

Abstract
Purpose
Plasma cell-free DNA (cfDNA) analysis is increasingly used clinically for cancer genotyping, but may lead to incidental identification of germline risk alleles. We studied EGFR T790M mutations in non-small cell lung cancer (NSCLC) toward the aim of discriminating germline and cancer-derived variants within cfDNA.
Experimental Design
Patients with EGFR-mutant NSCLC, some with known germline EGFR T790M, underwent plasma genotyping. Separately, deidentified genomic data and buffy coat specimens from a clinical plasma next-generation sequencing (NGS) laboratory were reviewed and tested.
Results
In patients with germline T790M mutations, the T790M allelic fraction (AF) in cfDNA approximates 50%, higher than that of EGFR driver mutations. Review of plasma NGS results reveals three groups of variants: a low AF tumor group, a heterozygous group (~50% AF), and a homozygous group (~100% AF). As the EGFR driver mutation AF increases, the distribution of the heterozygous group changes, suggesting increased copy number variation from increased tumor content. Excluding cases with high copy number variation, mutations can be differentiated into somatic variants and incidentally identified germline variants. We then developed a bioinformatic algorithm to distinguish germline and somatic mutations; blinded validation in 21 cases confirmed a 100% positive predictive value for predicting germline T790M. Querying a database of 31,414 patients with plasma NGS, we identified 48 with germline T790M, 43 with non-squamous NSCLC (p<0.0001).
Conclusion
With appropriate bioinformatics, plasma genotyping can accurately predict the presence of incidentally detected germline risk alleles. This finding in patients indicates a need for genetic counseling and confirmatory germline testing.
Keywords: NSCLC, EGFR T790M, germline genetics, plasma genotyping
Introduction
Genomic analysis of cancer specimens has been widely adopted as a tool for guiding precision cancer care. Many academic and commercial labs now offer next-generation sequencing (NGS) panels (1,2) to identify targetable somatic mutations. One limitation of tumor tissue NGS is the frequent inability to clearly distinguish whether detected variants represent somatic events, potentially driving tumor growth, or underlying germline events, potentially representing inherited risk alleles or benign inherited polymorphisms (3). Indeed, some have advocated for routine paired analysis of tumor DNA and germline DNA to better distinguish potentially targetable somatic alterations (3). However, sequencing of germline DNA requires collection of an additional germline biospecimen, increases cost, and may require additional patient consent due to ethical considerations (4).
One rapidly emerging alternative to tumor tissue genotyping is genomic analysis of plasma cell-free DNA (cfDNA), which can noninvasively identify targetable somatic genomic variants. Because there is variability from patient to patient in regards to how much tumor DNA is shed into the plasma, plasma cfDNA represents a dynamic mixture of germline cfDNA and circulating tumor-derived cfDNA (ctDNA). When ctDNA levels are high, plasma genotyping is technically similar to tumor tissue genotyping; when tumor DNA is absent, cfDNA genotyping is technically more similar to germline genotyping. In practice, ctDNA levels are typically between these extremes, which could theoretically allow for the segregation of somatic and germline cfDNA variants. We hypothesized that genomic analysis of plasma cfDNA could permit simultaneous tumor and germline genotyping, with accurate resolution of tumor-derived variants and germline variants.
To address this question, we chose to study somatic and germline variants in the EGFR gene, which include known germline variants and a well described group of oncogenic mutations present in 10–20% of NSCLC patients. One EGFR mutation, T790M, can be detected rarely as a germline variant where its presence has been associated with familial lung cancer (5). Families harboring germline EGFR T790M mutations are currently under investigation at our center as part of an ongoing multicenter clinical trial (NCT01754025) (6). EGFR T790M is more commonly seen as an acquired somatic mutation after a patient with NSCLC develops resistance to EGFR tyrosine kinase inhibitors (TKIs). Testing for EGFR T790M has become routine because lung cancers harboring T790M-mediated resistance after initial therapy exhibit exquisite sensitivity to a third-generation EGFR TKI, osimertinib (7). Due to the challenges of repeat biopsies in patients with advanced NSCLC, plasma genotyping to test for T790M has become common in patients with resistance to first-generation EGFR TKIs, and this is an area of active investigation at our center (8). The co-existence of two research programs studying both germline T790M as well as plasma genotyping for acquired T790M created an opportunity to study the behavior of these germline variants in plasma cfDNA.
Case report
A 49-year-old never smoker with a family history of lung cancer presented with metastatic lung adenocarcinoma; tissue genotyping showed EGFR L858R and T790M mutations. Given its preclinical activity against both EGFR L858R and T790M mutations (9), first-line afatinib was started; however, the patient returned with progressive brain metastases after only 2 months of therapy. Plasma NGS then demonstrated the previously observed EGFR L858R at an allelic fraction (AF) of 5.3%, while the EGFR T790M allele was detected at 50.9% AF (Supplemental Figure 1A). The patient was initiated on osimertinib, an EGFR TKI active in the setting of EGFR T790M-mediated resistance to initial EGFR TKI (7), and had clinical benefit lasting 9 months, at which time scans showed early progression in the lung. Repeat plasma NGS showed a reduced EGFR L858R at 0.6% AF but T790M relatively stable at 49.2% AF (Supplemental Figure 1B). After further disease progression, repeat NGS demonstrated increased levels of EGFR L858R at 18% AF, T790M at 54% AF, and a third EGFR mutation that mediates acquired resistance to osimertinib, C797S, at 1.3% AF (10). The presence of an EGFR T790M mutation at initial diagnosis, its high AF at cfDNA analysis, and the family history of lung cancer raised suspicion that the EGFR T790M mutation might have represented a germline risk allele.
