

Abstract
Acinetobacter baumannii, Acinetobacter nosocomialis, and Acinetobacter pittii are a frequent cause of multidrug-resistant, healthcare-associated infections. Our previous work demonstrated that A. nosocomialis M2 possesses a functional type II secretion system (T2SS) that is required for full virulence. Further, we identified the metallo-endopeptidase CpaA, which has been shown previously to cleave human Factor V and deregulate blood coagulation, as the most abundant type II secreted effector protein. We also demonstrated that its secretion is dependent on CpaB, a membrane-bound chaperone. In this study, we show that CpaA expression and secretion are conserved across several medically relevant Acinetobacter species. Additionally, we demonstrate that deletion of cpaA results in attenuation of A. nosocomialis M2 virulence in moth and mouse models. The virulence defects resulting from the deletion of cpaA were comparable with those observed upon abrogation of T2SS activity. The virulence defects resulting from the deletion of cpaA are comparable with those observed upon abrogation of T2SS activity. We also show that CpaA and CpaB strongly interact, forming a complex in a 1:1 ratio. Interestingly, deletion of the N-terminal transmembrane domain of CpaB results in robust secretion of CpaA and CpaB, indicating that the transmembrane domain is dispensable for CpaA secretion and likely functions to retain CpaB inside the cell. Limited proteolysis of spheroplasts revealed that the C-terminal domain of CpaB is exposed to the periplasm, suggesting that this is the site where CpaA and CpaB interact in vivo. Last, we show that CpaB does not abolish the proteolytic activity of CpaA against human Factor V. We conclude that CpaA is, to the best of our knowledge, the first characterized, bona fide virulence factor secreted by Acinetobacter species.
Keywords: bacterial pathogenesis, bacterial toxin, chaperone, metalloprotease, type II secretion system (T2SS)
Introduction
Bacterial protein secretion is one way in which cells interact with and impact their environment. For instance, toxins and hydrolytic enzymes secreted by the type II secretion system (T2SS)6 aid in nutrient acquisition and promote in vivo survival and virulence of several pathogens, including Vibrio cholerae, enterotoxigenic Escherichia coli, Pseudomonas aeruginosa, and Legionella pneumophila (1–7). The T2SS is composed of an outer membrane secretin, a periplasmic pseudopilus, an inner membrane platform, and a cytoplasmic ATPase, and it is widespread among pathogenic and non-pathogenic Gram-negative bacteria (8–11). Secretion through the T2SS is a two-step process. First, type II substrates are translocated from the cytosol to the periplasmic space by either the general secretory (Sec) pathway or the Twin-arginine translocation (Tat) system (10, 11). Then, folded type II substrates are pushed through the outer membrane secretin and into the extracellular space via pseudopilus polymerization. Although type II substrates associate with the secretion apparatus through interactions with the outer membrane secretin, components of the inner membrane platform, and/or components of the pseudopilus, including the major pseudopilin and the minor pseudopilins (12–14), it is not well-understood how type II substrates are targeted to the type II secretion apparatus and differentially recognized relative to other fully folded soluble periplasmic proteins (12).
Acinetobacter baumannii is among the most threatening multidrug-resistant nosocomial pathogens worldwide. In fact, the World Health Organization has recently categorized it as a critical priority for the research and development of new antibiotics (15). However, it is important to highlight that the entire Acinetobacter calcoaceticus–A. baumannii complex, which is comprised primarily of A. baumannii, Acinetobacter nosocomialis, A. calcoaceticus, and Acinetobacter pittii, is becoming an increasing medical concern (16, 17). In addition to being extensively drug-resistant, Acinetobacter species (spp.) are capable of forming biofilms and withstanding desiccation as well as a wide range of temperatures and pH values (18–22). Altogether, these features allow Acinetobacter to persist in healthcare facilities, where it is a frequent cause of infections such as ventilator-associated pneumonia and bacteremia (23–25).
The presence of T2SS components in A. baumannii was first reported in 2014 (26). Subsequently, Johnson et al. (27) showed that the T2SS of A. baumannii ATCC 17978, a commonly used laboratory strain, is active. Type II–dependent secretion of LipA is required for growth in minimal medium supplemented with long-chain fatty acids as the sole carbon source, indicating an important role in nutrient acquisition (27). Furthermore, the T2SS and T2SS-dependent lipid utilization are required for competitive colonization of a neutropenic murine model of infection (27).
Recently, through heterologous expression and secretion of A. nosocomialis M2 (formerly A. baumannii M2) T2SS substrates, we demonstrated that A. baumannii 19606, A. nosocomialis M2, A. pittii, A. calcoaceticus, and Acinetobacter junii also possess a functional T2SS (28). Proteomic comparison of supernatants from wild-type A. nosocomialis M2 and an outer membrane secretin gspD mutant revealed numerous putative substrate proteins, all with predicted N-terminal Sec signals (28). Three of these substrates, the protease CpaA and the lipases LipA and LipH, were confirmed as T2SS substrates. CpaA was shown previously to be a secreted zinc-dependent metallo-endopeptidase that is capable of degrading fibrinogen and Factor V, thus deregulating blood coagulation (29). Importantly, the T2SS of A. nosocomialis is required for full virulence in Galleria mellonella and pulmonary murine models of infection (28). Although the wild-type and complemented strains efficiently disseminate to the liver or spleen after intranasal infection, the T2SS-deficient strain does not disseminate as efficiently, suggesting that the T2SS of A. nosocomialis plays an important role in virulence. However, the contribution of specific T2SS substrates in virulence has not been investigated.
Similarly to the P. aeruginosa type II metallo-protease elastase (34), two Acinetobacter T2SS substrates, LipA and CpaA, were found to require chaperones, LipB and CpaB respectively, for their secretion (28). However, unlike elastase, which is produced as a preproelastase, where the prodomain functions as an intramolecular chaperone (30, 31), LipB and CpaB are encoded adjacently to their cognate effector proteins and are predicted to be membrane-bound. CpaB and LipB have no sequence or predicted structural domain similarities (28). Topological prediction servers and bioinformatic analysis of LipB and CpaB suggest that their N-terminal transmembrane domains are imbedded in the inner membrane, with the C-terminal globular portions present in the periplasm (28). CpaB and LipB appear to be specific for CpaA and LipA, respectively, and none of these chaperones are required for the secretion of LipH (28). LipB is functionally similar to the Burkholderia glumae type II secretion membrane-bound chaperone lipase-specific foldase (Lif), which is required for the production of enzymatically active lipase. The function of the transmembrane domain of Lif is unclear, as this domain was not required for the steric chaperone activity of Lif (32–34). It has been proposed that CpaB, LipB, and Lif belong to a novel class of chaperones collectively known as T2SS chaperones (28). The involvement of chaperones in type II secretion is reminiscent of type III secretion systems (T3SSs), where the chaperones are soluble cytoplasmic proteins that interact with a specific substrate or multiple substrates and aid in folding, stabilization, and/or regulation of the secretion of these substrates (35). However, unlike T3SS chaperones, T2SS chaperones do not present any sequence homology and are variable in size and isoelectric point. To our knowledge, CpaB is the first reported membrane-bound, periplasmic chaperone required for secretion of a type II protease.
In this study, employing a combination of mutational and biochemical methods, we investigated the role of CpaA in A. nosocomialis pathogenesis and characterized the interaction between CpaA and its T2SS chaperone CpaB. We demonstrate that CpaA is required for virulence against G. mellonella larvae and is required for dissemination of A. nosocomialis to the spleen in a murine pulmonary model of infection. Furthermore, we show that CpaA and CpaB strongly interact and that the C-terminal periplasmic domain of CpaB is sufficient for chaperone function and interaction with CpaA. Last, we demonstrate that CpaB binding is incapable of blocking the proteolytic activity of CpaA against human Factor V.
Results
CpaA is secreted by medically relevant Acinetobacter species
The metallopeptidase CpaA is the most abundantly secreted type II substrate in A. nosocomialis strain M2 (28). Although older isolates such as A. baumannii ATCC 17978 and ATCC 19606 do not encode cpaA, the cpaA gene is conserved across clinical isolates of A. baumannii (29). However, it is unknown whether CpaA is secreted by these strains. To determine the prevalence of CpaA secretion among recent clinical isolates of medically relevant Acinetobacter strains, we generated a specific polyclonal antibody against CpaA (see “Experimental procedures” and supplemental Fig. S1). The anti-CpaA antibody was employed to screen various A. baumannii, A. nosocomialis, and A. pittii strains for CpaA expression and secretion. CpaA is present in the supernatant of most of these recent isolates but not in the supernatant of older laboratory strains (Fig. 1). Furthermore, all strains that expressed CpaA secreted the protein. Interestingly, there seems to be strain-to-strain differences in the secretion efficiency of CpaA, suggesting possible differences in the regulation of CpaA secretion. The prevalence of this metallo-peptidase among medically relevant clinical isolates prompted us to investigate the contribution of CpaA to Acinetobacter pathogenesis.
