

Abstract
The ribose of RNA nucleotides can be 2′-O-methylated (Nm). Despite advances in high-throughput detection, the inert chemical nature of Nm still limits sensitivity and precludes mapping in mRNA. We leveraged the differential reactivity of 2′-O-methylated and 2′-hydroxylated nucleosides to periodate oxidation to develop Nm-seq, a sensitive method for transcriptome-wide mapping of Nm with base precision. Nm-seq uncovered thousands of Nm sites in human mRNA with features suggesting functional roles.
Chemical modifications of mRNA, collectively known as the epitranscriptome, regulate gene expression1. Current epitranscriptome studies concern only base modifications, but the ribose can be methylated at the 2′ position to form 2′-O-methylated nucleotides2. Nm is abundant in rRNA, tRNA, snRNA and microRNA; and it is essential for the biogenesis, metabolism and function of these molecules3–9. It is installed by either standalone methyltransferases that recognize their targets according to sequence and structure8 or by the enzyme fibrillarin (FBL), a part of a ribonucleoprotein complex that is guided to its targets by different C/D-box small nucleolar RNAs (snoRNAs)10.This modification also occurs at the 5′ cap11 and at internal positions of non-rRNA transcripts12. 2′-O-methylation endows nucleotides with greater hydrophobicity, protects them against nucleotlytic attack and stabilizes helices13,14.
Nm mapping traditionally relied on Nm’s property to pause reverse transcription in the presence of limited amounts of dNTPs15. This mapping method was recently adapted to a high-throughput sequencing format (2OMe-seq)16. It was joined by RiboMeth-seq17–19, a method that leverages the resistance of 2′-O-methylated ribose to alkaline hydrolysis, leading to under-representation of these positions among read starts or ends to provide a negative readout of the methylation landscape.
In higher eukaryotes, the 5′ penultimate and antepenultimate nucleotides in mRNA (m7GpppNmNm) can be 2′-O-methylated by standalone enzymes that recognize the cap. In addition to these Nm sites, accumulating evidence suggests that internal positions in mRNA could also be 2′-O-methylated20,21.
However, existing methods are underpowered to detect Nm positions present in relatively rare RNA molecules (e.g., mRNA) or at low stoichiometry. To address these challenges, we developed a conceptually distinct approach based on the different chemical properties of nucleosides with 2′-OH and 2′-OMe22–25, combining enrichment with detection of a positive signal (rather than the lack of signal17–19) to produce a sensitive method suited for discovery of Nm sites in rare RNA molecules or at low stoichiometry. Nm-seq leverages oxidative cleavage of ribose 2′,3′-vicinal diols by periodate to expose, enrich and map Nm sites in the transcriptome without bias and with single-nucleotide precision.
Nm-seq first exposes internal Nm sites in RNA fragments by iterative oxidation–elimination–dephosphorylation (OED) cycles that remove 2′-unmodified nucleotides (one per cycle) in the 3′-to-5′ direction. Vicinal diols in these nucleotides are readily oxidized by sodium periodate to yield a dialdehyde intermediate that undergoes spontaneous β-elimination under mildly basic conditions. With the removal of a nucleoside, the resulting 3′-mono-phosphate is enzymatically dephosphorylated to allow another cycle to take place. Once an Nm is encountered, the progressive shortening process comes to a halt, as lack of vicinal diols prevents Nm oxidation (Fig. 1a,b).
Figure 1.