Materials and Methods
Protection of human subjects
Patients at DFCI were enrolled to an institutional review board (IRB) approved correlative study allowing plasma collection for genomic analysis. Guardant Health separately received IRB approval for analysis of de-identified data for research purposes; clinical data such as age, stage, and diagnosis was submitted at time of testing on a requisition form. An exemption from IRB review was obtained to study de-identified plasma NGS results provided to DFCI by Guardant Health.
Droplet Digital PCR
Blood (6–10 mL) was collected into EDTA lavender capped vacutainer tubes and centrifuged for 10 min at 1200g. The plasma supernatant was further cleared by centrifugation for 10 min at 3000g. The second supernatant was stored in cryostat tubes at −80C until use. Cell free DNA was isolated using the QIAmp Circulating Nucleic Acid Kit (Qiagen) according to manufacturer’s protocol. Droplet digital PCR (ddPCR) was performed as previously described (11). Briefly, TaqMan PCR reaction mixtures were assembled from a 2× ddPCR Mastermix (Bio-Rad) and custom 40× TaqMan probes/primers made specific for each assay. Droplets were generated using an automated droplet Generator (Bio-RAD). PCR was performed to endpoint. After PCR, droplets were read on either a QX-100 or QX200 droplet reader (Bio-Rad). Analysis of the ddPCR data was performed with QuantaSoft analysis software (Bio-Rad). All ddPCR reagents were ordered from Bio-Rad. All primer and probes for were custom-ordered from Life Technologies.
PCR conditions:
EGFR L858R: Forward primer, 5′-GCAGCATGTCAAGATCACAGATT-3′, reverse primer, 5′-CCTCCTTCTGCATGGTATTCTTTCT-3′; probe sequences: 5′-VIC-AGTTTGGCCAGCCCAA-MGB-NFQ-3′, 5′-FAM-AGTTTGGCCCGCCCAA-MGB-NFQ-3′. Cycling conditions: 95 °C × 10 min (1 cycle), 40 cycles of 94 °C × 30 s and 58 °C × 1 min, and 10 °C hold.
EGFR del 19: Forward primer, 5′-GTGAGAAAGTTAAAATTCCCGTC-3′, reverse primer, 5′-CACACAGCAAAGCAGAAAC-3′; probe sequences: 5′-VIC-ATCGAGGATTTCCTTGTTG-MGB-NFQ-3′, 5′-FAM-AGGAATTAAGAGAAGCAACATC-MGB-NFQ-3′. Cycling conditions: 95 °C × 10 min (1 cycle), 40 cycles of 94 °C × 30 s and 55 °C × 1 min, followed by 10 °C hold.
EGFR T790M: Forward primer, 5′-GCCTGCTGGGCATCTG-3′, reverse primer, 5′-TCTTTGTGTTCCCGGACATAGTC-3′; probe sequences are: 5′-VIC-ATGAGCTGCGTGATGAG-MGB-NFQ-3′, 5′-FAM-ATGAGCTGCATGATGAG-MGB-NFQ-3′. Cycling conditions: 95 °C × 10 min (1 cycle), 40 cycles of 94 °C × 30 s and 58° C × 1 min, followed by 10 °C hold.
Plasma next-generation sequencing
All plasma sequencing was done using Guardant360 v2.9 (Guardant Health, Redwood City, CA), as described previously (12), as part of routine clinical patient care or through a dedicated research study. Briefly, cfDNA was isolated from 10ml of whole blood drawn in Streck cell-free DNA tubes, enriched by hybrid capture targeting exons of 70 genes and critical introns of 6 genes, and sequenced to an average depth of ~15,000X on an Illumina NextSeq500. Variant identification and origin assignment were done as per standard clinical testing operations.
Germline sequencing
For selected cases, de-identified buffy coat specimens from Guardant were provided to DFCI. With IRB approval, genomic DNA was extracted for Sanger sequencing of EGFR at the Laboratory for Molecular Medicine (LMM) at Harvard, using a sample ID that cannot be linked to the original participant in any way.
Statistical analysis
The relationship between AF of EGFR driver mutation and measures of copy number variation within the heterozygous group of variants was analyzed using a linear regression. The probability density function of the distribution of the standard deviation and mean AFs for individual cases was estimated using a Gaussian approximation, and outliers were identified using the Tukey method (13). Wilson/Brown’s method was used to determine the 95% confidence interval for EGFR T790M prevalence in each diagnosis of interest (14). The prevalence among the different diagnoses was compared using a two-tailed Fisher’s exact test.
Results
Plasma ddPCR of EGFR T790M reveals different behaviors of somatic and germline variants in cfDNA
We first studied 85 patients with advanced NSCLC who had an EGFR T790M mutation detected in plasma cfDNA using our previously described ddPCR platform (15). Of these patients, three were known to carry a germline EGFR T790M mutation based on prior germline sequencing, whereas the remainder had acquired EGFR T790M after TKI treatment. Studying the absolute concentration of T790M alleles in copies/mL of plasma, we found that some cases with somatic T790M carried an even higher concentration of mutant T790M alleles in plasma than the three cases with germline EGFR T790M (Figure 1A). In contrast, when we looked at the AF of T790M, calculated as the proportion of copies of mutant T790M out of all mutant or wildtype variants at that locus, the AF of the 3 germline cases hovered around 50%, higher than the AF of the somatic T790M cases (Figure 1A). We then studied changes in the levels of somatic versus germline EGFR mutations in plasma cfDNA upon treatment. In patients carrying acquired EGFR T790M after first-generation TKI resistance, the concentration of both the EGFR T790M mutations and the driver mutations (e.g. L858R or exon 19 deletion) decreased dramatically in response to therapy (Figure 1B). In contrast, in patients with germline EGFR T790M mutations, their EGFR driver mutation responded on therapy while the EGFR T790M levels stayed relatively stable (Figure 1B). These data provided proof-of-concept that quantitation of variant levels in plasma cfDNA can potentially be used to discriminate between somatic and germline origins of tumor-associated mutations.