Figure 1.

CpaA secretion is conserved among medically relevant Acinetobacter species. Whole cells (W) and supernatants (S) of A. baumannii (AB), Acinetobacter baylyi (AY), A. calcoaceticus (AC), A. pittii (AP), and A. nosocomialis (AN) strains were analyzed by Western blotting using anti-CpaA (green) and anti-RNA polymerase (red) antibodies. CpaA is detected in the supernatants of various AB, AP, and AN strains, all of which are recent clinical isolates. RNA polymerase was included as a lysis control.
CpaA is required for optimal virulence in G. mellonella larvae
We showed previously that the T2SS of A. nosocomialis M2 is required for virulence in G. mellonella larvae (28). However, we did not identify the specific substrate(s) responsible for this phenotype. Because CpaA is the most abundant T2SS substrate under laboratory conditions, and because it is secreted by many recently isolated strains, we tested the contribution of CpaA to virulence in G. mellonella. First, we generated a strain containing an unmarked deletion in cpaA (ΔcpaA) and its corresponding complemented strain by inserting a copy of the cpaAB locus under its native promoter into the chromosome (cpaA+) downstream of glmS2 (36, 37). We analyzed the supernatant fractions of wild-type A. nosocomialis M2, ΔcpaA, and cpaA+ by Western blot analysis. As expected, the strain devoid of cpaA did not secrete CpaA. However, CpaA secretion was restored upon complementation by inserting cpaAB in the chromosome of M2 ΔcpaA (supplemental Fig. S1). Deletion of cpaA and cpaB does not alter secretion of the type II substrate LipH, suggesting that CpaB is specific for CpaA and does not broadly impact type II secretion (supplemental Fig. S2).
G. mellonella larvae were infected with wild-type M2, ΔcpaA, or cpaA+ (see “Experimental procedures”), and survival was monitored over time (Fig. 2). The ΔgspD strain carries a deletion in the gspD gene that inactivates T2SS and therefore does not secrete any T2SS substrates (28). ΔgspD and its corresponding complemented strain (gspD+) were included for comparison purposes. Twenty-four hours post-infection, 50% of larvae infected with either ΔgspD or ΔcpaA survived, whereas infection with wild-type M2, gspD+, or cpaA+ resulted in less than 25% survival (Fig. 2). The difference in percent survival of larvae infected with either ΔgspD or ΔcpaA relative to the wild type or the complemented strains is statistically significant (Fig. 2). Therefore, deletion of cpaA results in a virulence defect in G. mellonella larvae comparable with the T2SS mutation, suggesting that CpaA is the most important T2SS substrate contributing to the virulence of A. nosocomialis M2.
Figure 2.
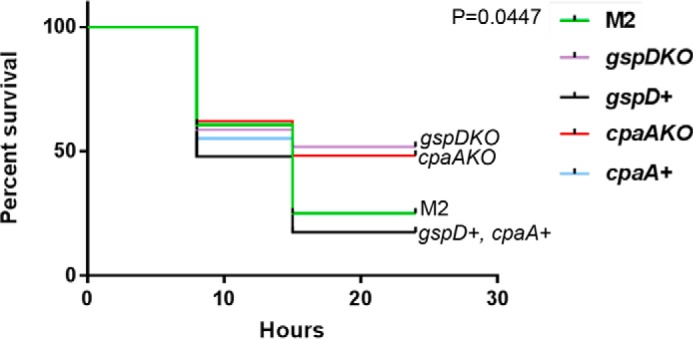
Type II secretion and CpaA are similarly required for full virulence in the G. mellonella infection model. G. mellonella larvae were injected with 10 μl of either M2, ΔgspD, ΔcpaA, or the complemented strains at an inoculum of ∼1 × 107. Infected larvae were incubated at 37 °C and monitored for viability over time. Viability was determined by melanin accumulation and mobility. Larvae infected with ΔgspD or ΔcpaA had ∼50% survival at 24 h. 25% of the larvae infected with M2 survived after 24 h. Larvae infected with the ΔgspD or ΔcpaA complemented strains had 17% survival at 24 h post-infection. The difference in percent survival of larvae infected with ΔgspD or ΔcpaA relative to the wild-type or complemented strains is statistically significant (Mantel Cox p = 0.0447).
CpaA aids in A. nosocomialis dissemination to the spleen in mice
To further confirm the role of CpaA in a mammalian model of infection, we infected mice intranasally with either wild-type, ΔcpaA, or the cpaA+ complemented strain. Thirty-six hours post-infection, the lungs and spleen were harvested and analyzed for colony-forming units. Although no significant difference was observed, comparison of the bacterial burden in the lungs of infected mice shows a trend toward decreased colonization by the ΔcpaA strain relative to the wild-type and complemented strains (Fig. 3A). In contrast, a statistically significant difference was observed in the bacterial burden in the spleens of mice infected with ΔcpaA compared with mice infected with the complemented strain (Fig. 3B). Furthermore, we confirmed that deletion of both CpaA and CpaB did not affect secretion of other T2SS substrates such as LipH (supplemental Fig. S2). Thus, CpaA appears to play a role in dissemination from the initial site of infection in the lungs to a distal site in the spleen.
Figure 3.
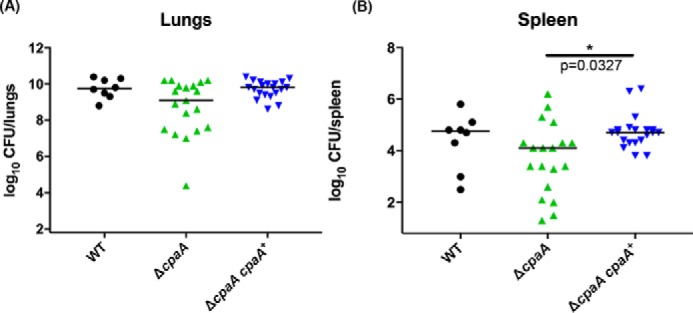
CpaA aids in colonization of the spleen in mice. Mice were intranasally inoculated with 1 × 109 cfus of either wild-type, ΔcpaA, or the cpaA complemented strain. 36 h post-infection, mice were sacrificed, organs were harvested, and colony-forming units were enumerated from homogenized tissue. A, total colony-forming units from the lung demonstrate a non-statistically significant trend toward decreased colonization by the ΔcpaA strain (Kruskal-Wallis non-parametric, p = 0.06). B, total colony-forming units from the spleen demonstrate a statistically significant difference between the complemented cpaA strain and the ΔcpaA mutant strain (Kruskal-Wallis non-parametric, *p = 0.0327).
CpaA physically interacts with its dedicated chaperone CpaB
We showed previously that CpaA secretion is dependent on an uncharacterized protein encoded immediately downstream of CpaA (28). We designated this protein CpaB because of its proximity to cpaA. Domain-enhanced lookup time accelerated (DELTA) BLASTp analysis identified a SRPBCC superfamily domain in CpaB (28). Because proteins with a SRPBCC domain are predicted to have a deep hydrophobic ligand-binding pocket and chaperone-like activity, we postulated that CpaB belongs to a new family of membrane-bound chaperones (T2SS chaperones) that mediate the secretion of specific T2SS substrates (28). T2SS chaperones could facilitate folding, inhibit self-damaging activities by functioning as immunity proteins, and/or pilot the secretion of their cognate substrates. All of these activities require the interaction of the chaperone with it is substrate. The physical interaction between CpaB and CpaA was addressed through a pulldown assay. cpaA containing a C-terminal FLAG tag and cpaB containing a C-terminal hexahistidine tag were both cloned into vector pWH1266 (pWH-cpaA-flag-cpaB-his) and expressed in M2 ΔcpaAB, which lacks the endogenous cpaA and cpaB genes. Nickel-NTA affinity chromatography was performed on cell lysate solubilized with 0.01% Triton X-100. CpaA-FLAG and CpaB-His were detected by Western blot analysis in the load fraction, indicating that both proteins were expressed (Fig. 4A, left). Despite not detecting CpaA-FLAG in the wash fractions, CpaA-FLAG was detected upon elution of immobilized CpaB-His (Fig. 4A). Moreover, nickel-NTA affinity chromatography on cell lysate from M2 ΔcpaAB-expressing cpaA-flag and untagged cpaB (pWH-cpaA-flag-cpaB) revealed that CpaA-FLAG is not eluted in the absence of hexahistidine-tagged CpaB (Fig. 4B), indicating that CpaA binds specifically to CpaB. Further, Coomassie staining of the elution from M2 ΔcpaAB pWH-cpaA-flag-cpaB-his lysate suggests that CpaA and CpaB associate specifically in an approximate 1:1 ratio (Fig. 4A, right).
