

Abstract
PAH1-encoded phosphatidate phosphatase, which catalyzes the dephosphorylation of phosphatidate to produce diacylglycerol at the endoplasmic reticulum membrane, plays a major role in controlling the utilization of phosphatidate for the synthesis of triacylglycerol or membrane phospholipids. The conserved N-LIP and haloacid dehalogenase–like domains of Pah1 are required for phosphatidate phosphatase activity and the in vivo function of the enzyme. Its non-conserved regions, which are located between the conserved domains and at the C terminus, contain sites for phosphorylation by multiple protein kinases. Truncation analyses of the non-conserved regions showed that they are not essential for the catalytic activity of Pah1 and its physiological functions (e.g. triacylglycerol synthesis). This analysis also revealed that the C-terminal region contains a previously unrecognized WRDPLVDID domain (residues 637-645) that is conserved in yeast, mice, and humans. The deletion of this domain had no effect on the catalytic activity of Pah1 but caused the loss of its in vivo function. Site-specific mutational analyses of the conserved residues within WRDPLVDID indicated that Trp-637 plays a crucial role in Pah1 function. This work also demonstrated that the catalytic activity of Pah1 is required but is not sufficient for its in vivo functions.
Keywords: diacylglycerol, glycerophospholipid, phosphatase, phosphatidic acid, triacylglycerol, yeast
Introduction
PAH1-encoded PAP,2 which catalyzes the dephosphorylation of PA to produce DAG at the ER membrane (Fig. 1A), is one of the most highly regulated enzymes of lipid metabolism in the yeast Saccharomyces cerevisiae3 (1–8). The enzyme is conserved in higher eukaryotes, including mice and humans (6, 7, 9–13). Pah1 plays a major role in the synthesis of TAG by producing its precursor DAG and regulates the de novo synthesis of membrane phospholipids derived from its substrate PA (1–8) (Fig. 1A). In an auxiliary pathway, the DAG produced by the Pah1 reaction may also be used to synthesize the phospholipids phosphatidylcholine and phosphatidylethanolamine, respectively, via the CDP-choline and CDP-ethanolamine branches of the Kennedy pathway when cells are supplemented with choline and ethanolamine (4, 5).
Figure 1.
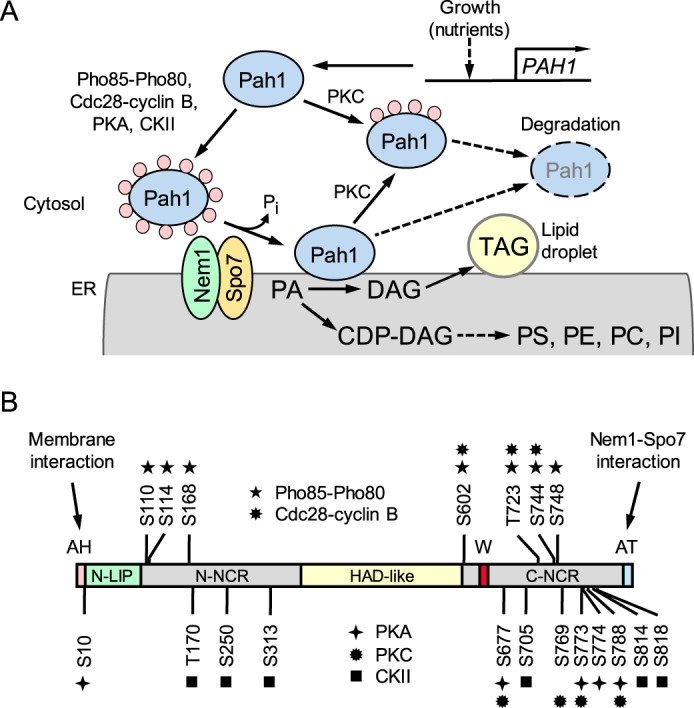
Model for the regulation of Pah1 by phosphorylation, dephosphorylation, and proteasomal degradation; domains and phosphorylation sites in Pah1. A, expression of PAH1 is regulated during growth by nutrients. After expression, Pah1 in the cytosol is phosphorylated by multiple protein kinases. Phosphorylated Pah1 (indicated by pink circles) translocates to the ER membrane through its dephosphorylation by the Nem1-Spo7 phosphatase complex. Dephosphorylated Pah1 associated with the membrane catalyzes the conversion of PA to DAG, which is then acylated to form TAG that is stored in lipid droplets. Dephosphorylated Pah1 or PKC-phosphorylated Pah1 that is not phosphorylated at the target sites for Pho85-Pho80/Cdc28-cyclin B is degraded by the proteasome (indicated by the dashed line arrows and ellipse). B, the diagram shows the positions of the amphipathic helix (AH, pink) required for ER membrane interaction; the N-LIP (green) and HAD-like (yellow) domains that are required for PAP activity; the acidic tail (AT, blue) required for interaction with Nem1-Spo7; the N-terminal non-conserved region (N-NCR, gray), the C-terminal non-conserved region (C-NCR, gray); the serine (S) and threonine (T) residues within the non-conserved regions that are phosphorylated by Pho85-Pho80, Cdc28-cyclin B, PKA, PKC and casein kinase II; and the tryptophan (W) residue within the newly identified C-terminal conserved WRDPLVDID domain (red) required for Pah1 function in vivo (this study). CKII, casein kinase II; PS, phosphatidylserine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PI, phosphatidylinositol.
Disturbing the Pah1-mediated control of the PA and DAG levels causes a drastic change in the metabolism of cellular lipids. Yeast cells lacking Pah1 PAP activity accumulate PA due to a defect in its conversion to DAG used for TAG synthesis (13–15). The increased level of PA in the pah1Δ mutant not only increases substrate availability for phospholipid synthesis but also induces the expression of phospholipid synthesis genes (e.g. INO1, CHO1, OPI3, and INO2), leading to a massive increase in the synthesis of membrane phospholipids and the irregular expansion of the nuclear/ER membrane; its defects in the synthesis of TAG and the resulting accumulation of fatty acids are related to an increase in the susceptibility to fatty acid–induced toxicity and a decrease in lipid droplet number (13–18). Pah1 deficiency also results in the reduction of energy levels, hypersensitivity to oxidative stress and a shortened chronological life span (19), the inability to grow at elevated temperatures (13, 16, 20) and on non-fermentable carbon sources (13, 20), and defects in cell-wall integrity (21, 22) and vacuole fusion and acidification (23, 24).
On a genetic level, PAH1 is transcriptionally regulated throughout growth by nutrient status, with maximum expression coinciding with the synthesis of TAG (24–26). On a biochemical level, the PAP activity of Pah1 is stimulated by negatively charged phospholipids (27) but inhibited by positively charged sphingoid bases (28) and by nucleotides (29). The posttranslational modification of Pah1 as mediated by phosphorylation and dephosphorylation plays a crucial role in the control of its catalytic activity, cellular location, and susceptibility to degradation by the proteasome (30–37). Pah1 in the cytosol is a phosphoprotein whose phosphorylation is carried out by multiple protein kinases that include Pho85-Pho80 (33), Cdc28-cyclin B (32), PKA (34), PKC (35), and casein kinase II (38) (Fig. 1). For its catalytic function, Pah1 localizes to the ER membrane through its dephosphorylation catalyzed by the organelle-associated Nem1 (catalytic subunit)-Spo7 (regulatory subunit) phosphatase (16, 30–34, 36, 39–41) (Fig. 1A). In the membrane localization of Pah1, its C-terminal acidic tail facilitates an interaction with the protein phosphatase complex (40), and its N-terminal amphipathic helix is required to associate with the membrane (31) (Fig. 1B). The phosphorylation of Pah1 tends to attenuate its cellular function by sequestering the enzyme to the cytosol apart from the location of its substrate PA as well as by inhibiting its PAP activity (30, 32–34). Overall, phosphorylation of Pah1 favors phospholipid synthesis at the expense of TAG synthesis, and its dephosphorylation favors TAG synthesis at the expense of phospholipid synthesis (3).
Pah1 contains conserved N-LIP and HAD-like domains (13, 42) that are required for PAP catalytic activity and the in vivo function of the enzyme (14) (Fig. 1B). The enzyme contains two non-conserved regions, one located between the conserved domains and the other at the C terminus. These non-conserved regions contain multiple sites of phosphorylation by different protein kinases (Fig. 1B). In this work, we performed a mutational analysis of the non-conserved regions to gain further insight into the role of the regions for Pah1 function. This analysis showed that the non-conserved N- and C-terminal regions are not essential for PAP activity and TAG production and for the function of Pah1 in the complementation of the pah1Δ temperature-sensitive phenotype. We identified a WRDPLVDID domain at the boundary of the C-terminal non-conserved region that is required for Pah1 function in vivo. In particular, the tryptophan residue of the domain, which is conserved in orthologous PAP enzymes (i.e. lipins) from mice and humans, is essential for Pah1 function in TAG synthesis. The mutations of the tryptophan residue did not abolish the catalytic activity of Pah1, demonstrating that PAP activity is required for, but not sufficient for, the physiological function of Pah1.
Results
Rationale and approach
We undertook an unbiased approach to examine the importance of the N- and C-terminal non-conserved regions of Pah1 (Fig. 1, gray) for its PAP activity and physiological function. A series of mutations (Table 1) were introduced into the coding sequence of PAH1, and the mutant alleles were expressed on a low-copy plasmid in the pah1Δ mutant that lacks the wild-type enzyme (Table 2). For experiments to evaluate the effects of the mutations on PAP activity, the mutant enzymes were expressed in pah1Δ app1Δ dpp1Δ lpp1Δ quadruple mutant cells (43), which lack all of the endogenous PAP activities encoded by PAH1 (13), APP1 (43), DPP1 (44), and LPP1 (45) (Table 2). The expression of Pah1 mutants was confirmed by immunoblot analysis with antibodies raised against N- and C-terminal peptides of the protein; the N-terminal antibody was used to detect the mutant enzymes with C-terminal truncations, and C-terminal antibody was used to detect those with the N-terminal truncations. Mutants defective in Pah1 function (e.g. gene deletion and catalytic site mutants) exhibit defects in lipid metabolism and cell physiology that are reflected in the loss of growth at the elevated temperature (13, 14, 16, 20). Thus, the in vivo function of Pah1 mutants was analyzed by their ability to complement the pah1Δ growth defect at 37 °C. As the major function of Pah1 is to catalyze the penultimate step of TAG synthesis (1–3, 13), the mutant enzymes were also examined for their effects on cellular TAG content.
Table 1.
Plasmids used in this study
| Plasmid | Relevant characteristics | Source or Reference |
|---|---|---|
| pRS415 | Single-copy E. coli/yeast shuttle vector with LEU2 | Ref. 56 |
| pGH315 | PAH1 inserted into pRS415 (for expression of full-length Pah1) | Ref. 32 |
| pGH315-ΔN-LIP | Pah1 lacking residues 18–104 | This study |
| pGH315-ΔHAD-like | Pah1 lacking residues 347–591 | This study |
| pGH315-ΔN-NCRa | Pah1 lacking residues 105–185 | This study |
| pGH315-ΔN-NCRb | Pah1 lacking residues 186–266 | This study |
| pGH315-ΔN-NCRc | Pah1 lacking residues 267–346 | This study |
| pGH315-ΔN-NCRab | Pah1 lacking residues 105–266 | This study |
| pGH315-ΔN-NCRbc | Pah1 lacking residues 186–346 | This study |
| pGH315-ΔN-NCRac | Pah1 lacking residues 105–185 and 267–346 | This study |
| pGH315-ΔN-NCR | Pah1 lacking residues 105–346 | This study |
| pGH315-C821 | Pah1 lacking residues 822–862 | This study |
| pGH315-C752 | Pah1 lacking residues 753–862 | This study |
| pGH315-C700 | Pah1 lacking residues 701–862 | This study |
| pGH315-C646 | Pah1 lacking residues 647–862 | This study |
| pGH315-C636 | Pah1 lacking residues 637–862 | This study |
| pGH315-C626 | Pah1 lacking residues 627–862 | This study |
| pGH315-C616 | Pah1 lacking residues 617–862 | This study |
| pGH315-C606 | Pah1 lacking residues 607–862 | This study |
| pGH315-C591 | Pah1 lacking residues 592–862 | This study |
| pGH315-ΔC-NCR1 | Pah1 lacking residues 647–834 | This study |
| pGH315-ΔC-NCR2 | Pah1 lacking residues 627–834 | This study |
| pGH315-ΔC-NCR3 | Pah1 lacking residues 592–834 | This study |
| pGH315-ΔC-CR | Pah1 lacking residues 629–645 | This study |
| pGH315-CR | Pah1 lacking residues 105–346, 592–628, and 646–834 | This study |
| pGH315-W637A | Pah1 with the W637A mutation | This study |
| pGH315-W637E | Pah1 with the W637E mutation | This study |
| pGH315-W637R | Pah1 with the W637R mutation | This study |
| pGH315-W637F | Pah1 with the W637F mutation | This study |
| pGH315-R638A | Pah1 with the R638A mutation | This study |
| pGH315-P640A | Pah1 with the P640A mutation | This study |
| pGH315-L641A | Pah1 with the L641A mutation | This study |
| pGH315-I644A | Pah1 with the I644A mutation | This study |
| pGH315-D645K | Pah1 with the D645K mutation | This study |
Table 2.
Strains used in this study
| Strain | Genotype or relevant characteristics | Reference |
|---|---|---|
| E. coli | ||
| DH5α | F− φ80dlacZΔΜ15Δ (lacZYA-argF)U169 deoR recA1 endA1 hsdR17 (rk− mk+) phoA supE44 λ−thi-1 gyrA96 relA1 | 53 |
| S. cerevisiae | ||
| W303-1A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 66 |
| GHY57 | pah1Δ::URA3 derivative of W303-1A | 13 |
| GHY66 | pah1Δ::URA3 app1Δ::natMX4 dpp1Δ::TRP1/Kanr lpp1Δ::HIS3/Kanr derivative of W303–1A | 43 |
| RS453 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-52 | 67 |
| SS1026 | pah1Δ::TRP1 derivative of RS453 | 16 |
Truncation analysis of the N- and C-terminal non-conserved regions of Pah1
The non-conserved region at the N terminus of Pah1 was divided into three segments of equal length (80 amino acids), and these segments were removed alone or in combination; the truncated enzymes were expressed in the pah1Δ mutant and tested for their ability to support growth at 37 °C. Like wild-type Pah1, its mutants with partial deletions of the N-terminal non-conserved region complemented the pah1Δ temperature sensitivity (Fig. 2). Similarly, Pah1 lacking the entire non-conserved region (ΔN-NCR) complemented the pah1Δ phenotype. Overall, these data indicate that the N-terminal non-conserved region of Pah1 is not essential for its in vivo function.
Figure 2.
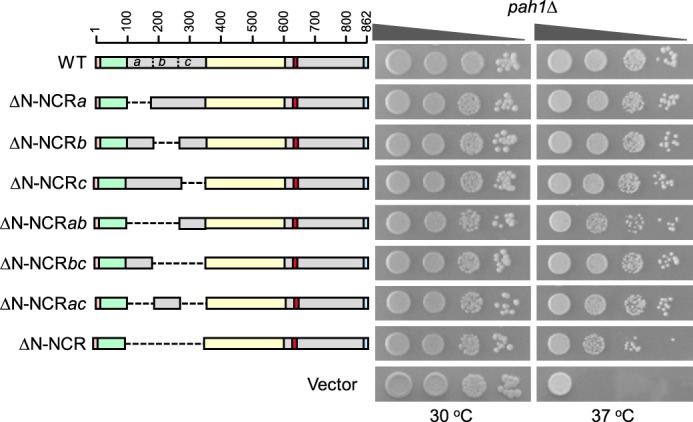
Effects of N-terminal region mutations of Pah1 on the complementation of the pah1Δ temperature-sensitive phenotype. The indicated wild-type and N-terminal mutant forms of Pah1 were expressed from pGH315-based plasmids (Table 1) in the pah1Δ mutant strain GHY57. The cells were grown to saturation in SC−Leu medium at 30 °C; serial dilutions (10-fold) of the cells were spotted onto SC−Leu agar plates, and growth was scored after 3 days of incubation at 30 and 37 °C. The data are representative of three independent experiments. Top left, positions of the amino acid residues of full-length Pah1. The color code for the domains of Pah1 is as indicated in the legend to Fig. 1.
For analysis of the C-terminal non-conserved region of Pah1, nested deletions were generated from the C-terminal end of the enzyme (Fig. 3) and examined for their effects on Pah1 function. Like wild-type Pah1, its mutants C821, C752, C700, and C646 complemented the temperature-sensitive phenotype of pah1Δ. However, the C591 mutant lacking residues 592–862 was not functional (Fig. 3). This indicated that a sequence between amino acids 592 and 646 was required for Pah1 function in vivo, but the rest of the C-terminal region was not required for function.
Figure 3.
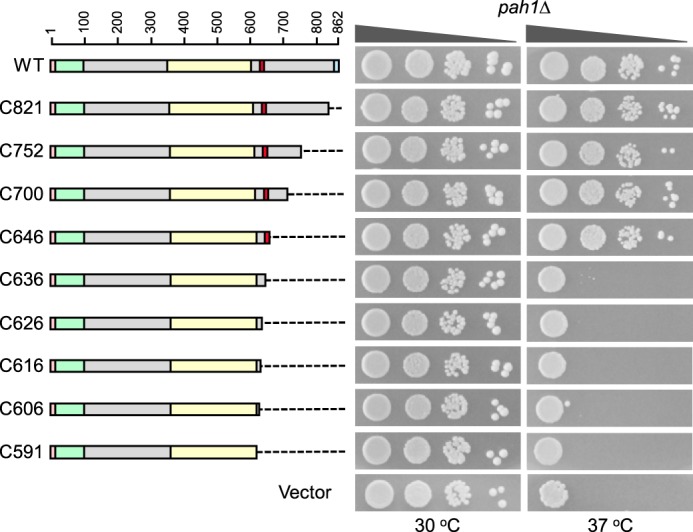
Effects of C-terminal region mutations of Pah1 on the complementation of the pah1Δ temperature-sensitive phenotype. The indicated wild-type and C-terminal mutant forms of Pah1 were expressed from pGH315-based plasmids (Table 1) in the pah1Δ mutant strain SS1026. The cells were grown to saturation in SC−Leu medium at 30 °C; serial dilutions (10-fold) of the cells were spotted onto SC−Leu agar plates, and growth was scored after 3 days of incubation at 30 and 37 °C. The data are representative of three independent experiments. The positions of the amino acid residues of full-length Pah1 are shown (top left). The color code for the domains of Pah1 is as indicated in the legend to Fig. 1.
The acidic tail of Pah1 (Fig. 1, blue), which is conserved among various yeast species, but not present in orthologous lipin proteins of higher eukaryotes (40), is required for the interaction with the Nem1-Spo7 phosphatase that dephosphorylates the enzyme (40). C-terminal truncation mutants were constructed that retained the acidic tail to examine its importance in this analysis. The growth phenotypes of cells expressing the mutants C646 and ΔC-NCR1 and mutants C591 and ΔC-NCR3 were similar (Figs. 3 and 4), indicating that loss of the acidic tail was not the basis for the inability of the C591 truncation mutant to complement the temperature-sensitive phenotype.
Figure 4.
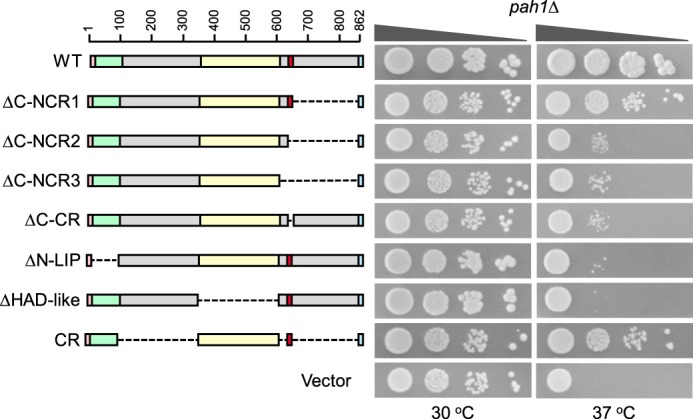
Effects of conserved and non-conserved region mutations of Pah1 on the complementation of the pah1Δ temperature-sensitive phenotype. The indicated wild-type and mutant forms of Pah1 were expressed from pGH315-based plasmids (Table 1) in the pah1Δ mutant strain SS1026. The cells were grown to saturation in SC−Leu medium at 30 °C; serial dilutions (10-fold) of the cells were spotted onto SC−Leu agar plates, and growth was scored after 3 days of incubation at 30 and 37 °C. The data are representative of three independent experiments. The positions of the amino acid residues of full-length Pah1 are shown (top left). The color code for the domains of Pah1 is as indicated in the legend to Fig. 1.
The sequence WRDPLVDID in the C-terminal region is essential for Pah1 function in vivo
To narrow down the region between residues 592 and 646 that is required for Pah1 function, a second set of mutations with 10-amino acid segments successively removed were constructed (e.g. C636, C626, C616, and C606) (Fig. 3). The analysis of these mutants indicated that the segment between amino acids 637 and 646 with the sequence WRDPLVDID was important for the in vivo function of Pah1 (Fig. 3). Likewise, the ΔC-NCR2 mutation, which is analogous to the C636 mutation but contains the acidic tail, did not complement the temperature-sensitive phenotype of pah1Δ cells (Fig. 4). To confirm that this region is required for Pah1 function, a mutant (i.e. ΔC-CR) that specifically lacks it (Fig. 1, red) was constructed and tested for its ability to support growth of pah1Δ cells at 37 °C. The ΔC-CR mutant was not functional in vivo (Fig. 4). In control experiments, we confirmed that the N-LIP and HAD-like domains are essential for Pah1 function in vivo (14); the mutants lacking these conserved domains failed to complement the temperature-sensitive phenotype of the pah1Δ mutant (Fig. 4). We also constructed a Pah1 mutant that contains the amphipathic helix, the N-LIP and HAD-like catalytic domains, the newly identified sequence WRDPLVDID, and the acidic tail (e.g. CR mutant). This mutant, which lacks both the N- and C-terminal non-conserved regions, was functional in vivo (Fig. 4).
TAG analysis of the N- and C-terminal region mutations of Pah1
Cells expressing Pah1 with the N- and C-terminal region deletions were examined for their cellular TAG content. As described previously for cells expressing wild-type Pah1 (13, 25), the amount of TAG in the stationary phase was nearly 4-fold greater when compared with that in the exponential phase, and the increase in TAG content in the stationary phase coincided with a decrease in phospholipids (Fig. 5A). In the exponential phase, the TAG content of cells expressing the mutant lacking the N-terminal non-conserved region (ΔN-NCR) was 1.6-fold greater when compared with cells expressing the wild-type enzyme (Fig. 5A). With respect to the stationary phase, the ΔN-NCR mutation caused a 15% decrease in TAG content (Fig. 5A).
Figure 5.

Effects of N- and C-terminal region mutations of Pah1 on TAG content and PAP activity. The indicated wild-type and mutant forms of Pah1 were expressed from pGH315-based plasmids (Table 1) in the pah1Δ mutant strain SS1026 (A) or the pah1Δ app1Δ dpp1Δ lpp1Δ quadruple mutant strain GHY66 (B and C). The cells were grown to the exponential and stationary phases of growth in SC−Leu medium with (A) or without (B) [2-14C]acetate (1 μCi/ml) at 30 °C. A, lipids were extracted and separated by TLC, and the phosphor images were subjected to ImageQuant analysis. The percentages shown for TAG and phospholipids were normalized to the total 14C-labeled chloroform-soluble fraction. B, cells were harvested by centrifugation, cell extracts were prepared, and PAP activity was measured by following the release of 32Pi from 32P-labeled PA. C, samples (50 μg of protein) of cell extracts from exponential phase cells were subjected to immunoblot analysis using 2 μg/ml anti-Pah1 antibodies raised against the C-terminal (top blot) or N-terminal (bottom blot) portions of Pah1 and anti-Pgk1 antibodies. The asterisks designate the positions of the wild-type and mutant forms of Pah1. The anti-Pah1 antibodies cross-react with proteins other than Pah1 (e.g. bands below (top blot) and above (bottom blot) the 75 kDa marker. The CR mutant was not detected with the anti-Pah1 antibodies. The data in A and B are means ± S.D. (error bars) from triplicate determinations of two independent experiments, whereas the immunoblots shown in C are representative of three independent experiments. The positions of the amino acid residues of full-length Pah1 are shown (A and B, top left). The color code for the domains of Pah1 is as indicated in the legend to Fig. 1. *, p < 0.05 versus WT exponential phase. *, p < 0.05 versus WT stationary phase.
We examined the TAG content of cells expressing the C-terminal region mutants retaining the acidic tail. The ΔC-NCR2 (2- and 4-fold, respectively) and ΔC-NCR3 (3.4- and 5.6-fold, respectively) mutations, which lack the region (Fig. 1, red) with the sequence WRDPLVDID, caused major reductions in TAG content in the exponential and stationary phases (Fig. 5A). Moreover, TAG content (2- and 3.9-fold, respectively, in the exponential and stationary phases) of cells expressing the mutant (e.g. ΔC-CR) that contains the C-terminal non-conserved region, but lacks the region containing WRDPLVDID, was very reduced when compared with the wild-type control (Fig. 5A). Cells expressing the C-terminal mutation that contains the sequence WRDPLVDID (e.g. ΔC-NCR1) had a TAG content similar to that of the wild-type control (Fig. 5A). The cells expressing the mutant Pah1 (e.g. CR), which contains the conserved regions but lacks the non-conserved regions, had a TAG content in the exponential phase similar to that found in cells expressing the wild-type enzyme. The amount of TAG in the stationary phase was 25% lower than that of cells expressing the wild-type Pah1 (Fig. 5A).
PAP activity analysis of the N- and C-terminal region mutations of Pah1
The PAP catalytic activity from cells expressing Pah1 with the N- and C-terminal region mutations was assessed by measuring the release inorganic phosphate from PA. As described previously (25), the cells expressing wild-type Pah1 exhibited 2.4-fold greater PAP activity in stationary phase when compared with exponential phase (Fig. 5B). The PAP activity of cells expressing the mutant lacking the N-terminal non-conserved region (ΔN-NCR) was 1.5-fold greater when compared with cells expressing the wild-type enzyme. However, with respect to the stationary phase, the ΔN-NCR mutation caused a dramatic decrease (2-fold) in the PAP activity when compared with the wild-type control (Fig. 5B).
The C-terminal mutations (e.g. ΔC-NCR2, ΔC-NCR3, and ΔC-CR), which failed to complement the temperature sensitivity of the pah1Δ mutant and exhibited reduced TAG content, did not have a negative effect on PAP catalytic activity (Fig. 5B). Instead, the mutations caused in an increase in the PAP activity. In particular, the ΔC-CR mutation, which lacks residues WRDPLVDID, resulted in an increase in activity of 2.3- and 1.6-fold, respectively, for exponential and stationary phase cells when compared with the wild-type control (Fig. 5B). The cells expressing Pah1 with the CR mutation, which lacks the non-conserved regions at both the N- and C-terminal regions, exhibited PAP activity (Fig. 5B). In the exponential phase, the PAP activity imparted by the CR mutant was 2-fold greater when compared with that of the wild-type enzyme. However, unlike cells expressing the wild-type enzyme where the PAP activity increased in stationary phase, the level of activity for cells expressing the CR mutant remained the same. Like the cells expressing Pah1 with the ΔN-NCR mutation, the PAP activity imparted by the CR mutant enzyme was reduced (20%) when compared with that of wild-type Pah1 (Fig. 5B).
The immunoblot analysis shown in Fig. 5C indicated that these mutations did not have a negative impact on the expression of Pah1 in the exponential phase of growth. We could not examine the expression of the Pah1 mutants from stationary phase cells because of the proteasomal degradation of the enzyme (46).
Trp-637 in the sequence WRDPLVDID is required for the in vivo function of Pah1, but not its PAP activity
We sought to identify the amino acid residue(s) within the sequence WRDPLVDID of Pah1 that is required for its function in vivo. A bioinformatics analysis revealed that several of the residues within the sequence are conserved or structurally conserved in Pah1/lipin proteins from other yeast, mice, and humans (Fig. 6). Accordingly, site-specific mutations for the conserved residues were constructed in Pah1; the mutants were expressed in pah1Δ cells and examined for their ability to complement the temperature-sensitive phenotype (Fig. 7). Of the mutations, only those for Trp-637 (e.g. W637A, W637E, and W637R) were unable to complement the temperature sensitivity of the pah1Δ mutant. The W637F mutant, however, did complement the pah1Δ growth defect at 37 °C.
Figure 6.

Amino acid residues within the sequence WRDPLVDID are evolutionarily conserved in yeast and mammalian Pah1/lipin proteins. Residues highlighted in yellow are 100% conserved, whereas those highlighted in blue are partially conserved.
Figure 7.
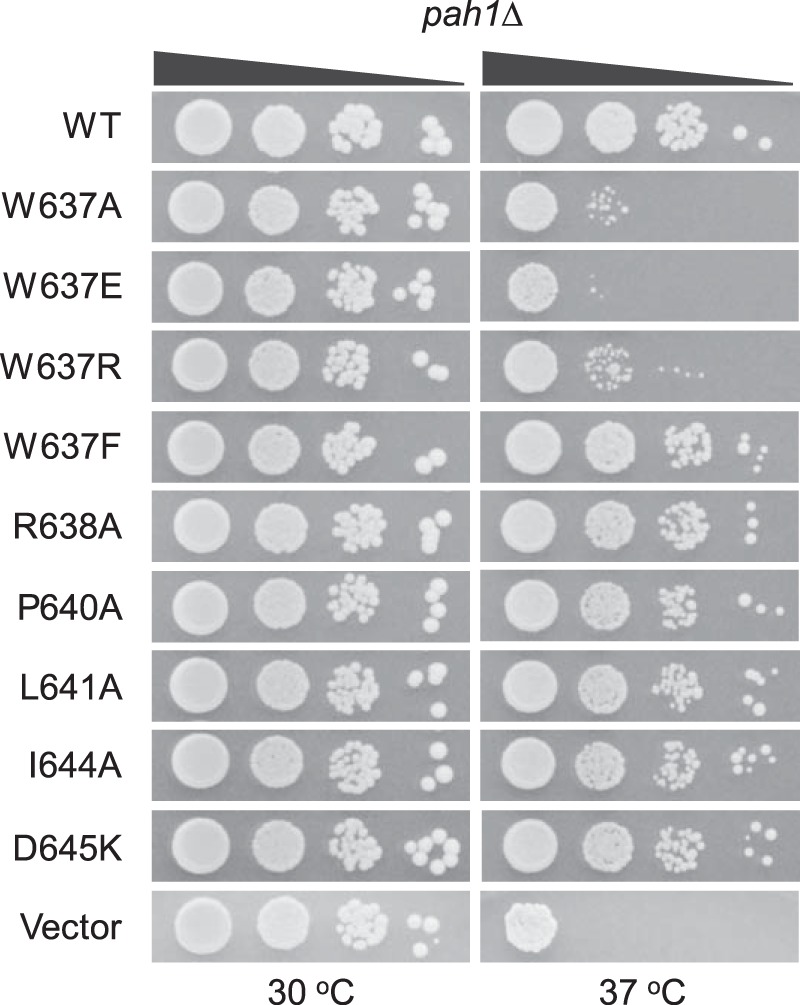
Effects of site-specific mutations of Pah1 within the sequence WRDPLVDID on the complementation of the pah1Δ temperature-sensitive phenotype. The indicated wild-type and site-specific mutant forms of Pah1 were expressed from pGH315-based plasmids (Table 1) in the pah1Δ mutant strain SS1026. The cells were grown to saturation in SC−Leu medium at 30 °C; serial dilutions (10-fold) of the cells were spotted onto SC−Leu agar plates, and growth was scored after 3 days of incubation at 30 and 37 °C. The data are representative of three independent experiments.
