

Abstract
In mitochondria, the sirtuin SIRT5 is an NAD+-dependent protein deacylase that controls several metabolic pathways. Although a wide range of SIRT5 targets have been identified, the overall function of SIRT5 in organismal metabolic homeostasis remains unclear. Given that SIRT5 expression is highest in the heart and that sirtuins are commonly stress-response proteins, we used an established model of pressure overload–induced heart muscle hypertrophy caused by transverse aortic constriction (TAC) to determine SIRT5's role in cardiac stress responses. Remarkably, SIRT5KO mice had reduced survival upon TAC compared with wild-type mice but exhibited no mortality when undergoing a sham control operation. The increased mortality with TAC was associated with increased pathological hypertrophy and with key abnormalities in both cardiac performance and ventricular compliance. By combining high-resolution MS-based metabolomic and proteomic analyses of cardiac tissues from wild-type and SIRT5KO mice, we found several biochemical abnormalities exacerbated in the SIRT5KO mice, including apparent decreases in fatty acid oxidation and glucose oxidation as well as an overall decrease in mitochondrial NAD+/NADH. Together, these abnormalities suggest that SIRT5 deacylates protein substrates involved in cellular oxidative metabolism to maintain mitochondrial energy production. Overall, the functional and metabolic results presented here suggest an accelerated development of cardiac dysfunction in SIRT5KO mice in response to TAC, explaining increased mortality upon cardiac stress. Our findings reveal a key role for SIRT5 in maintaining cardiac oxidative metabolism under pressure overload to ensure survival.
Keywords: cardiac hypertrophy, cardiac metabolism, metabolomics, proteomics, sirtuin
Introduction
SIRT5 (sirtuin 5) is one of three mitochondrial proteins that belong to the sirtuin family of NAD+-dependent deacylases. Mitochondrial sirtuins (SIRT3–5) control metabolism in physiological and pathophysiological conditions by their deacylation activity (1, 2). SIRT5 possesses demalonylase (3, 4), desuccinylase (4), and deglutarylase (5) activity, whereas SIRT3 is primarily a lysine deacetylase (2), and SIRT4 is a potent lysine deglutarylase, demethylglutarylase, and dehydroxymethylglutarylase (6). The protein acylation profile in SIRT5KO mice has been characterized by several proteomic studies with the goal of identifying SIRT5 targets (proteins with increased sites of lysine acylation in the SIRT5KO mouse compared with wild-type controls) and the role of SIRT5 in regulating metabolism. For example, analyses of SIRT5KO mouse liver succinyl-proteomic data sets identified many proteins in metabolic pathways enriched with protein lysine succinylation, including enzymes involved in fatty acid oxidation, branched-chain amino acid (BCAA)4 degradation, and the TCA cycle (7, 8). Within these pathways, specific targets of SIRT5 include pyruvate dehydrogenase (PDH), succinate dehydrogenase (SDH) (7), and 3-hydroxy-3-methlyglutaryl-CoA synthase 2 (HMGCS2) (8), to name a few. SIRT5-mediated desuccinylation has been reported to repress activity of PDH (7) and enhance activity of HMGCS2 (8). SDH activity has been reported to be both repressed by desuccinylation (8, 9) and enhanced (10). Aside from these targets, hundreds of sites of lysine succinylation (8), malonylation (11), and glutarylation (5) lack functional characterization. Thus, further work needs to be done to fully understand how sirtuins regulate their cellular targets. Studies investigating the precise mechanisms by which SIRT5 regulates metabolism and function are mainly focused on analyzing the consequence of protein acylation and deacylation on identified SIRT5 targets (12, 13). However, despite the discovery of several SIRT5 substrates in mouse liver tissue, no strong, liver-specific phenotypes in SIRT5KO mice have been found. Indeed, overall phenotypes in mice lacking SIRT5 are generally subtle.
SIRT5 protein and mRNA is highly expressed in mouse and human heart compared with other tissues (11). The heart is a highly metabolic tissue and relies predominately on oxidative metabolism to produce sufficient ATP for cardiac muscle contraction. Previous studies demonstrated that cardiac hypersuccinylation occurs in the absence of SIRT5 and that enzymes in several metabolic pathways are primarily enriched with succinylation (9, 14). Despite strong hyperacylation signals, characterization of the SIRT5KO mouse reveals no substantial metabolic phenotypes (15) and only a mild decrement in cardiac function that is apparent in early adulthood (14). Based on current knowledge, many metabolic enzymes are hyperacylated in the absence of SIRT5, but few physiological consequences of SIRT5 ablation under normal conditions have been found.
Consistent with the notion that sirtuins are stress-response proteins, studies to elucidate the role of SIRT5 in cardiac physiology have revealed that physiological stress is necessary to reveal a phenotype in SIRT5KO mice. For example, the acute stress of cardiac ischemia-reperfusion results in an increased infarct area in mice lacking SIRT5 (9). Additionally, aging SIRT5KO mice to 39 weeks results in increased cardiac hypertrophy and reduced cardiac function (14). Increased activity of SDH and decreased activity of hydroxyacyl-CoA dehydrogenase α subunit via increased hypersuccinylation in the SIRT5KO mouse heart were identified as mechanisms contributing to these cardiac phenotypes. Together, these studies demonstrate that SIRT5 regulates cardiac metabolism via its desuccinylase activity. Despite this progress, the overall role of SIRT5 in cardiac metabolism and function requires further study. Thus, we considered interrogating the role of SIRT5 in the heart by using a model of chronic cardiac stress, namely a pressure overload–induced hypertrophy model.
Pressure overload causes an increase in ventricular wall stress and cardiomyocyte growth (16, 17) that results in maladaptive cardiac hypertrophy (18). This form of remodeling precedes the development of cardiac dysfunction, which is closely associated with shifts in cardiac metabolism. Classically, these metabolic shifts include both a decrease in the contribution of fatty acid oxidation to ATP production and an increase in the contribution of glycolysis to ATP production. Ultimately, oxidative metabolism decreases and ATP production is not sufficient to meet the energetic demands of the heart, and failure occurs. Given the decline in oxidative metabolism in cardiac hypertrophy and that identified targets of SIRT5 are enzymes in oxidative metabolic pathways, we predicted that SIRT5 might play a role in controlling metabolism during the transition to cardiac dysfunction. Thus, we set out to characterize cardiac metabolism and physiology in whole-body SIRT5KO mice.
Results
Chronic transverse aortic constriction (TAC) results in significantly increased mortality in SIRT5KO mice
TAC is a well-characterized animal model of pressure overload–induced hypertrophy (19), wherein metabolic shifts away from oxidation occur that are associated with the onset of metabolic dysfunction. SIRT5KO mice and WT controls (10–12 weeks of age) underwent TAC and were monitored for 16 weeks. In response to TAC, median survival was starkly decreased in SIRT5KO mice compared with WT controls (2.4 versus 10.9 weeks post-TAC; Fig. 1A). No death was observed in the sham control groups for either genotype (data not shown). These findings suggest that SIRT5KO mice progress to cardiac dysfunction at an accelerated rate upon TAC-induced cardiac stress compared with controls.
Figure 1.
