secretion by Intestinal Epithelial Cell Lines
Studies have shown that TNF-α stimulation of IECs increases the production of various cytokines, including the chemokine CXCL8 [3, 16]. IL-22 has also been shown to stimulate the production of CXCL8, TNF-α, and antimicrobial proteins by IECs [9]. Since both IL-22 and TNF-α play a role in inflammation and are upregulated in IBD [1, 8], we investigated whether IL-22 could potentiate the TNF-α-induced secretion of CXCL8 by IECs.
Studies in our laboratory found that treatment of Caco-2 [17] or HT-29 [unpublished results] human colonic epithelial cells with TNF-α at 20ng/ml was sufficient to induce cytokine production by IECs. Therefore, using these cell lines as models for IECs, the cells were treated with TNF-α at 20ng/ml with or without IL-22 at 2, 20 or 100ng/ml. When the values for CXCL8 secretion by cells treated with IL-22 alone were compared by ANOVA to unstimulated controls only without the TNF-α stimulated values (Fig. 1A), IL-22 at both 20 and 100ng/ml significantly increased CXCL8 production by HT-29 cells over the untreated controls by up to a 5.6-fold (p<0.05 and p<0.01, respectively). This is consistent with previous studies [9]. Next, as expected, HT-29 cells cultured with TNF-α alone (20ng/ml) resulted in a significant 82-fold increase in CXCL8 production by over that of the untreated control (p<0.0001). Treatment of the HT-29 cells with both TNF-α and IL-22 at all concentrations also resulted in significant increases in CXCL8 production compared to cells treated with TNF-α alone with a maximum of a 1.9-fold increase with 100ng/ml of IL-22 (Fig. 1A). Treatment of the cells with both TNF-α and 100ng/ml IL-22 resulted in a 153-fold increase in CXCL8 secretion compared to the unstimulated controls. This suggests that IL-22 can significantly enhance TNF-induced CXCL8 secretion by IECs. Of note, these culture supernatants were also tested for levels of CCL20, and although TNF-α did stimulate CCL20 secretion by the HT-29 cells, IL-22 had no additional effect on CCL20 secretion (data not shown).
Fig. 1.
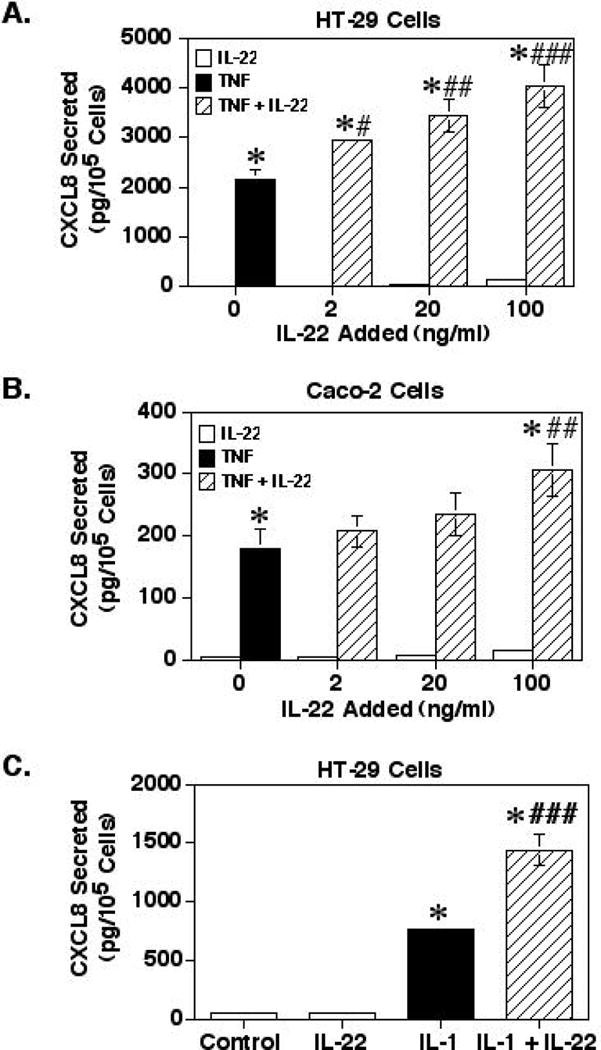
IL-22 enhances TNF-α- or IL-1-induced levels of secreted CXCL8. (A) HT-29 or (B) Caco-2 cells were cultured for 24 hours before adding 20ng/ml of TNF-α with or without IL-22 added as indicated. (C) HT-29 cells were cultured for 24 hours before adding 1ng/ml of IL-1β with or without 100ng/ml IL-22. The cells were then incubated for 24 hours. The supernatants were then collected for determining secreted CXCL8 levels by ELISA and the cells were removed and counted with a hemocytometer. Shown are the means ± SEM from three separate experiments. *Significant difference from unstimulated control cells (p<0.0001). # (p<0.05), ## (p<0.01), or ### (p<0.001) indicates a significant difference from cells treated with TNF-α alone.