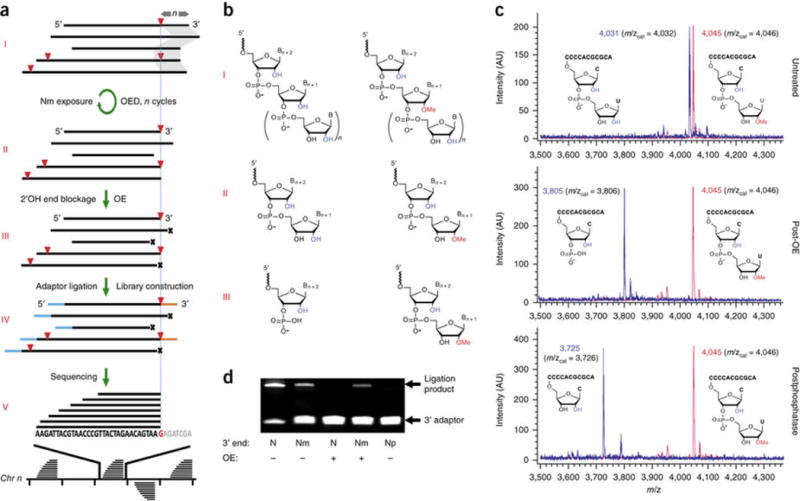
Nm-seq, a method based on oxidative cleavage for mapping 2′-O-methylation with base precision. (a) Schematic illustration. Fragmented RNA (I) is subjected to iterative oxidation–elimination–dephosphorylation (OED) cycles that remove 2′-hydroxylated nucleotides in the 3′-to-5′ direction to expose internal Nm sites at 3′ ends of fragments (II). Fragments ending with 2′-hydroxyl are then blocked by an incomplete cycle (OE, III), followed by adaptor ligation and library construction (IV). Paired-end sequencing (V) produces an asymmetric coverage profile, the uniform 3′ end of which corresponds to an Nm site shown in red below (G). Red triangles, Nm sites; n and gray shadow, number of OED cycles equals number of removed nucleotides, except when Nm is encountered; x, 3′-monophosphates representing blocked ends; blue and orange lines, 5′ and 3′ adaptors, respectively. (b) Chemical structures of 2′-O-methylated and 2′-hydroxylated RNA polymers at intermediate steps (I, II, III) of the method. (c) MALDI-TOF spectra of model RNA oligonucleotides, 3′-modified (red) and unmodified (blue), produced at intermediate steps listed on the right. AU, arbitrary units. (d) OE effectively blocks 3′ adaptor ligation to 2′-hydroxylated but not to 2′-O-methylated 3′ ends. N, RNA model ending with 2′-hydroxyl; Nm, RNA model ending with 2′-O-methyl; Np, RNA model ending with 3′ monophosphate.
The net result of the iterative exposure process is an enrichment of fragments ending with Nm, although those ending with N still constitute the vast majority of 3′ termini. A final round of oxidation–elimination (OE) reaction, excluding dephosphorylation, is then performed to generate two types of 3′ ends that differ in their ligation compatibility; while 2′-unmodified ends produce an unligatable 3′-monophosphate, 2′-O-methylated ends are resistant to oxidation and retain their ligatable 3′-hydroxyl. In this way, fragments ending with Nm are preferentially ligated to the 3′ adaptor and further enriched by PCR amplification (Fig. 1a–d and Supplementary Note 1).
This exposure and enrichment strategy, when coupled to a dual-ligation and directional library construction protocol, produces an asymmetric read pileup signature: a uniform 3′ end corresponding to the exact Nm position and a variable 5′ end mirroring the random fragmentation of RNA used in the process (Fig. 1a). In contrast, libraries prepared from untreated RNA fragments exhibit variability at both ends.
The sensitivity of the method is proportional to the number of OED cycles, the initial average size of RNA fragments and the starting amount of RNA. As each cycle removes one nucleotide, the minimum number of cycles required to expose an Nm position is equal to its nucleotide distance from the 3′ end. Increasing the number of OED cycles or decreasing the size of RNA fragments increases the chance of exposing more Nm sites. The specificity of the method depends on the efficiency of the last oxidation-elimination step and the incompatibility of RNA fragments ending with a 3′-monophosphate with 3′ adaptor ligation. Like other recently introduced approaches, Nm-seq is limited in its ability to detect the 5′ of two adjacent modified positions as well as positions too close to the 5′ end of transcripts as to preclude read alignment. Additionally, since Nm-seq uses enrichment to achieve high sensitivity, stoichiometric information is lost.
