

Abstract
Background
Occupational vinyl chloride (VC) exposures have been associated with toxicant-associated steatohepatitis and liver cancer. Metabolomics has been used to clarify mode of action in drug-induced liver injury but has not been performed following VC exposures.
Methods
Plasma samples from 17 highly exposed VC workers without liver cancer and 27 unexposed healthy volunteers were obtained for metabolite extraction and GC/MS and LC/MS2 analysis. Following ion identification/quantification, Ingenuity pathway analysis was performed.
Results
613 unique named metabolites were identified. Of these, 189 metabolites were increased in the VC exposure group while 94 metabolites were decreased. Random Forest analysis indicated that the metabolite signature could separate the groups with 94% accuracy. VC exposures were associated with increased long chain (including arachidonic acid) and essential (including linoleic acid) fatty acids. Occupational exposure increased lipid peroxidation products including monohydroxy fatty acids (including 13-HODE); fatty acid dicarboxylates; and oxidized arachidonic acid products (including 5,9, and 15-HETE). Carnitine and carnitine esters were decreased, suggesting peroxisomal/mitochondrial dysfunction and alternate modes of lipid oxidation. Differentially regulated metabolites were shown to interact with extracellular-signal-regulated kinase 1/2 (ERK1/2), Akt, AMP-activated protein kinase (AMPK), and the N-Methyl-D-aspartate (NMDA) receptor. The top canonical pathways affected by occupational exposure included tRNA charging, nucleotide degradation, amino acid synthesis/degradation and urea cycle. Methionine and homocysteine was increased with decreased cysteine, suggesting altered 1-carbon metabolism.
Conclusions
Occupational exposure generated a distinct plasma metabolome with markedly altered lipid and amino acid metabolites. ERK1/2, Akt, AMPK, and NMDA were identified as protein targets for vinyl chloride toxicity.
Keywords: Vinyl Chloride, Toxicant Associated Steatohepatitis, TASH, Metabolomics, Hemangiosarcoma, Occupational Health
1. Introduction
Vinyl chloride (VC) is an odorless, colorless gas polymerized to produce the ubiquitous plastic polyvinyl chloride (PVC). VC is associated with several pathologies. VC exposure is generally via inhalation in an occupational setting. Over 80,000 chemical workers have been exposed to vinyl chloride. VC may enter the environment via airborne industrial emissions, but may also leech into ground water as a solvent degradation product thus exposing surrounding populations (Kielhorn et al., 2000; Anon., 2015). The Agency for Toxic Substances & Diseases Registry lists VC as #4 on its Substance Priority List, a list prioritized by a combination of a substance’s frequency, toxicity, and potential for human exposure at National Priorities List sites (Anon., n.d.). Occupational VC exposure was first associated with the development of hepatic hemangiosarcoma at a Louisville, KY B.F. Goodrich Plant in 1974 (Makk et al., 1974). In response to this outbreak, a large occupational hepatology database and specimen bank was formed at the University of Louisville as part of a cancer surveillance project. More recently, we reported an 80% prevalence of toxicant-associated steatohepatitis (TASH) in highly exposed VC workers from this plant (Cave et al., 2010). Although those workers had much higher cumulative exposures than allowed under the Occupational Safety and Health Administration Vinyl Chloride Standard, a more recent ultrasound study of 347 asymptomatic current PVC workers reported a 38.9% prevalence of steatosis with a normal mean body mass index (Hsiao et al., 2004). This suggests TASH is a relevant health issue which may also occur in individuals with much lower level exposures to vinyl chloride monomer. The mechanisms leading to the disruption of hepatic lipid metabolism and TASH have recently been reviewed (Wahlang et al., 2013; Joshi-Barve et al., 2015). TASH appears mechanistically different from obesity or alcohol induced steatohepatitis. The metabolic disruption resulting from known VC exposure has never been characterized.
Metabolomics is the study of low molecular weight molecules (i.e., metabolites) found within cells and biological systems. With the advantages of high sensitivity and accuracy, wide dynamic range, and the ability to identify metabolites from complex samples, high-resolution mass spectrometry is an attractive option for metabolomics research. Metabolomics has been utilized for the evaluation of mechanistic disease processes and biomarker development of non-alcoholic fatty liver disease, non-alcoholic steatohepatitis (Kalhan et al., 2011; Puri et al., 2009), alcoholic steatohepatitis (Raszeja-Wyszomirska et al., 2012), and drug-induced liver injury (O’Connell and Watkins, 2010), but has not been performed for industrial chemical exposure, which is the purpose of this study. The goals of the present study are to describe the alterations of the plasma metabolome following chemical exposure at a polyvinyl chloride production facility compared to healthy controls. The changes in these metabolites could contribute to adverse health outcomes in chemical workers.
2. Methods
2.1. Subjects
De-identified data and plasma samples were obtained for highly exposed vinyl chloride workers (n = 17) from the University of Louisville Occupational Toxicology Specimen Bank. Samples were stored at − 80 °C in polypropylene microfuge tubes. They were thawed, aliquoted, and sent on dry ice to Metabolon for analysis. There were no other freeze-thaw cycles. These workers were employed at a single B.F. Good-rich plant in Louisville, KY, and were selected (inclusion criteria) based on exceptionally high cumulative VC exposures prior to implementation of the Occupational Safety and Health Administration (OSHA) Vinyl Chloride Standard in 1975; however, workers were also simultaneously exposed to a variety of other chemicals (Supplemental Table) in addition to VC. These workers came from the same cohort of “poly cleaners” that developed hepatic hemangiosarcoma (n = 26 to date) and toxicant-associated steatohepatitis (TASH)(Cave et al., 2010). These 17 workers never developed hepatic hemangiosarcoma during a prolonged follow-up period - now approximately 40 years in duration. Their TASH status was unknown. Cumulative VC exposures were quantified by the previously described cumulative exposure rank month (CERM) (Greenberg and Tamburro, 1981). Subjects were selected so that their CERMS matched as closely as possible to workers with hemangiosarcoma to allow for future comparison. By doing so, we hoped to gain mechanistic metabolic information about high-level exposures previously linked to liver disease. Unfortunately, healthy unexposed controls with a similar storage time were not available from the biorepository; therefore, 27 unexposed volunteers without a history of liver disease matched by age and gender as closely as possible to the chemical exposed group were recruited as controls. In order to obtain a relatively homogenous population (apart from exposure) for metabolomics analysis, the following exclusion criteria were used for all groups: age > 65 years old, body mass index (BMI) > 35 kg/m2, alcohol consumption >30 g per day, and history of malignancy. The protocol was approved by the University of Louisville Institutional Review Board and informed consent was obtained. Potential differences in demographics and exposure variables were determined by student’s t-test using GraphPad Prism 6 (La Jolla, CA). Statistical significance was set at p ≤ 0.05.
2.2. Metabolomics
Metabolon, Inc. (Durham, NC) performed gas chromatography/mass-spectrometry (GC/MS) (Thermo-Finnigan Trace DSQ fast-scanning single-quadruple mass spectrometer) and liquid chromatography-tandem mass spectrometry LC/MS/MS (Waters ACQUITY UPLC, Thermo-Finnigan LTQ mass spectrometer) following metabolite extraction. Software was used to match ions to a library of standards for metabolite identification and for metabolite quantitation by peak area integration. Statistical significance was determined using Welch’s t-tests, and p ≤ 0.05 was considered statistically significant.