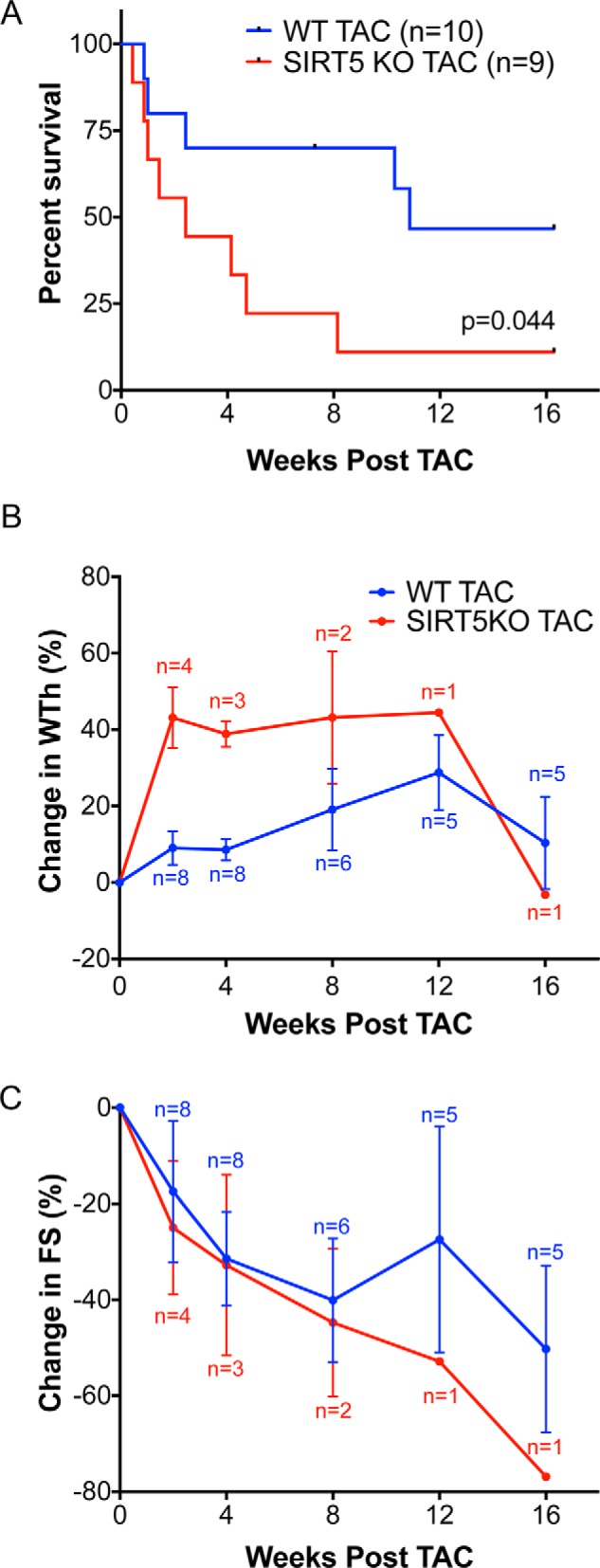
SIRT5KO mice have decreased survival after TAC. A, survival of the SIRT5KO mice was significantly reduced (Mantel-Cox test). B, percent change in wall thickness appeared to be increased in the SIRT5KO in the early stages of a 16-week chronic TAC study. C, percent change in fractional shortening decreased in both wild-type and SIRT5KO animals over the course of the study with no apparent genotypic differences. n = 10 for WT TAC, and n = 9 for SIRT5KO TAC at the start of the study. Mice were males and were 10–12 weeks of age at the start of the study. Error bars, S.E.
Serial echocardiography was used to measure wall thickness (interventricular septum wall thickness + posterior wall thickness) and fractional shortening ((EDD − ESD)/EDD × 100)) (where EDD represents end-diastolic diameter and ESD is end-systolic diameter) before TAC surgery and 2, 4, 8, 12, and 16 weeks after TAC surgery. Due to the significant, progressive mortality in the SIRT5KO group, it was not possible to perform appropriate statistical analyses of this experiment. However, a trend appeared for greater wall thickness in the SIRT5KO mice at 2 and 4 post-after TAC (Fig. 1B). No differences were measured in cardiac fractional shortening between the WT and SIRT5KO mice at any time point (Fig. 1C). Based on these findings, and to overcome the progressive mortality, further examination of cardiac physiology was restricted to the 4-week post-TAC time point in WT and surviving SIRT5KO mice.
Wall thickness and fractional shortening were measured by echocardiogram pre-TAC and 4 weeks post-TAC, and these data were combined with data at the same time points from the initial 16-week study to assess cardiac morphology and function. Concentric hypertrophy was increased in SIRT5KO TAC mice compared with WT TAC mice after 4 weeks of TAC (Fig. 2, A and B). As expected, wall thickness (including both measurements of interventricular septum wall (IVSW) and posterior wall (PW) thickness) increased with TAC in both genotypes compared with their respective sham controls (Fig. 2A). No significant differences in fractional shortening were seen between WT and SIRT5KO mice 4 weeks post-TAC (Fig. 2, C and D). Further, we found no differences in wall thickness or fractional shortening between the WT and SIRT5KO sham conditions (Fig. 2, A–D). Consistent with echocardiographic measurements of concentric hypertrophy, we observed an increase in left ventricle (LV) weight/body weight with TAC in both genotypes compared with their sham controls, indicative of the development of left ventricle cardiac hypertrophy (Fig. 2E). Although the LV weight/body weight ratio trended higher in the SIRT5KO TAC group compared with WT TAC group, the difference was not statistically significant. The SIRT5KO TAC group develops primarily concentric hypertrophy, as shown by increased wall thickness and maintained EDD (Fig. 2, A and C). In contrast, the WT TAC group shows both concentric and eccentric hypertrophy, as evidenced by increased wall thickness and a trend toward increased EDD (Fig. 2, A and C). Thus, whereas the LV weight/body weight is not different between genotypes with TAC, it appears that the morphology of hypertrophy is different.
Figure 2.

SIRT5KO mice develop exaggerated hypertrophy 4 weeks post-TAC. Shown are measurements of cardiac morphology at 4 weeks post-TAC. A, wall thickness (WTh), interventricular septum wall (IVSW) thickness, and posterior wall (PW) thickness. B, percent change of values in A. C, fractional shortening (FS), EDD, and ESD. D, percent change of values in C. n = 16 for WT sham, n = 13 for WT TAC, n = 13 for SIRT5KO sham, and n = 16 for SIRT5KO TAC. Shown are combined results from six independent experiments, including the 4-week time point from Fig. 1. Mice were males and were 12–21 weeks of age at 4 weeks TAC (A–D). Box plots depict the interquartile range with whiskers plotted to the minimum and maximum values. The horizontal line within the box is the median value, and the plus sign is the mean value. E, left ventricle weight normalized to body weight at 4 weeks post-TAC. n = 12 for WT sham, n = 14 for WT TAC, n = 10 for SIRT5KO sham, and n = 18 for SIRT5KO TAC. Combined results from five independent experiments are shown. Mice were males and were 12–21 weeks of age at 4 weeks TAC. F, cardiac mRNA expression of Anf, Bnp, Col3a1, and Col1a1 relative to 36B4. Values are mean ± S.E. (error bars). G, Rcan1.4 relative to 36B4. Values are mean ± S.E. n = 3 for WT sham, n = 5 for SIRT5KO sham, n = 4 for WT TAC, and n = 7 for SIRT5KO TAC. Mice were males and were 18–19 weeks of age at 4 weeks post-TAC (two-way ANOVA with multiple comparisons, Bonferroni correction). *, p ≤ 0.05; effect of TAC if no significance in multiple-comparison tests. +, p ≤ 0.05.