Figure 4.
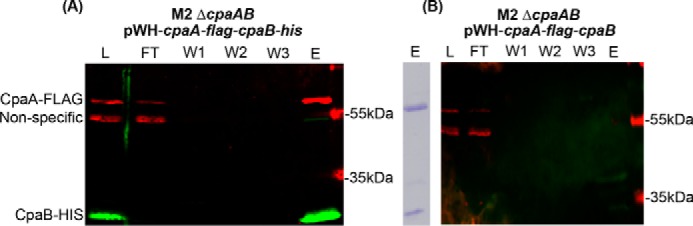
CpaA and CpaB strongly interact physically. A and B, Western blot analysis of nickel affinity purifications from cell lysates of M2 ΔcpaAB expressing either pWH-cpaA-flag-cpaB-his (A) or pWH-cpaA-flag-cpaB (B). Note that, in B, CpaB does not contain a C-terminal hexahistidine tag. CpaA-FLAG was detected using an anti-FLAG monoclonal antibody, whereas CpaB was detected using anti-histidine polyclonal antibodies. The right panel in A shows Coomassie-stained SDS-PAGE of the elution from A and thus represents the CpaA–FLAG–CpaB–His complex. L, loaded cell lysate, FT, flow-through, W1–3, washes one to three; E, elution.
We evaluated the stoichiometry of the CpaA–CpaB complex by size-exclusion chromatography and sedimentation equilibrium analytical centrifugation (Fig. 5). Size-exclusion chromatography of the co-purified complex of CpaA and a transmembrane-truncated CpaB resulted in an aggregate peak starting at a void volume of 40 ml and a complex peak at a volume of 85 ml (Fig. 5A). An SDS-PAGE gel of the complex peak showed two bands corresponding to CpaA and CpaB and was consistent with a 1:1 complex (Fig. 5B). To unambiguously determine the molecular weight of the complex in solution, we used sedimentation equilibrium analytical centrifugation, as this method can determine the mass of complexes in solution independent of shape. Sedimentation equilibrium analytical centrifugation of the complex peak (Fig. 5C) revealed a molecular mass of 83.7 kDa ± 4.7 kDa using a one-component ideal model to assess the molecular mass of the complex in solution (38). The theoretical molecular masses of CpaA and truncated CpaB are 63.1 kDa and 20.4 kDa, respectively. A 1:1 complex of the CpaA and CpaB therefore has a theoretical molecular mass of 83.5 kDa, and the sedimentation equilibrium experiment aligns well with the theoretical molecular mass of a 1:1 complex.
Figure 5.

CpaA and CpaB form a 1:1 complex in solution. A, size-exclusion chromatography of co-purified CpaA and CpaB reveals a single peak for the CpaA–CpaB complex. mAu, milli-absorbance unit. B, SDS-PAGE gel of the CpaA–CpaB complex peak reveals two bands that correspond to CpaA and CpaB. C, sedimentation equilibrium analytical centrifugation of the size-exclusion–purified CpaA–CpaB complex reveals a 1:1 complex of 83.7 kDa ± 4.7 kDa. Top panel, one-component ideal model fits for one representative experiment of three. Bottom panel, the residuals for the one-component ideal model fits for each individual scan.
The C-terminal domain of CpaB faces the periplasmic space, and its N-terminal transmembrane domain is not required for CpaA secretion
Topological prediction servers and bioinformatic analysis suggest that CpaB has an N-terminal transmembrane domain (28, 29). Therefore, it is likely the C-terminal domain of CpaB that interacts with CpaA. In vivo, this interaction could occur in either the cytoplasm or the periplasm. To determine the cell localization of the C-terminal domain of CpaB, we used limited proteolysis of either whole cells or spheroplasts containing CpaB-His, where the hexahistidine tag was added to the C terminus (32, 39). Samples were visualized by Western blot analysis probing for CpaB-His and RNA polymerase as a cytoplasmic control. Whole cells treated with proteinase K have consistent levels of CpaB-His and RNA polymerase over time (Fig. 6A), indicating that CpaB-His is inaccessible to the protease. In contrast, spheroplasts treated with proteinase K show decreasing levels of CpaB-His over time, suggesting that the C-terminal domain is accessible to the protease (Fig. 6B). Furthermore, the consistent levels of RNA polymerase over time indicate that cytoplasmic proteins were sheltered from proteolysis (Fig. 6B). Altogether, these results show that the C terminus of CpaB is periplasmic and suggest that CpaB interacts with CpaA in the periplasm.
Figure 6.
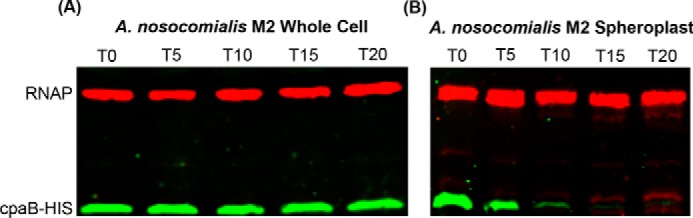
The C-terminal domain of CpaB is exposed to the periplasmic space. Shown is a Western blot analysis probing for RNA polymerase as a cytoplasmic control and CpaB-His of samples treated with 0.5 μg/ml proteinase K for 0–20 min at 56 °C. A, limited proteolysis of M2 whole cells expressing pWH-cpaB-his. B, limited proteolysis of spheroplasts isolated from M2 cells expressing pWH-cpaB-his.
To determine whether the N-terminal transmembrane domain of CpaB is required for CpaA secretion, we constructed a soluble periplasmic version of CpaB (CpaBpelB-His) by replacing the putative N-terminal transmembrane domain of CpaB-His with the Sec secretion signal of the periplasmic protein PelB from Escherichia coli. As expected, CpaBpelB-His localized to the periplasm (supplemental Fig. S3). We then transformed M2 ΔcpaAB with FLAG-tagged CpaA and either full-length CpaB-His or CpaBpelB-His and probed for the presence of CpaA in cell-free supernatants. CpaA secretion was complemented by either full-length CpaB-His or soluble periplasmic CpaBpelB-His (Fig. 7), indicating that CpaB does not need to be membrane-bound to mediate CpaA secretion. Interestingly, soluble CpaB-His was also detected in cell-free supernatants of cells expressing CpaA-FLAG (Fig. 7). To determine whether secretion of the soluble periplasmic chaperone CpaB was CpaA-dependent, we compared secretion of soluble CpaB-His in the presence or absence of CpaA by Western blotting (supplemental Fig. S4). In the absence of CpaA, secretion of soluble CpaB was abrogated, suggesting that soluble periplasmic CpaB is secreted in complex with CpaA (Fig. 6 and supplemental Fig. S4). The minimal amount of soluble CpaB that was secreted in the absence of CpaA could be a consequence of overexpression of soluble CpaB.
Figure 7.