In order to evaluate the performance of Nm-seq, we tested the method’s ability to detect known Nm sites in human rRNA18. An excellent overlap was found between the 106 known Nm sites in 18S and 28S rRNA and experimentally identified sites (Supplementary Fig. 1a–d and Supplementary Note 2).
We first determined the total levels of Nm in HeLa mRNA by LC-MS/MS and obtained Nm/N molar ratios ranging from 0.012% for Am/A to 0.15% for Um/U (Fig. 2a). With increasing mRNA purity (and reduced rRNA content), the molar ratio dropped precipitously until there was no measurable change between two consecutive rRNA-depletion steps, which indicated that the measured quantities were derived from mRNA and not from rRNA contamination.
Figure 2.
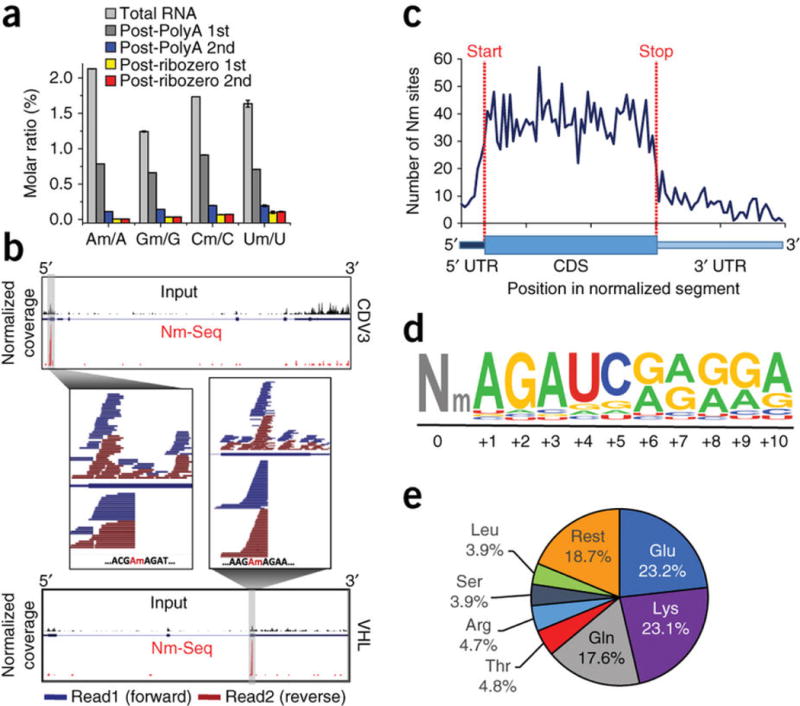
Nm sites in HeLa mRNA. (a) LC-MS/MS quantification of Nm in total and rRNA-depleted RNA. The level of each modified nucleoside is presented as a percentage of the unmodified one. Mean values ± s.e.m. are shown; n = 3. (b) Typical Nm-seq plots of methylated transcripts. Normalized summed sequence coverage of Nm-seq and input are shown below and above the transcript, respectively. Individual paired-end reads within the Nm site window are shown in magnification. (c) Metagene profile of Nm sites distribution along a normalized mRNA transcript illustrated below. (d) Sequence logo of the most enriched motif identified by HOMER in 33% of all Nm sites. It appears on average immediately downstream to the methylated position. (e) Distribution of Nm sites among different amino acid codons.
Nm-seq (based on eight OED cycles) was applied to total RNA from HeLa cells in four biological replicates; and the HOMER software package26 was used to identify sharp increases in signal from non-rRNA species, based on paired-end R2 reads, to denote Nm positions. We identified 7,412 sites in three out of four replicates (fold change (FC) ≥ 4, false discovery rate (FDR) ≤ 0.001), 3,515 of which had a minimal read coverage of ten reads and were considered for further analysis (typical examples are shown in Fig. 2b and Supplementary Fig. 2; data set provided in Supplementary Data Set 1).