Figure 1. Germline and tumor-derived EGFR mutations within plasma cell-free DNA.
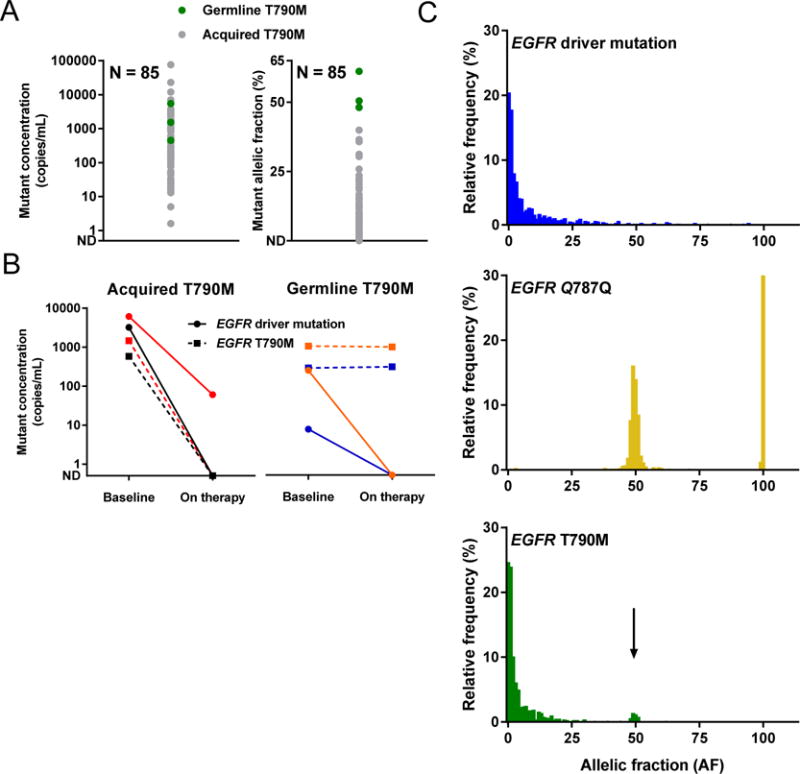
(A) Across ddPCR results for 85 patients, germline T790M mutations (green) are present at a similar concentration as somatic T790M mutations (gray), but at a higher allelic fraction (AF). (B) Studying ddPCR results for 4 patients on treatment, each represented by a different color, the concentrations of somatic EGFR mutations decrease while the concentration of germline EGFR T790M remains constant. (C) Across plasma NGS results for 950 cases, the AF distribution for EGFR T790M (green) includes a somatic peak also seen with EGFR driver mutations (blue), as well as a heterozygous peak (arrow) also seen more clearly with a common SNP (EGFR Q787Q, gold).
Plasma NGS identifies three populations of variants detected within cfDNA
While quantitative plasma genotyping is feasible with ddPCR, it is difficult to multiplex PCR assays across multiple genes. In contrast, next-generation sequencing (NGS) has the potential to capture a broad range of variants across a number of cancer-related genes. To further investigate the behavior of germline and somatic EGFR mutations in plasma cfDNA, we utilized the Guardant360 version 2.9 plasma NGS assay (Guardant Health Inc, Redwood City, CA; Supplemental Table 1). This assay sequences exonic regions of 70 oncogenes and tumor suppressor genes, and intronic regions of 6 genes in which oncogenic rearrangements occur (12). We first queried the Guardant database of clinical plasma NGS results to study the distribution of somatic and germline EGFR mutations. We identified test sets of 950 consecutive NSCLC samples for each of the following: known somatic mutations (L858R and exon 19 deletions), a common germline single nucleotide polymorphism (SNP) within the EGFR tyrosine kinase domain (Q787Q)(16), and T790M, and plotted the AF distribution of each (Figure 1C). The distribution of the known SNP comprised two discrete, normally distributed probability distributions centered at AFs of 50% and 100%, compatible with heterozygosity and homozygosity of the Q787Q allele. The distribution of the known somatic alterations, L858R and exon 19 deletions, in contrast, demonstrated an exponential decay distribution beginning at the assay’s limit of detection with the long tail extending to AFs exceeding 90%, compatible with somatic AFs that varied substantially but that were generally low (<5%). The distribution of T790M conformed predominantly to this same somatic distribution; however, there was a minor but discrete, normally-distributed sub-population centered at 50% AF (Figure 1C). This pattern supports study of variant AF in cfDNA as a method for categorizing variants like EGFR T790M, which may be of either germline or somatic origin.