The enhancing effect of IL-22 on TNF-α stimulated CXCL8 secretion was next verified in a second experiment using the Caco-2 cell line. Although IL-22 stimulation by itself did result in a 5-fold increase in CXCL8 levels, the levels were not significantly different from the untreated controls even with 100ng/ml IL-22. As shown in Fig. 1B, when Caco-2 cells were cultured with TNF-α alone, CXCL8 secretion was significantly increased by 60-fold compared to that of the untreated control (p<0.01). Stimulation of the cells with both TNF-α and IL-22 also significantly enhanced CXCL8 secretion by 102-fold compared to the controls. Yet more importantly, TNF-α and IL-22 stimulation resulted in a 1.7-fold increase in CXCL8 secretion as compared to cells treated with TNF-α only, but only with the highest concentration of IL-22 of 100ng/ml. This confirms that IL-22 can significantly enhance TNF-α induced CXCL8 secretion by IECs.
Since IL-1 can signal through several intracellular pathways similar to TNF-α, the effect of IL-22 co-stimulation with IL-1β on HT-29 cells was determined. The results in Fig. 1C show that IL-22 stimulation at 100ng/ml could significantly enhance IL-1β-stimulated CXCL8 secretion by 1.9-fold compared to cells treated with IL-1β only, indicating that IL-22 could also enhance IL-1-stimulated CXCL8 secretion by IECs.
Effect of TNF-α and IL-22 on CXCL8 mRNA Expression in IECs
Next, the effect of IL-22 on TNF-α-stimulated mRNA levels was determined using reverse transcription-real-time PCR analysis (Fig. 2). Treatment of the cells with IL-22 alone had no significant effect on CXCL8 mRNA levels in both the HT-29 or Caco-2 cells (p>0.7). However, stimulation of the cells with both IL-22 and TNF-α resulted in a significant 1.7- and 1.5-fold increase in CXCL8 mRNA levels in HT-29 and Caco-2 cells, respectively, as compared to cells treated with TNF-α alone at 6 hours. This suggests that the enhancing effect of IL-22 may be on the intracellular signaling events occurring prior to transcription.
Fig. 2.
