

Abstract
Chimeric antigen receptors (CARs) are synthetic molecules that provide new specificities to T cells. Although successful in treatment of hematologic malignancies, CAR T cells are ineffective for solid tumors to date. We found that the cell-surface molecule c-Met was expressed in ~50% of breast tumors, prompting the construction of a CAR T cell specific for c-Met, which halted tumor growth in immune-incompetent mice with tumor xenografts. We then evaluated the safety and feasibility of treating metastatic breast cancer with intratumoral administration of mRNA-transfected c-Met-CAR T cells in a phase 0 clinical trial (NCT01837602). Introducing the CAR construct via mRNA ensured safety by limiting the non-tumor cell effects (on-target/off-tumor) of targeting c-Met. Patients with metastatic breast cancer with accessible cutaneous or lymph node metastases received a single intratumoral injection of 3 × 107 or 3 × 108 cells. CAR T mRNA was detectable in peripheral blood and in the injected tumor tissues after intratumoral injection in two and four patients, respectively. mRNA c-Met-CAR T cells cell injections were well tolerated, as none of the patients had study drug–related adverse effects greater than grade 1. Tumors treated with intratumoral injected mRNA c-Met-CAR T cells were excised and analyzed by immunohistochemistry, revealing extensive tumor necrosis at the injection site, cellular debris, loss of c-Met immunoreactivity, all surrounded by macrophages at the leading edges and within necrotic zones. We conclude that intratumoral injections of mRNA c-Met-CAR T cells are well tolerated and evoke an inflammatory response within tumors.
Introduction
Chimeric antigen receptor modified T cells (CAR T cells) are redirected effector immune cells genetically modified to deliver tumoricidal functions upon recognition of antigen. CAR T cells are effective in the treatment of several hematologic malignancies [1–3]. However, the effectiveness of CAR T cells in the treatment of solid tumors remains modest. Barriers include the fact that most tumor antigens are expressed, albeit, at lower levels in normal tissues, which when targeted by CAR T cells, may lead to on-target/off-tumor effects. In addition, the microenvironment of solid tumors is immunosuppressive, which may limit the potency of CAR T cells [4].
Hepatocyte growth factor receptor, or c-Met, is a cell-surface protein tyrosine kinase expressed in a variety of solid tumors including breast cancer [5, 6]. A monovalent anti-c-Met antibody, onartuzumab, has been tested in a variety of patients with advanced stage solid cancers in clinical trials [7–10]. To determine whether c-Met might serve as a target for CAR T cells, we replaced the single chain variable fragment (scFv) portion of the CD19 binding domain of our previously established CD19-CAR construct [1] with that of onartuzumab so that we could evaluate the activity of CAR T cells directed against c-Met (c-Met-CAR T cells) in patients with metastatic breast cancer.
We have previously published our experience of a phase I clinical trial (NCT01355965) to evaluate the safety and feasibility of the use of systemic and intratumoral injection of mRNA mesothelin directed CAR T (mRNA meso-CAR T) cells to treat two patients with metastatic mesothelioma and one with pancreatic cancer respectively [11]. We noted that the mRNA meso-CAR T transgene was detectable in the ascites fluid of the patient with metastatic mesothelioma 3 days after systemic infusion of the study drug suggesting that systemically infused mRNA meso-CAR T cells had trafficked into the tumor microenvironment. In the treated patient with metastatic pancreatic cancer, we were able to detect mRNA meso-CAR T transgene within the pancreas in subsequent tumor biopsy following intratumoral injection of mRNA meso-CAR T cells. No serious adverse effects were noted in any of the three patients. These results support evaluating the mRNA CAR T cell platform, “in a controlled manner, the potential off-tumor on-target toxicities” [12], against other tumor antigens, e.g. c-Met, in the clinical setting.
We hypothesize that intratumoral injection of mRNA c-Met-CAR T cells into breast tumors is safe and feasible. After confirming the in vitro and in vivo effectiveness of c-Met-CAR T cells against breast cancer cells and c-Met expressing tumor xenografts in mice, we initiated a phase 0 clinical trial (NCT01837602) to assess safety and feasibility of c-Met-CAR T cells in the treatment of metastatic breast cancer [13]. The trial contained several safety features, including: 1) the use of electroporation of mRNA-encoded CAR transcripts directed against c-Met in T cells to ensure transient CAR expression; 2) intratumoral injection instead of systemic delivery of CAR T cells to limit systemic exposure to CAR T cells; and 3) excision of intratumorally injected tumor tissues 2 days after intratumoral injection, which further limits the potential extravasation of residual CAR T cells from the injected tumor. Finally, resection of the injected tumor provided the opportunity to evaluate the direct effects of mRNA c-Met-CAR T cells in breast tumor parenchyma.
Materials and Methods
Immunohistochemistry (IHC) staining protocol
Expression of c-Met was evaluated on formalin fixed paraffin embedded (FFPE) tissue sections by immunohistochemistry (IHC) staining with a rabbit monoclonal antibody specific for c-Met (SP44, Ventana), CD3, CD4, CD8, CD68, CD56 and S100 using a fully-automated Leica Bond™ Ultra with Polymer Refine Detection System. Slides were pre-treated with Bond ER2 solution for 20 minutes at 100oC.
To evaluate if c-Met expression might be associated with various breast cancer subtypes, we performed c-Met IHC as described [14] using unstained tissue sections obtained from archival FFPE tumor blocks of 59 patients with primary operable breast cancer treated consecutively between 2009 and 2011 at our institution after obtaining approval from our institutional review board (IRB). Because c-Met staining is heterogeneous within and across tumor sections, we used H-score ≥ 30 determined as the product of % positively stained tumor cells and IHC stain intensity (1, 2, or 3 with 3 being the most intense) in three separate high power fields [14, 15], to define (+) c-Met expression.
Cell lines
The human cancer cell lines, BT20 (ATCC HTB-19), MDA-MB-231 (ATCC HTB-26), and SK-OV-3 (ATCC HTB-77), were purchased from ATCC and maintained in media as recommended by ATCC. c-Met expression in SK-OV-3 was confirmed by flow cytometry. SK-OV-3 was transduced to express Click beetle green luciferase (SK-OV-3/luc) as described [16]. TB129, a primary human breast cancer cell line, was derived from a recurrent breast tumor and was maintained and stored in medium as described [14]. All experiments were conducted using cells kept in the lowest possible passage number after recovery from their respective frozen stocks and were confirmed mycoplasma negative. Flow cytometry studies using antibodies against mesothelin and c-Met were performed as described [14].