We further studied AF distribution by performing plasma NGS on pre-treatment and on-treatment plasma specimens from 3 cases with germline EGFR T790M known to harbor EGFR driver mutations in their cancer (two with L858R, one with L861Q). Studying the AFs of all coding and non-coding variants identified on plasma NGS, we were able to clearly visualize three groups of variants (Figure 2A). The lowest AF group of variants includes the EGFR driver and TP53 mutations, representing cancer-derived variants. The highest AF group of variants was centered around 100% AF, representing homozygous germline variants. Finally, a middle group of variants centered around 50%, includes the known germline EGFR T790M mutations and represents heterozygous germline variants. On treatment with a third-generation EGFR TKI (two with osimertinib, one with ASP7283), the low AF cancer-derived variants decreased or became undetectable (24% 0.2%, 3.7% ND, 1.1% ND). In contrast, the middle group of heterozygous germline variants changed only modestly and remained centered around 50% AF (56% 49%, 52% 49%, 49% 50%). Interestingly, some of these heterozygous variants appeared to have an increase in AF as the cancer responded to therapy while others had a decrease in AF. We hypothesized that these changes in the heterozygous group on treatment could represent a reduction in tumor-derived copy number variation, leading to a change in the variant AF in cfDNA.
Figure 2. Discriminating germline and somatic variants in plasma cell-free DNA.
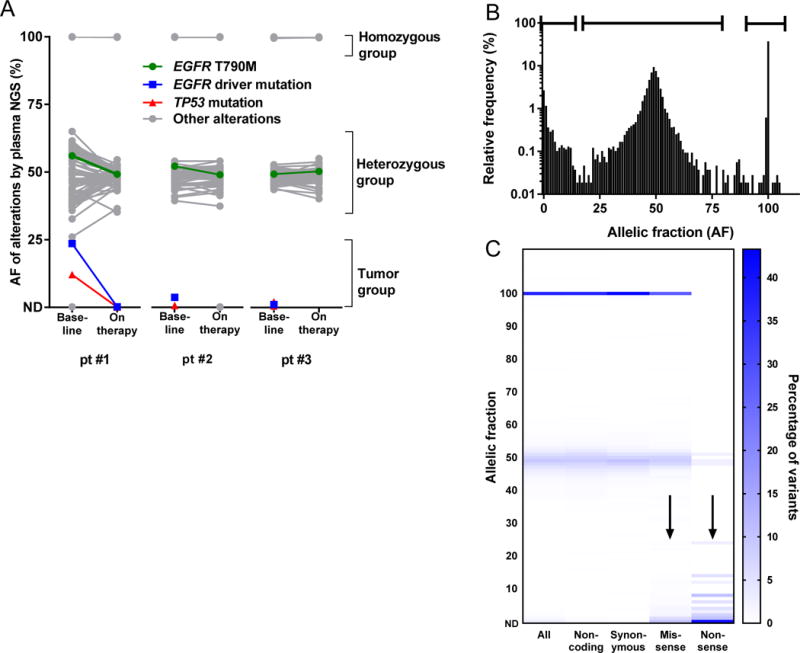
(A) Pre-treatment and on-treatment plasma specimens from three initial cases, all positive for germline EGFR T790M, were studied using plasma NGS. Studying all coding and non-coding variants detected, three groups of variants are evident, corresponding to the expected AF of homozygous, heterozygous, and tumor-derived variants. Variants in the tumor-derived group respond on therapy while variants in the homozygous and heterozygous groups remain at a relatively constant AF. (B) An additional 102 cases, for a total of 105 cases, were then studied using plasma NGS. Studying all coding and non-coding variants detected across 105 cases, a trimodal distribution is seen with peaks near 0% (likely tumor-derived), 49% (likely heterozygous), and 100% (likely homozygous). (C) For missense and nonsense variants, there is enrichment at low AF (arrows), where tumor-derived variants would be expected to be found. In contrast, synonymous variants, likely reflecting benign germline polymorphisms, are enriched around 50% and 100% AF.
We then similarly studied all coding and noncoding variants from plasma NGS of the initial case presented above (Supplemental Figure 1A). This revealed a pattern similar to the germline EGFR T790M cases studied, where the patient’s EGFR T790M mutation fell within the heterozygous group of variants, and the AF changed minimally on therapy as compared to the EGFR L858R mutation (Supplemental Figure 1B). This pattern seemed suspicious for an incidentally detected germline EGFR T790M mutation detected with plasma NGS.
Increased AF of driver mutation in cfDNA is associated with increased copy number variation in heterozygous variants
To further study the relationship between tumor content in cfDNA and heterozygous copy number variation, we queried the Guardant database for an additional 63 plasma NGS cases which were positive for EGFR T790M and 39 plasma NGS cases positive for an EGFR driver mutation without T790M. These 105 cases each had a median of 107 coding and noncoding variants detected. Looking at the AF distribution of all 10,702 variants in total (Figure 2B), one can clearly identify a trimodal distribution with three AF peaks at ~0%, 49%, and 100%. Compared to noncoding exonic and intronic variants, coding missense and nonsense variants were enriched in the low AF group of variants (Figure 2C), consistent with this representing a group of cancer-derived variants.
To study the relationship between potential germline and somatic variants, we plotted each plasma NGS case individually in order of low to high AF of the EGFR driver mutations (Figure 3A). While AF of the driver mutation is not a perfect measure of tumor content in cfDNA (due to the presence or absence of EGFR gene amplification in some cases) it can serve as a rough estimate of tumor content in cfDNA across a cohort (17). Studying the distribution of variant AFs in the heterozygous group, which we designated as all variants with an AF between 25% and 75%, we found that the distribution changed as the AF of the EGFR driver mutation increased. An increase in EGFR driver AF was associated with an increased standard deviation of the heterozygous group (Figure 3B) as well as an increase in the absolute difference between the case and the population mean, both of which suggest the presence of cancer-derived copy number variation. Studying the standard deviation of the AF of the variants in the heterozygous group, we fit a normal distribution to 94 cases while 11 cases had outlier characteristics (Supplemental Figure 2A). Similarly, studying the median of the AF for the variants in the heterozygous group, we fit a normal distribution to 94 cases while 11 cases had outlier characteristics (Supplemental Figure 2B). Because these outlier populations overlapped, 16 cases in total exhibited one of these two outlier characteristics with evidence of high copy number variation in cfDNA, thought to be due to high levels of tumor DNA causing variability in the AF of germline variants.