Consistent with these morphological changes, two molecular markers of hypertrophy, namely Anf and Bnp, trended toward increased in both WT and SIRT5KO animals after 4 weeks of TAC (Fig. 2F). Additionally, we found markers of cardiac fibrosis (Col3a1 and Col1a1) to be increased in the SIRT5KO TAC compared with SIRT5KO sham mice (Fig. 2F). Col1a1 was also higher in the SIRT5KO TAC compared with WT TAC animals. The mechanical stress of TAC activates several primary pathways, including the MAPK pathway and activation of calcineurin (20). A main effect of hypertrophic signaling is increased phosphorylation of NFAT that leads to its translocation to the nucleus and transcription of hypertrophic genes. We interrogated the expression of Rcan1.4, a target gene of NFAT signaling (21), and found that expression of this gene trended toward increased in both TAC conditions, with a significant increase in the SIRT5KO TAC compared with sham condition (Fig. 2G). This finding is consistent with our other findings of exacerbated hypertrophy in SIRT5KO mice with TAC. Together, these observations suggest that development of concentric cardiac hypertrophy is more exaggerated in the SIRT5KO mouse heart in response to TAC with evidence of accelerated heart failure based on survival and expression of hypertrophic markers.
SIRT5KO mice have impaired systolic function 4 weeks post-TAC
To further investigate the functional impact of SIRT5 ablation on the heart, we performed pressure-volume loop analyses after 4 weeks of TAC (Fig. 3, A–D). Systolic function worsened in both TAC conditions, as indicated by a decrease in cardiac output and trends of decreased stroke volume and dP/dtmax (Table 1). With the development of cardiac hypertrophy, ejection fraction was maintained in WT TAC mice and trended toward decreased in SIRT5KO TAC compared with SIRT5KO sham mice (p = 0.07) (Fig. 3E). Load-independent measures of contractility, specifically end-systolic pressure-volume relationship linear slope and maximum elastance, showed increases in the SIRT5KO TAC animal compared with their sham controls (Table 2). Increased contractility in the SIRT5KO TAC mouse is consistent with the exaggerated hypertrophy observed in the SIRT5KO TAC mouse. Measures of active relaxation, such as Tau and dP/dtmin (Table 1), and passive relaxation, such as the end-diastolic pressure-volume relationship (Table 2), suggest both prolonged active relaxation and elevated ventricular stiffness in SIRT5KO mice. Overall, SIRT5KO mice demonstrate enhanced pathologic concentric hypertrophy in response to pressure overload, with key abnormalities in both cardiac relaxation and performance.
Figure 3.

SIRT3KO mice have decreased ejection fraction and preserved cardiac energetics 4 weeks post-TAC. A–D, pressure-volume loops with inferior vena cava occlusion. Shown are representative images for wild-type sham (A), SIRT5KO sham (B), wild-type TAC (C), and SIRT5KO TAC (D). Red trace, end-systolic pressure-volume relationship (ESPVR); green trace, end-diastolic pressure-volume relationship (EDPVR). E, ejection fraction. F, pressure-volume area analysis. G, cardiac efficiency (stroke work/pressure-volume area). n = 6 for WT sham, n = 9 for SIRT5KO sham, n = 7 for WT TAC, and n = 11 for SIRT5KO TAC. Shown are the combined results from four independent experiments. Mice were males and were 12–21 weeks of age at the end of the study. Two-way ANOVA with multiple comparisons, Bonferroni correction, was performed. *, p ≤ 0.05; effect of TAC if no significance in multiple-comparison tests. +, p ≤ 0.05. Error bars, S.E.
Table 1.
Baseline hemodynamic parameters
Statistical comparisons were made using a two-way ANOVA with Bonferroni correction for multiple comparisons. Date are presented as mean ± S.E. *, p < 0.1 versus sham of same genotype; **, p < 0.05 versus sham of the same genotype. LVESP, left ventricle end systolic pressure; LVEDP, left ventricle end diastolic pressure.
| Parameters | Wild-type sham (n = 6) | SIRT5KO sham (n = 9) | Wild-type TAC (n = 7) | SIRT5KO TAC (n = 11) |
|---|---|---|---|---|
| Heart rate (beats/min) | 460.0 ± 5.9 | 451.6 ± 18.1 | 378.6 ± 22.6** | 392.6 ± 41.3** |
| LVESP (mm Hg) | 118.2 ± 16.1 | 120.4 ± 8.6 | 166.4 ± 9.5** | 197.0 ± 7.3** |
| LVEDP (mm Hg) | 8.78 ± 2.02 | 6.60 ± 1.55 | 11.57 ± 1.16 | 12.28 ± 2.43 |
| Systolic function | ||||
| Stroke volume (μl) | 27.72 ± 1.74 | 21.35 ± 2.78 | 19.29 ± 2.46 | 15.25 ± 1.74 |
| Ejection fraction (%) | 62.12 ± 5.75 | 56.97 ± 6.47 | 48.97 ± 9.29 | 37.16 ± 3.53* |
| Cardiac output (μl/min) | 12747 ± 813.0 | 9828 ± 1532.0 | 7451 ± 1190** | 6036 ± 748.4* |
| Stroke work (mm Hg × μl) | 2522 ± 203.4 | 2085 ± 287.3 | 2527 ± 396.6 | 2045 ± 301.8 |
| dP/dtmax (mm Hg/s−1) | 10,259 ± 1099 | 10,377 ± 812.1 | 8152 ± 830.0 | 8476 ± 745.5 |
| Diastolic function | ||||
| dP/dtmin (mm Hg/s−1) | −8502 ± 841.6 | −8520 ± 486.8 | −7865 ± 694.7 | −8845 ± 831.34 |
| Tau (ms) (Weiss) | 7.88 ± 0.38 | 7.44 ± 0.38 | 9.41 ± 0.76 | 9.47 ± 1.05 |
| Tau (ms) (Glantz) | 12.77 ± 1.22 | 11.81 ± 0.96 | 14.29 ± 0.95 | 14.31 ± 1.32 |
Table 2.
Load-independent hemodynamic measures
Shown are parameters of LV compliance (linear and quadratic derived end-diastolic pressure-volume relationship) and LV contractility (linear and quadratic derived end-systolic pressure-volume relationship, preload recruitable stroke work, dP/dtmax versus end-diastolic volume, and Emax). Data are represented as mean ± S.E.; statistical comparisons made with a two-way ANOVA with Bonferroni's multiple-comparison test. *, p ≤ 0.1 versus sham of the same genotype; **, p ≤ 0.05 versus sham of the same genotype. EDPVR, end-diastolic pressure-volume relationship; ESPVR, end-systolic pressure-volume relationship; EDV, end-diastolic volume; a, coefficient of curvilinearity; Vo, volume intercept; E′max, maximum slope of quadratic end-systolic pressure-volume relationship; PRSW, preload recruitable stroke work; Emax, maximal elastance.