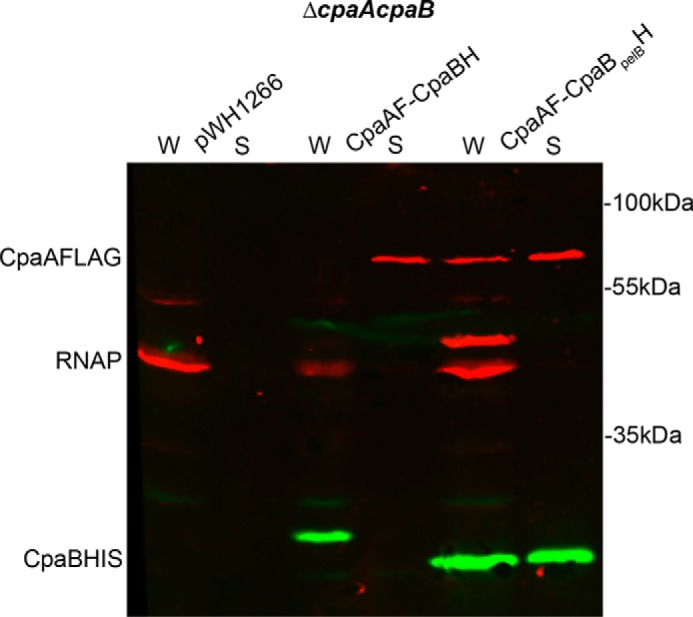
Soluble periplasmic CpaB complements a ΔcpaB mutant and is secreted with CpaA. Shown is a Western blot analysis on whole-cell (W) and cell-free supernatant (S) fractions of ΔcpaAB M2 expressing empty vector. pWH-cpaB-his and pBAV-cpaA-flag or pWH-cpaBpelB-his and pBAV-cpaA-flag were probed for CpaB-His. All fractions were probed for RNA polymerase as a lysis control. CpaB-His was detected in all cells carrying pWH-cpaB-his or pWH-cpaBpelB-his. Secretion of CpaB-His was detected in cells carrying pWH-cpaBpelB-his. CpaA* is a degradation product from CpaA-FLAG.
CpaA is degraded in the absence of CpaB
CpaA is hard to detect in the periplasmic fraction because its secretion is extremely efficient in wild-type A. nosocomialis M2. In a ΔcpaB strain, CpaA was not secreted and, instead, was degraded in the periplasm, as indicated by a CpaA-derived band of ∼30 kDa present under these conditions (supplemental Fig. S5). CpaB complementation restored CpaA secretion with the subsequent disappearance of the CpaA proteolytic fragment. This experiment demonstrates that CpaA is not stable in the absence of CpaB.
CpaB interaction with CpaA does not completely abolish proteolytic activity against Factor V
CpaA physically interacts with CpaB, and this interaction is required for CpaA secretion (Fig. 4) (28). However, the specific role of CpaB in CpaA secretion remains unclear. CpaA was shown previously to be a secreted metallo-endopeptidase with the ability to degrade human Factor V and thus deregulate blood coagulation (29). One possibility is that this interaction inactivates CpaA, preventing self-intoxication of the cell by the proteolytic activity of CpaA (40). To explore the possibility that CpaB is an immunity protein, we compared the proteolytic activity of CpaA alone and in complex with the soluble version of CpaB against human Factor V.
CpaA-His was purified from the cell-free supernatant of mid-log phase A. nosocomialis M2, whereas the complex CpaA-FLAG-CpaBpelB-His was purified from the supernatant of A. nosocomialis M2ΔcpaAB. For the complex, the hexahistidine tag was placed in CpaB and not in CpaA to ensure that all of the purified CpaA was complexed to CpaB. Cleavage of human Factor V was analyzed by Western blotting, probing for human Factor V and CpaA. Interestingly, CpaA was able to cleave Factor V irrespective of the presence of CpaB (Fig. 8). In contrast, neither heat-inactivated CpaA nor CpaB alone could cleave Factor V (Fig. 7). Although we have not followed the kinetics of this reaction, it can be concluded that CpaB does not completely inhibit CpaA activity against Factor V.
Figure 8.
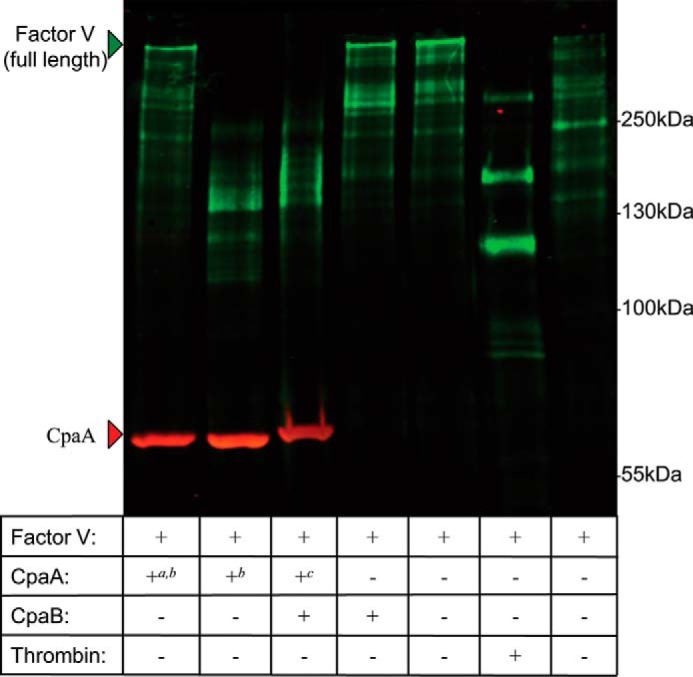
CpaA remains catalytically active when complexed with CpaB. Human Factor V was incubated with purified heat-inactivated CpaA-His, secreted CpaA-His, the secreted CpaA–FLAG–CpaB–His complex, CpaB-His, or buffer (20 mm HEPES and 150 mm NaCl (pH 7.4)) for 30 min at 37 °C. As a positive control, Factor V was mixed with thrombin in 25 mm Tris-HCl, 50 mm NH4Cl, and 5 mm CaCl2 (pH 7.4) at 37 °C for 1 h. The rightmost lane represents the buffer control for the thrombin reaction. Full-length Factor V is detectable in the buffer control samples as well as the heat-inactivated CpaA, indicating a lack of proteolysis. The low-molecular-weight bands corresponding to Factor V present in the thrombin lane are indicative of Factor V proteolysis. a, heat-inactivated; b, CpaA-His; c, CpaA-FLAG.
Discussion
CpaA is a zinc-dependent metallo-endopeptidase and T2SS substrate of several medically relevant Acinetobacter strains (28, 29). Although it is the most abundantly secreted type II substrate in A. nosocomialis M2, CpaA is not present in the genomes of typical Acinetobacter laboratory strains such as ATCC17978 and ATCC19606 (28). Acinetobacter often presents as pneumonia; therefore, a murine pulmonary infection was used to study the role of CpaA in lung colonization and dissemination. We showed previously that the T2SS of A. nosocomialis M2 was required for full virulence in G. mellonella and pulmonary murine models of infection (28). The T2SS-deficient strain did not disseminate to the liver or spleen as efficiently as the wild-type or complemented strains, suggesting that substrates secreted by the T2SS play an important role in Acinetobacter virulence. Here we show that CpaA is secreted by multiple clinical isolates of A. baumannii, A. pittii, and A. nosocomialis. Deletion of cpaA results in a virulence defect in G. mellonella larvae and decreased splenic colonization in a murine pulmonary infection model. The virulence defects resulting from the deletion of cpaA are comparable with those observed from the deletion of the T2SS. To our knowledge, CpaA is the first characterized, bona fide virulence factor secreted by Acinetobacter spp.
Numerous proteins are secreted by A. nosocomialis M2 in a type II-dependent manner (28). As mentioned, deletion of cpaA has a similar impact on murine splenic colonization and virulence against G. mellonella as the loss of type II secretion (ΔgspD) (Fig. 3). However, unlike the ΔgspD strain, the ΔcpaA strain did not show decreased murine lung colonization (Fig. 3) (28). This discrepancy could be attributed to the loss of secretion of all type II substrates in the ΔgspD strain compared with the single deletion of CpaA in the ΔcpaA strain. LipA, a confirmed Acinetobacter type II substrate, is required for A. baumannii competitive murine colonization in a bacteremia model (27). Therefore, it is plausible that LipA plays a role in murine lung colonization by A. nosocomialis and, thus, may partly account for the difference in colonization defect between the ΔgspD and ΔcpaA strains. Moreover, CpaA may play a role in dissemination from the initial site of infection in the lungs to a distal site in the spleen. The activity of CpaA against the coagulation factors fibrinogen and Factor V likely contributes to the defect in ΔcpaA to migrate from the initial infection site.