2′-O-methylation was found on all four bases, typically within less structured regions, with Um sites relatively less abundant (Supplementary Figs. 3a and 4a–c). The vast majority of Nm sites (95.7%) occurred in 2,398 RefSeq annotated genes, 95.9% of which were protein coding (Supplementary Fig. 3b). Among noncoding genes (4.1%), sites were found in snRNA, snoRNA, lincRNAs and antisense RNAs. RNA molecules <150 nt (like tRNAs and microRNAs) were depleted during the purification of total RNA used for this study and are consequently underrepresented in the data sets.
On average, each methylated gene harbors 1.4 Nm sites, with over 77% of those genes methylated only once (Supplementary Fig. 3c). Additional features are summarized in Supplementary Note 3, Supplementary Figure 3d and Supplementary Figure 4d,e.
In protein-coding transcripts, most of the sites occurred in the CDS (70.3%), and the rest occurred in 5′ and 3′ untranslated regions (3% and 10.6%, respectively) as well as in introns (16.2%), suggesting that Nm is installed cotranscriptionally in the nucleus. The ability of Nm-seq to identify sites in introns, which are largely undetectable in the total RNA input, attests to its high sensitivity. Only a small fraction of sites (6.7%) occurred in alternatively spliced regions (Supplementary Fig. 5a).
Metagene profiles generated by proportional rescaling of gene segments did not reveal any discrete location of enrichment or depletion, although they did show the tendency of Nm sites to broadly concentrate in the CDS mentioned above (Fig. 2c and Supplementary Fig. 5b). Plotting the distribution relative to the nearest splice site reveals that 50% of sites are contained within a 100-nt window centered on the splice site and so raises the possibility that these phenomena are linked (Supplementary Fig. 5c,d). Unbiased motif search using HOMER revealed a significantly enriched (P = 1 × 10−313) consensus sequence of 10 nt that consists of an AGAUC sequence followed by a 5-nt-long AG-rich stretch (Fig. 2d). This motif was present in 45% of CDS sites and in 33% of all sites; and, on average, it appeared immediately downstream to the modified position. Nm sites in rRNA are known to be installed by snoRNPs. The existence of a dominant motif suggests that this subset of mRNA Nm sites could be installed by a specific methyltransferase that may or may not involve snoRNPs (Supplementary Note 4).
We examined the distribution of Nm sites among different codons and found that 64% of sites occurred in only six codons corresponding to three amino acids—glutamate (23.2%), lysine (23.1%) and glutamine (17.6%)—commensurate with the identified motif (Fig. 2e). While Nm was found in all three codon positions, the 1st position had more methylation than expected by chance, and the 3rd had less (51% and 15%, respectively; P < 1 × 10−200, hypergeometric test) (Supplementary Table 1). The functional implication could be found in a recent study that showed a codon-position-dependent effect of 2′-O-methylation on translation21.
We applied Nm-seq to total RNA purified from HEK293 cells and obtained results recapitulating the features observed in HeLa (Supplementary Figs. 6 and 7, Supplementary Table 2 and Supplementary Data Set 1); these results underscored the validity of the method and singled out Nm as a conserved and fundamental phenomenon.
In summary, we have uncovered thousands of Nm sites for the first time in mammalian mRNA using an effective Nm-seq approach that relies on differential reactivity of 2′-OMe versus 2′-OH nucleotides toward periodate oxidation. Most Nm sites were found in CDS, almost half of which near splice sites, and a notable fraction of Nm sites were found in introns. A consensus sequence was present in a third of all Nm sites and, intriguingly, Nm was enriched in codons of three amino acids and unequally distributed between the three codon positions (Supplementary Note 5).
ONLINE METHODS
A step-by-step protocol is available as a Supplementary Protocol and an open resource in Protocol Exchange27.
Cells and reagents
Human HeLa (cervical adenocarcinoma) and HEK293 (human embryonic kidney) cells (purchased from the ATCC) were maintained in DMEM (Thermo Fisher Scientific) containing 25 mM glucose, 4 mM l-glutamine, supplemented with 100 U ml−1 penicillin, 100 µg ml−1 streptomycin and 10% fetal bovine serum (FBS). Cells were found negative for mycoplasma contamination.
