

Abstract
Compartmentalization and spatial control of biochemical reactions is the foundation of cell-based life on earth. The lipid bilayer system employed by eukaryote cells not only keeps them separate from the environment but also provides a platform for key receptors to sense and interact with outside factors. Arguably one of the cell types most reliant on interactions of this kind, immune cells depend on their membrane to keep functioning properly. In this review, the influence of variation in cholesterol levels, a key component of lipid bilayer stability, on T cells will be discussed in detail. In comparison to other cells, T cells must be able to undergo rapid activation followed by proliferation. Furthermore, receptor colocalization is an important mechanism in this activation process. The impact of cholesterol availability on the processes of T cell proliferation and receptor sensitivity, as well as its potential for immunomodulation in disease treatment will be considered.
Keywords: cholesterol, metabolism, T cell, disease, immunomodulation
Cholesterol Metabolism
In the cell, cholesterol is largely responsible for being able to maintain a cell membrane with a certain degree of stiffness to it, removing the need for a rigid cell wall (1). This is due to its molecular tetracyclic structure, making it planar and rigid, as well as to the attached hydroxyl group which allows for orientation toward a polar solvent. It also serves as the substrate for a multitude of steroid-based hormones. Since it is so vital to every cell, the involved synthesis and regulatory pathways are nearly ubiquitous across cell populations (1).
Biosynthesis
There are two major ways for a cell to acquire cholesterol: it can scavenge cholesterol from its surroundings (blood stream, etc.) or it can synthesize cholesterol itself. While a sizable amount of cholesterol is indeed ingested by most humans every day via animal tissue, the ratio of scavenged cholesterol vs. synthesized cholesterol favors the internally produced version (2). To preserve homeostasis, a transport mechanism from regions of high cholesterol to regions of low cholesterol, as well as regions where excess cholesterol is disposed of, must exist. Since cholesterol cannot be transported via the blood stream, it is bound up in clusters of lipids and proteins, either in the form of high-density lipoprotein (HDL) or low-density lipoprotein (LDL). Both categories have distinct regulatory mechanisms (2).
In the cell, cholesterol is synthesized from acetyl-coenzyme A, which is soon converted to hydroxymethylglutaryl-coenzyme A (HMG-CoA). This precursor goes through a multistep enzymatic reaction via intermediaries like mevalonate, squalene, and lanosterol until it eventually yields cholesterol. The biosynthesis is very well understood and documented by now. A less well-understood topic is the regulation of the metabolic pathway.
Regulatory Mechanisms
The primary signaling molecule at work in the cholesterol pathway is without a doubt cholesterol itself. Synthesis of more cholesterol is regulated via a feedback mechanism. Exogenous cholesterol therefore also decreases synthesis and vice versa. The main regulatory mechanisms will be showcased based on signaling in either a low-oxysterol or a high-oxysterol state (3).
The key protein for cholesterol sensing is sterol response element-binding protein (SREBP). This protein is situated in the ER membrane, bound to a second protein called SREBP cleavage-activating protein (SCAP). Normally, these two proteins move from the ER and to the Golgi apparatus, where SREBP is processed further. SCAP functions as mediator of this transport (4).
In high-oxysterol situations, however, another protein called insulin-induced gene protein (INSIG) is present in the ER and binds to SCAP. This binding keeps the SREBP–SCAP pair rooted in the ER membrane. Only in a low-oxysterol environment does INSIG disassociate from SCAP, enabling further processing. Once transported to the Golgi apparatus, SREBP is cleaved by site-1 and 2 proteases (S1P and S2P), yielding its active transcription factor (TF) form (5). This TF fragment then migrates to the nucleus and modulates expression of enzymes responsible for the biosynthesis pathway (HMG-CoA synthase, HMG-CoA reductase, farnesyl diphosphate synthase, squalene synthase, etc.), chief among them the rate-limiting enzyme HMG-CoA reductase (1). This manifold expression profile is divided between the different variants of SREBP, such as SREBP1a, SREBP1c, and SREBP2. SREBP1c and SREBP2 can be roughly categorized as regulators of fatty acid metabolism and cholesterol metabolism, respectively (4). In addition, SREBP2 also mediates lipid transport and lipoprotein uptake. SREBP1a targets genes in both categories, also including transport. There is a certain degree of redundancy to the signaling of SREBP1 and 2, and it has previously been demonstrated that this redundancy makes it possible for dysregulation of one component to be compensated by the other.