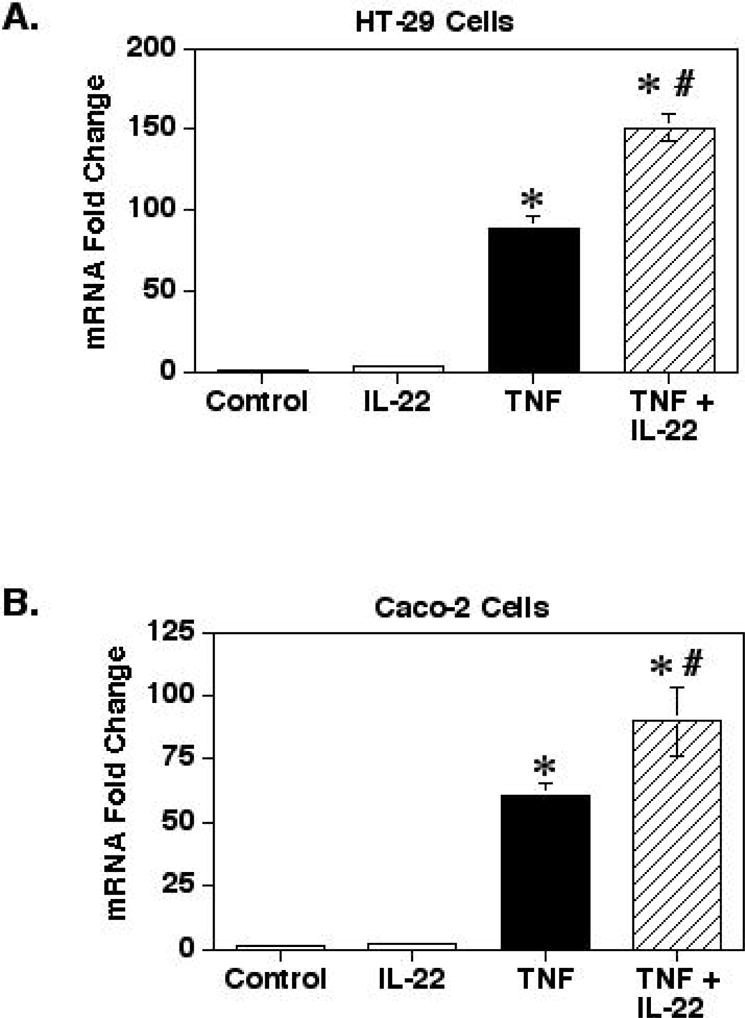
IL-22 enhances TNF-α induced increases in CXCL8 mRNA levels. HT-29 (A) and Caco-2 (B) cells were cultured for 24 hours before adding TNF-α (20ng/ml) and/or IL-22 (100ng/ml). The cells were then incubated for 6 hours and total RNA was isolated for reverse transcription and real-time PCR analysis with primers for CXCL8 and GAPDH as the control. Shown are the means ± SEM (n=3). *Significant difference from unstimulated control cells (p<0.0001). # Significant difference from cells treated with TNF-α alone (p<0.01).
IL-22 and TNF-α Effect on Intracellular Signaling Pathways
The binding of TNF-α to TNFR1 leads to the intracellular recruitment of MAPK pathway signal transducers, inducing the c-Jun N-terminal kinases (JNK), p38 MAPK, and ERK1/2 kinases, as well as signaling transducers in the IKK/IкBα/NFкB signaling pathway. Signaling through these pathways can then activate several transcription factors such as AP-1, NF-κB and others [18, 19]. The IL-22 receptor has been shown to signal through Akt/PKB, JNK, ERK1/2 and p38 MAPK pathways [8, 9, 13, 14], which have been shown to control CXCL8 responses [20]. However, a major signaling pathway associated with this IL-22 is the JAK/STAT pathway to activation of STAT3 and STAT1 in IECs [9].
To determine the signaling pathways involved, a variety of well-known chemical inhibitors specific for the various signaling pathways were used prior to treatment with IL-22 and/or TNF-α. As shown in Table 1, the enhancing effect of IL-22 on TNF-α-stimulated HT-29 CXCL8 secretion was not significantly affected by inhibiting signaling through JAK2/STAT3, STAT1, Tyk2, Phosphatidylinositol-4, 5-bisphosphate 3-kinase (PI3K), Akt (PKB), JNK, or ERK1/2. This suggests that these kinase signaling pathways were not involved in the enhancing effect of IL-22.
TABLE 1.
Pathways not contributing to the enhancing effect of IL-22 on TNF-α-induced CXCL8 secretion.
| Signaling Pathway | Inhibitor | Effect of Inhibitor on IL-22-Induced Enhancement* |
|---|---|---|
| JAK2/STAT3 | WP1066 | None |
| STAT1 | Fludarabine | None |
| Tyk2 | Bayer-18 | None |
| PI3K | Wortmanin | None |
| Akt/PKB | Triciribine | None |
| JNK | SP600125 | None |
| ERK1/2 | PD98059 | None |
Cultures of HT-29 cells were cultured with the appropriate concentrations of the inhibitors, with or without IL-22 and/or TNF-α as in Fig. 3.