Figure 3. Increased tumor content is associated with increased copy number variation within heterozygous group of variants.
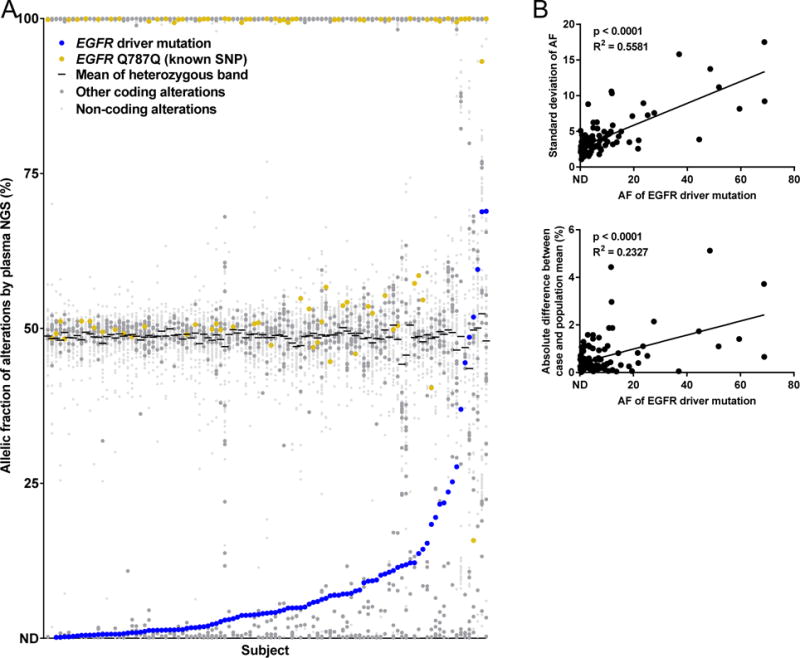
(A) AF of all variants found on plasma NGS of 105 cases positive for EGFR mutations, in increasing order of EGFR driver mutation AF (blue), with a common EGFR SNP shown (gold). (B) Studying the variant AFs between 25% and 75%, the standard deviation and absolute difference between case and population mean were calculated; both increased with an increase in EGFR driver AF.
Because the high copy number variation can result in substantial deviation of the AF of germline variants from the expected 50%, we hypothesized that germline-somatic discrimination would be impaired in these outlier cases. We thus segregated these 16 outlier cases from the 89 cases without outlier characteristics (Figure 4). Visual review of the coding variants of outlier cases (Figure 4A), it is challenging to distinguish a clear separation between germline heterozygous variants and somatic cancer-derived variants. In contrast, visual review of the coding variants of cases without these characteristics of high copy number variation (Figure 4B), one can clearly distinguish a group of heterozygous variants with AFs in the range of 35 to 60%, which are non-overlapping with a group of cancer-derived variants with AFs below 30%. We thus propose that by excluding plasma NGS cases with high copy number variation (and thus high tumor content), plasma NGS results can be accurately differentiated into somatic variants within the cancer-derived group and incidentally identified germline risk alleles within the heterozygous group.
Figure 4. Distinguishing heterozygous and tumor-derived coding variants in cases with low copy number variation.
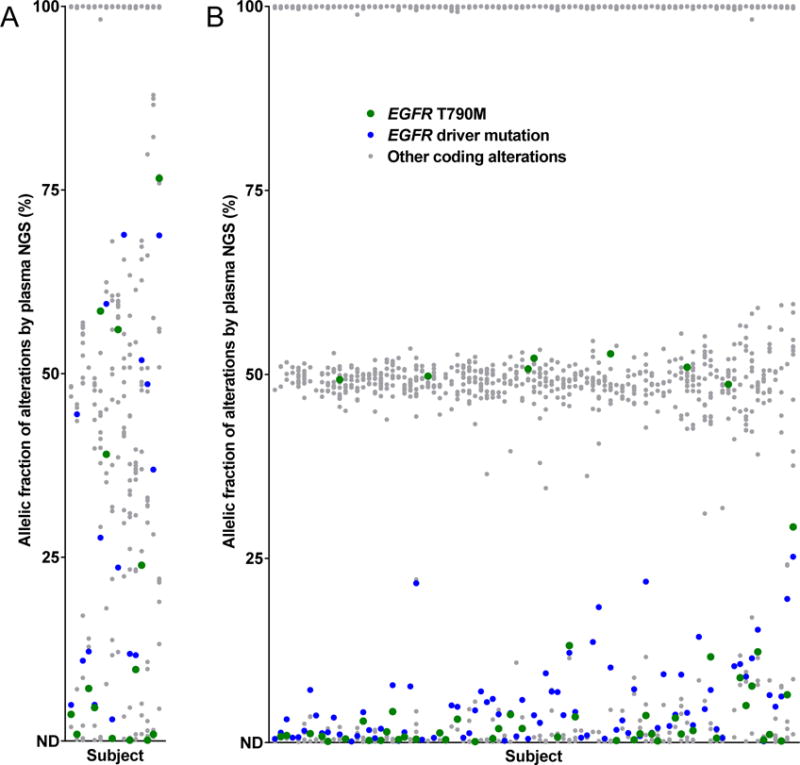
In outlier cases with high copy number variation (A), it is difficult to distinguish germline and somatic variants. When there is lower copy number variation (B), it is possible to visually distinguish which cases of germline EGFR T790M (green) are likely germline.