| Parameters | Wild-type sham (n = 6) | SIRT5KO sham (n = 7) | Wild-type TAC (n = 9) | SIRT5KO TAC (n = 11) |
|---|---|---|---|---|
| Compliance | ||||
| EDPVR (linear) slope | 0.19 ± 0.07 | 0.27 ± 0.06 | 0.36 ± 0.06 | 0.63 ± 0.18 |
| EDPVR (linear) intercept | −0.41 ± 0.99 | −1.40 ± 1.01 | −7.28 ± 5.00 | −8.27 ± 6.82 |
| EDPVR (quadratic) β coefficient | 0.043 ± 0.006 | 0.071 ± 0.011 | 0.076 ± 0.012 | 0.104 ± 0.018 |
| EDPVR (quadratic) α coefficient | 1.606 ± 0.542 | 0.866 ± 0.111 | 0.619 ± 0.315 | 0.482 ± 0.244 |
| Contractility | ||||
| ESPVR (linear) slope | 3.16 ± 0.55 | 5.46 ± 1.11 | 6.93 ± 1.01 | 10.13 ± 1.59** |
| ESPVR (linear) intercept | −19.63 ± 5.77 | −8.54 ± 3.92 | 3.35 ± 7.88** | 6.65 ± 4.47 |
| ESPVR (quadratic) a | −0.10 ± 0.06 | −0.23 ± 0.08 | −0.22 ± 0.07 | −0.38 ± 0.15 |
| ESPVR (quadratic) V0 | −10.27 ± 4.61 | −2.12 ± 2.95 | 10.96 ± 6.34** | 11.76 ± 4.23* |
| ESPVR (quadratic) E′max | 6.20 ± 1.77 | 9.50 ± 1.70 | 12.51 ± 1.19 | 19.02 ± 3.84* |
| PRSW slope | 52.66 ± 4.78 | 61.51 ± 4.81 | 89.52 ± 7.94* | 89.12 ± 12.95 |
| PRSW intercept | −4.01 ± 4.72 | 2.52 ± 2.98 | 16.49 ± 6.57* | 16.36 ± 5.20 |
| dP/dtmax versus EDV slope | 243.91 ± 29.47 | 277.42 ± 50.95 | 161.99 ± 17.53 | 252.92 ± 44.91 |
| dP/dtmax versus EDV intercept | 0.58 ± 4.05 | −2.79 ± 3.64 | 0.57 ± 5.73 | 1.30 ± 5.10 |
| Emax | 4.79 ± 0.76 | 6.50 ± 0.69 | 11.03 ± 1.40 | 15.59 ± 2.49** |
Next, we analyzed signaling pathways that are activated with cardiac hypertrophy to determine whether aberrant signaling may be responsible for the accelerated trajectory of cardiac dysfunction in the SIRT5KO TAC condition. We analyzed activation of mTOR by probing for phosphorylation of 4EBP1 and found a trend of increased phosphorylation of 4EBP1 in both WT and SIRT5KO TAC condition with no differences in genotype (supplemental Fig. 1, A and B). β-Adrenergic receptor signaling is often a secondary signaling response with pressure overload–induced hypertrophy (20) and is dysfunctional in later stages of heart failure (22). During activation of β-adrenergic receptor signaling, autophosphorylation of CaMKII occurs at Thr-286 (23). We examined phosphorylation of CaMKII and found that there was an increase in phosphorylation of CaMKII in the SIRT5KO TAC compared with sham condition (supplemental Fig. 1, C and D). Whereas we observed high variability in the WT TAC condition, we found a trend of increased CaMKII phosphorylation. Additionally, we looked at the mRNA expression of β-adrenergic receptor 1 (Adbr1) and β-adrenergic receptor 2 (Adbr2) because down-regulation of β-adrenergic receptors occurs with heart failure (24). We found a decrease in Adbr1 in the SIRT5KO TAC compared with sham condition and a trend of decreased Adbr1 in the WT TAC compared with sham condition (supplemental Fig. 1E). These findings are consistent with dysfunctional β-adrenergic receptor signaling in the WT and SIRT5KO TAC conditions. However, we found no marked differences between the WT and SIRT5KO TAC condition.
Decreased cardiac energy production is a common feature that coincides with developing cardiac dysfunction. To determine whether SIRT5KO mice had altered cardiac energetics in response to TAC, we measured cardiac efficiency from the hemodynamic data using pressure-volume area (PVA) analysis (Fig. 3, F and G). PVA is the sum of energy generated for mechanical work (stroke work) and energy that is generated but not used (potential work), whereas cardiac efficiency is defined as the ratio of stroke work to pressure-volume area (stroke work + potential work). PVA in the SIRT5KO heart trended lower compared with the WT heart in both sham and TAC conditions (Fig. 3F), which was driven mainly by slightly lower stroke work in the SIRT5KO hearts compared with WT hearts (Table 1). We observed no differences in potential work across all four conditions. Although we found no significant differences in cardiac efficiency (Fig. 3G), a subset of animals in the SIRT5KO TAC group appeared to have decreased cardiac efficiency compared with the other conditions tested (Fig. 3G), consistent with the idea that cardiac dysfunction develops at an accelerated rate in SIRT5KO mice. Together, these data show that after 4 weeks of TAC, both WT and SIRT5KO maintain energy generation compared with their sham controls. Overall, the SIRT5KO mice have a trend of decreased energy production compared with the WT mice. These data suggest that despite decrements in systolic and diastolic function, both WT and surviving SIRT5KO mice have compensatory metabolic pathways for energy generation to maintain cardiac efficiency.
Protein succinylation is increased in SIRT5KO hearts and abundant on enzymes in oxidative metabolism
To determine whether WT and SIRT5KO mice have similar metabolic compensatory mechanisms in response to TAC-induced cardiac stress, we combined proteomic and metabolomic high-resolution mass spectrometry profiling in whole-tissue cardiac lysates. Because SIRT5 is a potent demalonylase, desuccinylase, and deglutarylase, we first measured the corresponding post-translational modifications by Western blotting (Fig. 4, A–E). We found that lysine succinylation was significantly elevated across a wide range of proteins in whole-tissue lysates of SIRT5KO hearts under basal conditions (Fig. 4A). Other acyl-lysine modifications regulated by SIRT5 had a small (malonylation; Fig. 4B) or no (glutarylation; Fig. 4C) increase on proteins in SIRT5KO hearts under basal conditions, indicating that succinylation is the primary modification regulated by SIRT5 in the heart. Consistent with SIRT5 having weak deacetylase activity, we found no changes in protein acetylation in the SIRT5KO heart (Fig. 4D).
Figure 4.

Succinylation is dramatically increased in the SIRT5KO heart and abundant on enzymes in oxidative metabolism. Shown are Western blots of succinyl-lysine (A), malonyl-lysine (B), glutaryl-lysine (C), and acetyl-lysine (D) in WT and SIRT5KO mouse hearts at baseline. E, quantification of acyl-lysine blots. Acylation signal was normalized to total protein using Bio-Rad stain-free technology. Values are mean ± S.E. (error bars). n = 3 for WT, and n = 3 for SIRT5KO; Student's t test. *, p ≤ 0.05. Mice were males and were 8 weeks of age (A–E). F, Western blot of succinyl-lysine in WT and SIRT5KO mouse hearts after 4 weeks of TAC. G, quantification of succinyl lysine blot. Succinylation signal was normalized to total protein using Bio-Rad stain-free technology. Values are mean ± S.E. n = 3 for WT sham, n = 5 for SIRT5KO sham, n = 4 for WT TAC, and n = 7 for SIRT5KO TAC. Two-way ANOVA with multiple comparisons, Bonferroni correction, was performed. *, p ≤ 0.05. Mice were males and were 18–19 weeks of age at the end of the study (F and G). H, summary of succinylated proteins and peptides identified via succinyl-proteomics. I, volcano plot of log2 -fold change in succinylation on peptides identified in SIRT5KO heart relative to wild-type heart and their associated −log10 p values. J, comparison of the number of SIRT5 targets identified in previous studies. K, IPA summary of top pathways enriched with succinylated proteins. n = 3 for wild type, and n = 3 for SIRT5KO. Mice were males and were 11 weeks of age (H, I, and K). L, Venn diagram illustrating overlap of top pathways enriched with lysine succinylation in our study compared with previously published studies. Sadhukhan et al. (14) used DAVID to perform pathway analysis, and Boylston et al. (9) used IPA to perform pathway analysis.