Since IL-22 signaling through STAT3 has been shown to be a major signaling pathway for this cytokine, we sought to confirm that this signaling pathway was not involved. Fig. 3A and Fig. 3B show that IL-22, but not TNF-α, induced a strong activation of STAT-3 phosphorylation within 5 minutes that remained elevated for at least 30 minutes. However, treatment of the cells with the JAK2 inhibitor WP1066 had no effect on the enhancing effect of IL-22 on TNF-α-induced CXCL8 secretion (Table 1). To further confirm this result, HT-29 cells were treated with the STAT3 specific inhibitor, STAT3 Inhibitor VI. Treatment of the cells with this specific inhibitor had no effect on the enhancement of TNF-induced CXCL8 secretion induced by IL-22 (Fig. 3C). These experiments strongly confirm that the enhancing effect of IL-22 was not due to STAT3 signaling.
Fig. 3.
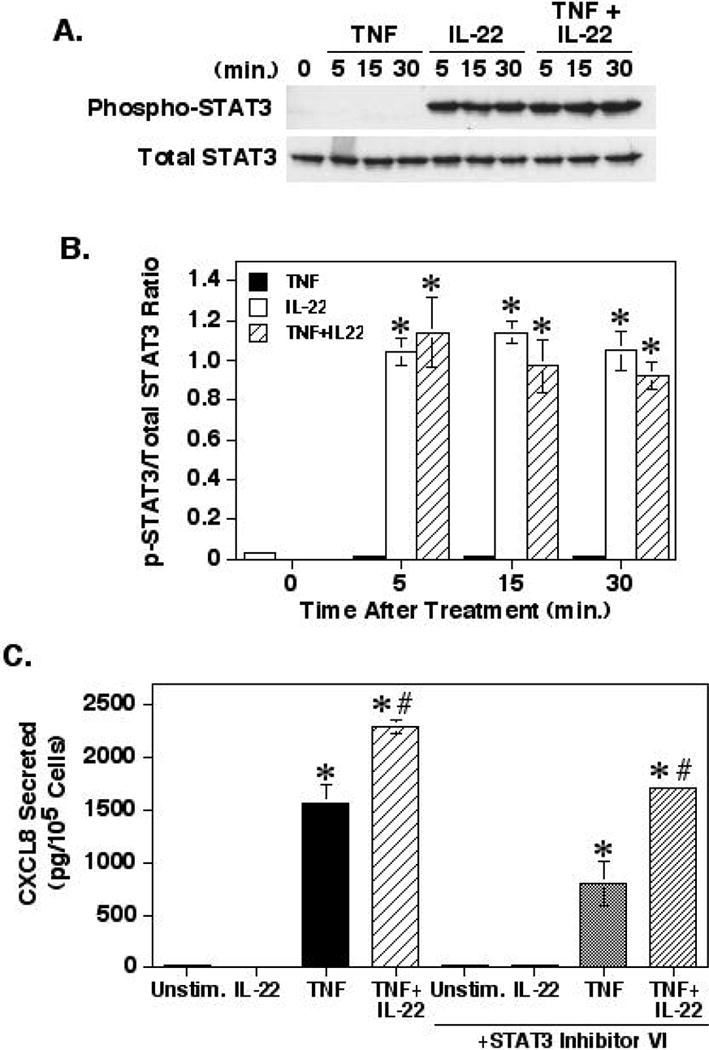
The role of STAT3 in the enhancement of TNF-α-induced CXCL8 responses by IL-22. (A) HT-29 cells were cultured for 24 hours before adding TNF-α (20ng/ml) and/or IL-22 (100ng/ml) in ITS-DMEM. At the times indicated, whole cell lysates were collected, separated by SDS-PAGE and were Western blotted for Tyr705 phosphorylated and total STAT3. Shown is a representative blot from 3 separate experiments. (B) Graph shows the means ± SEM of the band densities of phosphorylated Tyr705 STAT3 normalized to total STAT3 for three separate experiments. *Significant difference from unstimulated control cells (p< 0.001). (C) HT-29 cells were prepared as in Fig. 1 except the cells were pre-treated with or without the STAT3 Inhibitor-VI S31-201 (100μM) for 1 hour before treatment with TNF-α and IL-22. Shown are the means ± SEM (n=3). *Significant difference from the unstimulated cells (p<0.0001). #Significant difference from cells treated with the appropriate TNF-α control (p< 0.01).
Effect of IL-22 on IкBα Phosphorylation and Degradation
One of the major mechanisms for activation of CXCL8 gene transcription is through the activation of NF-κB [20]. Activation of NF-κB requires the phosphorylation and degradation of its inhibitor, IкBα [19]. Therefore, we next determined the effect of IL-22 on TNF-α-induced IкBα phosphorylation and degradation. As shown in Figs. 4A and B, IL-22 alone had no effect on IкBα phosphorylation. However, treatment of the cells with IL-22 and TNF-α resulted in a significant suppression of IкBα phosphorylation at 30 minutes, yet this suppression was still significantly elevated compared to the untreated controls. Furthermore, treatment of the HT-29 cells with TNF-α or IL-22 and TNF-α resulted in a significant degradation of total IкBα levels at 30 minutes (Figs. 4A and C). These results could indicate that IL-22 may delay IкBα phosphorylation, but ultimately may have little overall effect on this signaling pathway as secreted CXCL8 levels were not suppressed.
Fig. 4.
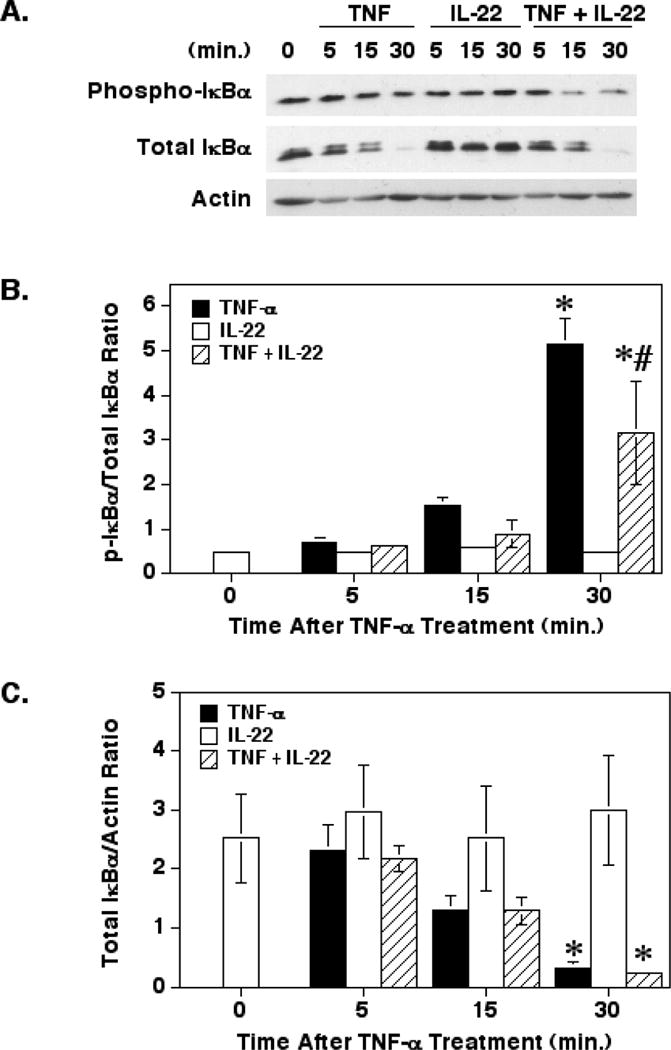
Effect of IL-22 on TNF-α-induced IкBα phosphorylation and degradation. (A) HT-29 cells were cultured and treated with TNF-α and/or IL-22 as in Fig. 3 before preparing cell extracts for SDS-PAGE and Western blotting for phosphorylated and total IкBα and actin. (B) Graph of the mean (±SEM) band density ratios of phosphorylated IкBα to total IкBα from three separate experiments. (C) Graph of the mean (±SEM) band density ratios of total IкBα to β-actin from three separate experiments. *Significant difference from unstimulated control cells (p< 0.05). # Significant difference from cells treated with TNF-α alone (p<0.001)
Effect of IL-22 and TNF-α on p38 MAPK Signaling
Both IL-22 and TNF-α are also known to signal through the p38 MAPK pathway [8, 13, 19]. Signaling though p38 MAPK can enhance CXCL8 responses by activating mechanisms that enhance the stability of mRNA and indirectly through affecting histone phosphorylation [20, 21]. As expected since TNF-α signals through p38 MAPK, pre-treatment of the HT-29 cells with the p38 MAPK inhibitor SB203580 resulted in a suppression of TNF-α-induced CXCL8 secretion (Fig. 5). More importantly, pre-treatment with the p38 MAPK inhibitor resulted in a complete abrogation of the enhancing effect of IL-22 on TNF-α-stimulated CXCL8 secretion. Therefore, the enhancing effect of IL-22 must involve signaling through the p38 MAPK.
Fig. 5.
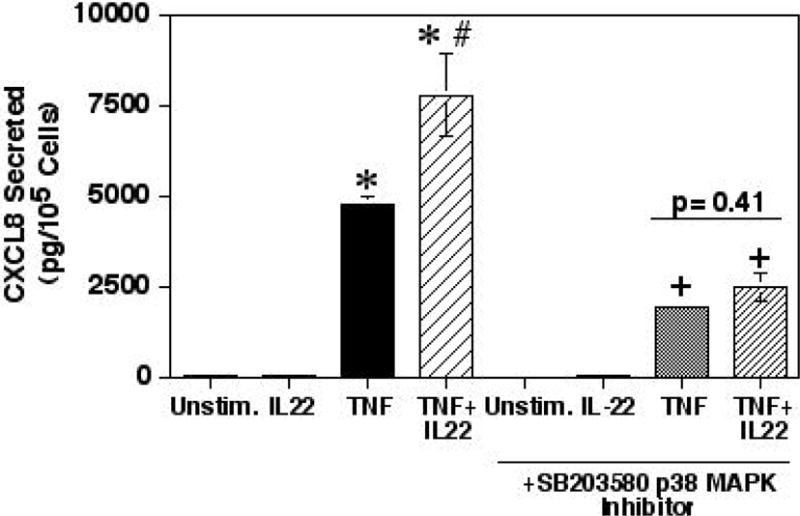
The enhancement of TNF-α-induced CXCL8 secretion by IL-22 was neutralized by inhibiting p38 MAPK. HT-29 cells were prepared as in Fig. 1 except the cells were pre-treated with or without the SB203580 p38 MAPK inhibitor (1μM) for 1 hour followed by treatment with TNF-α and/or IL-22 for 24 hours. Shown are the means ± SEM (n=3). *Significant difference from unstimulated control cells (p<0.01). #Significant difference from cells treated with the TNF-α only (p< 0.001). +Significant difference from cells treated with the SB203580 inhibitor only (p<0.01).
DISCUSSION
During an inflammatory response, cells are exposed to a network of cytokines. Recent reports have shown that IL-22 plays an important role in IBD [11], and as such belongs in this network of cytokines present during mucosal inflammatory responses. Yet, the well-characterized pro-inflammatory cytokines, TNF-α and IL-1, are major cytokines known to be important in chronic inflammatory diseases and TNF-α is a major driver for the production of pro-inflammatory cytokines by IECs in mucosal inflammatory diseases [4]. Therefore, any discussion on the role of IL-22 in mucosal inflammation must consider its effect in the context of the potent pro-inflammatory cytokines like TNF-α and IL-1.
IL-22 is expressed in IBD tissues [9] and IECs express IL-22 receptors, making them a major target for IL-22 [7]. Indeed, IL-22 has been shown to induce IEC proliferation, survival signals and wound healing along with inducing IECs to produce antimicrobial peptides [11]. IL-22 may also play a role in inflammation by inducing IECs to produce CXCL8 [9]. Since IL-22, TNF-α and IL-1 can all induce IECs to produce CXCL8, we examined the co-stimulatory effect of IL-22 with these pro-inflammatory cytokines on CXCL8 responses in IECs cell lines.
Although IL-22 alone did induce low levels of CXCL8 secretion from the HT-29 cell line, this level was relatively insignificant compared to the level of CXCL8 secreted after TNF-α or IL-1β stimulation. This suggests that by itself, IL-22 had little effect on CXCL8 responses by IECs. However, co-stimulation of IECs with both TNF-α and IL-22 or IL-1β and IL-22 resulted in an enhanced level of CXCL8 secretion. This co-stimulatory enhancing effect was also seen with increased levels of CXCL8 mRNA levels. This suggests that the true effect of IL-22 on chemokines may be to enhance TNF- or IL-1-stimulated CXCL8 responses by IECs in mucosal tissues.
Experiments were next performed to determine the mechanism through which IL-22 enhanced pro-inflammatory cytokine responses, focusing on TNF-α stimulation as both TNF-α and IL-1 signal through similar downstream pathways. IL-22 has been shown to induce intracellular signaling through the JAK/STAT signaling pathway to activate STAT1 and STAT3 [9], as well as the Akt/PKB, JNK, ERK1/2, and p38 MAPK pathways [8, 9, 13, 14]. Although all of the latter pathways have been linked to regulating CXCL8 responses [20], using well-known inhibitors of these signaling kinases revealed that only the p38 MAPK appeared to be involved in the enhancing effect of IL-22 on TNF-α-stimulated CXCL8 secretion. Indeed, even though STAT3 is a major signaling pathway for the IL-22 receptor, that STAT3 was not involved in the effect was confirmed with two different inhibitors as well as inhibitors of JAK2 and Tyk2. Furthermore, even though IL-22 has been shown to enhance NF-κB responses through an effect on IкBα in colonic subepithelial myofibroblasts [10], IL-22 had no enhancing effect on IкBα phosphorylation or degradation with TNF-α stimulation of the HT-29 cells. Indeed, a suppressive effect of IL-22 on IкBα phosphorylation after 30 minutes was actually found. Yet since TNF-α-induced CXCL8 secretion levels were enhanced with IL-22 co-stimulation, this suppressive effect of IL-22 on IкBα phosphorylation was apparently not important and may have been reflective of a delay in IкBα phosphorylation. Still, the significance of this suppressive effect of IL-22 on IкBα phosphorylation remains unknown.
Many reports have shown that p38 MAPK can greatly enhance CXCL8 responses by post-transcriptionally prolonging the half-life of CXCL8 mRNA [22]. This effect occurs through phosphorylation of the downstream MK2/3 kinases by p38 MAPK [21]. MK2/3 then phosphorylate the tristetraprolin (TTP) AU-rich element-binding protein, leading to an inhibition of the TTP-dependent degradation of CXCL8 mRNA [23]. The p38 MAPK can also phosphorylate histone H3, resulting in chromatin modulation to enhance the availability of NF-κB binding sites on cytokine gene promoters [21]. This provides two powerful mechanisms by which IL-22 could enhance TNF-α-mediated CXCL8 responses in IECs.
Studies suggest that IL-22 has several pro- and anti-inflammatory effects. An enhancing effect of IL-22 on TNF-α or IL-1-stimulated CXCL8 responses could lead to increased neutrophil migration into inflamed mucosal tissues in diseases like IBD, thus exacerbating the detrimental inflammation. However, a role for CXCL8 in wound healing responses exists in that CXCL8 can enhance angiogenesis in several tissues and cancer [24]. Thus, a CXCL8-enhanced angiogenic effect would be consistent with the wound healing aspects of IL-22. Therefore, our studies could support both a pro-inflammatory and wound healing effect of IL-22 in a cytokine network with TNF-α and IL-1, suggesting caution when targeting this cytokine for future anti-inflammatory therapies.
Acknowledgments
This work was supported by U.S. PHS Grant DK089459 and a grant from the Binghamton University Harpur College.
Abbreviations
- DMEM
Dulbecco’s Modified Eagle’s Medium
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IBD
inflammatory bowel disease
- IEC
intestinal epithelial cell
- JNK
c-Jun N-terminal kinase
- IL
interleukin
- ITS
insulin, transferrin, selenium
- TBS
Tris buffered saline
- TNF
tumor necrosis factor
References
- 1.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annual Review of Immunology. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson P, van Limbergen JE, Schwarze J, Wilson DC. Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflammatory Bowel Disease. 2011;17:382–395. doi: 10.1002/ibd.21379. [DOI] [PubMed] [Google Scholar]
- 3.Jung HC, Eckmann L, Yang S-K, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. Journal of Clinical Investigation. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onyiah JC, Colgan SP. Cytokine responses and epithelial function in the intestinal mucosa. Cellular and Molecular Life Sciences. 2016;73:4203–3212. doi: 10.1007/s00018-016-2289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murch SH, Braegger CP, Walker-Smith JA, MacDonald TT. Location of tumour necrosis factor α by immunohistochemistry in chronic inflammatory bowel disease. Gut. 1993;34:1705–1709. doi: 10.1136/gut.34.12.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1β by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clinical and Experimental Immunology. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annual Review of Immunology. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 8.Dudakov JA, Hanash AM, van den Brink MRM. Interleukin-22: Immunobiology and pathology. Annual Review of Immunology. 2015;33:747–785. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beigel F, Brand S, Olszak T, Zitzmann K, Eichhorst S, Otte J, Diepolder H, Marquardt A, Jagla W, Popp A, Leclair S, Herrmann K, Seiderer J, Ochsenkuhn T, Goke B, Auernhammer CJ, Dambacher J. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2006;290:G827–G838. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 10.Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A, Shimizu N, Fujiyama Y. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 11.Gong C, Li L-J, Zhao M-H, Feng B-S. Role of interleukin-22 in inflammatory bowel diseases. World Journal of Gastroenterology. 2014;20:18177–18188. doi: 10.3748/wjg.v20.i48.18177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Ota N, Manzanillo P, Kates L, Zvala-solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, Diehl L, van Bruggen L, Kolumam G, Ouyang W. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–241. doi: 10.1038/nature13564. [DOI] [PubMed] [Google Scholar]
- 13.Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa J, Renauld J. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line - Pathways that are shared with and distinct from IL-10. Journal of Biological Chemistry. 2002;277:33676–33682. doi: 10.1074/jbc.M204204200. [DOI] [PubMed] [Google Scholar]
- 14.Wolk K, Witte E, Wallace E, Docke W, Kunz S, Asadullah K, Volk H, Sterry W, Sabat R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. European Journal of Immunology. 2006;36:1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 15.Rafferty BJ, Unger BL, Perey AC, Tammariello SP, Pavlides S, McGee DW. A novel role for the Rho-Associated Kinase, ROCK, in IL-1-stimulated intestinal epithelial cell responses. Cellular Immunology. 2012;280:148–155. doi: 10.1016/j.cellimm.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treede I, Braun A, Jeliaskova P, Giese T, Fuellekrug J, Griffiths G, Stremmel W, Ehehalt R. TNF-alpha-induced up-regulation of pro-inflammatory cytokines is reduced by phosphatidylcholine in intestinal epithelial cells. BMC Gastroenterology. 2009;9:53–64. doi: 10.1186/1471-230X-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitkus SJD, Hanifin SA, McGee DW. Factors affecting Caco-2 intestinal epithelial cell interleukin-6 secretion. In Vitro Cellular and Developmental Biology-Animal. 1998;34:660–664. doi: 10.1007/s11626-996-0017-7. [DOI] [PubMed] [Google Scholar]
- 18.Baud V, Lie Z-G, Bennett B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: Oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes and Development. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu WM. Tumor necrosis factor. Cancer Letters. 2013;328:222–225. doi: 10.1016/j.canlet.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. Journal of Leukocyte Biology. 2002;72:847–855. [PubMed] [Google Scholar]
- 21.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signaling. Biochemistry Journal. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton T, Li X, Novotny M, Pavicic PG, Jr, Datta S, Zhao C, Hartupee J, Sun D. Cell type- and stimulus-specific mechanisms for post-transcriptional control of neutrophil chemokine gene expression. Journal of Leukocyte Biology. 2012;91:377–383. doi: 10.1189/jlb.0811404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiology and Molecular Biology Reviews. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, Chen Y, Han X, Wu K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine and Growth Factor Reviews. 2016;31:61–71. doi: 10.1016/j.cytogfr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]