RNA purification
Total RNA from cells in culture was purified using PerfectPure RNA Cultured Cell Kit (5 Prime) and DNase treated. Enrichment of polyadenylated RNA (PolyA RNA) from total RNA was carried out using GenElute mRNA Miniprep Kit (Sigma-Aldrich). Ribo-Zero Gold rRNA Removal Kit (Illumina) was used to deplete rRNA from PolyA RNA before LC-MS/MS.
Detection and quantitation of Nm in mRNA
Total RNA and mRNA samples (after polyA selection and rRNA depletion) were subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) for detection and accurate quantitation of Am, Cm, Gm and Um, essentially as previously described28. 200–400 ng of purified RNA was digested by P1 nuclease (Wako, 2 U) in 40 µl of buffer containing 25 mM NaCl and 2.5 mM ZnCl2 for 2 h at 37 °C. Subsequently, 5 units (1 µl) of Antarctic Phosphatase (New England BioLabs) and 1× Antarctic Phosphatase reaction buffer were added, and the sample was incubated for another 2 h at 37 °C. The sample was then filtered (0.22 µm, Millipore) and injected into a C18 reverse-phase column coupled on-line to Agilent 6410 QQQ triple-quadrupole LC mass spectrometer in positive electrospray ionization mode. Quantitation was performed based on nucleoside-to-base ion transitions (268-to-136 for A; 282-to-136 for Am; 245-to-113 for U; 259-to-113for Um; 284-to-152 for G; 298-to-152 for Gm; 244-to-112 for C; 258-to-112 for Cm) using standard curves of pure nucleosides as previously described29.
OED of synthetic model RNA oligonucleotides
Two synthetic 13-mer RNA oligonucleotides (5 µM), 2′-O-methylated or 2′-hydroxylated at their 3′ end (5′-ACGCGCACCCCCU-3′ and 5′-ACGCGCACCCCCUm-3′), were oxidized/eliminated using 10 mM NaIO4 (Sigma-Aldrich) in 200 mM lysine–HCl buffer (pH 8.5, Sigma-Aldrich) in a total volume of 40 µl at 37 °C for 30 min. The reaction was quenched by ethylene glycol, and the oligonucleotides were column purified (RNA Clean & Concentrator, Zymo) and analyzed by MALDI-TOF MS. Products were further dephosphorylated by Shrimp Alkaline Phosphatase (New England BioLabs, 40 units ml−1) at 37 °C for 30 min, column purified (RNA Clean & Concentrator, Zymo) and analyzed by MALDI-TOF MS.
3′-end ligation of synthetic model RNA oligonucleotides
Three synthetic 27-mer RNA oligonucleotides—2′-O-methylated, 2′-hydroxylated and 3′-phosphorylated at their 3′ end (5′-CGGUA CUGCAGCUGACCUCGGCUUGUG-3′, 5′-CGGUACUGCAGC UGACCUCGGCUUGUGm-3′, 5′-CGGUACUGCAGCUGACC UCGGCUUGUG-p-3′)—were ligated to a fluorescently tagged 3′ adaptor (5′-rAppAGATCGGAAGAGCACACGTCT-Alexa488-3′) using the 3′ ligation reagents supplied in NEBNext Small RNA Library Prep Set for Illumina (New England Biolabs) at 16 °C overnight. Reaction products were column purified (New England BioLabs), PAGE separated (4–12%, Thermo Fischer Scientific) and imaged directly.