Blinded validation of a bioinformatic algorithm for discriminating between germline and somatic alterations in plasma NGS data
Following the logic of these proof-of-concept studies, we developed and evaluated an integrated bioinformatics algorithm to segregate germline and somatic alterations across the 70 genes assayed using plasma NGS. This algorithm first assigns variants a presumed germline or somatic origin using a priori knowledge, including both internal and external databases of known germline and somatic variants (pathogenic and benign). For example, the EGFR Q787Q alteration is a benign polymorphism present in ~52% of germline exomes in the ExAC database (http://exac.broadinstitute.org/), allowing this to be designated as of presumed germline origin regardless of allelic fraction (16). Conversely, the EGFR L858R alteration is a relatively common oncogenic mutation in NSCLC but does not appear in germline databases, allowing this to be designated as of presumed somatic origin. Such a priori binning usually results in a median of 78 variants per case being assigned as germline, which allows construction of a heterozygous probability distribution by variant AF as described in the studies above. If all presumed somatic mutations (which are generally fewer in number) are present below the lower limit of this heterozygous germline distribution, germline-somatic discrimination of the remaining unassigned variants proceeds according to their AF relative to the germline distribution described by a priori variant classification. However, if the AF of presumed somatic variants exceed the AF of the lower limit of the heterozygous germline distribution, or if extreme chromosomal instability is detected (as assessed by the apparent diploid fraction of the genome), germline/somatic discrimination is deemed uncertain for the variants remaining within that region of overlap, and variants are presumed to be somatic in origin and reported as such. This approach is intended to allow identification of suspected germline variants with a high positive predictive value, understanding that sensitivity for variants of germline origin will be reduced in settings of high tumor DNA content.
This algorithm was then applied to 21 prospectively collected clinical samples with high AF (30–75%) EGFR T790M mutations detected on plasma NGS (Supplemental Figure 3). Cases were segregated into two cohorts based upon the germline-somatic segregation of EGFR T790M described above. Cohort A included 11 cases in which the distribution of somatic vs. germline derived variants led to the prediction of a germline T790M mutation being present. Cohort B included 10 cases in which determination of germline vs. somatic was complicated by high copy number variation and a broad heterozygous group. The genomic DNA-containing cellular fractions of each sample were then irreversibly de-identified and, with IRB approval, submitted to a CLIA-certified clinical laboratory for EGFR sequencing in a double-blinded manner such that no germline result was traceable to any individual patient. All 11 cases in cohort A were confirmed to harbor a germline EGFR T790M (positive predictive value 100%, 11/11). Of 10 cases in cohort B, one was found to be germline, resulting in a sensitivity of 92% (11/12) and an overall accuracy of 95% (20/21). The presence of a germline sample in Cohort B is suspected to be a case with high tumor content such that the AF of presumed somatic mutations overlapped with heterozygous germline distribution, making it impossible to discriminate germline variants with certainty.
Using a plasma NGS database to study the epidemiology of germline EGFR T790M
Having validated a method for identifying plasma NGS cases which carry germline EGFR T790M, we hypothesized that we could use existing plasma NGS data to learn about the association of germline variants with specific cancer types. We again queried the Guardant clinical testing database of 31,414 consecutive unique patients representing a wide variety of adult solid tumor types to identify 911 cases positive for EGFR T790M, of which 48 were of germline origin as adjudicated by the above methodology. Though non-squamous NSCLC was the cancer diagnosis in a minority of the overall patient cohort (41%), this was the cancer diagnosis in 43 of the 48 patients with germline EGFR T790M (90%, Figure 5A). Of the remaining 5 patients with germline EGFR T790M, three had a related diagnosis (squamous NSCLC, small cell lung cancer, carcinoma of unknown primary). The population frequency of germline EGFR T790M in patients with nonsquamous NSCLC (43/12774, 0.34%) was substantially higher than was seen in patients with another cancer diagnosis (5/18640, 0.03%, Figure 5B), the latter being only moderately higher than that reported by general population sequencing efforts (e.g. ExAC’s median allele frequency of 0.0082%) (16). These calculations are imperfect given the sub-perfect accuracy of determining germline status described above, but these observations are nonetheless congruent with existing hypotheses that patients with germline T790M are at increased risk specifically for NSCLC, and suggest that this allele does not confer substantially increased risk of other cancers aside from lung cancer.
Figure 5. Prevalence of germline T790M.
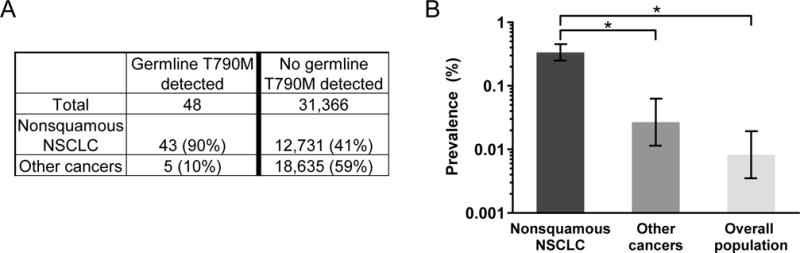
(A) Querying a database of 31,414 unique cancer patients with plasma NGS results, 48 (0.15%) were found to carry a germline EGFR T790M mutation. Non-squamous non-small cell lung cancer (NSCLC) was the dominant diagnosis in these patients. (B) As compared to the population prevalence of germline EGFR T790M in a reference cohort (0.008%), there is a higher prevalence in subjects with nonsquamous NSCLC (0.34%) but not in subjects with other cancers (0.03%, p = 0.06), suggesting germline EGFR T790M is a risk variant for lung cancer.