Metabolic stressors can lead to changes in protein acylation. For example, increases in lysine acetylation have been observed in both mouse models of heart failure (25, 26) and in human patients in end-stage heart failure (27). Therefore, we profiled changes in cardiac protein acylation in WT and SIRT5KO mice in sham and TAC conditions. We observed that TAC in WT mice leads to a slight reduction in global protein succinylation in whole-tissue lysates (Fig. 4, F and G). Like WT mice, TAC in SIRT5KO mice also leads to a reduction in global protein succinylation (Fig. 4, F and G), suggesting that shifts in succinyl-CoA metabolism, and therefore succinylation, are occurring in both WT and SIRT5KO mice under TAC-induced cardiac stress. Western blot profiling showed no changes in malonylation, glutarylation, or acetylation with TAC (Western blot image quantification summarized in Fig. 4G; images not shown).
Because succinylation was the primary modification regulated by SIRT5 in the heart and because basal conditions captured the largest breadth of protein succinylation, we performed succinyl-proteomics using label-free quantitative mass spectrometry of WT and SIRT5KO whole-heart tissue. Cardiac tissue was homogenized and digested, and peptides containing succinyl-lysine modifications were enriched using an anti-succinyl-lysine affinity matrix. Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) was used to identify specific sites of lysine succinylation across the cardiac proteome and then normalized to protein abundance (please see supplemental Table 1 for list of all succinylated peptides). Overall, we identified 1448 unique proteins containing one or more sites of lysine succinylation (Fig. 4H). In total, 6124 unique peptides were identified, with the majority (93.8%) of peptides containing a single succinylated lysine. Most peptides identified displayed an increase in lysine succinylation upon SIRT5 ablation (Fig. 4I). We defined potential SIRT5 targets as sites of lysine succinylation that increased at least 2-fold with a p value of ≤0.05 (Fig. 4I, orange dots). Using this criterion, we identified 2,457 peptides that mapped to 766 unique proteins. Comparing the number of SIRT5 targets identified in our SIRT5KO cardiac succinyl-proteomics study with previously published similar studies (9, 14), we find that we have greatly increased the number of identified sites of lysine succinylation with the potential to be regulated by SIRT5 (Fig. 4J). Next, we used Ingenuity Pathway Analysis (IPA) to determine pathways that were enriched in proteins containing lysine succinylation. The top pathways with succinylated proteins were primarily involved in oxidative metabolism, including oxidative phosphorylation, TCA cycle, BCAA degradation, and fatty acid β-oxidation (Fig. 4K). The proteins identified in these pathways tended to have increased succinylation in the SIRT5KO compared with WT heart. Whereas some instances of pathways with overall decreased succinylation were observed in the SIRT5KO heart, these pathways tended to represent minor or questionable metabolic pathways in the heart, such as gluconeogenesis (28). We again compared the results of the pathway analysis from our data set with the results of pathway analyses from previously published data sets (9, 14) (Fig. 4L). The three pathways that were common to all succinyl-proteomic data sets were oxidative phosphorylation, TCA cycle, and fatty acid metabolism/oxidation. We found additional overlap between pathways enriched for succinylation in our data set and the data set generated by Boylston et al. (9), which included BCAA degradation, glutaryl-CoA degradation, and ketogenesis. Together, these data confirm that sites of increased lysine succinylation in SIRT5KO mouse hearts occur on proteins involved in major oxidative metabolic pathways.
Oxidative metabolism is impaired in the SIRT5KO mouse 4 weeks post-TAC
Because pathways with a high number of proteins can be overrepresented in pathway analyses and artificially enhance p values, we integrated the proteomics with unbiased metabolomic profiling in an effort to identify the primary metabolic lesions in SIRT5KO mouse hearts. Metabolite data collected from high-resolution mass spectrometry were uploaded to MetaboAnalyst version 3.0 (29, 30) and analyzed for relevant groups of metabolites that were changing across conditions and genotypes. This analysis revealed that multiple acyl-carnitines were decreased in the SIRT5KO TAC compared with SIRT5KO sham heart (Fig. 5A), whereas few metabolites changed >2-fold when comparing SIRT5KO sham versus WT sham, SIRT5KO TAC versus WT TAC, and WT TAC versus WT sham (data not shown). Therefore, we extracted and analyzed the acyl-carnitine data and found that overall, these lipid metabolites were decreased after TAC in the SIRT5KO heart compared with the sham condition (Fig. 5B). Although acyl-carnitines trended toward decreased in the WT TAC compared with WT sham heart, the differences were not significant. Furthermore, we found that long-chain fatty acids (palmitate and stearate) accumulated in both TAC conditions (Fig. 5C). However, the metabolites of the oxidation of these long-chain fatty acids (C10:0, C12:0, and C14:0) are increased in the WT TAC condition yet decreased in the SIRT5KO TAC condition compared with WT sham. Together, this metabolic signature suggests that long-chain fatty acids are not metabolized to the same extent in the SIRT5KO TAC compared with the WT TAC condition and further suggest that fatty acid oxidation could be impaired in SIRT5KO TAC hearts.
Figure 5.

Fatty acid oxidation is impaired in SIRT5KO TAC hearts. A, volcano plot of metabolites detected comparing SIRT5KO TAC with SIRT5KO sham. B, metabolite profiles of acyl-carnitines (*, p ≤ 0.05 compared with SIRT5KO sham). C, metabolite profile of free fatty acids. D, cardiac mRNA expression of CD36, Cpt1, Acadvl, Acadl, Acadm, Pparα, Pparδ, and Pgc1α relative to 36B4. Values are mean ± S.E. (error bars) (B–D). n = 3 for WT sham, n = 5 for SIRT5KO sham, n = 4 for WT TAC, and n = 7 for SIRT5KO TAC. Mice were males and were 18–19 weeks of age at the end of the study (A–D). Two-way ANOVA with multiple comparisons, Bonferroni correction, was performed. *, p ≤ 0.05; effect of TAC if no significance in multiple comparisons tests. +, p ≤ 0.05. E, -fold change of all succinylated peptides identified on enzymes in fatty acid oxidation. The horizontal maroon bar represents the mean -fold change of all peptides identified on a single protein. n = 3 for wild type, and n = 3 for SIRT5KO. Mice were males and were 11 weeks of age.
To further interrogate these findings, we examined known mechanisms that regulate fatty acid oxidation. First, we performed RT-qPCR on key genes involved in fatty acid oxidation to determine whether decreased acyl-carnitines in the SIRT5KO heart could be due to differences in transcriptional regulation of the fatty acid oxidation machinery. Indeed, reduced expression (two-way ANOVA, p ≤ 0.05; variation due to TAC) of Acadm and of Pppara in TAC hearts compared with sham hearts is consistent with reduced fatty acid oxidation in early stages of cardiac hypertrophy (Fig. 5D) (31, 32). However, we found no significant differences between genotypes, suggesting that the significant decrease of acyl-carnitines observed in the SIRT5KO TAC heart compared with the SIRT5KO sham heart was not due to differences in transcriptional regulation. In contrast, consistent with other sirtuin knock-out models, several enzymes in the fatty acid oxidation pathway were hyperacylated (Fig. 5E). Thus, fatty acid oxidation might be impaired in this model due to changes in protein acylation of the oxidation machinery directly. However, analyses of the metabolomic data sets revealed additional biochemical abnormalities in the SIRT5KO TAC heart compared with the SIRT5KO sham heart.
Because glucose is the other main fuel metabolized by the heart, we inspected shifts in metabolites of glucose oxidation in the WT and SIRT5KO hearts. We found a trend toward increased glycolytic metabolites (Fig. 6A) and pentose phosphate pathway metabolites (Fig. 6B), suggesting an increase in glycolysis and pentose phosphate pathway flux. Importantly, the metabolic shifts observed here are consistent with several models of cardiac hypertrophy (28).