Nm-seq
See Supplementary Protocol and Protocol Exchange27 for detailed information regarding Nm-seq. The results of this study are based on eight OED cycles performed on total RNA; this number can be adjusted according to the desired sensitivity in the context of a biological system under study. Total RNA was fragmented using RNA Fragmentation Reagents (Thermo Fischer Scientific) at 95 °C for 5 min and column purified (RNA Clean & Concentrator, Zymo). RNA fragments (10 µg) were 3′-end repaired using Antarctic phosphatase (New England BioLabs, 200 units ml−1) at 37 °C for 30 min followed by inactivation at 65 °C and column purification (RNA Clean & Concentrator, Zymo). Eight cycles of OED were performed as described above for the model oligonucleotides, except dephosphorylation was performed directly on the quenched reaction without an intervening cleanup step. A final round of oxidation/elimination reaction, excluding dephosphorylation, followed by column purification (RNA Clean & Concentrator, Zymo) was performed. Purified fragments were 5′ phosphorylated by T4 polynucleotide kinase 3′ phosphatase minus (New England BioLabs, 200 units ml−1) at 37 °C for 60 min and column purified (RNA Clean & Concentrator, Zymo). Libraries were constructed from treated RNA fragments and untreated input fragments using NEBNext Small RNA Library Prep Set for Illumina (New England BioLabs) following the manufacturer’s protocol, with the exception that 3′ adaptor ligation was performed overnight. Sequencing was carried out on Illumina HiSeq2500 according to the manufacturer’s instructions, using 10 pM template per sample for cluster generation, TruSeq Rapid PE Cluster Kit - HS (Illumina), TruSeq Rapid SBS Kit - HS (Illumina) and TruSeq Multiplex Sequencing primer kit (Illumina).
Identification of Nm sites in rRNA
Human rRNA sequences were deduced from the human ribosomal DNA complete repeating unit (Genbank accession U13369, version GI:555853). To avoid any bias due to adaptor trimming state, reads were aligned to the rRNA sequence using the local mode of Bowtie2 aligner30. Duplicates were removed using the MarkDuplicates tool of the Picard tool suite (http://broadinstitute.github.io/picard). To reduce background levels resulting from the extremely high rRNA coverage, rRNA site detection analysis was restricted to reads that were properly mapped in pair (R1 and R2) to the reference rRNA sequence with forbidding mismatches and soft clipping. Only the 3′ end (5′ terminal base of R2 reverse read in each pair) was used to compute the coverage map of the sequences. Coverage was calculated for both treatment and input libraries. Library size was normalized, and background levels were subtracted from Nm-Seq reads. Performance of the Nm-Seq method was estimated by calculating the ratio of previously known 2′-O-methylations sites that have been detected to previously unknown detected Nm sites at increasing coverage threshold to construct receiver operating characteristic (ROC) curves. Matthews correlation coefficient (MCC) was then calculated for each point and used for defining the optimal coverage conditions.
Identification of Nm sites in the transcriptome
Adaptors and low-quality bases were trimmed from raw sequencing reads using Trim_galore (a wraparound program for cutadapt31). The trimming was conducted in two steps: first, the adaptor sequences AGA TCGGAAGAGCACACGTCTGAACTCCAGTCA and GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATT were removed from R1 (forward) and R2 (reverse) reads of each pair, respectively, followed by a second round of adaptor removal of the sequence CTCAGGACCGACTGA from the second pair only. Reads were aligned to the human genome (version hg19) using Bowtie230 global mode, and duplicates were removed using the MarkDuplicates tool of the Picard tool suite (http://broadinstitute.github.io/picard). R2 (reverse) reads that contained no more than two mismatches to the reference, and that were not soft clipped at their beginning, were subjected to 5′ RNA TSS Analysis tool in the HOMER package to identify Nm sites26. Only sites that were identified independently in at least three out of four replicates, with coverage above ten reads in at least one experiment, and localized to a known RefSeq transcript, were considered for further analysis. Sites were assigned to the most expressed transcript containing the site, based on expression data calculated using HtSeq32 from input HeLa RNA-Seq data, as previously described26. Base distribution was calculated according to the identified position in the hg19 genome.
Motif search
Sequences of 101 nt centered on the identified Nm sites were subjected to unbiased de novo motif search using the FindMotifs tool from the HOMER package26. Compatible background sequences were used—taken, if possible, from a remote location on the same part of the same transcript or from a matching location on another transcript.