2.3. Liver injury assessment
Plasma cytokeratin 18 (CK-18) whole (M65®) and caspase-cleaved fragments (M30 Apoptosense®) were measured by ELISA (diaPharma, Columbus, OH). Clinical chemistry analysis was performed at the University of Louisville Hospital laboratory. Values were measured by clinical chemistry analyzer. Statistical analysis was performed by student’s t-test using GraphPad Prism. Statistical significance was set as p ≤ 0.05. Results are reported ad means ± standard deviation.
2.4. Ingenuity pathway analysis
We investigated interactions between metabolites using the Ingenuity Pathway Analysis (IPA) software (QIAGEN; Redwood City, CA) and found 354 of our 613 metabolites could be mapped using the Human Metabolomic Database (HMDB).
3. Results
3.1. Demographics
The mean age of the exposed group (50 ± 6.3, n = 17) was significantly higher (p = 0.0002) than the mean age of the control group (38 ± 12, n = 26). All members of the exposed group were Caucasian males and this is reflective of plant hiring practices at the time. At the time of metabolomics analysis, sample storage age in years was significantly longer for exposed vs. unexposed group (35 ± 5.1 vs. 0.18 ± 0.0 years, p < 0.0001). Mean BMI (27 ± 3.4 vs. 26 ± 4.3 kg/m2, p = 0.46) was similar between VC-exposed and control groups. Alcohol consumption (5.0 ± 5.9 vs. 2.2 ± 2.5 drinks per week, p = 0.0342) was significantly higher in exposed group; however, their alcohol consumption was still less than one drink per day on average. There were 6 smokers in VC-exposed group and 1 smoker in the control group. VC exposures were determined by cumulative exposure rank month (CERM). The mean CERM was 1156 ± 420 and this corresponds to a mean duration of employment and high-level VC exposures (often exceeding 1000 PPM) of 25.50 ± 5.688 years. The demographic data are presented in Table 1. Results are reported as mean ± standard deviation.
Table 1.
Demographic information of control versus VC-exposed individuals.
| Control | VC-Exposed | |
|---|---|---|
| Number | 27 | 17 |
| Age (years) | 38 ± 12 | 50 ± 6.3a |
| BMI (kg/m2) | 26 ± 4.3 | 27 ± 3.4 |
| Alcohol consumption (drinks per week) | 2.2 ± 2.5 | 5.0 ± 5.9b |
| Duration of employment at time of sampling (years) | 0 | 25.50 ± 5.688 |
| CERM (cumulative exposure rank month) | 0 | 1156 ± 420 |
p = 0.0002 compared to control.
p < 0.05 compared to control.
3.2. Liver injury assessment
Routine liver chemistry results are demonstrated in Table 2. There were no significant differences between the groups in albumin (4.6 ± 2.3 vs. 4.5 ± 3.8 g/dL Reference: 3.5–5.0), total bilirubin (0.58 ± 0.25 vs. 0.57 ± 0.72 mg/dL Reference: 0.2–1.0), and ALT (23 ± 19 vs. 22 ± 3.6 U/L, Reference 20–70). There were significant differences in AST (24 ± 16 vs. 15 ± 3.5 U/L, Reference: 10–50, p < 0.0087) and alkaline phosphatase between VC-exposed group vs. control group (71 ± 15 vs. 50 ± 13 U/L Reference: 38–126, p < 0.0001); however, both of these laboratory values are still within the reference ranges. Mean cytokeratin 18 (CK18) fragment levels were higher in the control group vs. the VC-exposed group: CK18 M65 (458 ± 225 vs. 244 ± 119 U/L, p < 0.0004) and CK18 M30 (119 ± 50.3 U/L vs. 111 ± 82.8 vs., p = 0.737). The M30/M65 ratio of the VC-exposed group (0.463 [range: 0.229–0.860]) was significantly higher (p < 0.001) than the control group (0.271 [range: 0.153–0.589]).
Table 2.
Clinical laboratory values of control and VC-exposed individuals.
| Control | VC-Exposed | |
|---|---|---|
| Albumin | 4.5 ± 0.38 | 4.5 ± 0.23 |
| Total Bilirubin | 0.6 ± 0.7 | 0.6 ± 0.3 |
| Alkaline phosphatase | 50 ± 13 | 71 ± 15a |
| AST | 15 ± 3.5 | 24 ± 16b |
| ALT | 22 ± 3.6 | 23 ± 19 |
| CK18 M30 (U/L) | 119 ± 50.3 | 111 ± 82.8 |
| CK 18 M65 (U/L) | 458 ± 225 | 244 ± 119c |
p < 0.0001.
p < 0.05.
p = 0.0004.
3.3. Random Forest analysis
Random Forest analysis was performed between VC-exposed subjects and healthy controls to determine if the metabolic data could successfully separate the two groups. Sixteen of seventeen (94%) VC-exposed subjects could be separated from healthy controls with a class error of 0.059 (Fig. 1A). The importance plot displays the thirty metabolites that most effectively separated the groups. Twenty of these metabolites were lipids (Fig. 1B). An importance plot including unnamed metabolites was also performed. (Supplemental Figure).
Fig. 1.
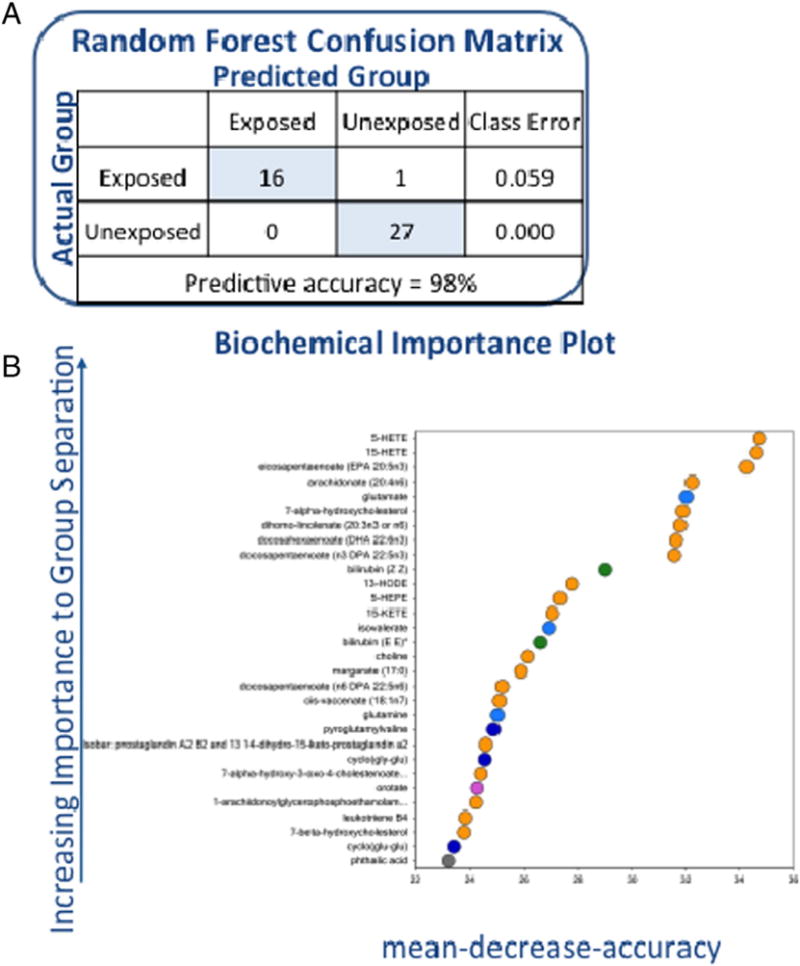
A (1 column) Confusion matrix for controls versus VC-exposed individuals and 1B (2 columns) Random Forest importance plot for controls versus VC-exposed individuals.