Figure 6.

Glucose oxidation is uncoupled from glycolysis in SIRT5KO TAC hearts. Metabolite profiles of glycolytic metabolites (A), pentose phosphate metabolites (B), and TCA cycle metabolites (C). Values are mean ± S. E. D, Western blot of phospho-PDH and total PDH. E, quantification of blot, normalized to total protein (boxplots). F, ratio of lactate to pyruvate normalized to WT sham (boxplots). G, Glut4, Glut1, Ldha, and Pdk4 relative to 36B4. Values are mean ± S. E. (error bars) n = 3 for WT sham, n = 5 for SIRT5KO sham, n = 4 for WT TAC, and n = 7 for SIRT5KO TAC. Mice were males and were 18–19 weeks of age at the end of the study. Two-way ANOVA with multiple comparisons, Bonferroni correction, was performed. *, p ≤ 0.05; effect of TAC if no significance in multiple comparisons tests. +, p ≤ 0.05 (panels A–G). Shown is -fold change of all succinylated peptides identified on enzymes in glycolysis (H), and TCA cycle (I). The horizontal maroon bar represents the mean -fold change of all peptides identified on a single protein. n = 3 for wild type and n = 3 for SIRT5KO. Mice were males and were 11 weeks of age (panels H–I).
Next, we sought to determine whether a signature of elevated glucose metabolism corresponded with an increase in TCA cycle metabolites, as would be expected for complete glucose oxidation. However, TCA cycle metabolites downstream of pyruvate were generally reduced in response to TAC (Fig. 6C) and malate was further decreased in the SIRT5KO hearts; together, the directionality of these metabolic shifts suggests that glucose-derived carbon is not entering the TCA cycle and further suggests a mismatch between TCA cycle anaplerosis and cataplerosis. To further characterize regulation of carbon entry into the TCA cycle, we examined the phosphorylation of PDH because this is a primary control mechanism for pyruvate entry into the TCA cycle. Phosphorylation of PDH at Ser-293 was increased in the SIRT5KO TAC heart compared with the SIRT5KO sham condition (Fig. 6, D and E), a mark indicative of uncoupled glycolysis from glucose oxidation (33) via less PDH activity (34). Supporting these findings, the lactate/pyruvate ratio in the SIRT5KO TAC heart compared with the SIRT5KO sham heart was elevated (Fig. 6F), suggesting that glucose carbon is shunted from elevated glycolysis into lactate and not pyruvate.
We again interrogated known mechanisms of glycolytic and TCA cycle control. RT-qPCR of key genes involved in glucose metabolism showed that Glut1 (the main glucose transporter in the hypertrophic heart) is modestly increased in the SIRT5KO TAC heart compared with the SIRT5KO sham condition (Fig. 6G); however, differences in transcription were subtle. Interestingly, succinyl-proteomics indicated that few enzymes in glycolysis were hypersuccinylated (Fig. 6H), whereas, like other oxidative pathways, enzymes in the TCA cycle were hypersuccinylated in the SIRT5KO heart (Fig. 6I). Most of the glycolytic enzymes identified had an average log2 -fold change less than zero, which indicated overall hyposuccinylation of glycolytic proteins in SIRT5KO hearts compared with WT. Together, these data support the overall notion that hypersuccinylation occurs primarily on enzymes in mitochondrial oxidative pathways. Overall, our findings demonstrate that although flux through glycolysis appears elevated in both WT and SIRT5KO TAC hearts, complete glucose oxidation is reduced and TCA cycle carbon is lower in the SIRT5KO TAC heart.
Given that both decreases in fatty acid oxidation and TCA cycle metabolism are observed in SIRT5KO TAC hearts, we hypothesized that overall mitochondrial oxidative metabolism might be impaired. To assess the mitochondrial redox state in WT and SIRT5KO TAC hearts, we extracted the β-hydroxybutyrate/acetoacetate ratio from the metabolomic data set, because it is a reliable indicator of the mitochondrial NAD+/NADH ratio (35). The SIRT5KO TAC mice have a strong trend of decreased β-hydroxybutyrate/acetoacetate ratio (BOHB/Acac) (p = 0.110) (Fig. 7A), indicative of increased mitochondrial NADH. Increased NADH could be due to lower NADH oxidation and OXPHOS activity. To assess the status of OXPHOS in the WT and SIRT5KO hearts, we first measured representative OXPHOS complex subunits by Western blotting and observed equal amounts of mitochondrial protein content between the WT and SIRT5KO hearts (Fig. 7B), suggesting that complex formation is normal. This is consistent with previously published findings in SIRT5KO livers (10).
Figure 7.

Oxidative phosphorylation may be a primary lesion of impaired oxidative metabolism. A, ratio of β-hydroxybutyrate to acetoacetate normalized to WT sham (box plots). n = 3 for WT sham, n = 5 for SIRT5KO sham, n = 4 for WT TAC, and n = 7 for SIRT5KO TAC. Mice were males and were 18–19 weeks of age at the end of the study. Two-way ANOVA with multiple comparisons, Bonferroni correction, was performed. B, Western blot of OXPHOS mixture antibody in WT and SIRT5KO heart tissue. C, -fold change of all succinylated peptides identified on enzymes in oxidative phosphorylation. The horizontal maroon bar represents the mean -fold change of all peptides identified on a single protein. n = 3 for wild type, and n = 3 for SIRT5KO. Mice were males and were 11 weeks of age. D, working model. Cardiac hypertrophy is accompanied by shifts in metabolism, including increased glycolysis and decreased fatty acid oxidation, that ultimately fail to meet the energetic requirements of the heart (left). In the SIRT5KO heart (right), the conical shifts in metabolism are observed with hypertrophy; however, there is an earlier reduction in oxidative metabolism. Succinylation of oxidative pathways may contribute to this phenotype. We suggest that impairment of OXPHOS leads to inhibition of glucose oxidation via phosphorylation of PDH and subsequent decreases in the TCA cycle and fatty acid oxidation. Both pathways appear to be more decreased in the SIRT5KO hypertrophic heart compared with WT hypertrophic heart. This ultimately results in an energetic stress that leads to early death in the SIRT5KO mouse after TAC. Error bars, S.E.
Interestingly, the SIRT5KO hearts appear to be in an energetically stressed state, as shown by AMPK phosphorylation (supplemental Fig. 2). Therefore, we propose a model where that electron flow through OXPHOS is impaired in the absence of SIRT5. Consistent with hypersuccinylation of other metabolic pathways, we found that many protein subunits of OXPHOS contained multiple sites of lysine succinylation, with many having at least one site of succinylation with a -fold change of >20 in SIRT5KO hearts compared with WT (Fig. 7C). Further supporting this model, impairments in OXPHOS could explain a lower NAD+/NADH; given the energetic observations and metabolite profiles, we conclude that mitochondrial dehydrogenase activity is not responsible for the reductive mitochondrial environment.
Overall, our data reveal metabolic abnormalities in fatty acid oxidation and TCA cycle function, which are coincident with protein hyperacylation in SIRT5KO hearts under chronic stress (Fig. 7D). These metabolic and biochemical deficiencies probably lead to an energetic crisis in the heart, which could explain the increased mortality in SIRT5KO mice with pressure overload.