Conversely, there is a TF limiting intracellular cholesterol levels: liver X receptor. This receptor forms a heterodimer with retinoid X receptor and binds cholesterol (and other oxysterols) as well as 9-cis retinoic acid; hence, it is active in a high-cholesterol environment. There are two isoforms of the receptor, LXRα and LXRβ with distinct expression profiles. While LXRα is primarily expressed in adipocyte and liver tissue, LXRβ is expressed more broadly, with enrichment in immune cells as well as heart and lungs. LXR has many downstream targets, most of them related to lipid metabolism. In reaction to sensing an excess of cholesterol, transferases such as cholesteryl ester transfer protein and ATB-binding cassette transporter A1 or G1 (ABCA1/ABCG1) mediate the efflux of cholesterol into HDL. Excess cholesterol is then transported to the liver either to be eventually excreted or converted to hormones and other molecules such as bile acid (6). The low-density lipoprotein receptor (LDLR) and the cholesterol esterification enzymes SOAT1/2 (alternative name ACAT1/2) also regulate intracellular cholesterol levels by changing the uptake rate or catalyzing conversion of cholesterol into cholesterol esters.
Impact of Cholesterol Metabolism Defects
Recalling the emphasis on the importance of cholesterol to every cell in the body, it is clear, that defects in the system will be reflected in drastic disorder in the body. Cholesterol is implicated in several diseases, particularly those prevalent in modern society such as atherosclerosis and Alzheimer’s disease. While it is true that nutrition is a significant risk factor for many diseases involving cholesterol (particularly atherosclerosis), it is also important not to neglect potential disorders on a molecular level. At the elementary level, all cholesterol (as well as lipid metabolism in general) disorders are categorized either as hyperlipidemia or hypolipidemia, depending on which direction the dysregulation is exhibited (7, 8).
Hypolipidemia
Hypolipidemia is only rarely observed clinically. While this does not necessarily imply low prevalence in the population, it suggests that most cases are asymptomatic (as are those who are discovered in routine lipid screenings). While genetic disorders exist, which directly cause hypolipidemia (abetalipoproteinemia, hypobetalipoproteinemia, and chylomicron retention disease), it is most often observed as a symptom of a mostly unrelated systemic disorder, such as chronic inflammation, acute or chronic infection, undernutrition and hyperthyroidism among others (9).
Hyperlipidemia
Hyperlipidemia, also referred to as hypercholesterolemia or simply “high blood cholesterol,” has shown an increasing incidence as well as prevalence in modern society. It can be classified as either primary (familial) or secondary (acquired), depending on the underlying causes. While the primary variant shows a relatively stable incidence relative to total population size, secondary hyperlipidemia incidence has increased over the course of recent decades (7).
Most cases of primary hyperlipidemia can be traced back to mutations in LDLR, apolipoprotein B, or proprotein convertase subtilisin/kexin type 9 (10, 11). Secondary hyperlipidemia on the other hand is usually associated with a high calorie diet or other metabolic disorders, such as diabetes. The health consequences of hyperlipidemia can potentially be life-threatening. The most commonly known consequence is without a doubt the accumulation of fatty material in the form of xanthomas on the lining of blood vessels. If left unchecked, this can eventually cause blockage of the blood vessel and thus heart attacks or stroke. There are other consequences, however, which are not as obvious. These more insidious malignancies are usually tied to the specific impact of cholesterol on the function of cells. The impact on T cells will be discussed in this review.
Cholesterol Metabolism in T Cells
Considering that cholesterol is important in maintaining cell membrane stiffness, its importance to immune cells, one of the cell types relying the most on motility and membrane–membrane interactions with other cells, is easy to see. Immune cells need to be able to move from tissue to tissue, need to be able to form a synapse with other cells, and sometimes even engulf target cells. While cholesterol is thus important for all subsets of immune cells, its importance to T cells will be examined here.
Since cholesterol can also be considered a kind of nutrient for the cell, it is first important to talk about how T cells manage their nutrients. Usually, a cell would derive energy from glycolysis, and oxidative phosphorylation, depending on how much oxygen is available to them. In highly proliferative cells, however, a much higher priority is placed on glycolysis (12, 13). One can observe this not only in proliferating T cells but also as the Warburg effect in cancer cells. Since proliferation requires lipids for membrane synthesis, fatty acid catabolic pathways are mostly suppressed in proliferative cells. In contrast, the mechanisms of fatty acid biosynthesis as well as cholesterol synthesis are strongly upregulated in T-cells (13).
Results regarding the cholesterol metabolism in different T cell subsets have so far been inconclusive. In one study (14), higher levels of GM1 were measured in thymic CD8+ single positive T cells when compared to CD4+ single positive T cells and CD4+CD8+ double positive T cells. Splenic CD8+ T cells had higher levels of GM1 and total cholesterol than CD4+ T cells. No obvious difference of GM1 expression between naive and memory T cells was noted.
Impact of Cholesterol on T Cell Activation and Proliferation
Considering that T cell activation generally triggers a large-scale proliferation of the activated cells, anabolic pathways are critical during this process of activation. This has been well established in cancer cells, where high proliferation is the primary defining characteristic (15, 16). Treating a population of naïve T cells with Statin suppresses progression of their differentiation and cell cycle. Statin is an inhibitor of HMG-CoA reductase, the enzyme responsible for catalysis in the rate-limiting step of cholesterol biosynthesis. Furthermore, inhibition of this enzyme leads to decreased rates of DNA synthesis in many highly proliferative cell populations, such as lymphocytes, fibroblasts, and PDGF-treated smooth-muscle cells (17, 18). These downstream effects of statin treatment play a role in stagnating T cell activation. It is important to note, that not all statin-mediated changes in the cell are inherently tied to cholesterol levels. As statins inhibitory target is further upstream from cholesterol synthesis, cholesterol-independent effects may be at work as well.
Once a T cell receives an activation signal, sulfotransferase 2B1 is upregulated to reduce oxysterol level, SREBP processing is promoted, and LXR signaling is suppressed (19). Induction of LXR signaling leads to stagnating proliferation, while a knockout confers a proliferative advantage. This link of LXR signaling to proliferation is directly related to cholesterol transport, as inactivation of the transporter proteins (e.g., ABCG1) prevents the effect of LXR induction. This showcases the importance of LXR as a metabolic checkpoint in T cell activation and immune cell function in general (19–21). SREBP has been shown to be indispensable for CD8+ T cell activation and proliferation as well (21). Metabolism checkpoints for T cell activation and proliferation can be found in many of the subset populations and include not only lipid metabolism but also glycolysis. mTOR and TRAF6, for example, have been shown to impact the differentiation of CD8+ T cells via their downstream targets, regulating cell metabolism (22–26). Other important regulatory proteins, such as Myc and AMPK, have been implicated as well (27–29).
There is some evidence to suggest that the role of lipid metabolism may not be as simple as a binary toggle for proliferation. Since the rate of de novo fatty acid synthesis can impact differentiation either to Th17 or Treg cells (30), a complex underlying regulatory system is implied.
The importance of understanding this relationship becomes clear when looking at cancer cases. The inability of T cells to cope with a developing tumor is aggravated as the energy consuming tumor decreases available nutrients in the body. If it reaches a point where blood LDL and HDL levels drop, even initial T cell activation can be compromised (31).
Exogenous cholesterol levels can shift the T cell population balance on the level of an entire organ in the body, as was observed in increased Treg differentiation in hypercholesterolemic conditions in the liver (32).
Lipid Rafts, Membrane Dynamics, and Nanoclustering
Since the membrane is mostly made up of lipids, their dynamics impact the function of embedded proteins (33–35). Changing the charge of acidic phospholipids, for example, can directly alter TCR and CD28 activation (36–38).
These findings suggest one of the primary mechanisms by which cholesterol can change T cell activation: via changing dynamics of lipid rafts and the membrane in general and therefore increasing or decreasing the colocalization of crucial receptors. Lipid rafts can be categorized as heterogenous regions of lipid distribution across the membrane, which are distinct in their composition and fluidity. They represent one of the corralling mechanisms active in the cell membrane, since they allow for spatial control of membrane-associated proteins. The importance of lipid rafts, as well as their dependence on cholesterol concentration is well known (39–42). The immunological synapse has been considered a physiological form of a “lipid raft” (43). Using Miltefosine to disrupt normal lipid raft dynamics impacts T cell proliferation immensely, showing an over 50% reduced rate of proliferation.
Spatial control over receptors is especially crucial for receptors reliant on colocalization to achieve their active conformation (44). These receptors need to bind partner molecules and stay in stable association to function. Introducing a factor which promotes either the bound or unbound state modulates the overall sensitivity of the receptor. This kind of control over receptor clustering was shown to modulate sensitivity independent of the associated ligand (45). Maintaining precise spatial control over receptor nanoclustering partially determines sensitivity to external stimuli, as is the case for CD4 and the TCR (46). Lipids in general (and cholesterol in particular) can modulate receptor signaling via promotion of various conformational states of membrane receptors (37).