CpaA is likely translocated to the periplasm by the SecYEG translocation system in an unfolded state (41). When in the periplasm, CpaA must fold to be secreted by the T2SS (8, 42). CpaB is a membrane-anchored protein encoded next to CpaA and is required for CpaA secretion. This is reminiscent of the B. glumae type II substrate LipA, which requires a membrane-bound, lipase-specific foldase to be folded into its active form and secreted (33, 34). In this work, we identified additional functional similarities between CpaB and the Lif proteins. We demonstrated that CpaB physically interacts with CpaA. Furthermore, similarly to Lif, the C-terminal globular domain of CpaB is periplasmic and sufficient for CpaA secretion (Fig. 7) (34). Although both chaperones are required for secretion of their cognate substrates, the transmembrane domains of both Lif and CpaB are dispensable for the secretion and proper folding of their respective protein substrates. These results reinforce our suggestion that, although there is absolutely no sequence conservation between CpaB and Lif, these proteins belong to a unique class of type II secretion, membrane-bound chaperones that possess evident functional analogies (28, 33, 34).
We demonstrate that soluble periplasmic CpaB lacking its transmembrane domain is secreted by A. nosocomialis M2 complexed with CpaA. This result agrees with a previous study that showed that the T2SS chaperone Lif from B. glumae lacking its transmembrane domain (Lifpp) is secreted along with its target lipase. The study also showed that Lifpp secretion was dependent on the expression and secretion of its cognate lipase (34). Altogether, these results suggest that the T2SS substrate–chaperone complex does not dissociate unless the chaperone is anchored to the inner membrane. Dissociation of the T2SS substrate–membrane–anchored chaperone complex is expected to result in type II-dependent secretion of the substrate and retention of the chaperone within the periplasmic space. In the T3SS, the separation of the substrate–chaperone complex is mediated by the ATPase that powers the system (43). How this occurs in the T2SS has yet to be elucidated, but we hypothesize that the membrane anchor in the T2SS chaperone may be involved in retention of the chaperone and disassembly of the complex, possibly through an interaction with the T2SS machinery. The observation that a minimal amount of soluble CpaB is secreted in the absence of CpaA could be the consequence of a hypothetical interaction between CpaB and the T2SS.
Because CpaA is a protease, we hypothesized that CpaB binding might inhibit CpaA activity to protect periplasmic proteins from proteolysis. To this end, we assayed human Factor V cleavage by CpaA alone or in complex with CpaB. We found that CpaB binding to CpaA does not prevent Factor V cleavage, suggesting that CpaB is not primarily an immunity protein. However, we performed an end point assay and thus did not assess the impact of CpaB binding on the kinetics of CpaA activity. We have not experimentally measured the stability of the CpaA–CpaB complex, but the ability to co-purify the complex and the fact that the complex withstands ultracentrifugation in the analytical ultracentrifugation experiment suggest that the complex between CpaA and CpaB is very stable. However, CpaA and CpaB may still dissociate under our experimental conditions, and the activity we detected may be due to free CpaA. For this reason, we cannot conclude that CpaB binding does not impact CpaA activity but, rather, that the presence of CpaB does not fully prevent CpaA from cleaving human Factor V. Furthermore, CpaA was unstable in the absence of CpaB (supplemental Fig. S5), suggesting that CpaB is involved in folding CpaA into its active, stable, secretion-competent form.
Here we have characterized CpaA as the first bona fide virulence factor of the A. baumannii–A. calcoaceticus complex and provided insight into the molecular interactions between CpaA and its T2SS chaperone CpaB. We have demonstrated that the membrane anchor of CpaB is not required for its chaperone function and further characterized the novel class of T2SS chaperones CpaB represents. We propose that CpaB is involved in folding of CpaA into its active, stable, secretion-competent form. It is plausible that the purpose of the transmembrane domain of CpaB is to retain CpaB in the cell, thus preventing co-secretion of CpaB with CpaA. Future efforts will focus on dissecting whether the membrane anchor facilitates an interaction with either SecYEG or the type II apparatus, yielding more efficient secretion of CpaA. These findings would be beneficial to public health, as elucidation of T2SS substrates and the mechanism of secretion could encourage the development of novel antivirulence treatments for Acinetobacter infections, which are particularly troublesome because of increasing rates of multidrug resistance. For instance, specific inhibitors designed to bind and inhibit the activity and/or secretion of a virulence factor are a viable option.
Experimental procedures
Strains, plasmids, and growth conditions
The bacterial strains, plasmids, and primers used in this study are listed in supplemental Tables S1 and S2. All strains were grown in LB/agar at 37 °C unless specified otherwise. Antibiotics for E. coli selection were used at the following concentrations: 100 μg/ml carbenicillin, 5 μg/ml tetracycline, 12.5 μg/ml chloramphenicol, and 20 μg/ml kanamycin. Antibiotics for A. nosocomialis were used at the following concentrations: 20 μg/ml kanamycin, 5 μg/ml tetracycline, 12.5 μg/ml chloramphenicol, and 200 μg/ml carbenicillin. To select for the loss of sacB, supplementation with 10% sucrose was used.
Generation of bacterial mutants and complemented mutants
Unmarked mutations were made using a method published previously (37, 44). The In-Fusion HD EcoDry cloning kit was used in the creation of pGEM-cpaA::kansacB and pGEMcpaAB::kansacB knockout plasmids, similarly to Refs. 28, 44. Briefly, 1000 bp upstream of the target gene were amplified with primers 5510015bpkmsacBrev and 5510015bppgem1fw (or cpaABKOup15bppgemfw2 and cpaABKOup15bpkmsacBrev1), which produced a product with 15 bp of homology to pGEM at the 5′ end and 15 bp of homology to kmsacB at the 3′ end. 1000 bp downstream of the target gene were amplified with primers 3510015bpkmsacB2fw and 3510015bppgem2rev (or cpaABKOdw15bppgemrev4 and cpaABKOdw15bpkmsacBfw3), which produced a product with 15 bp of homology to pGEM at the 3′ end and 15 bp of homology to kmsacB at the 5′ end. The kanamycin resistance cassette and sacB gene were amplified using primers Kmsacb2rev and Kmsacb1fw. pGEM was amplified using primers pgem1rev and pgem2fw. The In-Fusion HD EcoDry cloning kit was used to fuse the upstream, downstream, kmsacB, and pGEM fragments together, generating pGEM-cpaA::kmsacB or pGEM-cpaAB::kmsacB. These plasmids were introduced to A. nosocomialis through natural transformation (36). The FLP recombinase plasmid pFLP2 was transiently introduced into M2 cpaA::kmsacB and M2 cpaAB::kmsacB by triparental mating (37) to replace the resistance cassette with an frt scar. Strains ΔcpaA and ΔcpaAB were verified by PCR and sequencing. The cpaA mutation was complemented with the cpaAB locus (amplified with primers kpnIcpaABrevlocus and pstIcpaABfwlocus and cloned into pRSM4063 at KpnI and PstI) under its natural putative promoter using the miniTn7 described in Ref. 44. The MiniTn7 complementation construct pRSM4063-pcpaAB was introduced into ΔcpaA through natural transformation as above. Consistent with the nomenclature used by Harding et al. (28), mutants designated with “::frt” contain an frt scar in place of the target gene (28).
Generation of pWH1266-based constructs
The In-Fusion EcoDry cloning kit was used to create the following constructs: pWH-cpaA-flag-cpaB-his, pWH-cpaA-flag-cpaB, pWH-cpaB-his, and pWH-cpaBpelB-his. pWH-cpaA-flag-cpaB-his was created by fusing cpaA-flag (amplified with primers 5cpaAFLAGrev and 3cpaApromfwpwh1), pWH1266 (amplified with 2pwhrev1 and 1pwhfw2), and cpaB-his (amplified with primers 8CpaBHisrevpwh and 11cpaBfw15bpcpaAF). pWH-cpaA-flag-cpaB was created by fusing cpaA-flag (amplified with primers 5cpaAFLAG rev and 3cpaApromfwpwh1) to pWH1266 (amplified with 2pwhrev1 and 1pwhfw2) and cpaB (amplified with CpaBrevNT15bppwh2 and 11cpaBfw15bpcpaAF). pWH-cpaB-his was created by fusing the putative promoter of cpaAB (amplified with primers 3cpaApromfwpwh1 and 6cpaABpromrev), pWH1266, and cpaB-his (amplified with primers 8CpaBHisrevpwh and 7CpaBfwpromoverhang). pWH-cpaBpelB-his was created by fusing pWH1266 to the cpaAB promoter to cpaBpelB-his, where the N-terminal transmembrane domain has been replaced with an N-terminal Sec secretion signal of PelB, (amplified with 8CpaBHisrevpwh and 9CpaBnoTMpelB15bppromfw). All constructs were verified by PCR and sequencing.