Discussion
The overall goal of this study was to define the major role of SIRT5 in cardiac metabolism by exposing whole-body SIRT5KO mice to the cardiac stress of chronic pressure overload and the subsequent cardiac hypertrophic response. Taken together, our data support a model where SIRT5 is required to maintain oxidative metabolism under chronic pressure overload; we discovered that ablation of SIRT5 results in significantly decreased survival upon TAC. Our data further demonstrate that reduced survival is due to accelerated progression of cardiac dysfunction, as evidenced by more pronounced cardiac systolic and diastolic dysfunction and metabolic remodeling in the SIRT5KO mice that is consistent with a trajectory of cardiac dysfunction. The mechanistic basis for these functional deficits could be explained by two discrete but non-mutually exclusive models.
First, our data, consistent with historical data, show that hypersuccinylation occurs on key enzymes in oxidative pathways. As such, hyperacylation could inhibit metabolic pathways that are required for energy generation under cardiac stress. Widespread hyperacylation across several enzymes and metabolic pathways could coordinately alter cardiac energy generation, thereby accelerating maladaptive alterations in response to pressure overload–induced hypertrophy and ultimately heart failure. Supporting this model, we investigated the proteomic profile of oxidative pathways, including fatty acid oxidation (Fig. 5), the TCA cycle (Fig. 6), and OXPHOS (Fig. 7), and found that proteins in these pathways had multiple sites of lysine succinylation. Some previous examples show that hypersuccinylation increases enzyme activity (7, 9), whereas other studies show that lysine acylation more often decreases enzyme activity (36) and impairs flux through metabolic pathways; together, hyperacylation could reroute carbon metabolism away from an oxidative, energy-producing state. Thus, in this model, multiple succinylation events across multiple proteins contribute to the metabolic and cardiac phenotypes.
Alternatively, our data also support a new model, in which a primary defect in OXPHOS in the SIRT5KO mouse heart leads to an elevation in mitochondrial NADH (reduced form). This proximal metabolic lesion in OXPHOS, probably by hypersuccinylation of multiple enzymes in OXPHOS and impaired electron flow through this pathway, coordinately alters mitochondrial oxidative metabolism (Fig. 7D). Supporting this model, SIRT5 was recently described to regulate oxidative phosphorylation via its desuccinylase activity in liver (10), which identified the electron transport chain as the top pathway targeted by SIRT5 and further found that complex II and ATP synthase activities were significantly decreased in mitochondria isolated from SIRT5KO mouse liver.
If these studies hold true in SIRT5KO hearts, impairment of OXPHOS in the SIRT5KO TAC heart could explain observations of metabolic derangements that occurred upstream of OXPHOS in this model. Specifically, a lower NAD+/NADH ratio (elevated NADH) activates pyruvate dehydrogenase kinase, which increases phosphorylation of PDH and inhibits its activity (Fig. 6, D and E). Inhibition of PDH will lead to an increase in lactate formation (Fig. 6F) and a concomitant decrease in the TCA cycle carbon in the SIRT5KO TAC mouse. Furthermore, because the rate of fatty acid oxidation is dependent on the rate of supply of TCA carbon (37), this model could also explain the decrease in fatty acid oxidation observed in the SIRT5KO TAC condition (Fig. 5, B and C). Although NAD+/NADH is decreased in the SIRT5KO TAC heart and may impact the activity of other mitochondrial sirtuins, acetylation does not increase in the SIRT5KO TAC condition (Fig. 4G), suggesting that SIRT3 activity may not be affected. However, the accumulation of metabolic intermediates (i.e. acetyl-CoA or succinyl-CoA) is known to drive protein lysine acylation (27, 38), and acetyl-CoA trends toward decreased in the SIRT5KO TAC compared with WT TAC condition (Fig. 6C). This is consistent with decreased PDH activity (Fig. 6D) and may explain the absence of increased acetylation even if SIRT3 is inactive.
Overall, this alternative model posits that a primary node of impaired oxidative metabolism could occur at OXPHOS, which ultimately causes an energetic crisis and explains the decreased survival in SIRT5KO mice with chronic pressure overload. Further supporting this concept, inborn errors of metabolism that affect the respiratory chain often cause decreased mitochondrial NAD+/NADH and increased glycolytic flux, which results in an increased lactate/pyruvate ratio and a clinical presentation of hypertrophic cardiomyopathy (39). However, given the multifaceted roles of NAD+ (40), a major challenge moving forward will be to disentangle these two competing models and the contribution of lysine acylation/sirtuin activity and mitochondrial redox state to alterations in metabolism (41).
Regardless of the model, we would expect an accelerated trajectory of cardiac dysfunction in the setting of impaired oxidative metabolism, which often precedes overt heart failure (33). Shifts in metabolism are well characterized in models of pressure overload–induced cardiac hypertrophy. Under normal conditions, most ATP (70–90%) produced in the heart is from the oxidation of fatty acids (28). However, with development of hypertrophy, the contribution of fatty acids to cardiac ATP is decreased, whereas increased rates of glycolysis contribute more ATP than non-hypertrophied hearts (42, 43). Eventually, the metabolic adaptations are not sufficient to maintain cardiac energetics as oxidative metabolism continues to decline and cardiac dysfunction progresses. Consistent with previous observations, both WT and SIRT5KO hearts showed expected shifts in metabolism with TAC (increased glycolysis and decreased fatty acid oxidation), with evidence of accelerated defects in oxidative metabolism uniquely in the SIRT5KO TAC condition (Figs. 5–7). Importantly, uncoupling of glycolysis from glucose oxidation, as observed in the SIRT5KO TAC animals, can cause decreased cardiac efficiency and is detrimental to heart function (33).
The most salient finding presented here is that upon chronic TAC-induced cardiac stress, SIRT5KO mice have significantly reduced survival compared with WT mice (Fig. 1A). After 4 weeks of TAC, SIRT5KO hearts had increased hypertrophy compared with WT hearts and had greater impairment of systolic function compared with WT mouse hearts (Tables 1 and 2 and Fig. 3). The apparent discordance between the observed subtle functional defects and significant mortality may be explained by survivor (or survivorship) bias (44). Over half of the SIRT5KO TAC mice had already died at the 4-week post-TAC time point, making the surviving group of animals more homogeneous and the cause of future mortality more difficult to predict (45). Characterizing cardiac function at multiple early time points after the TAC procedure and/or stratifying mice based on cardiac function may be required to detect a marked defect in cardiac function in the SIRT5KO mice. Finally, it is possible that the phenotypes described here are due to cell-non-autonomous roles of SIRT5 (i.e. cells or tissues beyond the cardiomyocyte). Indeed, a role for SIRT3 in preventing fibrosis by activity in fibroblasts has been described recently (46). A tissue-specific SIRT5KO mouse model will be required to determine the cardiomyocyte-intrinsic roles of SIRT5.
Together, our results suggest a model in which the SIRT5KO mouse has accelerated development of pressure overload–induced cardiomyopathy (Fig. 7D). We predict that hypersuccinylation of oxidative pathways, especially OXPHOS, reduces energy production in SIRT5KO mouse hearts under TAC. However, survivor bias makes it difficult to uncover the molecular cause of mortality. More frequent monitoring of cardiac function may be required to more accurately define the mechanistic step(s) preceding mortality. Overall, this study points to an accelerated cardiac dysfunction in the SIRT5KO TAC animals as an explanation for the increased mortality observed with chronic TAC. Future studies on SIRT5 will identify how succinylation of mitochondrial proteins influences energy homeostasis in response to cardiac stress.