Not only colocalization of receptors on the membrane at large is implicated in this mechanism. Membrane fluidity and spatial control of receptors is crucial in maintaining one of the main avenues of communication for immune cells: the immunological synapse (47), since many of the involved proteins have to remain in close association.
Cholesterol As Signaling Molecule in T Cells
Apart from modifying membrane dynamics, cholesterol influences cellular signaling as direct ligand as well. Synthetic agonists to the LXR receptor family were shown to mediate an anti-inflammatory response in macrophages and other cells of the immune system (48, 49). The importance of these regulators of lipid metabolism to the innate immune system in general has been shown as well (50–53).
There are also cases, where it is still unclear whether the involved mechanism is related to direct cholesterol binding, or whether it is a downstream effect of the previously discussed modulation of membrane dynamics. Administration of squalene, a cholesterol precursor, was shown to increase the resting population of CD4 T helper cells as well as predispose T cells toward higher inflammatory reactivity (54) and direct depletion of cholesterol in the growth medium of T cells as well as diet of mice led to reduced rates of T cell activation and proliferation (55). It has been previously shown that rapidly dividing cell populations react to a higher demand for cholesterol with increased scavenging of serum LDL mediated via an increase of the responsible receptors (56).
Recently, the direct ligand role has gained some more momentum, however. Actual direct cholesterol binding modifies TCR via allosteric signaling (57, 58) and a direct binding motif for cholesterol has been discovered and is present in a number of membrane proteins, such as the metabotropic glutamate receptor, adenosine receptor A1, and GABA type B receptor (59). Cholesteryl esters play a role as conformational stabilizers (or cofactor) in CD1c, allowing self-reactive T cells to bind the protein (60). Cholesterol sulfate (structurally similar to cholesterol) can disrupt TCR multimers, effectively inhibiting signal transduction. This most likely occurs due to displacing receptor bound cholesterol (61) (Figure 1).
Figure 1.

Cholesterol and metabolite regulate T cell receptor clustering. Cholesterol can directly bind to the transmembrane domain of TCR-β chain to mediate TCR clustering on T cell surface, which can increase the avidity of TCR to foreign antigens and therefore augment TCR signaling. Meanwhile, cholesterol can keep TCR at an inactive conformation to avoid spontaneous signaling. Another safety control mechanism of TCR activity is the sequestration of TCR signaling motifs within the membrane bilayer via the ionic interactions between acidic phospholipids and polybasic regions in CD3 cytoplasmic domains. Cholesterol sulfate, a negatively charged cholesterol metabolite, however, can disrupt TCR clustering via the interference of cholesterol–TCR interaction. The underlying mechanism remains to be further illustrated.
Modulating T Cell Function via the Cholesterol Pathway
Based on the demonstrated importance of cholesterol and its related regulatory proteins, it is easy to see the potential of some of these components as treatment targets. Potential applications as well as methods for making use of them will be discussed in the following.
Tools for Modulation
Statins are, of course, the most obvious and ubiquitous choice of drug intervention for modulating cholesterol metabolism. They have been widely used in atherosclerosis treatment for many years. While their actual effect has come under increasing scrutiny in recent years (62), statins remain an effective HMG-CoA reductase inhibitor. Atorvastatin can normalize T cell signaling and reduce the production of IL-10 and IL 6 by inhibiting cholesterol biosynthesis and reducing cholesterol levels in the T cell membrane (42). As a cholesterol-depleting reagent, methyl-β-cyclodextrin is used to disrupt lipid rafts and prevent their clustering, thus abrogating the heightened proximal TCR signaling and reversing the hyperactivity of T cells (63, 64). Synthetic agonists specific to a particular subset of the LXR receptor family have already demonstrated their effectiveness (65). By now, many drugs which target LXR specifically are available (66). The impact on T cells has also been shown with decreased cell proliferation and colony formation rates (67). Treatment options also include the precursors of cholesterol in the biosynthesis pathway, some of which have more favorable qualities in terms of bioavailability. Squalene treatment increases resting CD4 T cell population and desmosterol functions as an effective LXR agonist and SREBP repressor (54, 68).
As most signaling of T cells is mediated via surface proteins, these proteins form a very attractive target for modulation. There have already been studies trying to modulate T cell function via these receptors (69) in traditional, antibody-based ways. Modulating the receptor clustering to increase receptor sensitivity, however, is a promising approach for modulation without the need of expensive antibodies or other vehicles. For example, small molecule-mediated inhibition of ACAT1 can cause better TCR clustering in CD8 T cells (44).