In vitro cell killing assay
Frozen human T cells collected from a single health donor were activated with CD3/28 beads (ThermoFisher), expanded in vitro, thawed and electroporated with mRNA transcripts encoding c-Met BBZ CAR, mesothelin BBZ CAR and CD19 BBZ CAR as described [11]. We have previously demonstrated that the cytotoxic activity of mRNA c-Met-CAR T cells was specific against c-Met expressing cell lines such as M30, a human tumor cell line derived from mesothelioma [17] and cytotoxic activity was not observed against NCI-H522, a lung cancer cell line which lacks c-Met expression [16]. To confirm the cytolytic activity on c-Met expressing breast cancer cell lines, BT20 (ATCC HTB-19) and TB129 [14], tumor cells were loaded with 51Cr and incubated with mRNA electroporated CAR T cells (effector cells or E) at various E:T ratios in V-bottom plates for 4 hours. 51Cr released as a result of cell lysis was quantified as described [18]. All in vitro cell killing assays were performed in duplicates and repeated in at least three independent experiments.
Mouse xenograft studies
Mouse experiments were performed as previously described [12] following approval from the Institutional Animal Care and Use Committee at the University of Pennsylvania. Briefly, c-Met–expressing Click beetle green luciferase labeled ovarian cancer cells (SK-OV-3/luc) [16] were implanted into the flanks of NOD/scid/γc(-/-) (NSG) mice provided by the University of Pennsylvania Stem Cell and Xenograft Core as described [16]and observed for about 6 weeks until palpable tumors were present. NSG mice with pre-established tumors were then randomized into three treatment groups and received four intratumoral injections with 1) 1.5 × 107 mRNA c-Met-CAR T cells (n= 8; 3 female and 5 male mice); 2) mRNA CD19-CAR T (n= 7; 3 female and 4 male mice) [12](allogeneic negative control) prepared from one single healthy donor or 3) PBS (negative control) (n=8; 2 female and 6 male mice) beginning at weeks 6, 7, 9, and 11. Mouse xenograft studies were repeated in one independent experiment without the use of intraperitoneal administration of cyclophosphamide. In an earlier study when NSG mice with advanced disseminated intraperitoneal (IP) M108-luc tumors were treated on day 56 with a single intraperitoneal injection of 2 × 107 mRNA meso-CAR T cells [12], we observed the onset of graft vs. host disease (GVHD) due to nonspecific T-cell alloreactivity that necessitated termination of the experiments on day 84 [19]. To avoid GVHD, we pretreated our tumor bearing mice with low dose cyclophosphamide prior to the second mRNA c-Met-CAR T cell injection as described [19]. Therefore, the second and subsequent intratumoral injections were preceded by an intraperitoneal injection of low dose cyclophosphamide at 60 mg/kg at 24 hours prior to intratumoral injection to prevent GVHD. Tumor growth was quantified using bioluminescence imaging weekly until week 14. Differences of tumor growth between treatment groups were compared using the two-sided Student t-test.
Study design
After receiving University of Pennsylvania IRB approval, we initiated an open label phase 0 clinical trial (NCT01837602) to evaluate the safety and feasibility of treating metastatic c-Met–expressing breast cancer with a single intratumoral injection of mRNA c-Met-CAR T cells for women with metastatic breast cancer. The clinical trial schema is shown in Supplementary Fig. S1. Patients (n = 6) with metastatic breast cancer presenting with accessible cutaneous or lymph node metastases received a single intratumoral injection of mRNA c-Met-CAR T cells at 3 × 107 in 0.5 mL or 3 × 108 in 1.0 mL (n = 3 per cohort). The primary endpoint was to determine feasibility and safety of treating c-Met+ breast cancer patients with mRNA c-Met-CAR T cells by intratumoral injection. Toxicity was determined using the current Common Toxicity Criteria for Adverse Events. Secondary endpoints included assessment of c-Met–directed responses in resected or biopsied tumor tissue.
Patient eligibility and enrollment
As part of the informed consent, patients with metastatic breast cancer were asked for permission to test their tumor for c-Met expression as one of the eligibility criteria. If archived tumor tissue from any metastatic tumor site was unavailable, patients were offered a 2 or 4 mm punch or percutaneous core needle biopsy of the most accessible metastatic deposit. Staining of archival tissue of the primary breast cancer for initial screening was permitted to determine eligibility. Patients with confirmed c-Met expression in tumor were further screened for eligibility. Inclusion criteria included all patients with stable metastatic breast cancer with an accessible tumor (cutaneous, subcutaneous, or superficial) and/or a palpable adenopathy/mass, with c-Met H-score ≥ 30 (defined as product of % tumor cells staining positive for c-Met on IHC and IHC intensity classified as 1, 2, or 3). Other inclusion criteria included age ≥ 18, Eastern Cooperative Oncology Group clinical performance status 0 or 1, adequate hematologic function (white blood count ≥ 3.0 × 109/L; platelet count ≥ 75 × 109/L; Hemoglobin ≥ 10 g/dL); adequate renal function defined as serum creatinine < 1.5 times upper limit of normal; adequate hepatic function defined as total bilirubin < 1.5 times upper limit of normal and transaminases < 2.5 times upper limit of normal, and a negative pregnancy test. Patients tested positive for HIV-1/HIV-2, and patients with active hepatitis B or C infection, history of alcohol or illicit drug abuse within 12 months of intratumoral injection, significant co-morbid disease such as myocardial infarction and psychiatric disorder were excluded. Once enrolled, cytotoxic therapy was not allowed from 2 weeks prior to day of apheresis up to Day 14 after resection of the injected tumor site (Day 0). Targeted therapy such as endocrine therapy or trastuzumab was allowed. Cytotoxic therapy could resume after this 6–8 week window if treatment was deemed necessary by the clinical care team.
Protocol treatment and assessments
Surgical excision of the intratumoral injected tumor occurred on Day 0 (Fig. S1) and was preceded by intratumoral injection of the RNA CAR T c-Met on Day -2. Ten research visits were required for each enrolled patient starting at week -4 (enrollment visit) to Day +25 (week +4) (safety assessment visit). Apheresis occurred in week -3 (Day -21). A pre-injection safety assessment visit occurred in Day -5. Additional safety assessment research visits on Day -2 (intratumoral injection), Day -1, Day 0 (surgical excision), Day 5, week +2 (Day +11), and week +4 (Day +25) were mandatory. Physical exam and complete blood count and blood chemistry were evaluated in each of the 10 research visits. On Day -2, patients were monitored at least 2 hours after intratumoral injection with vital signs monitoring immediately before and following intratumoral injection, and every 15 min for the first hour after intratumoral injection. Research blood samples to assess pharmacokinetics of mRNA c-Met CAR T cells were collected on Day -5, Day -2 at several time points pre and post intratumoral injection, and in all subsequent research visits.