Metagene profiling
Nm genomic positions were converted to transcriptomic positions in their assigned transcript. Nm sites were assigned to the corresponding gene segment (5′ UTR, CDS or 3′ UTR); and their relative position with respect to the transcript’s transcription start site (TSS), AUG start codon, first and nearest splice site positions as well as transcript length were calculated as described26.
Amino acid and codon distribution
The genomic coordinates of the Nm sites were translated by Annovar33 to the equivalent cDNA and protein coordinates, and the amino acid at the Nm position was identified. The relevant codon and position in the codon were extracted from the data provided by TransVar34.
Overlap with fibrillarin CLIP-seq
Nm sites were overlapped with fibrillarin (FBL)-binding regions downloaded from GEO (GSM1067864 and GSM1067865)35. A Monte Carlo simulation was undertaken to assess the statistical significance of the overlap: 100 same-size sets of the randomly selected regions were chosen using BEDTOOLS36 and simple scripts written in awk and maintaining the same length distribution, proportion of sites within Refseq-annotated genes and distribution between CDS, 5′ UTR and 3′ UTR segments as the FBL databases. The number of regions that overlapped Nm sites was recorded for the FBL data sets and for each of the randomly chosen sets. Statistical significance was calculated using one sample t-test.
RNA structure
The secondary RNA structures of a 200 nt window centered on Nm sites were analyzed using the Structure Surfer tool37 in aggregate mode. The average scores for each position in three different methods—PAR-seq, DMS-seq and ds/ssRNA-seq—were obtained from the output table of the tool and graphed. s.e.m are shown.
Gene ontology
Methylated gene RefSeq IDs were uploaded to DAVID Bioinformatics Resources (https://david.ncifcrf.gov/), and functional enrichment analysis was performed using all adequately expressed genes (above the 1st quartile) as background. Resulting enriched gene ontology (GO) terms were restricted to fold enrichment ≥2, Bonferroni corrected P ≤ 0.005.
Statistics
All error bars represent s.e.m. Sample size (n) is indicated in the legend for each experiment. Results of high-throughput sequencing experiments were restricted to a false discovery rate (FDR) ≤ 0.001. A Monte Carlo simulation and one-sample t-test were used to assess the statistical significance of overlap between data sets, where indicated in Supplementary Note 4.
Data availability statement
The data that support the findings of this study are available from the corresponding authors upon request. Raw and processed data are available at NCBI Gene Expression Omnibus, accession number GSE90164.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health NHGRI RM1 HG008935 to C.H.; a grant from the Kahn Family Foundation to D.D. and G.R.; and grants from the Ernest and Bonnie Beutler Research Program, Flight Attendant Medical Research Institute (FAMRI) and the Israeli Centers of Excellence (I-CORE) Program (ISF grants no. 41/11 and no. 1796/12) to G.R. C.H. is an investigator of the Howard Hughes Medical Institute (HHMI). G.R. is a member of the Sagol Neuroscience Network and holds the Djerassi Chair for Oncology at the Sackler Faculty of Medicine, Tel-Aviv University, Israel. D.D. was supported by a Human Frontier Science Program (HFSP) long-term fellowship. Q.D. is supported by the National Institutes of Health grant K01 HG006699. We wish to thank M. Salmon-Divon for advice and help with bioinformatic analysis and R. Mashiach for help with chemical structure drawings.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