Potential Treatment Applications
Metabolism has long been a target for cancer treatment (70). Usually, the treatments target the metabolism of cancer cells to deprive them of nutrients. Targeting the metabolism of antitumor immune cells instead may provide new treatment options. It is clear that immune cells undergo significant changes in the tumor microenvironment. Some of these changes may be mediated by metabolism signaling (71, 72). ACAT1 inhibition, leading to higher levels of plasma membrane cholesterol, induces higher antitumor activity in CD8 T cells (44) via enhanced receptor clustering on the cell surface. Under the same principle, modulation of cholesterol levels can decrease colocalization of crucial receptors in autoimmune diseases such as systemic lupus erythematosus (42). As such, it could potentially take the role of a sliding scale for sensitivity of T cells in general. Whether it is applicable in other diseases, such as long-term treatment of atherosclerosis via atorvastatin to inhibit the inflammatory response of CD4 T cells remains controversial (73, 74).
Using targets in metabolism for cell modulation always carries a risk of off-target activity. Interestingly, Cre-lox-based knockout of SCAP (and therefore SREBP1 and 2) led to asymptomatic phenotypes in the observed T cell populations, as long as they remained in a quiescent state. This suggests that quiescent cells could remain largely untouched from modifying these response elements (21).
As has been demonstrated in numerous studies now, cholesterol is important in the activation and proliferation of T cells. From this conclusion, the thought of utilizing this for treatment of diseases arises. Initial studies examining the effect of modulating cholesterol levels on cancer and autoimmune diseases already show promising results (Figure 2). Counteracting the immunosuppressive tumor microenvironment, for example, could be an important aspect of all cancer therapies, potentially raising their chances of success across the board.
Figure 2.
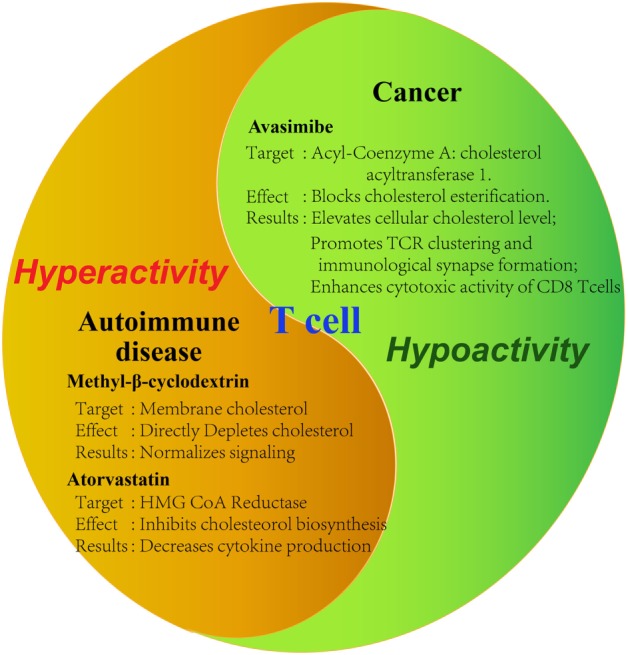
Tools for modulating cholesterol metabolism of T cells. Summary of demonstrated tools for the modulation of T cell activity by targeting cholesterol metabolism in disease contexts. Tumor-infiltrating T cells are characteristic of hypoactivity, whereas auto-reactive T cells are of hyperactivity. Therefore, different strategies should be applied to modulate cholesterol metabolism in different disease contexts. Specific reagents as well as the observed effects on the cell are summarized. Avasimibe: inhibitor of acyl-coenzyme A: cholesterol acyltransferase; GW3865: agonist of liver X receptor; methyl-β-cyclodextrin: strong cholesterol binder that can deplete membrane cholesterol; atorvastatin: inhibitor of hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase.
Conclusion and Perspectives
In recent years, the physiological and pathological importance of cholesterol metabolism in T cell immunity starts to be revealed, but we are still at the early stage to fully understand its roles and regulatory mechanisms. Modulating cholesterol metabolism appears to be a promising therapeutic direction to harness T cell activity in cancer and autoimmunity. Several key questions remain to be answered in the future: 1. What are the function of various cholesterol metabolites in T cells? 2. Why do different T cell subsets have different cholesterol metabolisms and what is the functional importance of this difference? 3. In different disease contexts, how does local environment affect cholesterol metabolism of T cells and how can this process be modified to rescue T cell activity? Answering these questions can eventually lead to the development of new cholesterol-based immunotherapies against various human diseases.