Patients were evaluated for dose-limiting toxicity (DLT) from the time of intratumoral injection (Day -2) to Day +25. DLT reporting period was up to the initiation of the standard of care therapy or protocol Day +25 whichever came first. DLT was defined as any new hematologic or non-hematologic toxicity (NCI Common Terminology Criteria for Adverse Events (CTCAE) v4.0) which developed following dosing and is at least possibly related to mRNA c-Met CAR T cells. A DLT was defined as any of the following: 1) Grade 3 or higher non-hematologic toxicity (except asymptomatic Grade 3 electrolytes or grade 3 nausea, vomiting, diarrhea, fatigue or other events that were pre-existing regardless of grading); 2) Grade 3 or higher hematologic toxicity (except asymptomatic lymphopenia or other blood counts that were pre-existing regardless of grading); 3) Grade 2 or higher autoimmune reaction; 4) Grade 2 or higher allergic reaction or reaction that involves bronchospasm or generalized urticaria (anaphylaxis); and 5) Grade 2 or higher cytokine release syndrome. The maximum tolerated dose (MTD) by intratumoral route of administration will be defined as the dose level at which 1 patient among 6 enrolled experienced DLT.
Study oversight
The Data Safety and Monitoring Committee (SMC) of our cancer center oversaw the data quality and adherence to safety rules of this clinical trial. A protocol-specific SMC comprising of two physicians and one statistician not involved with the study was created to ensure the safety of participants. The SMC reviewed all adverse events quarterly or more frequently as needed and made recommendations to the research team on whether to continue with the study, amend the study, and/or stop/pause the study as needed.
Sample collection and processing
Peripheral blood was collected in lavender and red top Vacutainer tubes (Becton Dickinson) as described [11]. All six patients underwent excision of the injected tumor in the operating room 2 days after intratumoral injection. The excised tumor was processed by standard surgical pathology procedures. A portion of the injected tumor and adjacent non-injected tumor tissues were collected, placed in ice-cold RPMI medium and transported to our core laboratory for correlative analyses within 2 hours after the tumor was excised. The remaining tumor was fixed in formalin and embedded in paraffin for standard pathological exam. Unstained sections from these archival FFPE tumor blocks were used for IHC examination of the tumor microenvironment described above.
mRNA c-Met-CAR T cell manufacturing
Clinical-grade mRNA was transcribed in vitro from the c-Met BBZ CAR construct as described [11]. Electroporation of mRNA transcribed from the c-Met BBZ CAR construct into expanded T cells was performed as described [11]. Autologous mRNA c-Met-CAR T cells were prepared as specified in FDA IND 15014 in the Cell and Vaccine Production Facility (CVPF) at the University of Pennsylvania according to standard operating procedures established for this purpose. The cells were not released from the CVPF until FDA-specified release criteria (e.g., cell purity, sterility, potency, pyrogenicity, etc.) were verified.
Polymerase chain reaction (PCR) analyses of mRNA transcripts and multiplex cytokine analyses
Blood samples were collected for all six patients at the following time points: 20 min, 2 hours, 1 day, 2 days, 7 days, and 14 days after intratumoral injection. mRNA was isolated directly from whole blood from each time point and fresh tumor tissues using Ribopure™ blood kits (Ambion). cDNA was synthesized and used in quantitative PCR (qPCR) assays to detect and quantify the abundance of transgene (CAR T construct) and CD3ε (total T-cell) transcripts. The proportion of CAR T cells to total T cells was determined using the transgene value/normalized CD3ε value as described [11].
Serum cytokines were examined as previously described [20]. Briefly, human cytokine magnetic 30-plex panel (catalog number LHC6003M) was purchased from Life Technologies (Carlsbad, CA). Serum samples cryopreserved at -80°C from day -5 or baseline to day +25 were thawed and batch analyzed in duplicate according to the manufacturers’ protocols. Assay plates were measured using a FlexMAP 3D instrument (Luminex, Austin, TX), and data acquisition and analysis were done using xPONENT software (Luminex).
Statistical considerations
We compared the clinical and tumor characteristics of our patient cohort as stratified by c-Met expression using a two-sided Student t-test for continuous variables (i.e. age, tumor size, and number of involved axilla nodes). We used the two-tailed Fisher’s Exact Test to determine if the distribution of breast cancer subtype, a categorical variable, was significantly different between c-Met+ and c-Met- tumors.
Results
c-Met is frequently expressed in breast cancer
Breast cancer is known to express c-Met [21, 22]. To confirm that c-Met is indeed a versatile tumor antigen in breast cancer irrespective of the various breast cancer subtypes, we first assessed the expression of c-Met in archived primary breast cancer specimens. We examined contemporary breast cancer tissue samples from patients treated at our institution in a pilot patient cohort with primary operable breast cancer. To evaluate whether c-Met expression may be associated with common prognostic variables such as tumor size, nodal status, and breast tumor subtype classifications, we evaluated c-Met expression by immunohistochemistry (IHC) staining. Expression of c-Met was heterogeneous within and across tumor samples in terms of the proportion of tumor cells staining positive for c-Met and the intensity of IHC staining (Supplementary Fig. S2). We stratified tumors into two subgroups by c-Met+ or c-Met- status. Results were summarized in Supplementary Table S1. Overall, c-Met was expressed in 46% of our tumor samples. Tumor size and nodal status were similar between the two subgroups. When we further stratified our tumor samples into tumor subtypes by estrogen (ER), progesterone (PR) and Her-2/neu (Her2) receptor expression status, we found that c-Met was expressed in all three breast cancer subtypes.
mRNA c-Met-CAR T cells kill c-Met+ breast cancer cells in vitro
As our data confirm a previous report that c-Met is commonly expressed in breast cancer [23], we hypothesized that c-Met-CAR T cells may be an effective targeted therapy for breast cancer. Because normal epithelial tissues such as hepatocytes also express c-Met at low levels, permanently transduced CAR T cells may lead to undesirable effects in which the correct molecule is recognized (on-target) but the wrong tissue would be affected (off-tumor). Therefore, our initial approach was to use mRNA-based CAR T cells as a safer alternative, since the resultant CAR expression would be transient and become negligible by day 7 after mRNA electroporation [12].