3.4. Metabolomic analysis
Using LC/MS and GC/MS analysis identified 613 named unique plasma metabolites along with 525 unique plasma metabolites that were not found in the reference library. Of the 613 identified named metabolites, 189 metabolites were significantly increased following VC exposure and 94 were significantly decreased (Table 3 and Supplemental Data).
Table 3.
Metabolites altered in VC-exposed individuals.
| Lipids | Carbohydrates | Amino acids | Bile acids | Peptides | Nucleotides | |
|---|---|---|---|---|---|---|
| Upregulated | 69 | 6 | 49 | 0 | 22 | 8 |
| Downregulated | 32 | 3 | 11 | 10 | 22 | 4 |
| No change | 77 | 23 | 83 | 4 | 54 | 13 |
| Total | 178 | 32 | 143 | 14 | 98 | 25 |
3.5. Linolenate and specific metabolites
Alpha-linolenate is an essential fatty acid in the human diet. This study did not differentiate between alpha or gamma linolenate. The metabolites classified as either alpha or gamma linolenate were upregulated 1.5-fold in VC-exposed individuals compared to healthy controls. A series of enzymatic reactions may convert linolenate to dihomo-linolenate. Dihomo-linolenate was upregulated 8-fold in VC-exposed individuals compared to healthy controls (Fig. 2).
Fig. 2.
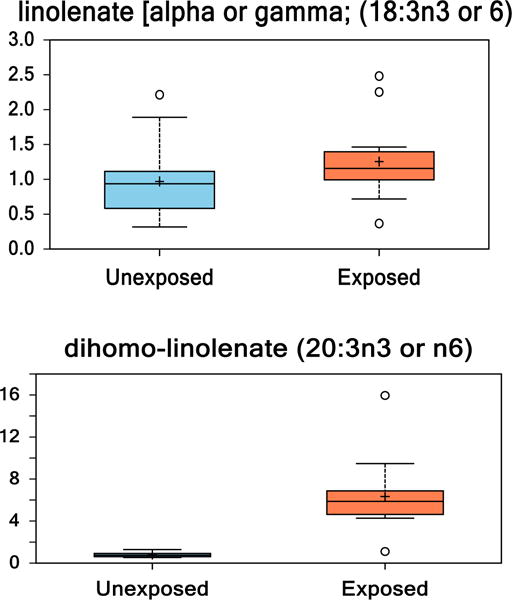
(1 column). Box and whisker plots of linolenate and its metabolite dihomolinolenate in healthy controls compared to VC-exposed individuals. (p < 0.05 unexposed vs. exposed.)
3.6. Lineoleate and specific metabolites
Lineoleate (LA) is an essential fatty acid. In this study, we noted a 1.5-fold increase of LA in VC-exposed individuals versus human controls. LA is metabolized via 15-lipoxygenase to 13-hydroxyoctadecadienoic (13-HODE), which was upregulated 211-fold in VC-exposed individuals compared to healthy, human controls (Fig. 3).
Fig. 3.
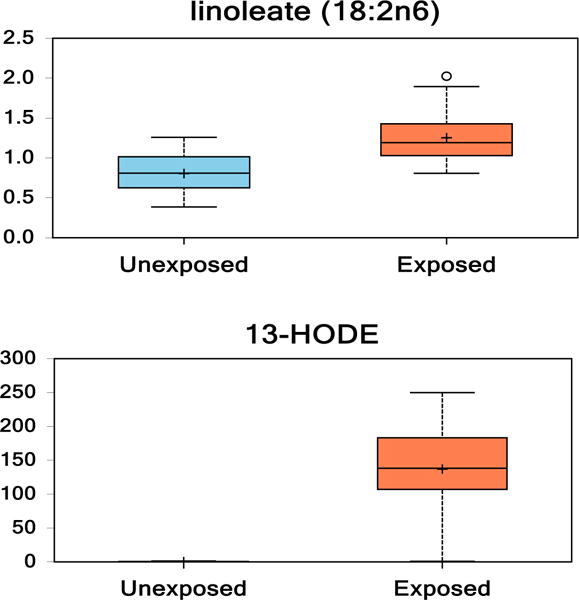
(1 column). Box and whisker plots of linoleate and its 12,15-lipoxygenase metabolite (13-HODE) in healthy controls compared to VC-exposed individuals. (p < 0.05 Unexposed vs. Exposed).
3.7. Arachidonic acid and specific metabolites
LA is metabolized via a series of enzymes to arachidonic acid (AA). AA was 6-fold increased in VC-exposed individuals compared to controls. AA is metabolized by lipoxygenases or P450 to hydroxyeicosatetraenoic acids (HETEs). Several HETEs were notably altered in the VC-exposed group compared to the healthy controls, including the lipoxygenase products 5-HETE (110-fold increase), 12-HETE (15-fold increase), and 15-HETE (615-fold increase), and the free radical-mediated arachidonic acid product 9-HETE (10-fold increase). Lipoxygenase may also metabolize AA to 15-oxo-5Z,8Z,11Z,13E-eicosatetraenoate (15-KETE) which was increased 12-fold in VC-exposed individuals compared to healthy controls. AA is metabolized via a series of enzymes to docosapentaenoate (n6 DPA; 22:5n6), which was upregulated 5-fold in VC-exposed individuals compared to healthy controls. AA is alternatively metabolized to leukotriene B4 via cyclooxygenase and leukotriene B4 was increased 52-fold in VC-exposed individuals compared to controls (Fig. 4).
Fig. 4.