Author Contributions
CX conceived the writing. AB wrote the first version. HZ revised the paper and made the figures. MX contributed to figure discussion.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Wei Wu for thoughtful discussion. CX is funded by NSFC grants (No. 31370860, No. 31425009, No. 31530022, and No. 31621003), and CAS grants (Strategic Priority Research Program No. XDB08020100).
References
- 1.Myant NB. Cholesterol metabolism. J Clin Pathol Suppl (Assoc Clin Pathol) (1973) 5:1–4. 10.1136/jcp.s1-5.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox RA, Garcia-Palmieri MR. Cholesterol, triglycerides, and associated lipoproteins. In: Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. Boston: Butterworth; (1990). p. 154–6. [Google Scholar]
- 3.Olkkonen VM, Beaslas O, Nissila E. Oxysterols and their cellular effectors. Biomolecules (2012) 2:76–103. 10.3390/biom2010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie (2004) 86:839–48. 10.1016/j.biochi.2004.09.018 [DOI] [PubMed] [Google Scholar]
- 5.Shao W, Espenshade PJ. Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). J Biol Chem (2014) 289:7547–57. 10.1074/jbc.M113.545699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li T, Chiang JY. Regulation of bile acid and cholesterol metabolism by PPARs. PPAR Res (2009) 2009:501739. 10.1155/2009/501739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson RH. Hyperlipidemia as a risk factor for cardiovascular disease. Prim Care (2013) 40:195–211. 10.1016/j.pop.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minicocci I, Santini S, Cantisani V, Stitziel N, Kathiresan S, Arroyo JA, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res (2013) 54:3481–90. 10.1194/jlr.P039875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elmehdawi R. Hypolipidemia: a word of caution. Libyan J Med (2008) 3:84–90. 10.4176/071221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lye SH, Chahil JK, Bagali P, Alex L, Vadivelu J, Ahmad WA, et al. Genetic polymorphisms in LDLR, APOB, PCSK9 and other lipid related genes associated with familial hypercholesterolemia in Malaysia. PLoS One (2013) 8:e60729. 10.1371/journal.pone.0060729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher WR, Zech LA, Stacpoole PW. ApoB metabolism in familial hypercholesterolemia. Inconsistencies with the LDL receptor paradigm. Arterioscler Thromb (1994) 14:501–10. 10.1161/01.ATV.14.4.501 [DOI] [PubMed] [Google Scholar]
- 12.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell (2017) 169:570–86. 10.1016/j.cell.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5:844–52. 10.1038/nri1710 [DOI] [PubMed] [Google Scholar]
- 14.de Mello Coelho V, Nguyen D, Giri B, Bunbury A, Schaffer E, Taub DD. Quantitative differences in lipid raft components between murine CD4+ and CD8+ T cells. BMC Immunol (2004) 5:2. 10.1186/1471-2172-5-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148:244–58. 10.1016/j.cell.2011.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal (2009) 2:ra82. 10.1126/scisignal.2000446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakrabarti R, Engleman EG. Interrelationships between mevalonate metabolism and the mitogenic signaling pathway in T lymphocyte proliferation. J Biol Chem (1991) 266:12216–22. [PubMed] [Google Scholar]
- 18.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. 10.1146/annurev-immunol-032712-095956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell (2008) 134:97–111. 10.1016/j.cell.2008.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bischoff ED, Daige CL, Petrowski M, Dedman H, Pattison J, Juliano J, et al. Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J Lipid Res (2010) 51:900–6. 10.1194/jlr.M900096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14:489–99. 10.1038/ni.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. 10.1038/nature08097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. 10.1038/nature08155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356:513–9. 10.1126/science.aal3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci (2013) 126:1713–9. 10.1242/jcs.125773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168:960–76. 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15. 10.1038/ni.2386 [DOI] [PubMed] [Google Scholar]
- 28.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42:41–54. 10.1016/j.immuni.2014.12.030 [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. 10.1016/j.immuni.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20:1327–33. 10.1038/nm.3704 [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Chi PD, Chen H, Xiang J, Xia ZJ, Zhang YJ. Low level of high-density lipoprotein cholesterol correlates with poor prognosis in extranodal natural killer/T cell lymphoma. Tumour Biol (2014) 35:2141–9. 10.1007/s13277-013-1284-z [DOI] [PubMed] [Google Scholar]
- 32.Mailer RKW, Gistera A, Polyzos KA, Ketelhuth DFJ, Hansson GK. Hypercholesterolemia induces differentiation of regulatory T cells in the liver. Circ Res (2017) 120:1740–53. 10.1161/CIRCRESAHA.116.310054 [DOI] [PubMed] [Google Scholar]
- 33.Wu W, Yan C, Shi X, Li L, Liu W, Xu C. Lipid in T-cell receptor transmembrane signaling. Prog Biophys Mol Biol (2015) 118:130–8. 10.1016/j.pbiomolbio.2015.04.004 [DOI] [PubMed] [Google Scholar]
- 34.Wu W, Shi X, Xu C. Regulation of T cell signalling by membrane lipids. Nat Rev Immunol (2016) 16:690–701. 10.1038/nri.2016.103 [DOI] [PubMed] [Google Scholar]
- 35.Li LY, Shi XS, Guo XD, Li H, Xu CQ. Ionic protein lipid interaction at the plasma membrane: what can the charge do? Trends Biochem Sci (2014) 39:130–40. 10.1016/j.tibs.2014.01.002 [DOI] [PubMed] [Google Scholar]
- 36.Shi X, Bi Y, Yang W, Guo X, Jiang Y, Wan C, et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature (2013) 493:111–5. 10.1038/nature11699 [DOI] [PubMed] [Google Scholar]
- 37.Guo X, Yan C, Li H, Huang W, Shi X, Huang M, et al. Lipid-dependent conformational dynamics underlie the functional versatility of T-cell receptor. Cell Res (2017) 27:505–25. 10.1038/cr.2017.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Guo X, Shi X, Li C, Wu W, Yan C, et al. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc Natl Acad Sci U S A (2017) 114:E5891–9. 10.1073/pnas.1701990114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honigmann A, Mueller V, Ta H, Schoenle A, Sezgin E, Hell SW, et al. Scanning STED-FCS reveals spatiotemporal heterogeneity of lipid interaction in the plasma membrane of living cells. Nat Commun (2014) 5:5412. 10.1038/ncomms6412 [DOI] [PubMed] [Google Scholar]
- 40.Hou TY, McMurray DN, Chapkin RS. Omega-3 fatty acids, lipid rafts, and T cell signaling. Eur J Pharmacol (2016) 785:2–9. 10.1016/j.ejphar.2015.03.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang H, Badralmaa Y, Yang J, Lempicki R, Hazen A, Natarajan V. Retinoic acid and liver X receptor agonist synergistically inhibit HIV infection in CD4+ T cells by up-regulating ABCA1-mediated cholesterol efflux. Lipids Health Dis (2012) 11:69. 10.1186/1476-511X-11-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jury EC, Isenberg DA, Mauri C, Ehrenstein MR. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J Immunol (2006) 177:7416–22. 10.4049/jimmunol.177.10.7416 [DOI] [PubMed] [Google Scholar]
- 43.Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol (2010) 11:688–99. 10.1038/nrm2977 [DOI] [PubMed] [Google Scholar]
- 44.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531:651–5. 10.1038/nature17412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molnár E, Swamy M, Holzer M, Beck-García K, Worch R, Thiele C, et al. Cholesterol and sphingomyelin drive ligand-independent T-cell antigen receptor nanoclustering. J Biol Chem (2012) 287:42664–74. 10.1074/jbc.M112.386045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roh KH, Lillemeier BF, Wang F, Davis MM. The coreceptor CD4 is expressed in distinct nanoclusters and does not colocalize with T-cell receptor and active protein tyrosine kinase p56lck. Proc Natl Acad Sci U S A (2015) 112:E1604–13. 10.1073/pnas.1503532112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dustin ML. The immunological synapse. Cancer Immunol Res (2014) 2:1023–33. 10.1158/2326-6066.CIR-14-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10:365–76. 10.1038/nri2748 [DOI] [PubMed] [Google Scholar]
- 49.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med (2003) 9:213–9. 10.1038/nm820 [DOI] [PubMed] [Google Scholar]
- 50.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell (2004) 119:299–309. 10.1016/j.cell.2004.09.032 [DOI] [PubMed] [Google Scholar]
- 51.Köberlin MS, Snijder B, Heinz LX, Baumann CL, Fauster A, Vladimer GI, et al. A conserved circular network of coregulated lipids modulates innate immune responses. Cell (2015) 162:170–83. 10.1016/j.cell.2015.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell (2012) 151:138–52. 10.1016/j.cell.2012.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell (2015) 161:459–69. 10.1016/j.cell.2015.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Surls J, Nazarov-Stoica C, Kehl M, Olsen C, Casares S, Brumeanu TD. Increased membrane cholesterol in lymphocytes diverts T-cells toward an inflammatory response. PLoS One (2012) 7:e38733. 10.1371/journal.pone.0038733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chyu KY, Lio WM, Dimayuga PC, Zhou J, Zhao X, Yano J, et al. Cholesterol lowering modulates T cell function in vivo and in vitro. PLoS One (2014) 9:e92095. 10.1371/journal.pone.0092095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho YK, Faust JR, Bilheimer DW, Brown MS, Goldstein JL. Regulation of cholesterol synthesis by low density lipoprotein in isolated human lymphocytes. Comparison of cells from normal subjects and patients with homozygous familial hypercholesterolemia and abetalipoproteinemia. J Exp Med (1977) 145:1531–49. 10.1084/jem.145.6.1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schamel WW, Alarcon B, Hofer T, Minguet S. The allostery model of TCR regulation. J Immunol (2017) 198:47–52. 10.4049/jimmunol.1601661 [DOI] [PubMed] [Google Scholar]
- 58.Swamy M, Beck-Garcia K, Beck-Garcia E, Hartl FA, Morath A, Yousefi OS, et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity (2016) 44:1091–101. 10.1016/j.immuni.2016.04.011 [DOI] [PubMed] [Google Scholar]
- 59.Fantini J, Di Scala C, Evans LS, Williamson PT, Barrantes FJ. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci Rep (2016) 6:21907. 10.1038/srep21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mansour S, Tocheva AS, Cave-Ayland C, Machelett MM, Sander B, Lissin NM, et al. Cholesteryl esters stabilize human CD1c conformations for recognition by self-reactive T cells. Proc Natl Acad Sci U S A (2016) 113:E1266–75. 10.1073/pnas.1519246113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang F, Beck-Garcia K, Zorzin C, Schamel WW, Davis MM. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol (2016) 17:844–50. 10.1038/ni.3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langsjoen PH, Langsjoen AM. The clinical use of HMG CoA-reductase inhibitors and the associated depletion of coenzyme Q10. A review of animal and human publications. Biofactors (2003) 18:101–11. 10.1002/biof.5520180212 [DOI] [PubMed] [Google Scholar]
- 63.Krishnan S, Nambiar MP, Warke VG, Fisher CU, Mitchell J, Delaney N, et al. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J Immunol (2004) 172:7821–31. 10.4049/jimmunol.172.12.7821 [DOI] [PubMed] [Google Scholar]
- 64.Deng GM, Tsokos GC. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J Immunol (2008) 181:4019–26. 10.4049/jimmunol.181.6.4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem (2002) 45:1963–6. 10.1021/jm0255116 [DOI] [PubMed] [Google Scholar]
- 66.Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov (2014) 13:433–44. 10.1038/nrd4280 [DOI] [PubMed] [Google Scholar]
- 67.Zhang R, Liu Z, Li Y, Wu B. LXR agonist regulates the proliferation and apoptosis of human T-cell acute lymphoblastic leukemia cells via the SOCS3 pathway. Int J Biochem Cell Biol (2016) 78:180–5. 10.1016/j.biocel.2016.07.007 [DOI] [PubMed] [Google Scholar]
- 68.Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem (2006) 281:27816–26. 10.1074/jbc.M603781200 [DOI] [PubMed] [Google Scholar]
- 69.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov (2013) 12:130–46. 10.1038/nrd3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov (2013) 12:829–46. 10.1038/nrd4145 [DOI] [PubMed] [Google Scholar]
- 71.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis (2015) 6:e1792. 10.1038/cddis.2015.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43:435–49. 10.1016/j.immuni.2015.09.001 [DOI] [PubMed] [Google Scholar]
- 73.DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol (2016) 16:149–63. 10.1038/nri.2015.18 [DOI] [PubMed] [Google Scholar]
- 74.Guasti L, Maresca AM, Schembri L, Rasini E, Dentali F, Squizzato A, et al. Relationship between regulatory T cells subsets and lipid profile in dyslipidemic patients: a longitudinal study during atorvastatin treatment. BMC Cardiovasc Disord (2016) 16:26. 10.1186/s12872-016-0201-y [DOI] [PMC free article] [PubMed] [Google Scholar]