Experimental procedures
Animals
SIRT5KO mice were obtained from Jackson Laboratory (Bar Harbor, ME) (stock number 012757) and backcrossed for 10 generations onto a C57BL/6J background obtained from Jackson Laboratory (stock number 000664). Mice backcrossed for 7–10 generations were used in these studies and had a mixed NNT background. Mice were group-housed on a 12-h light/dark cycle with free access to water and PicoLab Rodent Diet 20 (LabDiet (St. Louis, MO) catalog no. 5053). Age, sex, genotype, and number of animals used per study are provided in the appropriate figure legends. All in vivo procedures were performed on healthy animals in accordance with the Duke institutional animal care and use program.
Western blotting
Commercial antibodies were used as follows: anti-PDH E1α1 subunit from Cell Signaling (3205S), anti-phospho-PDH from Calbiochem (AP1062), anti-succinyl-lysine from PTM (catalog no. 401), anti-malonyl-lysine from Cell Signaling (catalog no. 14942), anti-acetyl-lysine from Cell Signaling (catalog no. 9441), anti-glutaryl-lysine from Cell Signaling (generous gift), anti-acetyl-CoA carboxylase from Cell Signaling (catalog no. 3662), anti-phospho-acetyl-CoA carboxylase (Ser-79) from Cell Signaling (catalog no. 3661), anti-AMPKα from Cell Signaling (catalog no. 2603), anti-phospho-AMPKα (Thr-172) from Cell Signaling (catalog no. 2535), anti-total OXPHOS mixture from MitoSciences (ab110413), anti-4EBP1 from Cell Signaling (9644S), anti-phospho-4EBP1 from Cell Signaling (2855S), anti-CaMKII-α from Cell Signaling (D10C11), and anti-phospho-CaMKII-α from Cell Signaling (D21E4). Anti-SIRT5 was provided as a generous gift from Leonard Guarente. Whole-cell protein extracts were resolved by SDS-PAGE using stain-free Bio-Rad gels and transferred to nitrocellulose membranes using Bio-Rad's Trans-Blot Turbo. Total protein was quantified with the GelDoc using stain-free technology. The membranes were blocked in 5% milk in TBS-T (TBS containing 0.1% Tween 20) for 1 h at room temperature and probed with primary antibodies in TBS-T. For anti-acyl-lysine blots, 3% BSA was added to the primary antibody solution. After incubation with infrared dye–conjugated antibodies, the blots were developed using the LI-COR Odyssey Infrared Imaging System.
RT-qPCR
RNA was extracted from tissues using the RNeasy minikit from Qiagen (catalog no. 74106). cDNA was made from 750 ng of RNA using the iScript cDNA synthesis kit from Bio-Rad (catalog no. 170-8890). Amplification was performed using iTaq Universal SYBR Green Supermix from BioRAD (catalog no. 1725121) on the QuantStudio 6 Flex (ThermoFisher Scientific). 36B4 was used as a reference gene, and relative expression was calculated using the ΔΔCt method.
Generation of pressure overload and serial echocardiography
Pressure overload in mice was induced using methods described previously (19), except that the suture was placed between the left carotid and the left axillary arteries. Serial echocardiography was performed on conscious mice from all groups with a Vevo 2100 high-resolution imaging system (VisualSonics) as described previously. Mice underwent either a sham or pressure overload procedure in a non-randomized fashion.
PV loop analysis
In vivo pressure-volume analysis was performed as described previously (47). Briefly, after bilateral vagotomy, the chest was opened, and the pericardium was dissected to expose the heart. A 7-0-suture ligature was placed around the transverse aorta to manipulate loading conditions. A 1.4-Fr pressure-conductance catheter (Millar Instruments, Houston, TX) was inserted retroaortically into the LV to record hemodynamics. Baseline hemodynamic parameters were obtained once the catheter recordings had achieved steady state, usually 3–5 min after conductance catheter placement. Load-independent parameters were established by generating a series of PV loops with decreasing preload through transient constriction of the inferior vena cava. Subsequently, parallel conductance was determined by a 10-μl injection of 15% saline into the right jugular vein to establish the parallel conductance of the blood pool. The derived parallel conductance was used to correct the PV loop data. Data were recorded digitally at 1000 Hz and analyzed with pressure-volume analysis software (PVAN data analysis software version 3.3, Millar Instruments) as described previously (47). Mice that died after receiving anesthesia or became hypotensive during the protocol, suggesting a surgical complication, were excluded. Pressure overload mice with a pressure gradient of <20 mm Hg were excluded from the study.
Metabolite extraction
10–20 mg of frozen crushed heart tissue was weighed in an Eppendorf tube, and 200 μl of ice-cold 80% methanol was added. A glass bead was added and was homogenized using the TissueLyzer for 2 min at 30 Hz. 300 μl of ice-cold 80% methanol was added, vortexed, and incubated on ice for 10 min. Tissue extract was centrifuged at 20,000 × g at 4 °C for 10 min. Supernatant containing 2 mg of tissue was transferred to a new Eppendorf tube and dried in a vacuum concentrator at room temperature. The dry pellets were reconstituted into 30 μl (per 2 mg of tissue) of sample solvent (water/methanol/acetonitrile, 2:1:1, v/v/v), and 3 μl was further analyzed by liquid LC-MS.
LC-MS method
An Ultimate 3000 UHPLC (Dionex) was coupled to a Q Exactive Plus-mass spectrometer (QE-MS, Thermo Scientific) for metabolite profiling. A hydrophilic interaction chromatography method employing an Xbridge amide column (100 × 2.1 mm inner diameter, 3.5 μm; Waters) was used for polar metabolite separation. The detailed LC method was described previously (48) except that mobile phase A was replaced with water containing 5 mm ammonium acetate (pH 6.8). The QE-MS is equipped with a HESI probe with related parameters set as follows: heater temperature, 120 °C; sheath gas, 30; auxiliary gas, 10; sweep gas, 3; spray voltage, 3.0 kV for the positive mode and 2.5 kV for the negative mode; capillary temperature, 320 °C; S-lens, 55; scan range (m/z), 70–900 for positive mode (1.31–12.5 min) and negative mode (1.31–6.6 min) and 100–1000 for negative mode (6.61–12.5 min); resolution, 70,000; automated gain control, 3 × 106 ions. Customized mass calibration was performed before data acquisition.
Metabolomics data analysis
LC-MS peak extraction and integration were performed using commercially available software, Sieve version 2.2 (Thermo Scientific). The peak area was used to represent the relative abundance of each metabolite in different samples. The missing values were handled as described in a previous study (48). Data were uploaded to Metaboanalyst for further downstream analysis (29, 30).
Proteomics sample preparation
Samples were analyzed using the PTMScan method as described previously (49, 50). Cellular extracts were prepared in urea lysis buffer, sonicated, centrifuged, reduced with DTT, and alkylated with iodoacetamide. 15 mg of total protein for each sample was digested with trypsin and purified over C18 columns for enrichment with the succinyl-lysine motif antibody (catalog no. 13764). Enriched peptides were purified over C18 STAGE tips (51). Enriched peptides were subjected to secondary digest with trypsin and second STAGE tip before LC-MS/MS analysis.
LC-MS/MS method
Replicate injections of each sample were run non-sequentially for each enrichment. Peptides were eluted using a 120-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nl/min. Tandem mass spectra were collected in a data-dependent manner with an LTQ Orbitrap VELOS mass spectrometer running XCalibur version 2.0.7 SP1 using a top 20 MS/MS method, a dynamic repeat count of one, and a repeat duration of 30 s. Real-time recalibration of mass error was performed using lock mass (52) with a singly charged polysiloxane ion m/z = 371.101237.
Proteomics data analysis
MS/MS spectra were evaluated using SEQUEST and the Core platform from Harvard University (53–55). