

Abstract
Background
The oxidative stress hypothesis links neurodegeneration in the later, progressive stages of multiple sclerosis (MS) to the loss of a major brain antioxidant, glutathione (GSH).
Objective
We measured GSH concentrations among major MS subtypes and examined the relationships with other indices of disease status including physical disability and MRI measures.
Methods
GSH mapping was performed on the fronto-parietal regions of patients with relapsing-remitting (RRMS, n=21), primary progressive (PPMS, n=20), and secondary progressive MS (SPMS, n=20), and controls (n=28) using GSH chemical shift imaging. Between-group comparisons were performed on all variables (GSH, T2-lesion, atrophy, EDSS).
Results
Patients with MS had substantially lower GSH concentrations than controls, and GSH was lower in progressive MS (PPMS and SPMS) compared with RRMS. GSH concentrations were not significantly different between PPMS and SPMS, or between RRMS and controls. Brain atrophy was significant in both RRMS and progressive MS compared with controls.
Conclusion
Markedly lower GSH in progressive MS than RRMS indicates more prominent involvement of oxidative stress in the progressive stage of MS than the inflammatory stage. The association between GSH and brain atrophy suggests the important role of oxidative stress contributing to neurodegeneration in progressive MS, as suggested in other neurodegenerative diseases.
Keywords: oxidative stress, glutathione, progressive multiple sclerosis, neurodegeneration, brain Search terms: Multiple sclerosis, MRI, MRS
Introduction
Although multiple sclerosis (MS) is generally considered to be an inflammatory disease, the role of inflammation in the later course of MS has been controversial. Contrast-enhanced MRI of patients with primary progressive MS (PPMS) and secondary progressive MS (SPMS) show very few acute inflammatory lesions 1, 2 and existing immunomodulatory therapies targeting inflammatory processes are much less effective for patients with progressive MS.3 Substantial brain atrophy in the absence of new lesions reported in progressive MS supports the involvement of neurodegenerative processes sustained to a greater extent by pathological mechanisms other than inflammation. Indirect evidence points to the role of oxidative stress in the pathogenesis of MS.4-8 For example, higher levels of oxidative stress markers and lower antioxidant capacity were found in plasma and saliva of patients with MS.9-11 Increases in intracellular enzymatic antioxidants 12 and products induced by oxidative stress including peroxynitrite and superoxide that depletes natural antioxidants13-15 have been detected in patents with MS, even in the absence of pathologically apparent inflammation.16
Glutathione (GSH) is the most concentrated non-enzymatic antioxidant scavenger of reactive oxygen species and thereby constitutes a front-line defense against cellular damage and eventual necrosis from oxidative stress.17 Loss of GSH occurs during the defense processes; thus reduced levels of GSH serve as a marker of oxidative stress.18 Until recently, the status of oxidative stress could only be evaluated indirectly using in vitro peripheral measures of GSH or glutathione reductase, an enzyme central to its synthesis. Conflicting results found in studies of oxidative stress in patients with MS 19, 20 are likely due to reliance on such indirect methods to evaluate a principal antioxidant system residing in the central nervous system. A suitable examination of oxidative stress in MS pathology hinges upon the development of noninvasive in vivo measures obtained directly within the living human brain. We have developed this capability using a specially designed, selective multiple quantum chemical shift imaging (CSI) technique.21
Our initial study 22 showed lower GSH in patients with SPMS than healthy controls, with GSH tending to be lower for more disabled patients. A small study also found lower GSH in patients without specified MS subtypes (7 patients and 6 controls), consistent with our findings.23 Following up on our initial study, we reexamined participants over the course of 3-5 years and found longitudinal changes in GSH concentrations were related to patients' clinical progression.24 In this study, we aimed to compare GSH in three major subtypes of MS with the contention that oxidative stress is a critical factor contributing to neurodegeneration in the later stages of MS. Differences in GSH concentrations between RRMS and progressive MS may indicate a shift in pathogenesis away from inflammatory mechanisms to ones more directly tied to oxidative stress. We also evaluated the relationships between GSH and other indices of disease status including physical disability and MRI measures of lesion volume and atrophy.
Subjects and Methods
Subjects enrollment and procedures
This study was approved by the Human Subjects Committee at the University of Kansas Medical Center. Written informed consent was obtained from all study participants. Study inclusion/exclusion criteria are listed in Table 1. The exclusion criteria were applied to all subjects. Sixty-one patients (48.6±10.6 years of age, range 18-65) with clinically definite MS 25 and 28 healthy controls (46.8±13.0 years of age, range 20-65) participated in the study. Patients and controls had comparable demographics, regarding age, sex and education (Table 2). All patients with MS were recruited from the University of Kansas MS Clinic. The neurologist reviewed patients' charts to confirm a definite diagnosis of MS and the subtype: RRMS (n=21), PPMS (n=20) and SPMS (n=20). The duration of MS diagnosis was 1 to 33 years (11.2±6.9 years). The Expanded Disability Status Scale (EDSS) 26 was performed at the time of recruitment; scores ranged from 1.0 to 8.5 (median=5.0). The healthy controls (n=28) were recruited in specific age ranges to ensure a close match with the age and sex of the patients with MS. MR scans were performed within two weeks of the recruitment of each subject.
Table 1. Subject recruitment criteria.
| Inclusion Criteria | Exclusion criteria |
|---|---|
|
| |
|
|
Note: The inclusion/exclusion criteria for controls are identical to those for patients except for MS diagnosis and requirements for kidney function because controls did not require Gd-enhanced MRI.
Table 2. Subject demographics and clinical variables.
| All patients | RRMS | PPMS | SPMS | Controls | |
|---|---|---|---|---|---|
| Number of subjects | 61 | 21 | 20 | 20 | 28 |
| Sex (Female:Male) | 33:28 | 14:7 | 7:13 | 12:8 | 18:10 |
| Age (years) (mean ± SD) | 48.6±10.6 | 40.5±10.9 | 53.7±7.4 | 52.0±7.8 | 46.7±13.0 |
| Education (years) (median; range) | 15; 12-20 | 15; 12-16 | 14; 12-20 | 16; 12-20 | 16; 12-20 |
| EDSS (median; range) | 5.0; 1.0-8.5 | 1.5; 1.0-4.0 | 5.75; 2.0-8.0 | 6.0; 2.0-8.5 | -- |
| Duration of Dx (years) (median; range) | 12.0; 1-33 | 8.0; 1-20 | 11.5; 1-22 | 16.5; 5-33 | -- |
Because these patients were evaluated during the normal course of clinical care, they were taking various disease modifying therapies. Seventeen of the 21 patients with RRMS were taking a disease modifying drug, including glatiramer acetate (n=10), Betainterferon (n=4), and dimethylfumarate (n=3). Twenty two of the 40 patients with SPMS were taking an immunomodulatory or immunosuppressive drug, including glatiramer acetate (n=6), betainterferon (n=2), methotrexate (n=5), fingolimod (n=1), mycophenolate (n=1), and azathioprine (n=8).
MR scan protocol
All MR scans were performed on a 3 T MR system (Skyra, Siemens, Erlangen, Germany) using a body coil transmit and 16-channel head coil receive set-up. Subjects were positioned supine in the magnet. Three-plane pilot MR images were acquired using a gradient echo sequence to position the subject's head at the iso-center of the magnet and to locate the volume of interest (VOI). GSH mapping was performed on a CSI slice positioned in the fronto-parietal regions above the corpus callosum using the selective multiple quantum CSI sequence (slice thickness=3 cm, matrix size=8×8, field of view (FOV)=200×200 mm2, echo time (TE)/repetition time (TR)=115/1500 ms, number of averages (NT)=10 and scan time=16 min).21 Creatine mapping was performed using a point resolved spectroscopy (PRESS) CSI sequence (slice thickness=1.5 cm, matrix size=12×12, FOV=16×16 cm2, VOI=8×8 cm2, TE/TR=30/2000 ms, NT=4 and scan time=5 min).
Structural MRI scans were performed for lesion and atrophy analyses using a series of MR sequences: First, T2 and proton density-weighted MRI were acquired using a dual-echo spin-echo sequence (TE=9, 90 ms, TR=4000 ms, slice thickness=3 mm, FOV=240×240 mm2, matrix=256×256, number of slices=60, and scan time=7 min). Second, three-dimensional high-resolution T1-weighted MRI were acquired using a magnetization-prepared rapid acquisition gradient echo (MPRAGE) sequence (TE/TR/TI=2.98/2300/900 ms, spatial resolution=1 mm3, flip angle=9 degree, and scan time=∼9 min). Third, pre- and post-contrast T1-weighted MRI were acquired before and after a standard dose of Gd-DTPA (0.1 mmol/kg body weight) administration using a gradient echo sequence (TE/TR=5.18/28 ms, slice thickness=3 mm, FOV=240×240 mm2, matrix=256×256, number of slices=60, flip angle=27 degree, and scan time=∼5 min). Fourth, fluid attenuated inversion recovery (FLAIR) T2-weighted MRI was acquired during a wait period after the Gd-DTPA administration (TE/TR/TI=78/8000/2500 ms, slice thickness=3 mm, FOV=250×250 mm2, matrix=256×256, number of slices=60, and scan time=8 min).
MR data analyses
GSH CSI data were processed using the previously described in-house software.22 GSH signals were analyzed and quantified using an internal reference method that uses simultaneously acquired creatine signals. The frequency and phase of the GSH signals were corrected based on those of the creatine signals from the corresponding voxels. Creatine signals also served as the internal concentration reference providing an automatic correction for the brain atrophy effect since both GSH and creatine are approximately 100 times more highly concentrated in brain tissues than in CSF.27, 28 GSH signals were quantified from the central portion of the CSI slab (5×5×3 cm3) that spans portions of both the frontal and parietal regions, where both the static magnetic field (B0) and the RF field (B1) homogeneity are the most optimized for the reliable estimation of GSH. Data quality criteria were used to automatically exclude voxels with poor spectral quality. The criteria consist of the linewidth of creatine signals less than 14 Hz and their frequency offset less than 8 Hz from those of the central voxel. The numbers of voxels excluded by the quality criteria for each group were 4, 6, 4, and 4 for control, RRMS, SPMS, and PPMS respectively and were not significantly different. The total area is labeled as “fronto-parietal”, with the anterior half (5×2.5×3 cm3) lying mostly in the frontal region (“frontal”), and the posterior half mostly in the parietal region (“parietal”). GSH concentrations were obtained for all three regions. Creatine signals were quantified using the LCModel analysis package 29 and unsuppressed water CSI signals as an internal concentration reference with the brain tissue volume correction as described previously.22 Creatine concentrations were obtained from the same position as GSH measurements in the fronto-parietal region.
MRI data were analyzed using a structural data processing pipeline to measure T2 hyper-intense lesion volumes (T2LV), and brain parenchymal fraction (BPF). The pipeline consisted of in-house written software and publicly available software packages including SPM8 (Wellcome Department of Imaging Neuroscience, London, UK) 30, FSL (FMRIB, Oxford University, Oxford, UK) 31, and Jim6 (Xinapse, Essex, UK). MRI data processing included the following: 1) preparation of MRI volumes by co-registering all MRI to a common volume, i.e., FLAIR MRI, using the FSL linear image registration tool (FLIRT), 32 and by cropping image volumes at the plane positioned 2-3 mm below the most inferior point of the cerebellum; 2) marking of T2 hyper-intense lesions on the FLAIR images by a neurologist and outlining lesion boundaries by a trained technician using the edge detection tool in Jim6, with MPRAGE, Post-Gd T1-weighted, and proton density images concurrently viewed to assist all lesion decisions; 3) semi-automatic delineation of the intracranial cavity (ICC) with manual correction by the technician; 4) lesion in-painting on MPRAGE images using FSL to remove the influence of lesions on tissue segmentation; and 5) brain tissue segmentation to ascertain volumes of GM, WM, and CSF on MPRAGE images using SPM8 with additional pre-aligned tissue probability maps. ICC volume was measured by automatic estimation using both MPRAGE and T2-weighted MRI in the brain extraction tool (FSL BET) followed by manually correcting errors in the estimated ICC boundaries. BPF, GMF and WMF were determined by dividing brain tissue volume (GM+WM), GM volume and WM volume by ICC volume, respectively.
Statistical analyses
The distributions for all variables were evaluated for normality using the Komolgorov-Smirnov test. Variables distributed asymmetrically (education, duration of diagnosis, and T2LV) or measured on an ordinal scale (EDSS) were subsequently analyzed with nonparametric statistics. Comparisons between patients with three MS subtypes and controls were performed using simple analyses of variance and covariance for parametric variables and the Kruskal-Wallis test for nonparametric variables. Significant overall outcomes were followed up with four planned comparisons: 1) all patients with MS vs. controls; 2) progressive MS (SPMS and PPMS) vs. RRMS; 3) RRMS vs. controls; and 4) PPMS vs. SPMS. Relationships between MR variables and clinical measures were evaluated for patients using Pearson correlations for parametric data and Spearman rank-order correlations for nonparametric data. Partial correlation was used to control for the variability in subjects' age.
Results
Demographic characteristics
The demographic variables for all four groups and disease-related variables for patients are summarized in Table 2. An overall comparison of all four groups (RRMS, PPMS, SPMS, and controls) showed no significant difference in years of education (χ2=3.92; df=3, p=0.264) or in sex (χ2=5.44; df=3, p=0.143), even though the ratio of females to males was substantially lower in PPMS. A significant overall difference in age (F=6.78, df=3,85, p<0.001) was due to the younger average age of the RRMS than the progressive MS groups (p<0.001). The three subtypes of patients differed in duration of diagnosis (χ2=12.82; df=2, p=0.002); SPMS had longer duration than RRMS (p=0.002) and PPMS (p=0.04). The three subtypes also differed in EDSS (χ2=35.80; df=2, p<0.001); PPMS and SPMS had higher EDSS ratings than RRMS (p<0.001 for both).
Between-group comparisons
Figure 1 shows GSH maps of a control subject and patients with three subtypes of MS (top row) and the corresponding GSH CSI of the patient with RRMS (bottom row). The primary outcome measures for GSH and all other variables are summarized in Table 3. Overall differences across the four groups were found in GSH concentrations for the frontal (F=8.62, df=3, 85, p<0.001), parietal (F=6.10, df=3, 85, p=0.001), and fronto-parietal (F=9.85, df=3, 85, p<0.001) regions (Fig. 2). The four planned comparisons used to follow up on these overall differences resulted in following significant outcomes: (1) all patients with MS had lower GSH concentrations compared with controls (frontal: 12.2%; parietal: 9.9%; fronto-parietal: 10.7%; all p's≤0.001); (2) patients with progressive MS had lower GSH concentrations than those with RRMS (frontal: 9.9%, p=0.006; parietal: 7.4%, p=0.02; fronto-parietal: 8.3%, p=0.006). GSH concentrations were not significantly different between RRMS and controls (p=0.11, 0.16, and 0.06, respectively) or between PPMS and SPMS (p=0.78, 0.81, and 0.77, respectively). When these analyses were repeated with age and sex entered as covariates, the results were unchanged. Creatine concentrations were not different among all MS subtype groups and controls (F=0.69, df=3, 70, p=0.56), thus providing validity of using creatine as a concentration reference for GSH quantification.
Figure 1. Chemical shift imaging of GSH in the brains of patients with three subtypes of MS.
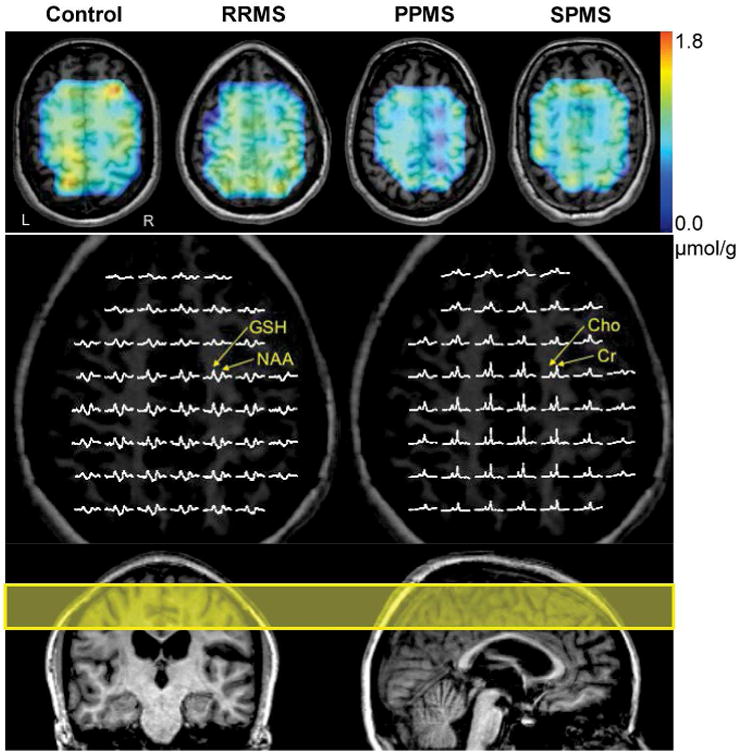
GSH concentration maps of a healthy control subject and patients with three subtypes of MS (RRMS, PPMS, and SPMS) are overlaid on T1-weighted MR images (top row). Low-resolution GSH concentration maps were resampled to match high-resolution anatomical images. The color bar (top right) indicates GSH concentration ranges from 0 to 1.8 μmol/g. Voxels with poor spectral quality and significant spectral fitting errors were excluded from the maps. Partial views of the in vivo GSH CSI (middle left) show clear detection of GSH signals throughout the CSI slice. The GSH CSI spectra (middle left) and the simultaneously acquired creatine (Cr) and choline (Cho) CSI spectra (middle right) are from the identical patient with RRMS shown in the color GSH concentration map (top row). Nominal voxel size for each CSI spectrum was 1.25 × 1.25 × 3 cm3 after 2× zero padding. Displayed spectral ranges of GSH and creatine/choline spectra are from 3.4 ppm to 2.2 ppm and from 3.6 ppm to 2.6 ppm, respectively. NAA: N-acetyl-aspartate. The yellow rectangle overlaid on sagittal and coronal MR images (bottom) indicates a 3-cm thick slice that was selected for GSH chemical shift imaging.
Table 3. Primary outcome measures of GSH and all other variables.
| All patients | RRMS | PPMS | SPMS | Controls | |
|---|---|---|---|---|---|
| Number of subjects | 61 | 21 | 20 | 20 | 28 |
| frontal GSH (μmol/g tissue) | 1.08±0.17 | 1.16±0.12 | 1.04±0.20 | 1.05±0.16 | 1.23±0.11 |
| parietal GSH (μmol/g tissue) | 1.09±0.16 | 1.15±0.10 | 1.06±0.20 | 1.07±0.14 | 1.21±0.11 |
| Fronto-parietal GSH (μmol/g tissue) | 1.09±0.15 | 1.15±0.11 | 1.05±0.18 | 1.06±0.14 | 1.22±0.08 |
| T2LV (ml) (median; range) | 17.76±16.60; 12.61; 1.56-68.26 | 15.52±14.20; 12.61; 1.56-49.84 | 14.68±16.35; 8.22; 1.75-65.16 | 23.19±18.54; 16.01; 2.78-68.26 | 0.65±0.99; 0.35; 0.00-3.70 |
| BPF | 0.73±0.04 | 0.76±0.04 | 0.71±0.03 | 0.71±0.04 | 0.78±0.04 |
| GMF | 0.45±0.03 | 0.46±0.03 | 0.43±0.03 | 0.44±0.03 | 0.48±0.05 |
| WMF | 0.28±0.03 | 0.30±0.03 | 0.28±0.02 | 0.28±0.03 | 0.30±0.03 |
Note: All data are shown in mean ± SD. T2 hyper-intense lesion volumes (T2LV) are shown in mean ± SD as well as median and range due to its asymmetrical distribution. BPF: brain parenchymal fraction; GMF: gray matter fraction; WMF: white matter fraction. GSH concentrations in progressive MS (PPMS and SPMS) were lower than those of controls and RRMS, while no GSH difference was found between RRMS and controls, and between PPMS and SPMS. Similarly, BPF, GMF, and WMF in progressive MS were lower than those in controls and RRMS, while no difference was found between PPMS and SPMS. Additionally GMF in RRMS was lower than that in controls.
Figure 2. Comparisons of brain GSH concentrations between patients with three subtypes of MS and healthy controls.
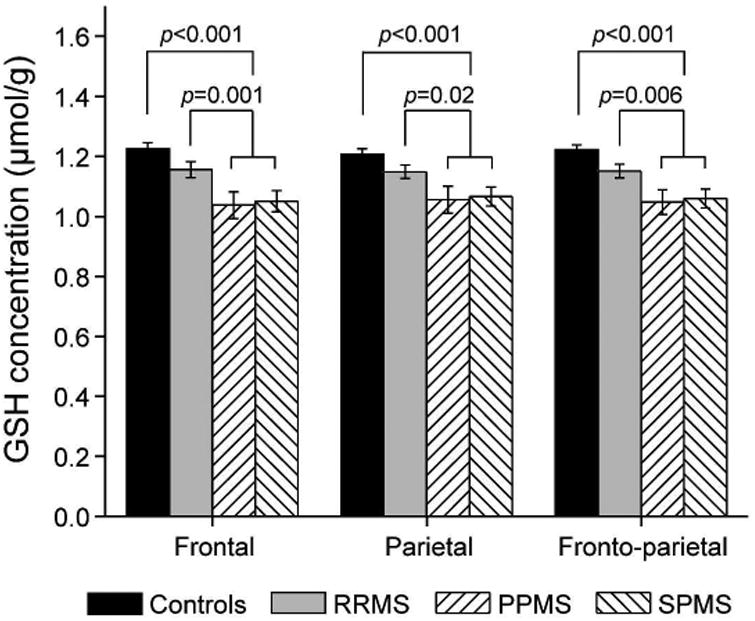
GSH concentrations were significantly lower for the full sample of patients with MS (all three subtypes combined) compared with healthy controls (p<0.001 for all brain regions), and for those with progressive MS (PPMS and SPMS) compared with RRMS (p=0.001, 0.02, 0.006) or healthy controls (p<0.001 for all brain regions) in frontal, parietal, and fronto-parietal regions, respectively.
The four groups also differed on each of the conventional MRI measures: T2LV (χ2=55.48; df=3, p<0.001); BPF (F=16.37, df=3, 85, p<0.001); GMF (F=13.90, df=3, 85, p<0.001); and WMF (F=6.04, df=3, 85, p=0.001). All MS patients, regardless of subtype, had larger T2LV than controls. The differences in T2LV among subtypes were not significant, although SPMS showed a trend toward larger T2LV than PPMS (p=0.06). When T2LV was estimated from the GSH CSI slab, overall group comparisons showed very similar results to the whole brain T2LV, except that the trend toward larger T2LV in SPMS than PPMS became significant (p=0.01). The planned comparisons applied to BPF, GMF, and WMF yielded similar findings: all patients had smaller values than controls (BPF: p<0.001; GMF: p<0.001; WMF: p=0.002) and progressive MS had smaller values than RRMS (BPF: p<0.001; GMF: p=0.001; WMF: p=0.005). PPMS and SPMS did not differ on any of these measures (BPF: p=0.77; GMF: p=0.85; WMF: p=0.80). RRMS had lower values than controls on GMF (p=0.04), but not on BPF (p=0.07) or WMF (p=0.51). When the analyses were repeated with age and sex entered as covariates, the outcome remained unchanged except that the difference between RRMS and controls on BPF also became significant (p=0.03).
Bivariate parametric or nonparametric correlations between MR variables and the other variables were examined for the full sample of patients. Point bi-serial correlations showed that patients' sex was unrelated to the MR variables, but age was related to frontal GSH (R=-0.28, p=0.03), fronto-parietal GSH (R=-0.23, p=0.08), BPF (R=-0.42, p=0.001), GMF (R=-0.48, p<0.001), and EDSS (rho=0.60, p<0.001). Therefore, all correlations in Table 4 are partial correlations (Rp) controlling for age. To simplify these results, duration of diagnosis was excluded from Table 4 because it was not significantly related to any of the MR variables. GSH concentrations were correlated with both BPF and GMF. Both parietal GSH (Rp=-0.27, p=0.04) and fronto-parietal GSH (Rp=-0.26, p=0.05) were correlated with EDSS, although the zero-order correlation between frontal GSH and EDSS (rho=-0.29, p=0.03) was no longer significant after controlling for age (Rp=-0.19, p=0.15).
Table 4. Relationships between GSH concentrations and other measures.
| frontal GSH | parietal GSH | fronto-parietal GSH | T2LV | BPF | GMF | WMF | ||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| EDSS | Rp | -0.19 | -0.27 | -0.26 | 0.26 | -0.26 | -0.28 | -0.13 |
| p | 0.15 | 0.04 | 0.05 | 0.04 | 0.04 | 0.03 | 0.30 | |
|
| ||||||||
| frontal GSH | Rp | 0.67 | 0.92 | -0.10 | 0.29 | 0.35 | 0.11 | |
| p | <0.001 | <0.001 | 0.44 | 0.02 | 0.01 | 0.41 | ||
|
| ||||||||
| parietal GSH | Rp | 0.91 | -0.17 | 0.29 | 0.31 | 0.14 | ||
| p | <0.001 | 0.18 | 0.03 | 0.02 | 0.30 | |||
|
| ||||||||
| fronto-parietal GSH | Rp | -0.19 | 0.31 | 0.36 | 0.13 | |||
| p | 0.19 | 0.02 | 0.005 | 0.33 | ||||
|
| ||||||||
| T2LV | Rp | -0.50 | -0.52 | -0.28 | ||||
| p | <0.001 | <0.001 | 0.03 | |||||
|
| ||||||||
| BPF | Rp | 0.74 | 0.79 | |||||
| p | <0.001 | <0.001 | ||||||
|
| ||||||||
| GMF | Rp | 0.17 | ||||||
| p | 0.20 | |||||||
Note: Rp: partial correlation controlling for age. Correlations involving T2LV or EDSS are nonparametric partial correlations. Significance is based on 58 degrees of freedom.
Discussion
The results of the between-group comparisons of GSH concentrations were consistent with the study hypotheses. Collectively, patients with MS had lower GSH than controls, although the differences between RRMS and controls were not significant. Furthermore, patients with progressive MS had lower GSH than patients with RRMS. The GSH concentrations for patients with progressive MS, i.e., PPMS and SPMS, did not differ. This pattern supports the notion that oxidative stress, estimated by neuroimaging measures of cerebral GSH, contributes to neurodegeneration primarily in the later, progressive stages of MS.
The results of lower GSH in patients with SPMS were consistent with our previous study, which also found lower GSH concentrations in frontal, parietal, and fronto-parietal regions of patients with SPMS relative to controls.22 In both studies, the disparity between patients and controls was greater for frontal as opposed to parietal GSH concentrations. This consistency attests to the reliability of our measurement of GSH using the selective multiple quantum CSI technique given that these two studies were separated by over 4 years and employed different MR scanners (Allegra vs. Skyra 3 T systems) and RF coils (in-house constructed helmet coil vs. Siemens head coil).
The disparity in outcomes between GSH and conventional MRI measures is noteworthy. While GSH did not differ between RRMS and controls, whole brain and gray matter volumes were lower in RRMS than in controls. These results suggest that neurodegeneration evident in RRMS via brain atrophy is less likely attributable to oxidative stress. In contrast, brain atrophy accompanied by significantly lower GSH in progressive MS may indicate that neurodegeneration in this stage is sustained by mechanisms other than inflammation, such as oxidative stress.
In vivo MRS provides measures of neurochemicals that can serve as specific markers of biochemical and pathophysiologic processes underlying neurological diseases.33-35 At present, GSH appears to be one of the most promising biomarkers for pathogenic shifts in MS. Unlike static biomarkers such as brain atrophy, which is a measure of accumulated neurodegeneration and irreversible upon a reversal of the disease process, GSH provides a dynamic measure of an ongoing process of oxidative stress that likely contributes to neurodegeneration. Dynamic biomarkers are conducive to longitudinal studies, showing the prognostic utility to predict the clinical status of patients, as indicated in our earlier study.24 Dynamic measures of pathologic processes are also conducive to intervention studies, especially those involving treatments targeting the pathology linked to the measure.35 For example, the reduction of oxidative stress has been proposed as one of the factors contributing to the efficacy of dimethyl fumarate. Assessment of cerebral GSH would be a valuable tool for monitoring the treatment effects of such a compound. Only three patients in our study were taking dimethyl fumarate, which was too few to draw any meaningful conclusions. Further studies with an adequate sample size are necessary to examine the effects of dimethyl fumarate on GSH concentrations in the brain. The impact of disease modifying therapies on GSH could not be measured in this study since we could not ethically withhold those therapies from patients with RRMS and the study was not powered to distinguish GSH concentration differences among different disease modifying therapies.
Given its dynamic nature, the lack of longitudinal measures of GSH is a shortcoming of the present study. Without longitudinal data, the study offers little reassurance concerning the degree and severity of ongoing oxidative stress in patients or the implications of changes in GSH for disease progression. Nonetheless, the results highlight the presence of oxidative stress in MS and its prominence in the later, progressive stages of MS. The absence of differences between PPMS and SPMS in all the variables suggests that the mechanisms underlying these two MS subtypes may be the same or very similar. In clinical trials directed at progressive MS, the use of a single group of patients with progressive MS may simplify subject recruitment and facilitate such clinical trials.
Conclusions
In this study, the quantitative assessment of regional distributions of cerebral GSH is demonstrated in three major subtypes of MS using the selective multiple quantum CSI of GSH. We demonstrate markedly lower GSH in the brains of progressive MS (PPMS and SPMS) compared with RRMS, indicating the prominent involvement of oxidative stress in the later, progressive stage of MS relative to the inflammatory stage.
Acknowledgments
The authors thank Mr. Allan Schmidt for his assistance in MR data acquisition and all the study participants. This study was supported in part by the National Multiple Sclerosis Society grant RG4495-A-4 (Lynch) and grants from the NIH (R03AG022193 (IYC), UL1TR000001 (PL), P20GM103418 (PL). The Hoglund Brain Imaging Center is supported by grants from the NIH (S10RR029577) and the Hoglund Family Foundation.
Dr. Belliston is supported by clinical fellowship awards from the National Multiple Sclerosis Society and Biogen.
Dr. Lynch has received research funding from National Multiple Sclerosis Society, and Kansas City Area Life Sciences Institute. She has participated in MS clinical trials sponsored from entities such as NIH, NMSS, Novartis, Biogen, TEVA Neurosciences, Genentech, Pfizer, Roche, Sun Pharma, Vaccinex, Acorda, Actelion, and Genzyme.
Study funding: This study was partly supported by National Multiple Sclerosis Society (RG 4495-A-4) and NIH (R03AG022193, UL1TR000001, P20GM103418). The Hoglund Brain Imaging Center is supported by the NIH (S10RR029577) and the Hoglund Family Foundation.
Footnotes
Declaration of Conflicting Interests: Dr. Choi reports no disclosures.
Dr. Lee reports no disclosures.
Dr. Denney reports no disclosures.
References
- 1.Rojas JI, Romano M, Ciapponi A, Patrucco L, Cristiano E. Interferon beta for primary progressive multiple sclerosis. Cochrane Database Syst Rev. 2009:CD006643. doi: 10.1002/14651858.CD006643.pub2. [DOI] [PubMed] [Google Scholar]
- 2.La Mantia L, Vacchi L, Rovaris M, et al. Interferon beta for secondary progressive multiple sclerosis: a systematic review. J Neurol Neurosurg Psychiatry. 2013;84:420–6. doi: 10.1136/jnnp-2012-303291. [DOI] [PubMed] [Google Scholar]
- 3.Markowitz CE. The current landscape and unmet needs in multiple sclerosis. Am J Manag Care. 2010;16:S211–8. [PubMed] [Google Scholar]
- 4.Ferreira B, Mendes F, Osorio N, Caseiro A, Gabriel A, Valado A. Glutathione in multiple sclerosis. British journal of biomedical science. 2013;70:75–9. doi: 10.1080/09674845.2013.11669939. [DOI] [PubMed] [Google Scholar]
- 5.Ferretti G, Bacchetti T, Principi F, et al. Increased levels of lipid hydroperoxides in plasma of patients with multiple sclerosis: a relationship with paraoxonase activity. Mult Scler. 2005;11:677–82. doi: 10.1191/1352458505ms1240oa. [DOI] [PubMed] [Google Scholar]
- 6.Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251:261–8. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- 7.Gonsette RE. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci. 2008;274:48–53. doi: 10.1016/j.jns.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 8.Rejdak K, Eikelenboom MJ, Petzold A, et al. CSF nitric oxide metabolites are associated with activity and progression of multiple sclerosis. Neurology. 2004;63:1439–45. doi: 10.1212/01.wnl.0000142043.32578.5d. [DOI] [PubMed] [Google Scholar]
- 9.Pasquali L, Pecori C, Lucchesi C, et al. Plasmatic oxidative stress biomarkers in multiple sclerosis: relation with clinical and demographic characteristics. Clinical biochemistry. 2015;48:19–23. doi: 10.1016/j.clinbiochem.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 10.Karlik M, Valkovic P, Hancinova V, Krizova L, Tothova L, Celec P. Markers of oxidative stress in plasma and saliva in patients with multiple sclerosis. Clinical biochemistry. 2015;48:24–8. doi: 10.1016/j.clinbiochem.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 11.Oliveira SR, Kallaur AP, Simao AN, et al. Oxidative stress in multiple sclerosis patients in clinical remission: association with the expanded disability status scale. J Neurol Sci. 2012;321:49–53. doi: 10.1016/j.jns.2012.07.045. [DOI] [PubMed] [Google Scholar]
- 12.Schreibelt G, van Horssen J, van Rossum S, Dijkstra CD, Drukarch B, de Vries HE. Therapeutic potential and biological role of endogenous antioxidant enzymes in multiple sclerosis pathology. Brain research reviews. 2007;56:322–30. doi: 10.1016/j.brainresrev.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Jack C, Antel J, Bruck W, Kuhlmann T. Contrasting potential of nitric oxide and peroxynitrite to mediate oligodendrocyte injury in multiple sclerosis. Glia. 2007;55:926–34. doi: 10.1002/glia.20514. [DOI] [PubMed] [Google Scholar]
- 14.Carlson NG, Rose JW. Antioxidants in multiple sclerosis: do they have a role in therapy? CNS drugs. 2006;20:433–41. doi: 10.2165/00023210-200620060-00001. [DOI] [PubMed] [Google Scholar]
- 15.Oleszak EL, Zaczynska E, Bhattacharjee M, Butunoi C, Legido A, Katsetos CD. Inducible nitric oxide synthase and nitrotyrosine are found in monocytes/macrophages and/or astrocytes in acute, but not in chronic, multiple sclerosis. Clinical and diagnostic laboratory immunology. 1998;5:438–45. doi: 10.1128/cdli.5.4.438-445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holley JE, Newcombe J, Winyard PG, Gutowski NJ. Peroxiredoxin V in multiple sclerosis lesions: predominant expression by astrocytes. Mult Scler. 2007;13:955–61. doi: 10.1177/1352458507078064. [DOI] [PubMed] [Google Scholar]
- 17.Orlowski M, Karkowsky A. Glutathione metabolism and some possible functions of glutathione in the nervous system. Int Rev Neurobiol. 1976;19:75–121. doi: 10.1016/s0074-7742(08)60702-3. [DOI] [PubMed] [Google Scholar]
- 18.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–60. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 19.Tasset I, Aguera E, Sanchez-Lopez F, et al. Peripheral oxidative stress in relapsing-remitting multiple sclerosis. Clinical biochemistry. 2012;45:440–4. doi: 10.1016/j.clinbiochem.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Calabrese V, Raffaele R, Cosentino E, Rizza V. Changes in cerebrospinal fluid levels of malondialdehyde and glutathione reductase activity in multiple sclerosis. International journal of clinical pharmacology research. 1994;14:119–23. [PubMed] [Google Scholar]
- 21.Choi IY, Lee P. Doubly selective multiple quantum chemical shift imaging and T1 relaxation time measurement of glutathione (GSH) in the human brain in vivo. NMR in biomedicine. 2013;26:28–34. doi: 10.1002/nbm.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi IY, Lee SP, Denney DR, Lynch SG. Lower Levels of Glutathione (GSH) in the Brains of Secondary Progressive Multiple Sclerosis Patients Measured by 1H Magnetic Resonance Chemical Shift Imaging at 3 T. Mult Scler. 2011;17:289–96. doi: 10.1177/1352458510384010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srinivasan R, Ratiney H, Hammond-Rosenbluth KE, Pelletier D, Nelson SJ. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn Reson Imaging. 2010;29:163–70. doi: 10.1016/j.mri.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Choi IY, Lee P, Hughes AJ, Denney DR, Lynch SG. Longitudinal changes of cerebral glutathione (GSH) levels associated with the clinical course of disease progression in patients with secondary progressive multiple sclerosis. Mult Scler. 2016 doi: 10.1177/1352458516669441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–52. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 27.Agren H, Niklasson F. Creatinine and creatine in CSF: indices of brain energy metabolism in depression. Short note. J Neural Transm. 1988;74:55–9. doi: 10.1007/BF01243575. [DOI] [PubMed] [Google Scholar]
- 28.Samuelsson M, Skogh E, Lundberg K, Vrethem M, Ollinger K. Taurine and glutathione in plasma and cerebrospinal fluid in olanzapine treated patients with schizophrenia. Psychiatry research. 2013;210:819–24. doi: 10.1016/j.psychres.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–9. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- 30.Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–51. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23:S208–S19. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 32.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage. 2002;17:825–41. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- 33.Bakshi R, Thompson AJ, Rocca MA, et al. MRI in multiple sclerosis: current status and future prospects. Lancet Neurol. 2008;7:615–25. doi: 10.1016/S1474-4422(08)70137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poloni G, Minagar A, Haacke EM, Zivadinov R. Recent developments in imaging of multiple sclerosis. Neurologist. 2011;17:185–204. doi: 10.1097/NRL.0b013e31821a2643. [DOI] [PubMed] [Google Scholar]
- 35.Rocca MA, Messina R, Filippi M. Multiple sclerosis imaging: recent advances. J Neurol. 2013;260:929–35. doi: 10.1007/s00415-012-6788-8. [DOI] [PubMed] [Google Scholar]