The site-specific mutants of Trp-637 were also examined for their ability to complement the pah1Δ mutant for its defect in TAG content. The W637A, W637E, and W637R mutants, which were not functional in vivo, did not complement the defect in TAG content. For example, the TAG content of exponential and stationary phase cells expressing the W637E mutant enzyme, respectively, was 2.8- and 8-fold lower when compared with cells expressing wild-type Pah1 (Fig. 8A). The W637F mutation, which could complement the pah1Δ growth defect at 37 °C, had lower TAG content (1.8- and 2.3-fold, respectively) in exponential and stationary phase cells when compared with the control, but these levels were not as low as those exhibited by cells expressing the other tryptophan mutations.
Figure 8.
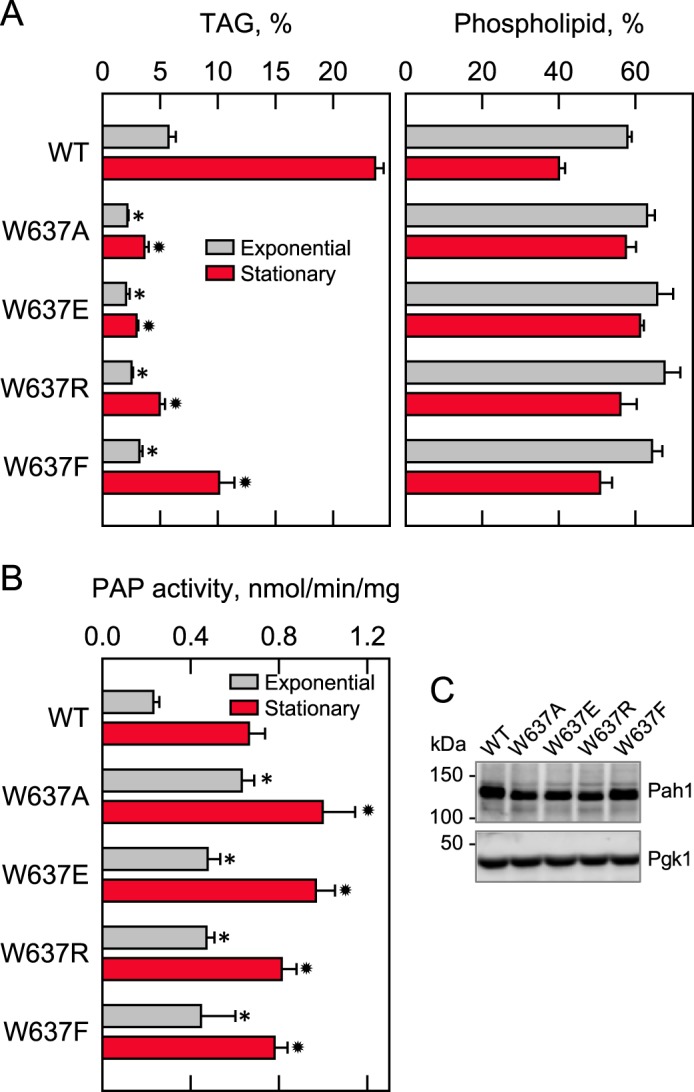
Effects of Trp-637 mutations of Pah1 on TAG content and PAP activity. The indicated wild-type and mutant forms of Pah1 were expressed from pGH315-based plasmids (Table 1) in the pah1Δ mutant strain SS1026 (A) or the pah1Δ app1Δ dpp1Δ lpp1Δ quadruple mutant strain GHY66 (B). The cells were grown to the exponential and stationary phases of growth in SC−Leu medium with (A) or without (B) [2-14C]acetate (1 μCi/ml) at 30 °C. A, lipids were extracted and separated by TLC, and the phosphor images were subjected to ImageQuant analysis. The percentages shown for TAG and phospholipids were normalized to the total 14C-labeled chloroform-soluble fraction. B, cells were harvested by centrifugation, cell extracts were prepared, and PAP activity was measured by following the release of 32Pi from 32P-labeled PA. C, samples (50 μg of protein) of cell extracts from exponential phase cells were subjected to immunoblot analysis using 2 μg/ml anti-Pah1 antibodies raised against the C-terminal portion of Pah1 and anti-Pgk1 antibodies. The data in A and B are means ± S.D. (error bars) from triplicate determinations of two independent experiments, whereas the immunoblots shown in C are representative of three independent experiments. *, p < 0.05 versus WT exponential phase. *, p < 0.05 versus WT stationary phase.
The effects of the site-specific Trp-637 mutations on PAP catalytic activity were examined. Whereas the W637A, W637E, and W637R mutations had strong negative effects on the physiological function of Pah1, they did not compromise the PAH1-encoded PAP activity (Fig. 8B). In fact, in the exponential phase cells, these mutations, along with the W637F mutation, caused increases in the PAP activity of 2–2.7-fold (Fig. 8B). Smaller increases in PAP activity (1.6-fold) were observed for the mutant enzymes from stationary phase cells. Immunoblot analysis indicated that the mutations of Trp-637 did not affect the expression of Pah1 in the exponential phase of growth (Fig. 8C).
Discussion
Pah1 has emerged as one of the most important enzymes in yeast lipid metabolism (1–8). It plays a major role in controlling the utilization of its substrate PA for the synthesis of TAG or membrane phospholipids. Loss of Pah1 results in devastating effects on several aspects of cell physiology (3). Accordingly, much attention has been paid to understanding the mode of action and regulation of the enzyme (1–3). Our laboratory has pursued mutagenic studies on Pah1 to gain insights into its structural features and related functions. Previous studies have shown that the conserved Asp-398 and Asp-400 of the DXDX(T/V) catalytic motif within the HAD-like domain and the conserved Gly-80 within the N-LIP domain are essential for PAP catalytic activity and enzyme function in vivo (14). It has been known that the amphipathic helix found at the N terminus is required for Pah1 interaction with the membrane (31), and the C-terminal acidic tail is required for Pah1 to interact with the Nem1-Spo7 phosphatase (40). We also know that the phosphorylation of Pah1 takes place at the non-conserved regions located at the N- and C-terminal regions of the protein. When these regions are phosphorylated, the association of Pah1 with the cytosol is favored, and its catalytic function is inhibited (30–37), yet these phosphorylations also protect Pah1 from degradation by the 20S proteasome (46, 47).
In this work, we undertook an unbiased approach to systematically remove portions of the non-conserved regions to gain additional information on their importance to enzyme function. The truncation analyses of the N- and C-terminal non-conserved regions indicated that neither region is required for the in vivo function of Pah1, as determined by the complementation of the pah1Δ temperature-sensitive phenotype. In fact, the CR mutant protein, which lacks both the N- and C-terminal non-conserved regions, had PAP activity and complemented the pah1Δ growth defect at 37 °C. Although the non-conserved regions were not essential for the in vivo function of Pah1, each region had a regulatory effect on PAP activity. On one hand, the ΔN-NCR mutation negated the increase in PAP activity that is normally observed in the stationary phase of growth. On the other hand, the C-terminal truncation mutants exhibited greater PAP activity than that normally observed in wild-type cells. Both the N- and C-terminal non-conserved regions contain phosphorylation sites for Pho85-Pho80, PKA, and casein kinase II (33, 34, 38) (Fig. 1B). Because the phosphorylations by these protein kinases normally cause the inhibition of PAP activity (33, 34, 38), the loss of these sites might be expected to result in enzyme activation. The elevated activity imparted by the C-terminal truncation mutants is consistent with this hypothesis, but the reduced activity imparted by the N-terminal truncation mutants is not. Elevated PAP activity was also observed in cells expressing the ΔC-CR mutant lacking the sequence WRDPLVDID. This mutant still possesses the C-terminal non-conserved region with its phosphorylation sites. Thus, the basis for elevated PAP activity by the C-terminal truncation mutations is not simply the loss of phosphorylation sites. However, it is not clear whether the C-terminal conserved sequence influences the phosphorylations by Pho85-Pho80, PKA, and casein kinase II that take place at the non-conserved regions. Also, we cannot rule out the possibility that the reduced or elevated PAP activity, respectively, imparted by the N- and C-terminal mutations, are due to differences in Pah1 abundance. We know that enzyme abundance was not majorly affected by the mutations in the exponential phase, but we could not address the question of abundance in the stationary phase because of the inability to detect Pah1 by immunoblotting due to the proteasomal degradation of the enzyme (46).
By whittling down the C-terminal region of Pah1, we discovered that it contains the conserved sequence WRDPLVDID, which is important for the in vivo function of Pah1. This sequence, which is a member of the protein domain family known as WRNPLPNID (48), contains residues conserved (or partially conserved) in Pah1/lipin proteins from yeast, mice, and humans. The WRNPLPNID domain is typically found in unstructured, intrinsically disordered regions of proteins (49), and indeed, this domain is present in a Pah1 region predicted to be unfolded (47). The tryptophan in the Pah1 sequence WRDPLVDID is contained within a putative WW-binding domain known to be involved in protein–protein interactions (50, 51), and the residues following the tryptophan in the sequence are contained within a putative destruction box (e.g. RXPLXXI) (48).
The analysis of the site-specific mutations of the conserved residues within the sequence WRDPLVDID revealed that Trp-637 is required for Pah1 function in vivo. The conserved residues that comprise the putative destruction box were not critical for the in vivo function, and we did not pursue a further analyses of those mutants in this work. Unlike other conserved residues (e.g. Gly-80, Asp-398, and Asp-400) that are essential for PAP activity and the in vivo function of Pah1, the mutations of Trp-637 did not compromise the enzyme activity. This may be explained by the fact that Gly-80, Asp-398, and Asp-400 are catalytic residues (14), whereas Trp-637 is presumably a regulatory residue. These findings revealed that the catalytic function of Pah1 is required, but not sufficient, for its in vivo function.
The mutation of Trp-637 to alanine, glutamate, or arginine resulted in much reduced levels of TAG and the loss of Pah1 function in vivo. Although the phenylalanine mutation caused a significant reduction in TAG content, it still afforded the complementation of the pah1Δ temperature-sensitive phenotype. This indicated that there is some minimal amount of TAG that will support cell function. Along these lines, previous studies have shown that loss of the acidic tail causes a reduction in TAG content (40), but as shown here, the acidic tail was not essential for the complementation of the pah1Δ growth defect at 37 °C. All of the phenotypes (e.g. inability to complement the growth defect of pah1Δ at 37 °C and much reduced TAG content, but elevated PAP activity) imparted by the ΔC-CR mutation, which lacks the sequence WRDPLVDID, were recapitulated by the W637A, W637E, and W637R mutations.
We showed the importance of Trp-637 in the in vivo function of Pah1, but the underpinnings of how this residue affects enzyme function are still unclear. This tryptophan residue was not important for the catalytic function of Pah1 because its mutations did not compromise PAP activity. As discussed above, the in vivo function of Pah1 is not only governed by its catalytic activity, but also by its cellular location as mediated by the phosphorylation and dephosphorylation of the enzyme (3). At this point, it is unclear whether Trp-637 mediates interactions with a protein kinase and/or the Nem1-Spo7 phosphatase. It is also possible that Trp-637 mediates interaction with some yet to be identified protein that affects enzyme location. Thus, the Trp-637 mutations will be useful in addressing these questions in future studies. Pah1 abundance is partially stabilized in the stationary phase of mutants defective in the ubiquitin degradation pathway (46). Because the destruction box is known to be important for the ubiquitin pathway of protein degradation (52), the mutations within the RXPLXXI motif should be useful in addressing the hypothesis that Pah1, in addition to being degraded by a ubiquitin-independent mechanism (47), is also degraded by a ubiquitin-dependent mechanism. Clearly, the revelation that Pah1 contains the conserved WRDPLVDID domain, which is important for the in vivo function of Pah1, is a major finding of this work and opens new avenues of investigation.
Experimental procedures
Materials
Growth medium components were obtained from Difco. Phusion high fidelity DNA polymerase and carrier DNA for yeast transformation were purchased from New England Biolabs and Clontech, respectively. The DNA gel extraction and plasmid purification kits were from Qiagen. DNA size ladders and protein assay reagents were obtained from Bio-Rad. Aprotinin, benzamidine, bovine serum albumin, leupeptin, pepstatin, and Triton X-100 were purchased from Sigma-Aldrich. Acrylamide solutions and scintillation counting supplies were from National Diagnostics. Radiochemicals and silica gel 60 plates were purchased from PerkinElmer Life Sciences and EM Science, respectively. Anti-Pah1 antibodies directed against the N terminus (residues 85–103) or C terminus (residues 778–794) of the protein were generated in rabbits at BioSynthesis, Inc. Alkaline phosphatase-conjugated goat anti-rabbit IgG antibodies, alkaline phosphatase-conjugated goat anti-mouse IgG antibodies, and mouse anti-phosphoglycerate kinase antibodies were from Thermo Scientific, Pierce, and Invitrogen, respectively. The Western blotting detection kit was from GE Healthcare. All other chemicals were reagent grade or better.
Strains and growth conditions
The yeast strains used in this study are listed in Table 2. Escherichia coli strain DH5α was used for the propagation of plasmids. E. coli cells were grown at 37 °C in LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.0); ampicillin (100 μg/ml) was added to select for cells carrying plasmids. Standard methods were used for culturing yeast (53, 54). Yeast cells were routinely grown at 30 °C in synthetic complete medium without leucine (SC−Leu) to select for cells carrying specific plasmids. The growth of yeast cells in liquid medium was measured by absorbance at 600 nm using a spectrophotometer. Solid media for the growth of E. coli and yeast contained agar at a concentration of 1.5 and 2%, respectively. For the measurement of growth on agar plates, serially diluted (10-fold) cell suspensions of stationary phase cells were spotted onto agar plates, and growth was scored after incubation for 3 days at 30 and 37 °C.
DNA manipulations and construction of mutants
The plasmids used in this study are listed in Table 1. Standard methods were used for isolation of chromosomal and plasmid DNA, for digestion and ligation of DNA, and for PCR amplification of DNA (53–55). Plasmid pGH315, which is a derivative of the E. coli/yeast shuttle vector pRS415 with LEU2 (56), directs the low-copy expression of Pah1 in yeast (32). The deletion of PAH1 codons was performed by overlap extension PCR with appropriate primers at the XbaI/XhoI sites in pGH315. pGH315-CR was generated from pGH315-ΔN-NCR by overlap extension PCR at the XbaI/XhoI sites in the plasmid. The derivatives of pGH315 that contained single-site mutations were constructed by PCR-mediated site-directed mutagenesis using appropriate primers as described previously (32). Plasmid transformations of E. coli (53) and yeast (57) were performed as described previously. All mutations were confirmed by PCR analysis and DNA sequencing.
Preparation of cell extracts
All steps to prepare cell extracts were performed at 4 °C. Yeast cultures were harvested in the exponential (A600 nm = 0.5) and the stationary (A600 nm = 3–4) phases by centrifugation at 1,500 × g for 5 min. The cells were washed with water and resuspended in lysis buffer (50 mm Tris-HCl (pH 7.5), 0.3 m sucrose, 10 mm 2-mercaptoethanol, 0.5 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine, 5 μg/ml aprotinin, 5 μg/ml leupeptin, and 5 μg/ml pepstatin). Glass beads (0.5-mm diameter) were added to cell suspensions and then subjected to five repeats of 1-min burst and 2-min cooling using a BioSpec Products Mini-Beadbeater-16 (58). The disrupted cells were centrifuged at 1,500 × g for 10 min to separate unbroken cells and cell debris (pellet) from cell extracts (supernatant). The protein concentration was determined by the method of Bradford (59) using bovine serum albumin as a standard.
SDS-PAGE and Western blot analysis
SDS-PAGE (60) using 8% polyacrylamide gels and Western blotting (61, 62) with PVDF membrane were performed by standard protocols. The samples for blotting were normalized to total protein loading. Ponceau S staining was used to monitor the protein transfer from the polyacrylamide gels to the PVDF membrane. Rabbit anti-Pah1 (32) antibodies and mouse anti-Pgk1 antibodies (for a loading control) were used at a concentration of 2 μg/ml. Alkaline phosphatase-conjugated goat anti-rabbit IgG antibodies and goat anti-mouse IgG antibodies were used at a dilution of 1:5,000. Immune complexes were detected using the enhanced chemifluorescence Western blotting detection kit. Fluorimaging was used to acquire images from Western blots, and the relative densities of the images were analyzed using ImageQuant software. Signals were in the linear range of detectability.
Preparation of 32P-labeled PA and measurement of PAP activity
[32P]PA was enzymatically synthesized from DAG and [γ-32P]ATP with E. coli DAG kinase (58). PAP activity was measured for 20 min at 30 °C by following the release of water-soluble 32Pi from chloroform-soluble [32P]PA (10,000 cpm/nmol) as described previously (58). The reaction mixture in a total volume of 100 μl contained 50 mm Tris-HCl (pH 7.5), 1 mm MgCl2, 0.2 mm PA, 2 mm Triton X-100, and enzyme protein. Enzyme assays were performed in triplicate, and all reactions were linear with time and protein concentration. A unit of PAP activity was defined as the amount of enzyme that catalyzed the formation of 1 nmol of product per minute.
Radiolabeling and analysis of lipids
The steady-state labeling of lipids with [2-14C]acetate was performed as described previously (63). Lipids were extracted (64) from the radiolabeled cells and then separated by one-dimensional TLC (65). The resolved lipids were visualized by phosphorimaging and quantified by ImageQuant software using a standard curve of [2-14C]acetate. The identity of radiolabeled lipids was confirmed by comparison with the migration of authentic standards visualized by staining with iodine vapor.
Analyses of data
Statistical analyses were performed with SigmaPlot software. The p values < 0.05 were taken as a significant difference.
Author contributions
Y. P., G.-S. H., and G. M. C. designed the study, analyzed the results, and prepared the manuscript. Y. P. and G.-S. H. performed the experiments.
Acknowledgments
We acknowledge Michael Airola for the observation that Pah1 contains the WRDPLVDID domain and that it is conserved in mammalian lipin proteins.
This work was supported, in whole or in part, by National Institutes of Health Grant GM028140 from the United States Public Health Service. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
In this paper, Saccharomyces cerevisiae is used interchangeably with yeast.
- PAP
- phosphatidate phosphatase
- PA
- phosphatidate
- DAG
- diacylglycerol
- TAG
- triacylglycerol
- ER
- endoplasmic reticulum
- HAD
- haloacid dehalogenase
- SC
- synthetic complete.
References
- 1. Carman G. M., and Han G.-S. (2006) Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem. Sci. 31, 694–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carman G. M., and Han G.-S. (2009) Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J. Biol. Chem. 284, 2593–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pascual F., and Carman G. M. (2013) Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta 1831, 514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carman G. M., and Han G.-S. (2011) Regulation of phospholipid synthesis in the yeast Saccharomyces cerevisiae. Annu. Rev. Biochem. 80, 859–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henry S. A., Kohlwein S. D., and Carman G. M. (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190, 317–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Siniossoglou S. (2009) Lipins, lipids and nuclear envelope structure. Traffic 10, 1181–1187 [DOI] [PubMed] [Google Scholar]
- 7. Siniossoglou S. (2013) Phospholipid metabolism and nuclear function: roles of the lipin family of phosphatidic acid phosphatases. Biochim. Biophys. Acta 1831, 575–581 [DOI] [PubMed] [Google Scholar]
- 8. Fernández-Murray J. P., and McMaster C. R. (2016) Lipid synthesis and membrane contact sites: a crossroads for cellular physiology. J. Lipid Res. 57, 1789–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brindley D. N., Pilquil C., Sariahmetoglu M., and Reue K. (2009) Phosphatidate degradation: phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim. Biophys. Acta 1791, 956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Csaki L. S., and Reue K. (2010) Lipins: multifunctional lipid metabolism proteins. Annu. Rev. Nutr. 30, 257–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Csaki L. S., Dwyer J. R., Fong L. G., Tontonoz P., Young S. G., and Reue K. (2013) Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog. Lipid Res. 52, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reue K., and Dwyer J. R. (2009) Lipin proteins and metabolic homeostasis. J. Lipid Res. 50, S109–S114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han G.-S., Wu W.-I., and Carman G. M. (2006) The Saccharomyces cerevisiae lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 281, 9210–9218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han G.-S., Siniossoglou S., and Carman G. M. (2007) The cellular functions of the yeast lipin homolog Pah1p are dependent on its phosphatidate phosphatase activity. J. Biol. Chem. 282, 37026–37035 [DOI] [PubMed] [Google Scholar]
- 15. Fakas S., Qiu Y., Dixon J. L., Han G.-S., Ruggles K. V., Garbarino J., Sturley S. L., and Carman G. M. (2011) Phosphatidate phosphatase activity plays a key role in protection against fatty acid-induced toxicity in yeast. J. Biol. Chem. 286, 29074–29085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santos-Rosa H., Leung J., Grimsey N., Peak-Chew S., and Siniossoglou S. (2005) The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J. 24, 1931–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han G.-S., and Carman G. M. (2017) Yeast PAH1-encoded phosphatidate phosphatase controls the expression of CHO1-encoded phosphatidylserine synthase for membrane phospholipid synthesis. J. Biol. Chem. 292, 13230–13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adeyo O., Horn P. J., Lee S., Binns D. D., Chandrahas A., Chapman K. D., and Goodman J. M. (2011) The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J. Cell Biol. 192, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park Y., Han G. S., Mileykovskaya E., Garrett T. A., and Carman G. M. (2015) Altered lipid synthesis by lack of yeast Pah1 phosphatidate phosphatase reduces chronological life span. J. Biol. Chem. 290, 25382–25394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irie K., Takase M., Araki H., and Oshima Y. (1993) A gene, SMP2, involved in plasmid maintenance and respiration in Saccharomyces cerevisiae encodes a highly charged protein. Mol. Gen. Genet. 236, 283–288 [DOI] [PubMed] [Google Scholar]
- 21. Lussier M., White A. M., Sheraton J., di Paolo T., Treadwell J., Southard S. B., Horenstein C. I., Chen-Weiner J., Ram A. F., Kapteyn J. C., Roemer T. W., Vo D. H., Bondoc D. C., Hall J., Zhong W. W., et al. (1997) Large scale identification of genes involved in cell surface biosynthesis and architecture in Saccharomyces cerevisiae. Genetics 147, 435–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruiz C., Cid V. J., Lussier M., Molina M., and Nombela C. (1999) A large-scale sonication assay for cell wall mutant analysis in yeast. Yeast 15, 1001–1008 [DOI] [PubMed] [Google Scholar]
- 23. Sasser T., Qiu Q. S., Karunakaran S., Padolina M., Reyes A., Flood B., Smith S., Gonzales C., and Fratti R. A. (2012) The yeast lipin 1 orthologue Pah1p regulates vacuole homeostasis and membrane fusion. J. Biol. Chem. 287, 2221–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherr G. L., LaMassa N., Li E., Phillips G., and Shen C. H. (2017) Pah1p negatively regulates the expression of V-ATPase genes as well as vacuolar acidification. Biochem. Biophys. Res. Commun. 491, 693–700 [DOI] [PubMed] [Google Scholar]
- 25. Pascual F., Soto-Cardalda A., and Carman G. M. (2013) PAH1-encoded phosphatidate phosphatase plays a role in the growth phase- and inositol-mediated regulation of lipid synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 288, 35781–35792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Soto-Cardalda A., Fakas S., Pascual F., Choi H. S., and Carman G. M. (2012) Phosphatidate phosphatase plays role in zinc-mediated regulation of phospholipid synthesis in yeast. J. Biol. Chem. 287, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu W.-I., and Carman G. M. (1996) Regulation of phosphatidate phosphatase activity from the yeast Saccharomyces cerevisiae by phospholipids. Biochemistry 35, 3790–3796 [DOI] [PubMed] [Google Scholar]