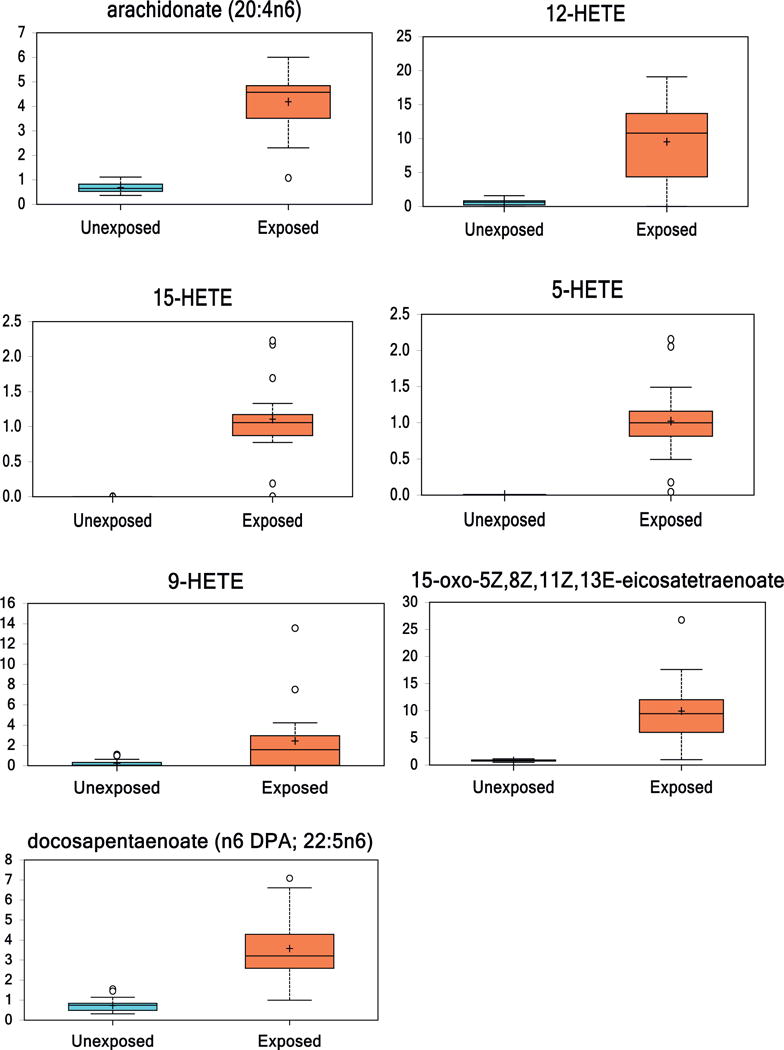
(2 columns). Box and whisker plots of fold change of arachidonate, its lipoxygenase metabolites (5-HETE, 12-HETE, 15-HETE, 15-KETE), P450 metabolite (9-HETE), COX metabolite (leukotriene B4), and other metabolite (n6 DPA) in healthy controls compared to VC-exposed individuals. (p < 0.05 Unexposed vs. Exposed).
3.8. Eicosapentaenoic acid and specific metabolites
LA is metabolized via a series of enzymes to eicosapentaenoic acid (EPA). EPA was 12-fold increased in VC-exposed individuals compared to healthy controls. Lipoxygenases metabolize EPA to hydroxyperoxyeicosapentaenoic acids (HEPEs). HEPEs were altered in the VC-exposed group compared to healthy controls, including 5-HEPE (8-fold increase) and 18-HEPE (11-fold increase). EPA is metabolized via a series of enzymes or via lipid peroxidation to docosapentaenoate (n3 DPA; 22:5n3), which was upregulated 3.5-fold in VC-exposed versus healthy controls. DPA (n3) is then metabolized to docosahexaenoate (DHA; 22:6n3), which is upregulated 3.5-fold in VC-exposed versus healthy controls. (Fig. 5).
Fig. 5.
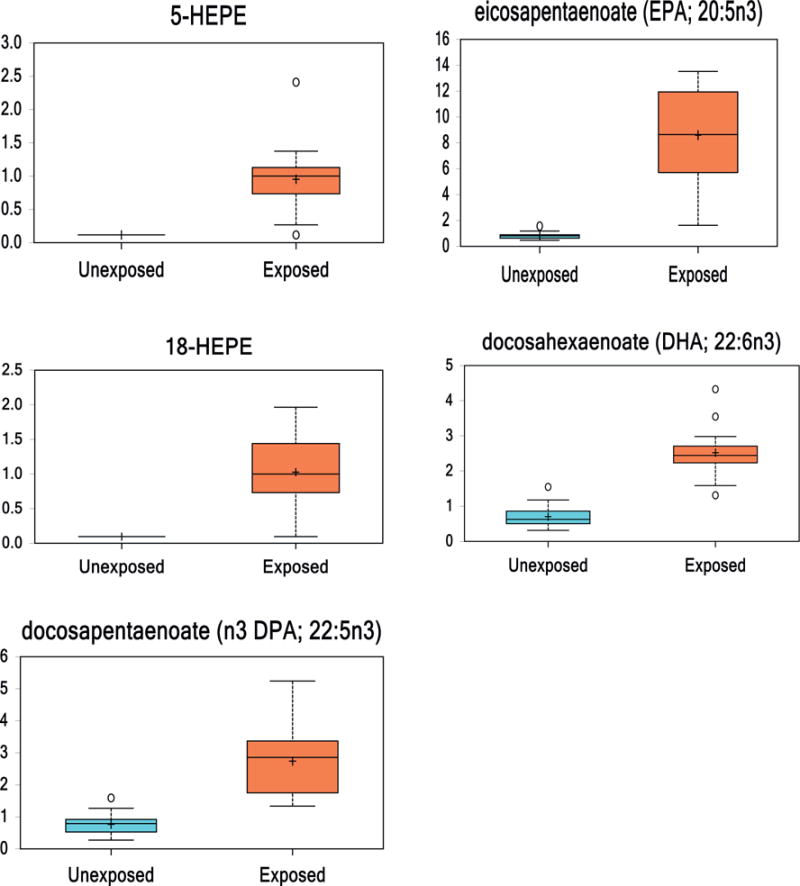
(2 columns). Box and whisker plots of eicosapentaenoate (EPA), EPA lipoxygenase metabolites (5-HEPE, 18-HEPE), and other EPA metabolites (DHA, n3 DPA; 22:5n3) in healthy controls and VC-exposed individuals. (p < 0.05 Unexposed vs. Exposed.)
3.9. Short, medium, and long chain fatty acids
One short chain, nine medium chain, and 19 long chain fatty acids were identified in the plasma of individuals. Valerate, the short chain fatty acid identified, was increased 3.32 fold in VC-exposed individuals. Of the nine medium chain fatty acids, only caproate (2.61-fold) was significantly increased in VC-exposed individuals (Supplemental Data) The other medium chain fatty acids were not significantly altered between groups. Eighteen of the 19 long chain fatty acids exhibited a significant increased fold-change in the VC-exposed group, notably margarate (3-fold increase) (Supplemental Data).
3.10. Other metabolites
Other metabolites were identified that could be used to separate VC-exposed versus healthy control subjects. Glutamate was upregulated 4-fold while glutamine was downregulated 3-fold in VC-exposed. The nutrient choline was upregulated 4.5-fold in VC-exposed individuals versus healthy controls. Additionally, the peptides pyroglutamylvaline (28-fold increase), cyclo(gly-glu) (14-fold increase), cyclo(glu-glu) (24-fold increase) were altered in VC-exposed individuals versus healthy controls. Vitamin C, an important antioxidant, was downregulated 6-fold in VC-exposed individuals compared to healthy controls (Supplemental Data).
3.11. IPA ingenuity analysis
Metabolite interactions were investigated using the Ingenuity Pathway Analysis (IPA) software. 354 of our 613 metabolites that could be mapped using the Human Metabolomic Database (HMDB). IPA Ingenuity generated two networks with scores >40 – one network identified as Amino Acid Metabolism, Small Molecule Biochemistry, Cellular Compromise (Fig. 6A) and the other identified as Increased Levels of Alkaline Phosphatase, Cell Death and Survival, Free Radical Scavenging (Fig. 6B). Multiple metabolites that were altered in our study were shown to interact with the proteins extracellular signal-regulated kinases 1/2 (ERK1/2) (9 metabolites), protein kinase B (Akt) (10 metabolites), AMP-activated protein kinase (AMPK) (3 metabolites), and N-methyl-D-aspartate (NMDA) receptor (6 metabolites).
Fig. 6.
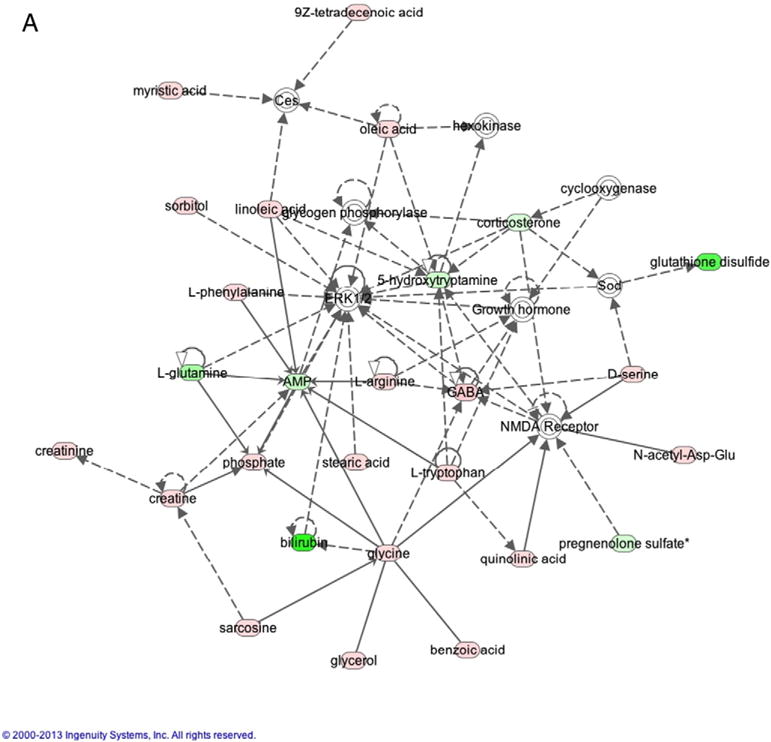
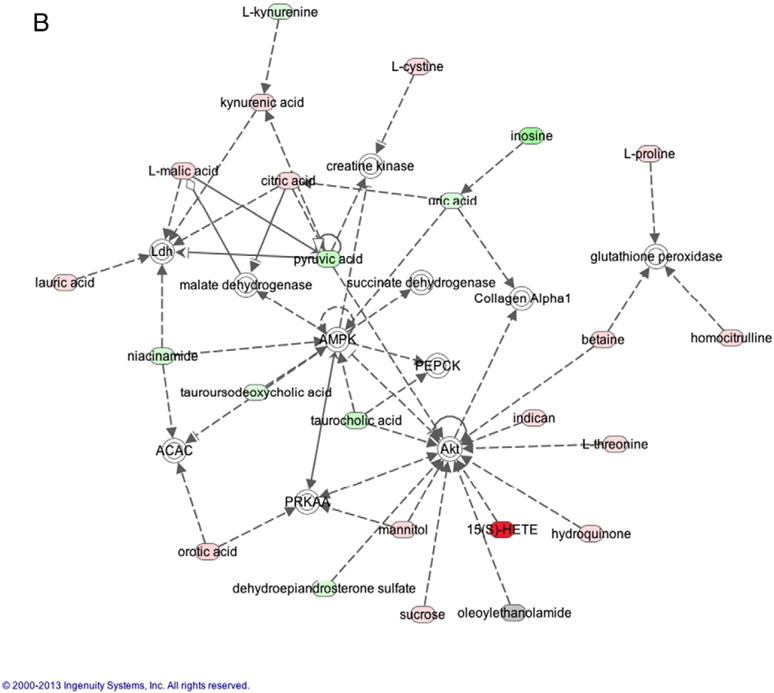
A (2 columns). Ingenuity Analysis: Network 1 - Amino Acid Metabolism, Small Molecule Biochemistry, and Cellular Compromise. This pathway demonstrates interactions with metabolites altered in this study and ERK1/2 and NMDA. B (2 columns). Ingenuity Analysis: Network 2 – Increased Levels of Alkaline Phosphatase, Cell Death and Survival, Free Radical Scavenging. This pathway demonstrates interactions with AMPK and Akt.
Table 4 lists the top ten canonical pathways identified by IPA Ingenuity that were affected by vinyl chloride exposure. These pathways include tRNA charging, urea cycle, nucleotide degradation, and amino acid synthesis and degradation.
Table 4.
Top ten canonical pathways affected by Vinyl Chloride exposure.
| Ingenuity canonical pathways | −log (p-value) |
Ratio | Molecules |
|---|---|---|---|
| tRNA charging | 2.01E01 | 2.35E-01 | AMP, L-phenylalanine, L-glutamine, L-asparagine, L-tryptophan, glycine, L-methionine, L-histidine, L-cysteine, L-valine, L-alanine, L-isoleucine, L-leucine, L-proline, L-threonine, L-aspartic acid, L-arginine, L-tyrosine, L-lysine |
| Superpathway of Citrulline Metabolism | 1.13E01 | 2.63E-01 | Citrulline, AMP, L-glutamine, L-proline, urea, phosphate, fumaric acid, L-aspartic acid,2-oxoglutaric acid, L-arginine |
| Urea Cycle | 9.05E00 | 3.5E-01 | Citrulline, AMP, urea, phosphate, fumaric acid, L-aspartic acid, L-arginine |
| Purine Nucleotides Degradation II (Aerobic) | 8.62E00 | 2.29E-01 | Xanthosine, AMP, inosine, uric acid, adenosine, phosphate, hypoxanthine, xanthine |
| Arginine Biosynthesis IV | 8.42E00 | 2.92E-01 | Citrulline, AMP, phosphate, fumaric acid, L-aspartic acid,2-oxoglutaric acid, L-arginine |
| Citrulline Biosynthesis | 8.15E00 | 2.69E-01 | citrulline,L-glutamine,L-proline,urea,phosphate,2-oxoglutaric acid, L-arginine |
| Adenosine Nucleotides Degradation II | 8.15E00 | 2.69E-01 | AMP, inosine, uric acid, adenosine, phosphate, hypoxanthine, xanthine |
| Purine Ribonucleosides Degradation to Ribose-1-phosphate | 7.68E00 | 3.33E-01 | Xanthosine, inosine, adenosine, phosphate, hypoxanthine, xanthine |
| Superpathway of Methionine Degradation | 7.66E00 | 1.41E-01 | Pyruvic acid,adenosine,betaine,phosphate,2-oxoglutaric acid, L-methionine, alpha-ketobutyric acid, dimethylglycine, L-cysteine |
| Phenylalanine Degradation IV (Mammalian, via Side Chain) | 7.46E00 | 2.05E-01 | AMP, l-phenylalanine, L-glutamine, alpha-N-phenylacetyl-L-glutamine, phenylpyruvic acid, glycine, 2-oxoglutaric acid, phenylacetic acid |
4. Discussion
VC exposure has been linked to several pathologies including a variety of cancers (Makk et al., 1974; Mastrangelo et al., 2004; Ruckart et al., 2015), liver disease (Cave et al., 2010), and autoimmune diseases (Powell et al., 1999). In the present study, we performed metabolomics analyses of plasma samples to obtain a comprehensive view of the alterations of metabolism in individuals who were exposed to VC and other chemicals, without developing cancer. Following VC exposure, a significant number of metabolites were altered in the plasma metabolome of individuals. The most striking differences in the present study were observed in the elevations of fatty acids and their oxidized metabolites, giving insight into the mechanisms of VC-induced toxicity and potential noninvasive biomarkers of exposure. Recently, Stachowska et al. demonstrated a positive correlation between 13-HODE, 9-HODE, 5-HETE and hepatic steatosis and observed a significant decrease in these lipid metabolites following dietary intervention and ultrasound-confirmed resolution of the patient group’s steatosis (Maciejewska et al., 2015). Our metabolomics data along with our assessment of liver injury demonstrated that VC-induced alterations in the plasma metabolome may be independent of the development of liver injury associated with VC-exposure. While VC-exposed subjects in our study had normal CK18 or ALT values and neither liver ultrasounds nor biopsies were available, we focused on lipid metabolites based on the results of our Random Forest analysis and confusion matrix; however, the Ingenuity analysis demonstrated that multiple additional pathways were also altered in VC exposure.
Polyunsaturated fatty acids (PUFAs) can be classified as either n-6 or n-3 and have opposing effects (Schmitz and Ecker, 2008). The n-6 PUFAs, linoleic acid and arachidonic acid, along with their oxidized metabolites were all significantly upregulated in our study. Increased levels of these metabolites, especially the ratio of n-6/n-3, are associated with systemic inflammation and hepatic injury (Araya et al., 2004; Lopez-Vicario et al., 2014). Linoleic acid is metabolized by lipoxygenases to oxidized linoleic acid metabolites (OXLAMs). Two specific OXLAMs 9-and 13-HODE have previously been shown to be elevated in non-alcoholic steatohepatitis (NASH) (Feldstein et al., 2010) and alcoholic-associated steatohepatitis (ASH) (Raszeja-Wyszomirska et al., 2012). In Feldstein’s study, the 25th percentile value for 9- and 13-HODE in NASH patients was above the median value in control patients. Additionally, chronic binge alcohol drinking has been shown to increase plasma OXLAM levels, specifically 9- and 13-HODE (Liu et al., 2015). Similarly here, 13-HODE was significantly elevated in the plasma metabolic profile. Comparing 13-HODE levels to linoleic acid levels may lend insight into the severity of NASH (Alkhouri et al., 2014). Indeed, in our study 13-HODE levels were increased 200-fold in VC-exposed individuals versus controls, while linoleic acid levels were increased only 1.6-fold.
Phospholipase A2 (PLA2) catalyzes the release of AA from phospholipids (Burke and Dennis, 2009). AA and its eicosanoid metabolic products are potent pro-inflammatory agents. AA is decreased in NASH and previous metabolomics studies have reported either increased (Puri et al., 2009) or unaltered (Feldstein et al., 2010) levels of AA lipoxygenase products 5-HETE and 15-HETE in NASH patients. Feldstein et al. demonstrated no changes in the arachidonic acid P450 products 9-HETE and 12-HETE in patients with NAFLD or NASH (Feldstein et al., 2010). Previous studies in patients with ASH reported plasma elevations of 5-HETE, 12-HETE, and 15-HETE (Raszeja-Wyszomirska et al., 2012). We measured levels of AA and its 5-, 9-, 12-, 15-HETE metabolites at levels that in some cases, to our knowledge, have never been reported in human subjects (up to 615-fold increase over controls). Soluble PLA2 has been shown to be released from cells undergoing necrosis. As necrotic cell death is the major player in VC-induced liver injury, the increase in AA may stem from the increased release of PLA2. Lysolipids, natural products formed by the hydrolysis of phospholipids, may also be cytotoxic (Park et al., 2010). Here, lysolipids were also elevated as expected with an increase in AA. AA and its eicosanoid metabolites are primary components of the phospholipid membrane of inflammatory cells (Gibney and Hunter, 1993; Kew et al., 2004; Faber et al., 2011). The increase in lipoxygenase (LOX) and P450 enzymatic products could be derived from an increase in substrate or, as observed in ASH (Raszeja-Wyszomirska et al., 2012), from increased expression of these enzymes. We postulate that lipotoxicity, pro-inflammatory lipid peroxidation products, and inflammatory oxidized lipid metabolites from AA may be important mechanisms for VC and other chemical-related organ injury. Additionally, the increased levels of pro-inflammatory lipids suggests that VC exposure may increase cardiovascular disease risk (Burhop et al., 1988; Kayama et al., 2009; Maayah and El-Kadi, 2016).
Although steatohepatitis biomarkers (ALT and CK18) were negative in workers exposed to VC, the pattern of the lipid peroxidation products is similar to those reported by Feldstein et al. These findings suggest that HODE and HETEs arise due to a chemical process, rather than an enzyme-mediated process. Lack of production of 20-hydroxy-HETE, EETs and dihydroxy-EETs suggests that the P450-dependent pathways don’t occur under these conditions; however, these oxidized lipids may activate receptors, such as PPAR-gamma, known to be activated by HODEs. The accumulation of long chain fatty acids suggests that PLA2 may be activated, releasing the precursors to HODES.
AMPK, ERK1/2, and Akt are protein kinases that were at the center of two of the top Networks specified by IPA Ingenuity Analysis. The dysregulation of ERK1/2, AMPK, and Akt has been implicated in insulin resistance and altered metabolism (Jiao et al., 2013; Wang et al., 2004). Akt is a serine/threonine-specific protein kinase involved in multiple pathways, including glucose metabolism, apoptosis, and cell proliferation. ERK1/2 are protein kinases involved in the insulin signaling. AMPK is an enzyme that plays a role in energy homeostasis – upregulation leads to hepatic fatty acid oxidation and ketogenesis, lipogenesis, etc. (Hardie, 2007). AMPK activation induces catabolic events and favors adenosine triphosphate (ATP) generation during energy depletion, e.g. glycolysis is enhanced and ATP-consuming processes are inhibited by AMPK (Krause et al., 2002). Both AMPK and Akt activity were increased in a mouse model of VC metabolite exposure (Anders et al., 2016). The observation that multiple metabolites known to regulate these enzymes have been differentially regulated is important and could have implications for future research in VC exposure-related diseases, such as TASH and hemangiosarcoma. Adiponectin leads to increased insulin sensitivity via AMPK activation (Yamauchi et al., 2002). The decreased levels of adiponectin seen in VC-exposed individuals with TASH (Cave et al., 2010) may lead to decreased activation of AMPK leading to the insulin resistance observed in that population. Tumor uptake of linoleic acid is directly proportional to 13-HODE production and 13-HODE was shown to be required for ERK1/2 phosphorylation in rat hepatomas (Sauer et al., 2007). Linoleic acid-dependent tumor growth in rats has been associated with increased ERK1/2 phosphorylation. Interaction of ERK1/2 with the altered metabolites in our study could provide mechanistic insight into the development of fatty liver in toxicant-associated steatohepatitis, hepatocellular carcinoma, and liver hemangiosarcoma seen in individuals exposed to VC. Moreover, ERK1/2 activation has been shown to be cytoprotective in oxidative stress-induced cell death and inhibition of the ERK1/2 pathway sensitized hepatocytes to apoptosis (Czaja et al., 2003; Rosseland et al., 2005). Indeed, in a mouse model of VC metabolite exposure, indices for oxidative stress (4-HNE adduct formation) were increased (Anders et al., 2016), while ERK1/2 phosphorylation was inhibited (Lang AL and Beier JI, unpublished observation).