Generation of pBAV-cpaA-flag
CpaA-flag was PCR amplified with 5100fwbamhI and 5100flagrevpstI and cloned into pBAVMCS at PstI and BamH1. pBAV-cpaA-flag was confirmed by PCR and sequencing.
Generation of pETDuet-cpaA-his-cpaB and pET28a-cytocpaB-his
CpaB was PCR-amplified with CpaBcytorevKpnI and CpaBcytofwduetNdeI and cloned into multiple cloning site 2 at KpnI and NdeI. CpaA was cloned into pETDuet using the In-Fusion HD EcoDry cloning kit using petDuetfwinfuse and petDuetrevinfuse to amplify pETDuet-cpaB and CpaAcytofwduet15bp and CpaAcytoHisrevduet15bp to amplify cpaA with 15 bp of homology to pETDuet. The pETDuet-cpaA-his-cpaB was confirmed through PCR and sequencing.
Cyto-cpaB was PCR-amplified with 15 bp of homology on the 5′ and 3′ ends to pET28a with CytocpaBrev and CytoCpaBfw. Cyto-cpaB was cloned into pET28a in-frame with the C-terminal histidine tag using the In-Fusion HD EcoDry cloning kit using Pet28ahisfw and Pet28auprev. pET28a-cyto-cpaB-his was confirmed by sequencing.
Generation of polyclonal rabbit sera against CpaA
The pETDuet vector was used to overexpress CpaA and CpaB in the cytoplasm of E. coli Rosetta 2 cells for purification. 1 liter of LB was inoculated from an overnight culture of Rosetta 2/pETDuet-cpaA-his-cpaB at 0.05 A/ml, grown to mid-log phase, and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. The culture was grown for an additional 4 h. Cells were harvested at 8000 rpm for 10 min. Cells were washed with 30 mm Tris (pH 8) and resuspended in 40 ml of 50 mm NaH2PO4, 300 mm NaCl, and 10 mm imidazole (pH 8). Cells were lysed with a cell disruptor using two rounds at 35,000 p.s.i (Constant System Ltd., Kennesaw, GA). Cell lysates were clarified at 11,000 rpm for 30 min. Cell lysates were passed over a nickel-NTA agarose column (Gold Bio, St. Louis, MO). The load fraction is the total cell lysate. The flow-through was collected as what passed through the column and did not bind the nickel-NTA resin. The column was washed with 20 ml of 50 mm NaH2PO4, 300 mm NaCl, and 25 mm imidazole (pH 8) and 20 ml of 50 mm NaH2PO4, 300 mm NaCl, and 50 mm imidazole (pH 8) (45). Proteins were eluted with 50 mm NaH2PO4, 300 mm NaCl, and 250 mm imidazole (pH 8). Elution fractions were analyzed by SDS-PAGE analysis and Coomassie staining. The polyacrylamide gel band corresponding to CpaA-His was sent to Abore Inc. (Ramona, CA) for peptide extraction and development of rabbit-derived polyclonal antibodies.
Nickel-NTA affinity purification of CpaB-His and CpaA-FLAG
Cultures of M2 ΔcpaAB carrying pWH-pCpaA-flag-cpaB-his, pWH-pCpaA-flag-cpaB, or pWH1266 were grown overnight. Cells were pelleted at 10,000 × g for 10 min. Cells were washed with 30 mm Tris (pH 8) and resuspended in 15 ml of 50 mm NaH2PO4, 300 mm NaCl, and 10 mm imidazole (pH 8). Cells were lysed with a cell disruptor using two rounds at 35,000 p.s.i. (Constant System Ltd.). Cell lysates were incubated with 0.05% Triton X-100 and rolling at 4 °C for 1 h to solubilize CpaB-His (46). Cell lysates were clarified at 11,000 rpm for 30 min. Cell lysates were passed over a nickel-NTA-agarose column (Gold Bio). The load fraction is the total cell lysate. The flow-through was collected as what passed through the column and did not bind the nickel-NTA resin. The column was washed with 20 ml of 50 mm NaH2PO4, 300 mm NaCl, and 25 mm imidazole (pH 8) and 20 ml of 50 mm NaH2PO4, 300 mm NaCl, and 50 mm imidazole (pH 8) (45). Proteins were eluted with 50 mm NaH2PO4, 300 mm NaCl, and 250 mm imidazole (pH 8). Load, flow-through, wash, and elution fractions were analyzed by Western blot analysis probing for CpaA-FLAG and CpaB-His.
Size-exclusion purification of CpaA and CpaB
CpaA-His and CpaB were co-purified from E. coli Rosetta 2 cells by nickel-NTA resin as described above. The nickel-NTA elution was further purified by a Superdex 200 Prepgrade size-exclusion column (GE Healthcare) with 10 mm HEPES (pH 7.4) and 150 mm NaCl. The fractions under the CpaA–CpaB complex peak were concentrated using an Amicon Ultra centrifugal filter (Millipore) with a 10-kDa molecular mass cutoff.
Analytical ultracentrifugation of the CpaA–CpaB complex
Sedimentation equilibrium experiments were performed in a Beckman/Coulter XL-A analytical ultracentrifuge using an An60Ti rotor at 10 °C with size-exclusion–purified CpaA–CpaB complex at concentrations of 4.2 μm, 3.4 μm, and 2.5 μm. Data were obtained at speeds of 7000, 8000, and 9000 rpm. A partial specific volume for CpaA–CpaB of 0.7321 was calculated with Sednterp (47), and a global fit analysis was performed in Ultrascan II version 9.9 (38). The molecular mass reported is the mean ± S.D. of the three independent experiments at all concentrations and speeds analyzed.
CpaB-His localization and limited proteolysis of A. nosocomialis M2 spheroplasts
Overnight cultures of A. nosocomialis M2 carrying pWH-cpaB-his or pWH-cpaBpelB-his were used to inoculate LB supplemented with tetracycline that were grown at 37 °C and 225 rpm. Periplasmic and spheroplast preparations were done as reported previously (39). Briefly, cultures grown to 0.5 A/ml were pelleted by centrifugation at 10,000 rpm for 10 min. Cells were resuspended at 1 A/50 μl in 20% sucrose, 30 mm Tris-HCl (pH 8.0), 1 mm EDTA, and 1 mg/ml lysozyme (Gold Bio) and incubated on ice for 2 h. Spheroplasts were pelleted at 16,000 × g for 5 min. The supernatant fraction was considered the periplasmic fraction. For CpaB-His localization, periplasmic and spheroplast fractions were analyzed by SDS-PAGE and immunoblotting. Proteolysis of spheroplasts was performed as follows. Spheroplasts were resuspended at 0.01 A/μl in 0.5 mg/ml proteinase K (Sigma) in 30 mm Tris-HCl (pH 8.0) and incubated at 56 °C for 0–20 min. Protease activity was stopped by the addition phenylmethylsulfonyl fluoride and 4× Laemmli buffer and boiling at 100 °C. The equivalent of 0.1 A of each sample was resolved by SDS-PAGE and analyzed by Western blotting, probing for CpaB-His and RNA polymerase as a cytoplasmic control.
Immunoblotting
Bacterial secretion whole-cell and supernatant samples were prepared as published previously (28, 48). Briefly, cultures were grown to 0.5 A/ml, and 0.5 A was pelleted by centrifugation and resuspended in 50 μl of 1× Laemmli buffer for the whole-cell samples. Supernatant samples were obtained by TCA-precipitating cell-free supernatants as published previously (48). Proteins were resolved by SDS-PAGE analysis and transferred to a nitrocellulose membrane by semidry transfer and probed with either monoclonal anti-FLAG (1:2000, Sigma), polyclonal anti-histidine (1:2000, Pierce), polyclonal anti-CpaA (1:1000, this study), and/or monoclonal anti-RNA polymerase (1:2000, Neoclone). Western blots were probed with IRDye-conjugated secondary antibodies and visualized with an Odyssey CLx imaging system (LI-COR Biosciences, Lincoln, NE).
G. mellonella infection
G. mellonella infections with A. nosocomialis M2, ΔgspD::frt, ΔcpaA::frt, and the complemented strains were done as published previously (28). Cultures were grown in LB to 0.5 A/ml. Cells were pelleted by centrifugation, washed with sterile PBS, and resuspended in PBS. G. mellonella were injected with 10 μl of either M2, ΔgspD::frt, ΔcpaA, or the complemented strains at an inoculum of 1 × 107. Infected larvae were incubated at 37 °C and monitored for viability over time. Larvae were considered dead when they did not respond to touch.