To demonstrate the effectiveness of mRNA c-Met-CAR T cells in mediating CAR T-directed cell lysis, we evaluated the cytolytic activity of CAR T cells on two breast cancer cell lines: BT20, a commercially available breast cancer cell line derived from TNBC, and TB129, a primary breast cancer cell line derived from a patient with Her2 amplified breast cancer as previously described [14]. Both breast cancer cell lines expressed c-Met at similar levels and mesothelin, another tumor antigen, at varying levels (Fig.1, A & B, lower panels). CAR constructs targeting c-Met (cMet.BBz) and mesothelin (SS1.BBz) [18] have been developed. Cytolytic activity of cMet.BBz, SS1.BBz, and CD19.BBz (a negative control CAR T directed against CD19, a B cell–specific tumor marker) was measured as described [18]. As shown in Fig. 1A (upper panel), c-Met directed lysis of BT20 increased from ~40% to ~100% when the effector cell (CAR T) to tumor cell (E:T) ratio increased from 1:1 to 5:1. A less robust cytolytic activity was noted when BT20 was treated with mRNA meso-CAR T cells. This was likely due to a lower mesothelin expression in BT20 as demonstrated on flow cytometry analysis (Fig. 1A lower panel). As expected, CD19-CAR T cells did not have cytolytic activity against BT20 since BT20 lacked CD19 expression, also demonstrating that the T cells did not exhibit non-self (allogeneic) MHC reactivity against the tumor. The cytolytic activities of mRNA c-Met-CAR T cells and meso-CAR T cells against BT20 were significantly higher than mRNA CD19-CAR T cells with P values = 0.0001 and 0.0015 respectively. Similar results were obtained when TB129 was treated with mRNA c-Met-CAR T cells (cMet.BBz). As the E:T ratio increased from 1:1 to 5:1, c-Met directed lysis increased from ~40 to 80% (Fig. 1B upper panel). mRNA meso-CAR T cells were also effective in mediating mesothelin-directed lysis of TB129 whereas mRNA CD19-CAR T cells did not have lytic activity on TB129 as expected. The cytolytic activities of mRNA c-Met-CAR T cells and meso-CAR T cells against TB129 were significantly higher than mRNA CD19-CAR T cells with P values = 0.0001 for both.
Fig. 1. Cytotoxicity of c-Met-CAR T cells.
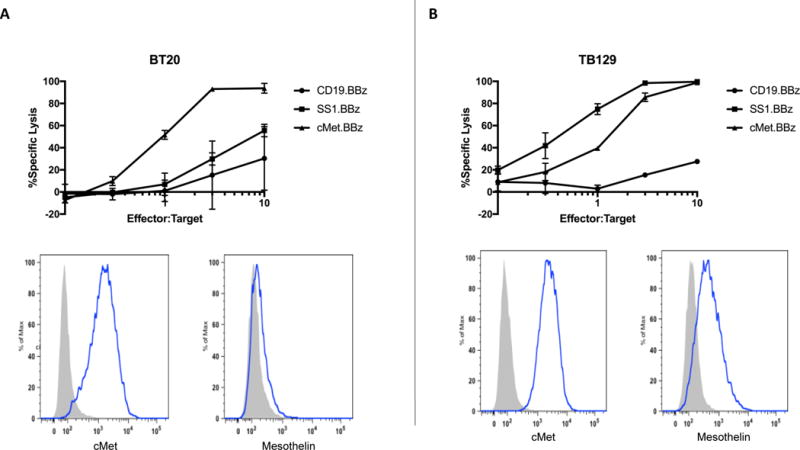
Chromium release cytolytic activity assays of mRNA CAR T cells in two breast cancer cell lines, BT20 and TB129. Each assay was performed in duplicates and repeated in at least three independent experiments. A) Upper panel, BT20 tumor cell lysis induced by CAR T directed against c-Met (cMet.BBz, solid triangles), mesothelin (SS1.BBz, solid squares) or CD19 (CD19.BBz, solid circles) at various effector T cell:tumor cell (E:T) ratios. mRNA c-Met-CAR T cells were more effective than mRNA meso-CAR T cells in inducing CAR T cell directed tumor cell lysis. Lower panel, histograms of flow cytometry analyses of c-Met and mesothelin expression on BT20 cells as stained by antibodies against c-Met and mesothelin. B) Upper panel, TB129 tumor cell lysis induced by CAR T directed against c-Met (cMet.BBz, solid triangles), mesothelin (SS1.BBz, solid squares) or CD19 (CD19.BBz, solid circles) at various E:T ratios. mRNA c-Met-CAR T cells cytolytic activity was similar to that of mRNA meso-CAR T cells in inducing CAR T directed tumor cell lysis. Lower panel, histograms of flow cytometry analyses of TB129 cells as stained by antibodies against c-Met and mesothelin.
Antitumor effects of mRNA c-Met-CAR T cells in a xenograft model
We have previously demonstrated the effectiveness of stably transduced c-Met-CAR T cells in shrinking pre-established c-Met expressing tumors in NSG mouse tumor models [16]. We have also performed a side-by-side comparison of the effectiveness of stably transduced vs. mRNA electroporated CAR T cell directed against another tumor antigen, mesothelin, in reducing tumor growth [12]. In disseminated intraperitoneal tumors established in NSG mice with 8×106 M108-Luc, a human mesothelioma cell line, tumor shrinkage was observed in mice treated with four consecutive intraperitoneal injections of mRNA meso-CAR T cells. mRNA meso-CAR T cells were effective but less so than stably transduced meso-CAR T cells in shrinking pre-established tumors [12]. We also noted that expression of CAR in these transiently modified mRNA meso-CAR T cells persisted for 4 days but disappeared by day 7 after electroporation. We extrapolated from these results and hypothesized that mRNA c-Met-CAR T cells may have activity in shrinking pre-established tumors in our xenograft tumor model and that the activity of these mRNA c-Met-CAR T cells would be transient and limit potential on-target off-tumor effects in our pilot study. To evaluate the effectiveness of intratumoral injections of mRNA c-Met-CAR T cells in controlling tumor growth in pre-established c-Met expressing tumors in vivo, SK-OV-3/luc, a Click beetle green luciferase-labeled c-Met expressing human ovarian cancer cell line [16], was implanted into the flanks of NSG mice as described [12]. Tumor bearing mice were treated with four intratumoral injections of mRNA c-Met-CAR T cells at weeks 6, 7, 9, and 11 (Fig. 2). Our results demonstrated that multiple injections with mRNA c-Met-CAR T cells were effective in controlling tumor growth when compared to intratumoral injections with mRNA CAR T CD19, given as a control for allogeneic effects. Tumor control continued until week 14, 3 weeks following the final mRNA c-Met-CAR T cell intratumoral injection (Fig. 2). Whether extravasation of these intratumorally injected mRNA c-Met-CAR T cells has occurred in the treated mice is unknown. However in a previous study, we were able to detect 1 × 103 – 1 × 104 human T cells in peripheral blood of treated mice 40 days after intraperitoneal injection of mRNA CAR T cells, indicating that extravasation of some of these intraperitoneal injected CAR T cells had occurred [19].
Fig. 2. Antitumor efficacy of mRNA c-Met-CAR T cells in NSG mice.