D.D., Q.D. and C.H. conceived the approach. D.D. and Q.D. developed the methods and performed experiments. D.D., S.M.-M., D.H. and N.K. analyzed the data. D.D., S.M.-M., N.A., G.R. and C.H. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhao BS, Roundtree IA, He C. Nat Rev Mol Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Machnicka MA, et al. MODOMICS: a database of RNA modification pathways-2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daffis S, et al. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deryusheva S, Choleza M, Barbarossa A, Gall JG, Bordonné R. RNA. 2012;18:31–36. doi: 10.1261/rna.030106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jöckel S, et al. J Exp Med. 2012;209:235–241. doi: 10.1084/jem.20111075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang XH, Liu Q, Fournier MJ. RNA. 2009;15:1716–1728. doi: 10.1261/rna.1724409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin J, et al. Nature. 2011;469:559–563. doi: 10.1038/nature09688. [DOI] [PubMed] [Google Scholar]
- 8.Somme J, et al. RNA. 2014;20:1257–1271. doi: 10.1261/rna.044503.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Züst R, et al. Nat Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shubina MY, Musinova YR, Sheval EV. Biochemistry (Mosc) 2016;81:941–950. doi: 10.1134/S0006297916090030. [DOI] [PubMed] [Google Scholar]
- 11.Byszewska M, Śmietański M, Purta E, Bujnicki JM. RNA Biol. 2014;11:1597–1607. doi: 10.1080/15476286.2015.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacoux C, et al. Nucleic Acids Res. 2012;40:4086–4096. doi: 10.1093/nar/gkr1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S, Mapa K, Maiti S. Biochemistry. 2014;53:1607–1615. doi: 10.1021/bi401677d. [DOI] [PubMed] [Google Scholar]
- 14.Yildirim I, Kierzek E, Kierzek R, Schatz GC. J Phys Chem B. 2014;118:14177–14187. doi: 10.1021/jp506703g. [DOI] [PubMed] [Google Scholar]
- 15.Maden BE, Corbett ME, Heeney PA, Pugh K, Ajuh PM. Biochimie. 1995;77:22–29. doi: 10.1016/0300-9084(96)88100-4. [DOI] [PubMed] [Google Scholar]
- 16.Incarnato D, et al. Nucleic Acids Res. 2017;45:1433–1441. doi: 10.1093/nar/gkw810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birkedal U, et al. Angew Chem Int Ed Engl. 2015;127:461–455. [Google Scholar]
- 18.Krogh N, et al. Nucleic Acids Res. 2016;44:7884–7895. doi: 10.1093/nar/gkw482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchand V, Blanloeil-Oillo F, Helm M, Motorin Y. Nucleic Acids Res. 2016;44:e135. doi: 10.1093/nar/gkw547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gumienny R, et al. Nucleic Acids Res. 2017;45:2341–2353. doi: 10.1093/nar/gkw1321. http://dx.doi.org/10.1093/nar/gkw1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoernes TP, et al. Nucleic Acids Res. 2016;44:852–862. doi: 10.1093/nar/gkv1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonenberg N, Shatkin AJ. Proc Natl Acad Sci USA. 1977;74:4288–4292. doi: 10.1073/pnas.74.10.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weith HL, Gilham PT. Science. 1969;166:1004–1005. doi: 10.1126/science.166.3908.1004. [DOI] [PubMed] [Google Scholar]
- 24.Weith HL, Gilham PT. J Am Chem Soc. 1967;89:5473–5474. doi: 10.1021/ja00997a042. [DOI] [PubMed] [Google Scholar]
- 25.Yu B, et al. Science. 2005;307:932–935. doi: 10.1126/science.1107130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinz S, et al. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai Q, et al. Nm-seq protocol Protocol Exchange. 2017 http://dx.doi.org/10.1038/protex.2017.088.
- 28.Dominissini D, et al. Nature. 2016;530:441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kellner S, et al. Nucleic Acids Res. 2014;42:e142. doi: 10.1093/nar/gku733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead B, Salzberg SL. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin M. EMBnet Journal. 2011;17:10–12. [Google Scholar]
- 32.Anders S, Pyl PT, Huber W. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou W, et al. Nat Methods. 2015;12:1002–1003. doi: 10.1038/nmeth.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishore S, et al. Genome Biol. 2013;14:R45. doi: 10.1186/gb-2013-14-5-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan AR, Hall IM. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berkowitz ND, et al. BMC Bioinformatics. 2016;17:215. doi: 10.1186/s12859-016-1071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon request. Raw and processed data are available at NCBI Gene Expression Omnibus, accession number GSE90164.