Mouse model of pneumonia
The Vanderbilt University Medical Center Institutional Animal Care and Use Committee approved the infection experiments. Jackson Laboratories wild-type C57BL/6 mice were infected with either the ΔcpaA or the cpaA+ complemented strain as published previously (28). Nine-week-old male mice were inoculated intranasally with 1 × 109 cfus. Thirty-six hours post-infection, mice were euthanized, and lung, liver, spleen, and heart tissues were homogenized and dilution-plated to determine colony-forming units. Median colony-forming unit counts from each mouse are reported and were analyzed by Mann Whitney non-parametric tests using GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA).
Factor V cleavage assay
CpaA-His or CpaA-FLAG-CpaBpelB-His was purified out of the supernatant of A. nosocomialis M2. Briefly, A. nosocomialis M2 carrying pWH-cpaA-his-cpaB or A. nosocomialis ΔcpaAB::frt carrying both pBAV-cpaA-flag and pWH-cpaBpelB-his was grown in LB supplemented with tetracycline or kanamycin and tetracycline to mid-log phase. CpaA-His or the CpaA-FLAG-CpaBpelB-His complex was purified from cell-free supernatants through nickel affinity chromatography as above. CpaB-His was purified from cell lysate of E. coli Rosetta 2 cells carrying pET28-cpaB-his. Cells were grown overnight at 30 °C and 225 rpm in autoinduction medium supplemented with kanamycin. Cells were harvested and lysed as described above. CpaB-His was purified by nickel affinity chromatography as above. The purified proteins were concentrated, and the buffer was changed to 20 mm HEPES, 150 mm NaCl, and 50% glycerol (pH 7.4) using Amicon Ultra centrifugal filter units.
The Factor V cleavage assay was carried out according to Tilley et al. (29) with modifications. Briefly, 1.6 μg of purified CpaA or CpaB (in 20 mm HEPES, 150 mm NaCl, and 50% glycerol (pH 7.4)) was mixed with 11 ng of human Factor V (Abcam, Cambridge, MA) in a total volume of 20 μl (20 mm HEPES and 150 mm NaCl (pH 7.4)). Heat-inactivated CpaA was incubated at 100 °C for 20 min prior to the addition of Factor V. Based on Coomassie staining indicating that CpaA and CpaB form a 1:1 complex, 3.2 μg of the CpaA–CpaB complex was used in the assay, which is estimated to contribute ∼1.6 μg of CpaA. All samples were incubated at 37 °C for 30 min.
α-Thrombin was used as a positive control in the proteolytic cleavage of Factor V (49). The thrombin-catalyzed digestion of FV was carried out as described previously (50) with modifications. Briefly, 0.6 mg/ml human Factor V and 2 μg/ml of α-thrombin (Thermo Fisher, Waltham, MA) in 25 mm Tris-HCl (pH 7.4), 50 mm NH4Cl, and 5 mm CaCl2 was incubated at 37 °C for 60 min. The buffer control for this reaction was carried out in the same way but in the absence of α-thrombin.
Factor V cleavage was monitored by Western blotting by resolving 8.2 ng of Factor V per lane on an 8% polyacrylamide gel. Factor V was detected using polyclonal sheep anti-human Factor V (Thermo Fisher, 1:1000), polyclonal rabbit anti-sheep Ig (Fc-specific, Sigma-Aldrich, St. Louis, MO, 1:12,000), and IRDYE®800CW goat anti-rabbit IgG (LI-COR, 1:15,000). CpaA-His was detected with monoclonal mouse anti-His6 (Thermo Fisher, 1:1000) and IRDYE®680RD goat anti-mouse IgG (LI-COR, 1:15,000), whereas CpaA-FLAG was detected using monoclonal mouse anti-FLAG M2 (Sigma-Aldrich, 1:1000) and IRDYE®680RD goat anti-mouse IgG (LI-COR).
Author contributions
The experiments were designed by R. L. K. under the guidance of M. F. F. The mouse infection experiment was designed and performed by L. D. P. under the guidance of E. P. S. The Factor V cleavage assay was performed by J. L. N. D. S. performed the size-exclusion chromatography and analytical ultracentrifugation experiments under the guidance of N. H. T. All other experiments were performed by R. L. K. The manuscript was written by R. L. K., J. L., and M. F. F. All authors edited the manuscript.
Supplementary Material
Acknowledgment
We thank M. Florencia Haurat for assistance with protein purification.
This work was supported by a startup fund to M. F. F. from the Department of Molecular Microbiology at Washington University School of Medicine. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S5 and Tables S1 and S2.
- T2SS
- type II secretion system
- spp.
- species
- T3SS
- type III secretion system
- nickel-NTA
- nickel-nitrilotriacetic acid
- AB
- Acinetobacter baumannii
- AY
- Acinetobacter baylyi
- AC
- Acinetobacter calcoaceticus
- AP
- Acinetobacter pittii
- AN
- Acinetobacter nosocomialis.
References
- 1. DebRoy S., Dao J., Söderberg M., Rossier O., and Cianciotto N. P. (2006) Legionella pneumophila type II secretome reveals unique exoproteins and a chitinase that promotes bacterial persistence in the lung. Proc. Natl. Acad. Sci. U.S.A. 103, 19146–19151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fujieda M., Aoyagi Y., Matsubara K., Takeuchi Y., Fujimaki W., Matsushita M., Bohnsack J. F., and Takahashi S. (2012) L-ficolin and capsular polysaccharide-specific IgG in cord serum contribute synergistically to opsonophagocytic killing of serotype III and V group B streptococci. Infect. Immun. 80, 2053–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCoy-Simandle K., Stewart C. R., Dao J., DebRoy S., Rossier O., Bryce P. J., and Cianciotto N. P. (2011) Legionella pneumophila type II secretion dampens the cytokine response of infected macrophages and epithelia. Infect. Immun. 79, 1984–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sandkvist M., Morales V., and Bagdasarian M. (1993) A protein required for secretion of cholera toxin through the outer membrane of Vibrio cholerae. Gene 123, 81–86 [DOI] [PubMed] [Google Scholar]
- 5. Sikora A. E., Zielke R. A., Lawrence D. A., Andrews P. C., and Sandkvist M. (2011) Proteomic analysis of the Vibrio cholerae type II secretome reveals new proteins, including three related serine proteases. J. Biol. Chem. 286, 16555–16566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ho T. D., Davis B. M., Ritchie J. M., and Waldor M. K. (2008) Type 2 secretion promotes enterohemorrhagic Escherichia coli adherence and intestinal colonization. Infect. Immun. 76, 1858–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jyot J., Balloy V., Jouvion G., Verma A., Touqui L., Huerre M., Chignard M., and Ramphal R. (2011) Type II secretion system of Pseudomonas aeruginosa: in vivo evidence of a significant role in death due to lung infection. J. Infect. Dis. 203, 1369–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Costa T. R., Felisberto-Rodrigues C., Meir A., Prevost M. S., Redzej A., Trokter M., and Waksman G. (2015) Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359 [DOI] [PubMed] [Google Scholar]
- 9. Nivaskumar M., and Francetic O. (2014) Type II secretion system: a magic beanstalk or a protein escalator. Biochim. Biophys. Acta 1843, 1568–1577 [DOI] [PubMed] [Google Scholar]
- 10. Sandkvist M. (2001) Biology of type II secretion. Mol. Microbiol. 40, 271–283 [DOI] [PubMed] [Google Scholar]
- 11. Sandkvist M. (2001) Type II secretion and pathogenesis. Infect. Immun. 69, 3523–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pineau C., Guschinskaya N., Robert X., Gouet P., Ballut L., and Shevchik V. E. (2014) Substrate recognition by the bacterial type II secretion system: more than a simple interaction. Mol. Microbiol. 94, 126–140 [DOI] [PubMed] [Google Scholar]
- 13. McLaughlin L. S., Haft R. J., and Forest K. T. (2012) Structural insights into the type II secretion nanomachine. Curr. Opin. Struct. Biol. 22, 208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan Z., Yin M., Xu D., Zhu Y., and Li X. (2017) Structural insights into the secretin translocation channel in the type II secretion system. Nat. Struct. Mol. Biol. 24, 177–183 [DOI] [PubMed] [Google Scholar]
- 15. World Health Organization (2017) Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. [Google Scholar]