Discussion
This analysis for the first time demonstrates the power of cfDNA genomics as a tool for investigation of germline cancer risk alleles. We developed and validated a bioinformatic algorithm to distinguish germline variants from cancer-derived somatic variants within cfDNA NGS profiles, suggesting that a single assay can both offer insight into tumor genotype for therapy selection as well as screen for hereditary risk alleles. We then queried a large clinical testing database to explore the rare germline allele, EGFR T790M, and observed enrichment for this mutation in patients with nonsquamous NSCLC. We believe that the application of cfDNA sequencing to study association of germline alleles with specific cancer mutations represents a novel approach deserving further investigation.
As is the case with tumor NGS, it is expected that plasma NGS will at times identify incidental germline mutations in cancer patients that have potential clinical implications for patients and their families. Here, we focused on germline EGFR mutations, but this approach can be applied to other germline mutations with potential implications both for therapy and inherited risk, such as mutations in TP53 (18), BRCA1/2 (19), and mismatch repair genes (20). Reporting clinically significant germline findings incidental to somatic genomic testing in cfDNA may provide clinical benefits to advanced cancer patients. In a large tissue-based pan-cancer NGS study, 2.3% of patients were found to have previously unrecognized pathogenic germline variants (21). Additionally, numerous reports in non-lung cancers have found significant rates of potentially targetable germline alterations in patients who would not be screened on the basis of family history, particularly in DNA repair genes (22–25).
In our validation, we found that presence of a suspected germline mutation using our bioinformatic algorithm predicted for a confirmed germline mutation with a high positive predictive value, a critical diagnostic performance characteristic for a rule-in test. However, in the setting of high tumor content and high copy number variation, germline variants will be extremely difficulty to accurately distinguish from somatic variants. Importantly, genotyping of cfDNA does not replace genetic testing of germline DNA – patients with incidentally detected germline mutations on plasma NGS should be referred for formal genetic counseling and confirmatory germline testing, as per guideline recommendation (26–28). Given the widespread use of plasma genotyping for lung cancer care, patients with high levels of EGFR T790M on plasma genotyping are now eligible for germline testing on our ongoing study of families with germline EGFR T790M mutations (NCT01754025) (6).
Discernment of germline and somatic variants in plasma cfDNA has potential impact on the understanding of cancer biology. With tumor NGS, it is difficult to determine if a variant of unknown significance in an oncogene represents a potential driver mutation or a germline polymorphism. In plasma NGS cases without high copy number variation, it now appears possible to differentiate these two types of genomic alterations using a single assay, reducing the risk of a germline polymorphism being therapeutically targeted in error. Additionally, in instances where serial plasma genotyping is being used over time to monitor response and resistance to therapy, the ability to distinguish germline and somatic variants in plasma cfDNA can make it easier to accurately track tumor DNA levels. Finally, our approach could aid accurate calculation of tumor mutation burden (TMB), an emerging biomarker for understanding sensitivity and resistance to immune checkpoint inhibitors (29), by reducing the chance of germline polymorphisms being mistaken for potentially antigenic somatic mutations.
One limitation of our analysis is the focus on a single rare germline variant, EGFR T790M; studies of other germline cancer risk alleles are planned. A second limitation is our use of peripheral white cells as our source of germline DNA, an approach which could be vulnerable to somatic mosaicism such as clonal hematopoiesis of indeterminate potential (CHIP) (30,31). However, the clear majority of mutations found in patients with clonal hematopoiesis are present at an AF in the range of 0–30%, and are unlikely to be mistaken for germline variants. As large studies have found no somatic EGFR mutations within peripheral white cells, clonal hematopoiesis would not interfere the analysis performed in this report. Additional validation studies could consider using an alternate source for germline DNA.
Our data highlights the need for laboratories offering plasma genotyping to be vigilant for germline variants and prepared to report to providers when suspicion for a pathogenic germline mutation is high. Moreover, clinicians ordering plasma genotyping must be prepared for the possibility of incidentally identifying germline risk alleles, and will need referral systems in place to care for these patients should such results be identified. Prospective studies are needed to better understand the frequency of such incidental germline findings in routine oncology care.
Supplementary Material
Statement of Translational Relevance.
While genomic analysis of plasma cell-free DNA (cfDNA) is increasingly performed to gain insight into cancer biology, cfDNA remains a poorly understood biospecimen made up of DNA from multiple sources. Here, we develop methods for differentiating germline and somatic variants within plasma cfNDA, using germline and somatic EGFR mutations as a clinical model. We then develop and validate a bioinformatic approach permitting us to identify and study incidental germline EGFR T790M mutations within a database of plasma next-generation sequencing results. Our approach suggests cfDNA genomics, currently used for somatic cancer genotyping, may also offer an untapped opportunity for germline genomic research.
Acknowledgments
This research was supported in part by the Damon Runyon Cancer Research Foundation, Anna Fuller Fund, Conquer Cancer Foundation of ASCO, Bonnie J. Addario Lung Cancer Foundation, National Cancer Institute (NCI) of the National Institutes of Health (R01 CA114465), NCI Cancer Clinical Investigator Team Leadership Award (P30 CA006516 supplement), and Stading-Younger Cancer Research Foundation Thoracic Oncology Fund.