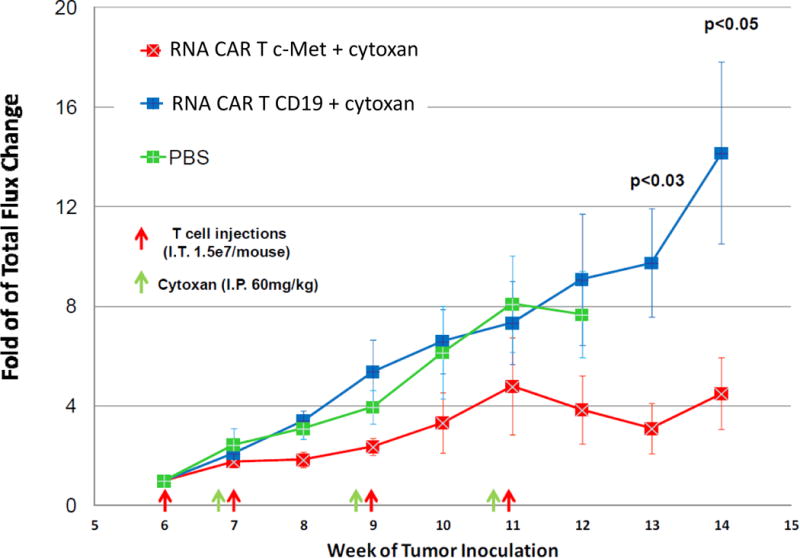
Each experiment was performed using groups of 7–8 female and male NOD/scid/γc(–/–) (NSG) mice and repeated in one independent experiment without the use of cyclophosphamide. Tumor growth curve as measured by bioluminescence imaging demonstrated effectiveness of multiple intratumoral injections of mRNA c-Met-CAR T cells into pre-established c-Met+ Click beetle green luciferase labeled tumor xenografts (SK-OV-3/luc) in controlling tumor growth in NSG mice. Red squares depicted bioluminescence fold change in tumor size of mRNA c-Met-CAR T cells treated mice; blue square depicted those of mRNA CD19-CAR T cell (allogeneic control) treated mice; and green squares depicted those of phosphate buffered saline (PBS, negative control) treated mice. Intratumoral injection time points were depicted by red arrows. Intraperitoneal (IP) injections of Cytoxan (cyclophosphamide) were given 24 hours prior to the intratumoral injections as depicted by green arrows to eliminate the previous T cells, preventing the development of graft-versus-host disease (21).
Phase 0 trial to test mRNA c-Met-CAR T cells
We conducted a phase 0 study to test the safety and feasibility of intratumoral injections of CAR T cells. We are leveraging our experience with mesothelin targeted CAR T cells to establish whether c-Met might serve as a target in patients with advanced breast cancer. Patients with metastatic breast cancer presenting with accessible cutaneous or lymph node metastases received a single intratumoral injection at one of two mRNA c-Met-CAR T cell dose levels: 3×107 and 3×108 (n = 3 per cohort). Clinical grade mRNA c-Met-CAR T cells were successfully manufactured for all patients (Table 1).
Table 1.
Apheresis products and cMet CAR T cell product characterization.
| Assay | Specification | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 |
|---|---|---|---|---|---|---|---|
| Apheresis Product | |||||||
| Flow Cytometry CD3+, CD45+ |
N/A | 63.20% | 31.40% | 42.40% | 65.90% | 56.90% | 44.50% |
| CD4/CD8 ratio | N/A | 1.57 | 1.45 | 1.57 | 2.87 | 1.34 | 1.11 |
| cMet CART Release Criteria | |||||||
| Total Cell Number Infused | 3 × 107 – 1st 3 subjects 3 × 108 – 2nd 3 subjects |
3 × 107 | 3 × 107 | 3 × 107 | 3 × 108 | 3 × 108 | 3 × 108 |
| Cell Viability | >= 70% | 73.60% | 91.20% | 90.00% | 85.00% | 95.50% | 96.30% |
| % CD3+ of CD45+ Cells (post-expansion)1 |
>= 80% | 97.60% | 96.80% | 93.50% | 93.60% | 97.20% | 75.0%2 |
| Residual Bead # | <= 100 beads/3 × 106 Cells | 4 | 2 | 1 | 1 | 0 | 0 |
| Endotoxin | <= 3.5 EU/mL | <1 EU/mL | <1 EU/mL | <0.5 EU/mL | <0.5 EU/mL | <0.5 EU/mL | <0.5 EU/mL |
| Mycoplasma | Negative | Negative | Negative | Negative | Negative | Negative | Negative |
| Sterility (Bactec) | No Growth | No Growth | No Growth | No Growth | No Growth | No Growth | No Growth |
| Fungal Culture | No Growth | No Growth | No Growth | No Growth | No Growth | No Growth | No Growth |
| Transfection Efficiency | >= 20% | 85.80% | 69.40% | 79.30% | 39.60% | 44.80% | 36.50% |
Release assay performed post expansion and prior to overnight incubation, electroporation, and formulation.
Following electroporation, the final product was assayed. CD3+ of CD45+ cells were 85.9%.
The clinical characteristics and outcome of the six subjects with median follow up of 10 months (range, 3 – 28 months) were summarized in Table 2. Four of six patients had metastatic triple negative breast cancer (TNBC), whereas the remaining two had ER+Her2− metastatic breast cancer. Intratumoral injection of mRNA c-Met-CAR T cells was well tolerated in all 6 patients. All adverse events (AE) and serious adverse events (SAE) were summarized in Supplementary Table S2. Grade I erythema occurred at the intratumoral injection sites in 3 of 6 patients. All 6 patients reported mild subjective myalgia, which resolved within 24 hours. One patient reported prolonged myalgia/arthralgia which resolved within 2 weeks after intratumoral injection. This patient’s subjective symptoms were not attributable to IL-6 release (Table S3). There were six grade 3 AE: 1) pain at skin graft donor site on right thigh of one patient which was attributed to skin graft procedure performed at the time of surgical resection of the injected tumor to provide skin coverage of the resected chest wall tumor; 2) four grade 3 AE for nausea and vomiting were reported from another patient who presented to the emergency department four times for severe nausea and vomiting between week +2 and week +4 and required hospital admission. This patient had prior radiation treatment to her known liver metastases and her symptoms were attributed to radiation enteritis. One episode of grade 2 hypotension was recorded in one patient and was attributed to dehydration from her nausea/vomiting; 3) Grade 3 anemia requiring transfusion was reported on day +1 in one patient which was attributed to blood loss from her surgical resection and skin graft procedure. All grade 3 SAE were deemed unrelated to the study drug.
Table 2.