A limitation of our study is the age of the samples of VC-exposed subjects versus the age of our control samples. The samples of VC-exposed subjects were collected and frozen in the 1970s. We have utilized old samples for previous studies of workers exposed to VC and acrylonitrile, 1,3-butadiene, and styrene (ABS) (Cave et al., 2010; Cave et al., 2011); however, this is the first time we have performed metabolomics on such samples. Being frozen for such an extended period of time could have altered the metabolites in these samples. Oxidized lipids were increased, which could have been due to storage time. Additionally, the workers were exposed to other chemicals besides VC, which could confound the results. However, due to the unusually high exposures of VC to the subjects – similar to workers who developed hemangiosarcoma, VC is still considered the major culprit in the current study. The ideal control group would have been unexposed subjects from the 1970s matched perfectly for demographic factors, unfortunately, those subjects were not available in our biorepository. While the modern day controls were matched well for BMI, they were younger and drank significantly less alcohol – differences that are potential confounders in our study. However, both groups consist of middle-aged adults and neither group drank excessively. Low-risk drinking is defined by National Institute on Alcohol Abuse and Alcoholism as no >14 drinks per week for men; therefore, both groups in our study are in the low risk range (Anon., n.d.). Additionally, our results demonstrate a different metabolic profile from that observed in alcoholic steatohepatitis (Raszeja-Wyszomirska et al., 2012). Thus, while statistically significant, the differences in mean age and alcohol consumption were probably not clinically significant; however, given the sensitivity of the metabolomics technique, these minor differences could have impacted our study.
5. Conclusion
This study is the first to examine the plasma metabolome following chemical exposure at a polyvinyl chloride production facility. The chemical mixture exposure altered the plasma metabolome in distinct ways and allowed for predictable group assignment with Random Forest analysis with 95.41% accuracy. We postulate that differences in biologically active metabolites including lipids contribute to the pathogenesis of VC-related diseases. In this study, workers were selected based on exposure and not disease. In the future, metabolomics studies should include workers selected for exposure-associated diseases. This study provides proof of concept that a metabolomics approach yields useful information when applied to exposed chemical workers. Thus, these data will be used for further research to identify potential targets for studying the mechanism of industrial chemical toxicity and potential targets for therapeutic interventions. Metabolomics analysis may also be a useful tool in the investigation of other industrial toxicants and their impact on human health.
Supplementary Material
Acknowledgments
We would like to acknowledge Russell Prough, PhD for his assistance with interpretation of biochemical pathways related to cytochrome P450s.
Financial support
This research was funded in part by the NIH (1R01ES021375 (MC), K23AA18399 (MC), T35ES014559 (MC,CJM), 1R13ES024661(MC), DK096042 (JB), and DK107912 (JB). Research reported in this publication was also supported by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under Award Number P50AA024337, and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM113226. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- 4-HNE
4-hydroxynonenal
- AA
arachidonic acid
- Akt
protein kinase B
- AMPK
AMP-activated protein kinase
- ASH
alcoholic steatohepatitis
- BMI
body mass index
- CK18
cytokeratin-18
- CERM
cumulative exposure risk month
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- ERK
extracellular-signal-regulated kinase
- GC-MS
gas chromatography-mass spectrometry
- HEPE
hydroxyeicosapentaenoic acid
- HETE
hydroxyicosatetraenoic
- HODE
hydroxyoctadecadienoic
- HMDB
human metabolome database
- LA
linoleic acid
- LC
liquid chromatography
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OXLAM
oxidized linoleic acid metabolite
- PUFA
poly-unsaturated fatty acid
- PVC
polyvinyl chloride
- TASH
toxicant-associated steatohepatitis
- VC
vinyl chloride
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.taap.2016.10.001.
Footnotes
Declarations
The authors declare they have no competing interests.
Authorship
JG performed statistical analysis, interpreted metabolomics data, and drafted the manuscript. JB interpreted metabolomics data and drafted the manuscript. KCF interpreted metabolomics data. BW assisted in interpreting data. CM and MC concieved the study. MC coordinated the investigation and drafted the manuscript. All authors approved and read the final manuscript.
Conflict of interest statement
The authors Drs. Guardiola, Beier, Falkner, Wheeler, McClain, and Cave have nothing to disclose and no conflicts of interest.
Transparency document
The Transparency document associated with this article can be found, in online version.
References
- Alkhouri N, Berk M, Yerian L, Lopez R, Chung YM, Zhang R, McIntyre TM, Feldstein AE, Hazen SL. OxNASH score correlates with histologic features and severity of nonalcoholic fatty liver disease. Dig Dis Sci. 2014;59:1617–1624. doi: 10.1007/s10620-014-3031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders LC, Lang AL, Anwar-Mohamed A, Douglas AN, Bushau AM, Falkner KC, Hill BG, Warner NL, Arteel GE, Cave M, McClain CJ, Beier JI. Vinyl chloride metabolites potentiate inflammatory liver injury caused by LPS in mice. Toxicol Sci. 2016;151:312–323. doi: 10.1093/toxsci/kfw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon. Medicine I of: Review of VA Clinical Guidance for the Health Conditions Identified by the Camp Lejeune Legislation. National Academies Press; Washington, D.C.: 2015. [PubMed] [Google Scholar]
- Anon. Agency for toxic substances and disease registry. d. http://www.atsdr.cdc.gov/spl/
- Anon. Rethinking drinking: alcohol and your health. d. http://rethinkingdrinking.niaaa.nih.gov/
- Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci. 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- Burhop KE, Selig WM, Malik AB. Monohydroxyeicosatetraenoic acids (5-HETE and 15-HETE) induce pulmonary vasoconstriction and edema. Circ Res. 1988;62:687–698. doi: 10.1161/01.res.62.4.687. [DOI] [PubMed] [Google Scholar]
- Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–S242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M, Falkner KC, Ray M, Joshi-Barve S, Brock G, Khan R, Bon Homme M, McClain CJ. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology. 2010;51:474–481. doi: 10.1002/hep.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M, Falkner KC, Henry L, Costello B, Gregory B, McClain CJ. Serum cytokeratin 18 and cytokine elevations suggest a high prevalence of occupational liver disease in highly exposed elastomer/polymer workers. J Occup Environ Med. 2011;53:1128–1133. doi: 10.1097/JOM.0b013e31822cfd68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology. 2003;37:1405–1413. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- Faber J, Berkhout M, Vos AP, Sijben JW, Calder PC, Garssen J, van Helvoort A. Supplementation with a fish oil-enriched, high-protein medical food leads to rapid incorporation of EPA into white blood cells and modulates immune responses within one week in healthy men and women. J Nutr. 2011;141:964–970. doi: 10.3945/jn.110.132985. [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, Zhang R, McIntyre TM, Hazen SL. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res. 2010;51:3046–3054. doi: 10.1194/jlr.M007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney MJ, Hunter B. The effects of short- and long-term supplementation with fish oil on the incorporation of n-3 polyunsaturated fatty acids into cells of the immune system in healthy volunteers. Eur J Clin Nutr. 1993;47:255–259. [PubMed] [Google Scholar]
- Greenberg RA, Tamburro CH. Exposure indices for epidemiological surveillance of carcinogenic agents in an industrial chemical environment. J Occup Med. 1981;23:353–358. [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hsiao TJ, Wang JD, Yang PM, Yang PC, Cheng TJ. Liver fibrosis in asymptomatic polyvinyl chloride workers. J Occup Environ Med. 2004;46:962–966. doi: 10.1097/01.jom.0000137722.66767.38. [DOI] [PubMed] [Google Scholar]
- Jiao P, Feng B, Li Y, He Q, Xu H. Hepatic ERK activity plays a role in energy metabolism. Mol Cell Endocrinol. 2013;375:157–166. doi: 10.1016/j.mce.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Barve S, Kirpich I, Cave MC, Marsano LS, McClain CJ. Alcoholic, nonalcoholic, and toxicant-associated steatohepatitis: mechanistic similarities and differences. Cell Mol Gastroenterol Hepatol. 2015;1:356–367. doi: 10.1016/j.jcmgh.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, Milburn M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60:404–413. doi: 10.1016/j.metabol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayama Y, Minamino T, Toko H, Sakamoto M, Shimizu I, Takahashi H, Okada S, Tateno K, Moriya J, Yokoyama M, Nojima A, Yoshimura M, Egashira K, Aburatani H, Komuro I. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J Exp Med. 2009;206:1565–1574. doi: 10.1084/jem.20082596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew S, Mesa MD, Tricon S, Buckley R, Minihane AM, Yaqoob P. Effects of oils rich in eicosapentaenoic and docosahexaenoic acids on immune cell composition and function in healthy humans. Am J Clin Nutr. 2004;79:674–681. doi: 10.1093/ajcn/79.4.674. [DOI] [PubMed] [Google Scholar]
- Kielhorn J, Melber C, Wahnschaffe U, Aitio A, Mangelsdorf I. Vinyl chloride: still a cause for concern. Environ Health Perspect. 2000;108:579–588. doi: 10.1289/ehp.00108579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause U, Bertrand L, Hue L. Control of p70 ribosomal protein S6 kinase and ace-tyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur J Biochem. 2002;269:3751–3759. doi: 10.1046/j.1432-1033.2002.03074.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Beier JI, Arteel GE, Ramsden CE, Feldstein AE, McClain CJ, Kirpich IA. Transient receptor potential vanilloid 1 gene deficiency ameliorates hepatic injury in a mouse model of chronic binge alcohol-induced alcoholic liver disease. Am J Pathol. 2015;185:43–54. doi: 10.1016/j.ajpath.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Vicario C, Gonzalez-Periz A, Rius B, Moran-Salvador E, Garcia-Alonso V, Lozano JJ, Bataller R, Cofan M, Kang JX, Arroyo V, Claria J, Titos E. Molecular interplay between Delta5/Delta6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2014;63:344–355. doi: 10.1136/gutjnl-2012-303179. [DOI] [PubMed] [Google Scholar]
- Maayah ZH, El-Kadi AOS. 5-, 12- and 15-Hydroxyeicosatetraenoic acids induce cellular hypertrophy in the human ventricular cardiomyocyte, RL-14 cell line, through MAPK- and NF-κB-dependent mechanism. Arch Toxicol. 2016;90:359–373. doi: 10.1007/s00204-014-1419-z. [DOI] [PubMed] [Google Scholar]
- Maciejewska D, Ossowski P, Drozd A, Ryterska K, Jamiol-Milc D, Banaszczak M, Kaczorowska M, Sabinicz A, Raszeja-Wyszomirska J, Stachowska E. Metabolites of arachidonic acid and linoleic acid in early stages of non-alcoholic fatty liver disease-a pilot study. Prostaglandins Other Lipid Mediat. 2015 doi: 10.1016/j.prostaglandins.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Makk L, Creech JL, Whelan JG, Jr, Johnson MN. Liver damage and angiosarcoma in vinyl chloride workers. A systematic detection program. JAMA. 1974;230:64–68. [PubMed] [Google Scholar]
- Mastrangelo G, Fedeli U, Fadda E, Valentini F, Agnesi R, Magarotto G, Marchi T, Buda A, Pinzani M, Martines D. Increased risk of hepatocellular carcinoma and liver cirrhosis in vinyl chloride workers: synergistic effect of occupational exposure with alcohol intake. Environ Health Perspect. 2004;112:1188–1192. doi: 10.1289/ehp.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell TM, Watkins PB. The application of metabonomics to predict drug-induced liver injury. Clin Pharmacol Ther. 2010;88:394–399. doi: 10.1038/clpt.2010.151. [DOI] [PubMed] [Google Scholar]
- Park S, Kim JA, Choi S, Suh SH. Superoxide is a potential culprit of caspase-3 dependent endothelial cell death induced by lysophosphatidylcholine. J Physiol Pharmacol. 2010;61:375–381. [PubMed] [Google Scholar]
- Powell JJ, Van de Water J, Gershwin ME. Evidence for the role of environmental agents in the initiation or progression of autoimmune conditions. Environ Health Perspect. 1999;107(Suppl):667–672. doi: 10.1289/ehp.99107s5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, Contos MJ, Sterling RK, Fuchs M, Zhou H, Watkins SM, Sanyal AJ. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50:1827–1838. doi: 10.1002/hep.23229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raszeja-Wyszomirska J, Safranow K, Milkiewicz M, Milkiewicz P, Szynkowska A, Stachowska E. Lipidic last breath of life in patients with alcoholic liver disease. Prostaglandins Other Lipid Mediat. 2012;99:51–56. doi: 10.1016/j.prostaglandins.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Rosseland CM, Wierød L, Oksvold MP, Werner H, Ostvold AC, Thoresen GH, Paulsen RE, Huitfeldt HS, Skarpen E. Cytoplasmic retention of peroxide-activated ERK provides survival in primary cultures of rat hepatocytes. Hepatology. 2005;42:200–207. doi: 10.1002/hep.20762. [DOI] [PubMed] [Google Scholar]
- Ruckart PZ, Bove FJ, Shanley E, Maslia M. Evaluation of contaminated drinking water and male breast cancer at Marine Corps Base Camp Lejeune, North Carolina: a case control study. Environ Health. 2015;14:74. doi: 10.1186/s12940-015-0061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer LA, Blask DE, Dauchy RT. Dietary factors and growth and metabolism in experimental tumors. J Nutr Biochem. 2007;18:637–649. doi: 10.1016/j.jnutbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Ecker J. The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res. 2008;47:147–155. doi: 10.1016/j.plipres.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, Cave MC. Toxicant-associated steatohepatitis. Toxicol Pathol. 2013;41:343–360. doi: 10.1177/0192623312468517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004;53:2735–2740. doi: 10.2337/diabetes.53.11.2735. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.