- 16. Park Y. K., Jung S. I., Park K. H., Kim S. H., and Ko K. S. (2012) Characteristics of carbapenem-resistant Acinetobacter spp. other than Acinetobacter baumannii in South Korea. Int. J. Antimicrob. Agents 39, 81–85 [DOI] [PubMed] [Google Scholar]
- 17. Chusri S., Chongsuvivatwong V., Rivera J. I., Silpapojakul K., Singkhamanan K., McNeil E., and Doi Y. (2014) Clinical outcomes of hospital-acquired infection with Acinetobacter nosocomialis and Acinetobacter pittii. Antimicrob. Agents Chemother. 58, 4172–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wendt C., Dietze B., Dietz E., and Rüden H. (1997) Survival of Acinetobacter baumannii on dry surfaces. J. Clin. Microbiol. 35, 1394–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomaras A. P., Dorsey C. W., Edelmann R. E., and Actis L. A. (2003) Attachment to and biofilm formation on abiotic surfaces by Acinetobacter baumannii: involvement of a novel chaperone-usher pili assembly system. Microbiology 149, 3473–3484 [DOI] [PubMed] [Google Scholar]
- 20. Howard A., O'Donoghue M., Feeney A., and Sleator R. D. (2012) Acinetobacter baumannii. Virulence 3, 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Espinal P., Martí S., and Vila J. (2012) Effect of biofilm formation on the survival of Acinetobacter baumannii on dry surfaces. J. Hosp. Infect. 80, 56–60 [DOI] [PubMed] [Google Scholar]
- 22. Cisneros J. M., and Rodríguez-Baño J. (2002) Nosocomial bacteremia due to Acinetobacter baumannii: epidemiology, clinical features and treatment. Clin. Microbiol. Infect. 8, 687–693 [DOI] [PubMed] [Google Scholar]
- 23. Bergogne-Bérézin E., and Towner K. J. (1996) Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin. Microbiol. Rev. 9, 148–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valencia R., Arroyo L. A., Conde M., Aldana J. M., Torres M.-J., Fernández-Cuenca F., Garnacho-Montero J., Cisneros J. M., Ortíz C., Pachón J., and Aznar J. (2009) Nosocomial outbreak of infection with pan–drug-resistant Acinetobacter baumannii in a tertiary care university hospital. Infect. Control Hosp. Epidemiol. 30, 257–263 [DOI] [PubMed] [Google Scholar]
- 25. Fournier P. E., Richet H., and Winstein R. A. (2006) The epidemiology and control of Acinetobacter baumannii in health care facilities. Clin. Infect. Dis. 42, 692–699 [DOI] [PubMed] [Google Scholar]
- 26. Eijkelkamp B. A., Stroeher U. H., Hassan K. A., Paulsen I. T., and Brown M. H. (2014) Comparative analysis of surface-exposed virulence factors of Acinetobacter baumannii. BMC Genomics 15, 1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson T. L., Waack U., Smith S., Mobley H., and Sandkvist M. (2015) Acinetobacter baumannii is dependent on the type II secretion system and its substrate LipA for lipid utilization and in vivo. J. Bacteriol 198, 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harding C. M., Kinsella R. L., Palmer L. D., Skaar E. P., and Feldman M. F. (2016) Medically relevant Acinetobacter species require a type II secretion system and specific membrane-associated chaperones for the export of multiple substrates and full virulence. PLoS Pathog. 12, e1005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tilley D., Law R., Warren S., Samis J. A., and Kumar A. (2014) CpaA a novel protease from Acinetobacter baumannii clinical isolates deregulates blood coagulation. FEMS Microbiol. Lett. 356, 53–61 [DOI] [PubMed] [Google Scholar]
- 30. Kessler E., and Safrin M. (1994) The propeptide of Pseudomonas aeruginosa elastase acts as an elastase inhibitor. J. Biol. Chem. 269, 22726–22731 [PubMed] [Google Scholar]
- 31. Shinde U., and Inouye M. (2000) Intramolecular chaperones: polypeptide extensions that modulate protein folding. Semin. Cell Dev. Biol. 11, 35–44 [DOI] [PubMed] [Google Scholar]
- 32. Frenken L. G., Bos J. W., Visser C., Müller W., Tommassen J., and Verrips C. T. (1993) An accessory gene, lipB, required for the production of active Pseudomonas glumae lipase. Mol. Microbiol. 9, 579–589 [DOI] [PubMed] [Google Scholar]
- 33. Rosenau F., Tommassen J., and Jaeger K. E. (2004) Lipase-specific foldases. ChemBioChem 5, 152–161 [DOI] [PubMed] [Google Scholar]
- 34. El Khattabi M., Ockhuijsen C., Bitter W., Jaeger K. E., and Tommassen J. (1999) Specificity of the lipase-specific foldases of gram-negative bacteria and the role of the membrane anchor. Mol. Gen. Genet. 261, 770–776 [DOI] [PubMed] [Google Scholar]
- 35. Page A. L., and Parsot C. (2002) Chaperones of the type III secretion pathway: jacks of all trades. Mol. Microbiol. 46, 1–11 [DOI] [PubMed] [Google Scholar]
- 36. Harding C. M., Tracy E. N., Carruthers M. D., Rather P. N., Actis L. A., and Munson R. S. Jr. (2013) Acinetobacter baumannii strain M2 produces Type IV Pili which play a role in natural transformation and twitching motility but not surface-associated motility. MBio 4, e00360–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carruthers M. D., Nicholson P. A., Tracy E. N., and Munson R. S. (2013) Acinetobacter baumannii utilizes a type VI secretion system for bacterial competition. PLoS ONE 8, e59388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Demeler B. (2005) in Modern Analytical Ultracentrifugation: Techniques and Methods (Scott D.J., Harding S.E., and Rowe A.J., eds.) pp. 210–219, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 39. Feldman M. F., Wacker M., Hernandez M., Hitchen P. G., Marolda C. L., Kowarik M., Morris H. R., Dell A., Valvano M. A., and Aebi M. (2005) Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weber B. S., Kinsella R. L., Harding C. M., and Feldman M. F. (2017) The secrets of Acinetobacter secretion. Trends Microbiol. 25, 532–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Green E. R., and Mecsas J. (2016) Bacterial secretion systems: an overview. Microbiol. Spectr. 4, 10.1128/microbiolspec.VMBF-0012-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Filloux A. (2004) The underlying mechanisms of type II protein secretion. Biochim. Biophys. Acta 1694, 163–179 [DOI] [PubMed] [Google Scholar]
- 43. Akeda Y., and Galán J. E. (2005) Chaperone release and unfolding of substrates in type III secretion. Nature 437, 911–915 [DOI] [PubMed] [Google Scholar]
- 44. Harding C. M., Nasr M. A., Kinsella R. L., Scott N. E., Foster L. J., Weber B. S., Fiester S. E., Actis L. A., Tracy E. N., Munson R. S. Jr., and Feldman M. F. (2015) Acinetobacter strains carry two functional oligosaccharyltransferases, one devoted exclusively to type IV pilin, and the other one dedicated to O-glycosylation of multiple proteins. Mol. Microbiol. 96, 1023–1041 [DOI] [PubMed] [Google Scholar]
- 45. Bornhorst B. J. A., and Falke J. J. (2000) Purification of proteins using polyhistidine affinity tags. Methods Enzymol. 326, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schnaitman C. A. (1971) Effect of ethylenediaminetetraacetic acid, Triton X-100, and lysozyme on the morphology and chemical composition of isolated cell walls of Escherichia coli. J. Bacteriol. 108, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laue T. M., Shah B. D., Ridgeway T. M., and Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., and Horton J. C., Horton, eds.), pp. 90–125, Cambridge Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 48. Weber B. S., Miyata S. T., Iwashkiw J. A., Mortensen B. L., Skaar E. P., Pukatzki S., and Feldman M. F. (2013) Genomic and functional analysis of the type VI secretion system in Acinetobacter. PLoS ONE 8, e55142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Camire R. M., and Bos M. H. (2009) The molecular basis of Factor V and VIII procofactor activation. J. Thromb. Haemost. 7, 1951–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzuki K., Dahlbäck B., and Stenflo J. (1982) Thrombin-catalyzed activation of human coagulation Factor V. J. Biol. Chem. 257, 6556–6564 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.