Clinical Characteristics of trial participants
| ID | Age | Race | Breast Cancer Subtype | C-met IHC status | Sites of Metastasis | IT site | Disease status |
|---|---|---|---|---|---|---|---|
| 1 | 58 | White | TNBC | 2+, 100% | Chest wall | chest wall | DOD |
| 2 | 47 | Black | ER+ Her2- | 2+, 100% | Left breast, Bone, liver, mediastinal nodes | Left Breast | DOD |
| 3 | 44 | White | TNBC | 2+, 90% | left supraclavicular, bilateral cervical nodes, right axilla nodes | right axilla node | DOD |
| 4 | 64 | White | ER+ Her2- | 1+, 60% | Right breast, Bone | right breast | SD |
| 5 | 55 | Black | TNBC | 1+, 90% | Right breast, Skin, liver, left axilla | chest wall | PD |
| 6 | 64 | Asian | TNBC | 2+, 70% | right mastectomy and left mastectomy Skin flaps | Right chest wall | PD |
Abbreviations used: TNBC - triple negative breast cancer; ER - estrogen receptor; Her2 - Her-2/ne; DOD - dead of disease; SD - stable disease; PD - Progressed disease;
No measurable clinical responses were observed (Table 2).
Pharmacokinetics of mRNA c-Met-CAR T cells
We performed qPCR to measure the amount of CAR T transgene relative to CD3ε transcripts in PBMC and tumor biopsies. Low levels of CAR mRNA were detected in peripheral blood or in tumor tissues in 5 of 6 patients (Table 3). Low levels of CAR mRNA were detected in the blood samples collected at 20 min after intratumoral injection of two patients and at 2 hours after intratumoral injection in one patient. This was likely due to extravasation of mRNA c-Met-CAR T cells in peripheral blood. CAR mRNA was not detected in blood samples collected at subsequent time points (at 1, 2, 7 and 14 days after intratumoral injection) from all six patients (Table 3). As one patient reported prolonged myalgia and had the highest level of CAR mRNA detected in the tumor biopsy, we performed multiplex cytokine analyses using serum collected from this patient before and after intratumoral injection. Elevated serum IL6 levels were not observed in this patient (Table S3).
Table 3.
qPCR to detect RNA CART c-Met in PBMC and tissues expressed as normalized ratio CART/CD3 × 100
| Patient ID | Pre-injection PBMC | 20 min post IT PBMC | 2 hours post IT PBMC | Day 4 post IT PBMC | Tumor tissue at IT injection site | Tumor tissue away from IT injection site |
|---|---|---|---|---|---|---|
| 1 | ND | ND | ND | ND | 0.02376 | ND |
| 2 | ND | 0.00441 | ND | ND | ND | ND |
| 3 | ND | ND | ND | ND | 0.000491 | ND |
| 4 | ND | ND | ND | ND | ND | ND |
| 5 | ND | 0.0059 | 0.0005 | ND | 0.278 | 3.46 |
| 6 | ND | ND | ND | ND | 1.6 | ND |
Abbreviations used: PBMC - peripheral blood mononuclear cells; ND - not detected
mRNA c-Met-CAR T cells induce necrosis and inflammation in the tumor microenvironment
As resection of the intratumorally injected tumors was part of the trial schema (Supplementary Fig. S1), we evaluated the effects of CAR T cells on tumor tissues two days after intratumoral injection. Pre-treatment tumor tissues and tumor tissues surrounding the intratumoral injection site and away from the intratumoral injection site from two patients (Fig. 3 and supplementary Fig. S3) were evaluated histologically by H&E stain. We noted necrosis, hemorrhage and inflammatory cell infiltration at the intratumoral injection site (Fig. 3). Tumor cells, which were intact in the pre-treatment and non-injection site, no longer displayed a discernable cell membrane or nuclei on H&E. The IHC staining also demonstrated loss of c-Met immunoreactivity at the injected site, in contrast to the bright staining for c-Met in the baseline tumor (Fig. 3). Overall, the intratumoral injection site was associated with polymorphonuclear (PMN) and mononuclear immune cell infiltration. The PMNs within the tumor microenvironment had the characteristics of neutrophils on H&E. Similar results were seen at the intratumoral injection site of the second patient (Supplementary Fig. S3). In addition, we compared the tissue effects of intratumoral injection of 1 mL of lidocaine (as negative control) and 1 mL of mRNA c-Met-CAR T cells in one patient and noted that intratumoral injection of mRNA c-Met-CAR T cells, not lidocaine, resulted in more immune cell infiltration (Supplementary Fig. S4).
Fig 3. Histology and IHC of tumor tissue pre and post intratumoral injection of mRNA c-Met-CAR T cells from one treated patient.
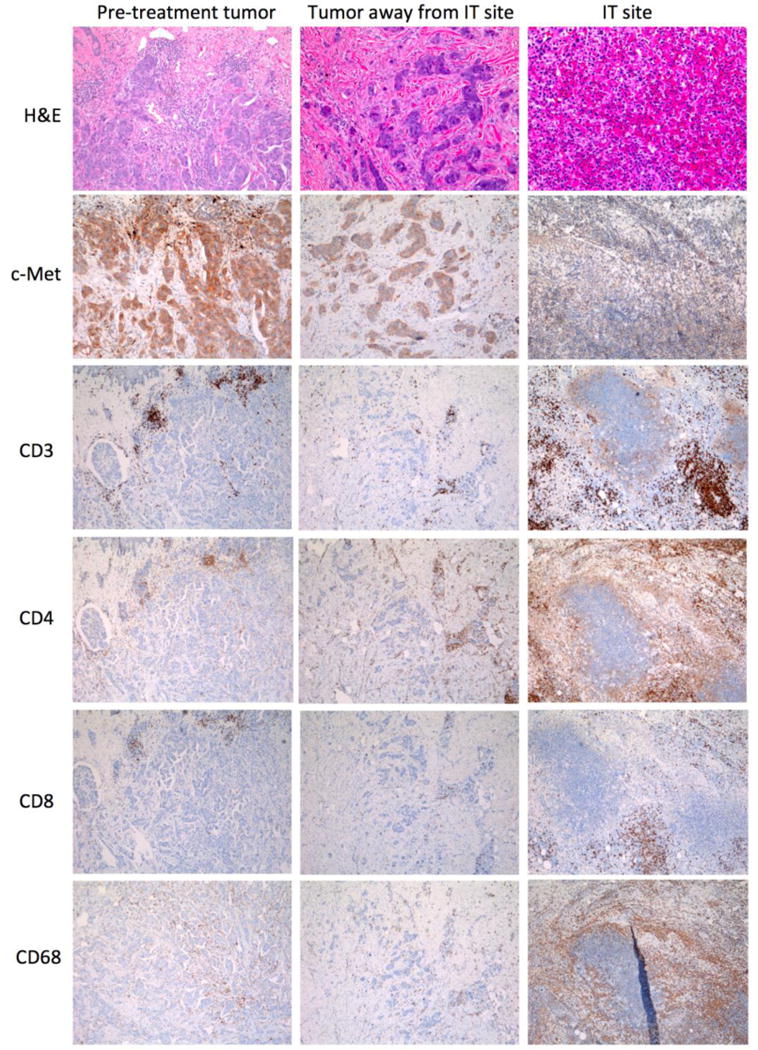
Histologic (H&E) and IHC evaluation of c-Met expression and T cell (CD3+, CD4+, CD8+) and macrophage (CD68+) infiltration in tumor tissues of pre-treatment, away from intratumoral injection site and at intratumoral injection site from a patient treated with 3 × 108 mRNA c-Met-CAR T cells.