Footnotes
Conflict of Interest Disclosures:
Odegaard, Fairclough, Nagy, and Lanman are employees of Guardant Health; Nagy and Lanman have ownership interest in Guardant Health. Garber receives research funding or has research collaborations with Myriad Genetics, AstraZeneca, and Ambry and receives consulting fees from Helix. Paweletz received consulting fees from Digital Bioanalytics and honoraria from BioRad. Oxnard received consulting fees from AstraZeneca and Inivata and honoraria from BioRad and Sysmex. All other authors have no potential conflicts of interest to report.
Author Contributions:
Study concept and design: Y. Hu, R.S. Alden, J.I. Odegaard, G.R. Oxnard
Acquisition of data: Y. Hu, R.S. Alden, J.I. Odegaard, S.R. Fairclough, R. Chen, J. Heng, N. Feeney, R.J. Nagy, J. Shah, B. Ulrich, M. Gutierrez, G.R. Oxnard
Analysis and interpretation of data: Y. Hu, R.S. Alden, J.I. Odegaard, S.R. Fairclough, R.J. Nagy, J.E. Gutierrez, C.P. Paweletz, G.R. Oxnard
Writing, review, and/or revision of the manuscript: Y. Hu, R.S. Alden, J.I. Odegaard, S.R. Fairclough, R. Chen, J. Heng, N. Feeney, R.J. Nagy, J. Shah, B. Ulrich, M. Gutierrez, R. Lanman, J.E. Garber, C.P. Paweletz, G.R. Oxnard
Administrative, technical, or material support: Y. Hu, R.S. Alden, R. Chen, J. Heng, C.P. Paweletz
Study supervision: G.R. Oxnard
References
- 1.Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2(1):82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagemann IS, Devarakonda S, Lockwood CM, Spencer DH, Guebert K, Bredemeyer AJ, et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer. 2015;121(4):631–9. doi: 10.1002/cncr.29089. [DOI] [PubMed] [Google Scholar]
- 3.Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7(283):283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schrijver I, Aziz N, Farkas DH, Furtado M, Gonzalez AF, Greiner TC, et al. Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology. J Mol Diagn. 2012;14(6):525–40. doi: 10.1016/j.jmoldx.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37(12):1315–6. doi: 10.1038/ng1671. [DOI] [PubMed] [Google Scholar]
- 6.Oxnard GR, Heng JC, Root EJ, Rainville IR, Sable-Hunt AL, Shane-Carson KP, et al. Initial results of a prospective, multicenter trial to study inherited lung cancer risk associated with germline EGFR T790M: INHERIT EGFR. Journal of Clinical Oncology. 2015;33(15_suppl):1505. doi: 10.1200/jco.2015.33.15_suppl.1505. [DOI] [Google Scholar]
- 7.Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–99. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 8.Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, Feeney N, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016;2(8):1014–22. doi: 10.1001/jamaoncol.2016.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–10. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–2. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paweletz CP, Sacher AG, Raymond CK, Alden RS, O’Connell A, Mach SL, et al. Bias-Corrected Targeted Next-Generation Sequencing for Rapid, Multiplexed Detection of Actionable Alterations in Cell-Free DNA from Advanced Lung Cancer Patients. Clin Cancer Res. 2016;22(4):915–22. doi: 10.1158/1078-0432.CCR-15-1627-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mack PC, Banks KC, Zill OA, Mortimer SA, Chudova DI, Odegaard J, et al. O02: Plasma Next Generation Sequencing of Over 5,000 Advanced Non-Small Cell Lung Cancer Patients With Clinical Correlations. J Thorac Oncol. 2016;11(10S):S168–S9. doi: 10.1016/j.jtho.2016.08.005. [DOI] [Google Scholar]
- 13.Tukey JW. Exploratory Data Analysis. Addison-Wesley Publishing Company; 1977. [Google Scholar]
- 14.Brown LD, Cai TT, DasGupta A. Interval Estimation for a Binomial Proportion. Statistical Science. 2001;16(2):101–17. [Google Scholar]
- 15.Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20(6):1698–705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34(28):3375–82. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.VanderLaan PA, Rangachari D, Mockus SM, Spotlow V, Reddi HV, Malcolm J, et al. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer. 2017;106:17–21. doi: 10.1016/j.lungcan.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somlo G, Frankel P, Arun B, Ma CX, Garcia A, Cigler T, et al. Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1- or BRCA2-Associated Metastatic Breast Cancer. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meric-Bernstam F, Brusco L, Daniels M, Wathoo C, Bailey AM, Strong L, et al. Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol. 2016;27(5):795–800. doi: 10.1093/annonc/mdw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol. 2017:JCO2017723502. doi: 10.1200/JCO.2017.72.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hampel H, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, et al. Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res. 2007;67(19):9603. doi: 10.1158/0008-5472.CAN-07-2308. [DOI] [PubMed] [Google Scholar]
- 24.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precision Oncology. 2017(1):1–16. doi: 10.1200/po.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Royer R, Li S, McLaughlin JR, Rosen B, Risch HA, et al. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol Oncol. 2011;121(2):353–7. doi: 10.1016/j.ygyno.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 26.Daly MB, Pilarski R, Berry M, Buys SS, Farmer M, Friedman S, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Canc Netw. 2017;15(1):9–20. doi: 10.6004/jnccn.2017.0003. [DOI] [PubMed] [Google Scholar]
- 27.Robson ME, Bradbury AR, Arun B, Domchek SM, Ford JM, Hampel HL, et al. American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J Clin Oncol. 2015;33(31):3660–7. doi: 10.1200/JCO.2015.63.0996. [DOI] [PubMed] [Google Scholar]
- 28.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yarchoan M, Johnson BA, 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–22. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.