- 28. Wu W.-I., Lin Y.-P., Wang E., Merrill A. H. Jr., and Carman G. M. (1993) Regulation of phosphatidate phosphatase activity from the yeast Saccharomyces cerevisiae by sphingoid bases. J. Biol. Chem. 268, 13830–13837 [PubMed] [Google Scholar]
- 29. Wu W.-I., and Carman G. M. (1994) Regulation of phosphatidate phosphatase activity from the yeast Saccharomyces cerevisiae by nucleotides. J. Biol. Chem. 269, 29495–29501 [PubMed] [Google Scholar]
- 30. O'Hara L., Han G.-S., Peak-Chew S., Grimsey N., Carman G. M., and Siniossoglou S. (2006) Control of phospholipid synthesis by phosphorylation of the yeast lipin Pah1p/Smp2p Mg2+-dependent phosphatidate phosphatase. J. Biol. Chem. 281, 34537–34548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karanasios E., Han G.-S., Xu Z., Carman G. M., and Siniossoglou S. (2010) A phosphorylation-regulated amphipathic helix controls the membrane translocation and function of the yeast phosphatidate phosphatase. Proc. Natl. Acad. Sci. U.S.A. 107, 17539–17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choi H.-S., Su W.-M., Morgan J. M., Han G.-S., Xu Z., Karanasios E., Siniossoglou S., and Carman G. M. (2011) Phosphorylation of phosphatidate phosphatase regulates its membrane association and physiological functions in Saccharomyces cerevisiae: identification of Ser602, Thr723, and Ser744 as the sites phosphorylated by CDC28 (CDK1)-encoded cyclin-dependent kinase. J. Biol. Chem. 286, 1486–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi H.-S., Su W.-M., Han G.-S., Plote D., Xu Z., and Carman G. M. (2012) Pho85p-Pho80p phosphorylation of yeast Pah1p phosphatidate phosphatase regulates its activity, location, abundance, and function in lipid metabolism. J. Biol. Chem. 287, 11290–11301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su W.-M., Han G.-S., Casciano J., and Carman G. M. (2012) Protein kinase A-mediated phosphorylation of Pah1p phosphatidate phosphatase functions in conjunction with the Pho85p-Pho80p and Cdc28p-cyclin B kinases to regulate lipid synthesis in yeast. J. Biol. Chem. 287, 33364–33376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Su W.-M., Han G.-S., and Carman G. M. (2014) Cross-talk phosphorylations by protein kinase C and Pho85p-Pho80p protein kinase regulate Pah1p phosphatidate phosphatase abundance in Saccharomyces cerevisiae. J. Biol. Chem. 289, 18818–18830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu Z., Su W.-M., and Carman G. M. (2012) Fluorescence spectroscopy measures yeast PAH1-encoded phosphatidate phosphatase interaction with liposome membranes. J. Lipid Res. 53, 522–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Su W.-M., Han G.-S., and Carman G. M. (2014) Yeast Nem1-Spo7 protein phosphatase activity on Pah1 phosphatidate phosphatase is specific for the Pho85-Pho80 protein kinase phosphorylation sites. J. Biol. Chem. 289, 34699–34708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsieh L.-S., Su W.-M., Han G.-S., and Carman G. M. (2016) Phosphorylation of yeast Pah1 phosphatidate phosphatase by casein kinase II regulates its function in lipid metabolism. J. Biol. Chem. 291, 9974–9990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Siniossoglou S., Santos-Rosa H., Rappsilber J., Mann M., and Hurt E. (1998) A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J. 17, 6449–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karanasios E., Barbosa A. D., Sembongi H., Mari M., Han G.-S., Reggiori F., Carman G. M., and Siniossoglou S. (2013) Regulation of lipid droplet and membrane biogenesis by the acidic tail of the phosphatidate phosphatase Pah1p. Mol. Biol. Cell 24, 2124–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barbosa A. D., Sembongi H., Su W.-M., Abreu S., Reggiori F., Carman G. M., and Siniossoglou S. (2015) Lipid partitioning at the nuclear envelope controls membrane biogenesis. Mol. Biol. Cell 26, 3641–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Péterfy M., Phan J., Xu P., and Reue K. (2001) Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 27, 121–124 [DOI] [PubMed] [Google Scholar]
- 43. Chae M., Han G.-S., and Carman G. M. (2012) The Saccharomyces cerevisiae actin patch protein App1p is a phosphatidate phosphatase enzyme. J. Biol. Chem. 287, 40186–40196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Toke D. A., Bennett W. L., Dillon D. A., Wu W.-I., Chen X., Ostrander D. B., Oshiro J., Cremesti A., Voelker D. R., Fischl A. S., and Carman G. M. (1998) Isolation and characterization of the Saccharomyces cerevisiae DPP1 gene encoding for diacylglycerol pyrophosphate phosphatase. J. Biol. Chem. 273, 3278–3284 [DOI] [PubMed] [Google Scholar]
- 45. Toke D. A., Bennett W. L., Oshiro J., Wu W.-I., Voelker D. R., and Carman G. M. (1998) Isolation and characterization of the Saccharomyces cerevisiae LPP1 gene encoding a Mg2+-independent phosphatidate phosphatase. J. Biol. Chem. 273, 14331–14338 [DOI] [PubMed] [Google Scholar]
- 46. Pascual F., Hsieh L.-S., Soto-Cardalda A., and Carman G. M. (2014) Yeast Pah1p phosphatidate phosphatase is regulated by proteasome-mediated degradation. J. Biol. Chem. 289, 9811–9822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hsieh L.-S., Su W.-M., Han G.-S., and Carman G. M. (2015) Phosphorylation regulates the ubiquitin-independent degradation of yeast Pah1 phosphatidate phosphatase by the 20S proteasome. J. Biol. Chem. 290, 11467–11478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C. J., Lu S., Chitsaz F., Derbyshire M. K., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Lu F., Marchler G. H., Song J. S., et al. (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davey N. E., and Morgan D. O. (2016) Building a regulatory network with short linear sequence motifs: lessons from the degrons of the anaphase-promoting complex. Mol. Cell 64, 12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Staub O., and Rotin D. (1996) WW domains. Structure 4, 495–499 [DOI] [PubMed] [Google Scholar]
- 51. Sudol M. (1996) Structure and function of the WW domain. Prog. Biophys. Mol. Biol. 65, 113–132 [DOI] [PubMed] [Google Scholar]
- 52. Glotzer M., Murray A. W., and Kirschner M. W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 [DOI] [PubMed] [Google Scholar]
- 53. Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 54. Rose M. D., Winston F., and Heiter P. (1990) Methods in Yeast Genetics: A Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 55. Innis M. A., and Gelfand D. H. (1990) in PCR Protocols: A Guide to Methods and Applications (Innis M. A., Gelfand D. H., Sninsky J. J., and White T. J., eds) pp. 3–12, Academic Press, Inc., San Diego [Google Scholar]
- 56. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ito H., Fukuda Y., Murata K., and Kimura A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carman G. M., and Lin Y.-P. (1991) Phosphatidate phosphatase from yeast. Methods Enzymol. 197, 548–553 [DOI] [PubMed] [Google Scholar]
- 59. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 60. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 61. Burnette W. (1981) Western blotting: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112, 195–203 [DOI] [PubMed] [Google Scholar]
- 62. Haid A., and Suissa M. (1983) Immunochemical identification of membrane proteins after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Methods Enzymol. 96, 192–205 [DOI] [PubMed] [Google Scholar]
- 63. Morlock K. R., Lin Y.-P., and Carman G. M. (1988) Regulation of phosphatidate phosphatase activity by inositol in Saccharomyces cerevisiae. J. Bacteriol. 170, 3561–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 65. Henderson R. J., and Tocher D. R. (1992) in Lipid Analysis (Hamilton R. J., and Hamilton S., eds) pp. 65–111, IRL Press, New York [Google Scholar]
- 66. Ostrander D. B., O'Brien D. J., Gorman J. A., and Carman G. M. (1998) Effect of CTP synthetase regulation by CTP on phospholipid synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 273, 18992–19001 [DOI] [PubMed] [Google Scholar]
- 67. Wimmer C., Doye V., Grandi P., Nehrbass U., and Hurt E. C. (1992) A new subclass of nucleoporins that functionally interact with nuclear pore protein NSP1. EMBO J. 11, 5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]