To further characterize the mononuclear cells within the tumor microenvironment, we stained consecutive tumor sections from two patients with markers of T cells, T-helper cells, cytotoxic T cells and monocytes/macrophages with anti-CD3, CD4, CD8, and CD68, respectively. We noted that the T cells within the intratumoral injection site were predominantly CD4+ and that some of the infiltrated immune cells were CD68+ macrophages (Supplementary Fig. S3 and Fig. 3). We further characterized these CD68+ tumor infiltrated immune cells by multiplex IHC as described in the methods section. Comparison of the CD68+ cells within the pre-treatment and post intratumoral injection sites demonstrated heterogeneity in the infiltrated myeloid cells which have phenotypes associated with immune suppression, e.g. an increase in the ratio of CD163/CD68 at the intratumoral injection site (Supplementary Fig. S5).
The RT-PCR results comparing CAR T with CD3ε transcripts in tumor tissue at the intratumoral site in one patient two days after intratumoral injection demonstrated that CAR T transcripts comprised 1.6% of total CD3ε transcripts, indicating that most of the CD3+ cells in the tumor tissues were native T cells, not mRNA c-Met-CAR T cells. However, it is not possible to determine whether the tumor infiltrating T cells were previously expressing the CAR and whether the mRNA and CAR protein had since been metabolized. Alternatively, it is possible that an influx of T cells was triggered after the initial injection of mRNA c-Met-CAR T cells.
In addition, we also performed IHC with CD56, a marker for natural killer cells, and S100, a marker of dendritic cells or Langerhans histiocytes, and the results of the staining indicated no contribution of natural killer cells or dendritic cells within the tumor microenvironment at the intratumoral injection site.
Discussion
This study reports the development of a CAR T cell targeting c-Met and the results of our Phase 0 clinical trial evaluating the safety and feasibility of intratumoral injection of mRNA c-Met-CAR T cells for patients with metastatic breast cancer expressing c-Met. The study met the feasibility endpoint as clinical grade CAR T-cell products were successfully manufactured for all subjects. In addition, the protocol met the safety endpoint as the mRNA c-Met-CAR T-cell injections were well tolerated. Specifically, no patient experienced cytokine release syndrome.
Two dose levels of CAR T cells were administered. There was no obvious dose-response relationship; however, the patient with prolonged myalgia and extensive tumor necrosis on biopsy was in the higher dose cohort. The c-Met-CAR T cell mRNA was detectable at low levels in peripheral blood in 2 of 6 patients without any adverse effects.
One of our objectives was to evaluate the direct effects of mRNA c-Met-CAR T cells on breast cancer tumor tissues. Two observations suggest that the mRNA c-Met-CAR T cells had on-target effects after intratumoral injection. First, there was a loss of c-Met expression in the post injection biopsy compared to the pre-injection tumor sample from two evaluable patients. The loss of c-Met expression was confined to the biopsy obtained from the injection site, whereas c-Met expression was retained in the biopsy sample from non-injected tumor. Second, there was necrosis in the injected tumor tissue and not at the non-injected tumor biopsy suggesting that some the observed tumor cell necrosis or lysis may be a consequence of cytolytic activity mediated by mRNA c-Met-CAR T cells. However, in the absence of a side-by-side comparison between intratumoral injection of activated/unmodified T cells vs. mRNA c-Met-CAR T cells, we could not ascertain whether mRNA c-Met-CAR T cells were solely responsible for the observed tissue effects since activated unmodified T cells could also contribute to tumor necrosis. As we had previously demonstrated that stably transduced meso-CAR T cells but not CD19-CAR T cells (a control CAR T cell) were effective in shrinking mesothelin+ tumors in mice whereas CD19-CAR T cells did not have antitumor effect [18], we concluded that the observed tumor necrosis was at least in part attributable to specific effects from mRNA c-Met-CAR T cells.
In addition, c-Met-CAR T mRNA was detected in peripheral blood after intratumoral injection from two of six patients suggesting that extravasation of mRNA c-Met-CAR T cells had occurred (Table 3). Since no serious adverse effects were noted in these two patients, we postulate that systemic infusion of mRNA c-Met-CAR T cells may be tolerated. We will evaluate this in a future clinical trial based on present results that demonstrate that mRNA c-Met-CAR T cells induce necrosis in tumors of patients with advanced metastatic breast cancer.
Given the heterogeneous expression of c-Met in breast tumors, we anticipate that antigen-directed tumor lysis may spare tumor cells that do not express c-Met. However, antigen-directed tumor lysis may result in the release of other tumor antigens. An immune response against these additional tumor specific antigens may be elicited, a process known as epitope spreading. We have demonstrated this phenomenon of epitope spreading in a previous publication describing two patients treated by systemic infusion and intratumoral injection of mRNA meso-CAR T cells [11]. Furthermore, it is possible to speculate that tumors that lose the benefit of c-Met signaling may be less aggressive. The recruitment of macrophages into the intratumoral site coupled with tumor necrosis further raises the possibility that some of these cells may serve as antigen-presenting cells, which when appropriately stimulated, could induce epitope-spreading and enhance anti-tumor immunity against tumor neoantigens through cross-presentation. In addition to the macrophages, it is also possible that the T cells themselves could serve as antigen presenting cells [24, 25]. Such vaccine response is, however, unlikely to result from a single intratumoral injection of mRNA c-Met-CAR T cells. An attractive strategy may be to combine CAR T-cell injections with injections of a stimulator of interferon gene (STING) agonist to provoke or prime T-cell responses to neoantigens [26].
In conclusion, intratumoral injections of mRNA c-Met-CAR T cells are well tolerated and elicit an inflammatory response within tumors. This proof of concept study will pave the way for the use of intratumoral injection of mRNA CAR T as a platform to evaluate CAR T or other immunotherapy strategies in the treatment of tumors by intratumoral injection. As the expression of CARs in mRNA CAR T cells is transient, mRNA CAR T cells serve as a platform to allow us to evaluate the safety of CAR T directed against tumor antigens in clinical trial settings by avoiding the potential unrelenting on-target off-tumor effects of stably transduced CAR T. A phase 1 trial (NCT03060356) to assess the safety of systemically administered mRNA c-Met-CAR T cells in the treatment of metastatic breast cancer or melanoma is underway with emphases on vigilant monitoring and supportive management of potential toxicities through procedures such as the use of tocilizumab in the management of cytokine release syndrome [27, 28]. In the event of other toxicity, a method to attenuate or eliminate c-Met-CAR T cells will be introduced.
Supplementary Material
Acknowledgments
The authors acknowledge the contributions of Alexander Malykhin and members of the Clinical Cell and Vaccine Production Facility, the Translational and Correlative Studies Laboratory, regulatory support from the Center for Cellular Immunotherapies, and the Tumor Tissue and Biospecimen Bank (TTAB) of the Abramson Cancer Center. The authors also acknowledge Kimberly S. Smythe (FHCRC Experimental Histopathology) for performing the multiplex immunohistochemistry, supported in part by the FHCRC/UW Cancer Consortium Cancer Center Support Grant of the National Institutes of Health (P30 CA015704).
Grant Support
This research was, in part, funded by the NCI Cancer Center Support Grant (2-P30-CA-016520-35) (JT), NCI 5R01CA120409 (YZ and CHJ), the Breast Cancer Alliance Research Foundation (JT), the Breast Cancer Immunotherapy Funds (JT), the Breast Cancer Research Foundation (RHV and AM), and the Pennsylvania Department of Health Cure Grant #0972501 (CHJ). CHJ and RHV are members of the Parker Institute for Cancer Immunotherapy, which supports the University of Pennsylvania Cancer Immunotherapy Program.
Footnotes
Disclosure of Potential Conflicts of Interest
The University of Pennsylvania has a strategic alliance with Novartis for the development of chimeric antigen receptors. This arrangement is managed in accordance with the University of Pennsylvania’s Conflict of Interest Policy. The authors are in compliance with this policy. Y.Z., B.L.L, and C.H.J. are co-founders of Tmunity Therapeutics.
References
- 1.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, Ambrose D, Grupp SA, Chew A, Zheng Z, Milone MC, Levine BL, Melenhorst JJ, June CH. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beatty GL, O’Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol Ther. 2016;166:30–39. doi: 10.1016/j.pharmthera.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 6.Ghoussoub Ra, Dillon Da, D’Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82:1513–20. doi: 10.1002/(sici)1097-0142(19980415)82:8<1513::aid-cncr13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, Carano RAD, Kasman I, Mai E, Young J, Zha J, Zhang Z, Ross S, Schwall R, Colbern G, Merchant M. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68:4360–8. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 8.Martens T, Schmidt N-O, Eckerich C, Fillbrandt R, Merchant M, Schwall R, Westphal M, Lamszus K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. 2006;12:6144–52. doi: 10.1158/1078-0432.CCR-05-1418. [DOI] [PubMed] [Google Scholar]
- 9.Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, Huang A, Yang N, Nishimura M, Greve J, Santell L, Zhang Y-W, Su Y, Kaufman DW, Billeci KL, Mai E, Moffat B, Lim A, Duenas ET, Phillips HS, Xiang H, Young JC, Vande Woude GF, Dennis MS, Reilly DE, Schwall RH, Starovasnik MA, Lazarus RA, Yansura DG. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110:E2987–96. doi: 10.1073/pnas.1302725110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pérol M. Negative results of METLung study: an opportunity to better understand the role of MET pathway in advanced NSCLC. Transl lung cancer Res. 2014;3:392–4. doi: 10.3978/j.issn.2218-6751.2014.09.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, Kalos M, June CH. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, Albelda SM, June CH. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kummar S, Rubinstein L, Kinders R, Parchment RE, Gutierrez ME, Murgo AJ, Ji J, Mroczkowski B, Pickeral OK, Simpson M, Hollingshead M, Yang SX, Helman L, Wiltrout R, Collins J, Tomaszewski JE, Doroshow JH. Phase 0 clinical trials: conceptions and misconceptions. Cancer J. 2008;14:133–137. doi: 10.1097/PPO.0b013e318172d6f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tchou J, Wang LC, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, Kalos M, Vondeheide RH, Albelda SM, June CH, Zhang PJ. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133:799–804. doi: 10.1007/s10549-012-2018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li YR, Xian RR, Ziober A, Conejo-Garcia J, Perales-Puchalt A, June CH, Zhang PJ, Tchou J. Mesothelin expression is associated with poor outcomes in breast cancer. Breast Cancer Res Treat. 2014;147:675–84. doi: 10.1007/s10549-014-3077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD, Ang S, Cooper LJN, Platt JM, Johnson FB, Paulos CM, Zhao Y, Kalos M, Milone MC, June CH. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crisanti MC, Wallace AF, Kapoor V, Vandermeers F, Dowling ML, Pereira LP, Coleman K, Campling BG, Fridlender ZG, Kao GD, Albelda SM. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol Cancer Ther. 2009;8:2221–2231. doi: 10.1158/1535-7163.MCT-09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Jiang S, Fang C, Li H, Zhang X, Zhang F, June CH, Zhao Y. Novel T cells with improved in vivo anti-tumor activity generated by RNA electroporation. Protein Cell. 2017;8:514–526. doi: 10.1007/s13238-017-0422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, Barrett DM, Weiss SL, Fitzgerald JC, Berg RA, Aplenc R, Callahan C, Rheingold SR, Zheng Z, Rose-John S, White JC, Nazimuddin F, Wertheim G, Levine BL, June CH, Porter DL, Grupp SA. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho-Yen CM, Jones JL, Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res. 2015;17:52. doi: 10.1186/s13058-015-0547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho-Yen CM, Green AR, Rakha EA, Brentnall AR, Ellis IO, Kermorgant S, Jones JL. C-Met in Invasive Breast Cancer Is There a Relationship With the Basal-Like Subtype? doi: 10.1002/cncr.28386. [DOI] [PubMed] [Google Scholar]
- 23.Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15:2207–14. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- 24.Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science. 2005;309:264–8. doi: 10.1126/science.1110267. [DOI] [PubMed] [Google Scholar]
- 25.Foster AE, Leen AM, Lee T, Okamura T, Lu A, Vera J, Atkinson R, Bollard CM, Dotti G, Rooney CM. Autologous designer antigen-presenting cells by gene modification of T lymphocyte blasts with IL-7 and IL-12. J Immunother. 2007;30:506–16. doi: 10.1097/CJI.0b013e318046f3b1. [DOI] [PubMed] [Google Scholar]
- 26.Corrales L, McWhirter SM, Dubensky TW, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126:2404–11. doi: 10.1172/JCI86892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DCG, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia HHS Public Access. Sci Transl Med Febr. 2014;19:224–25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.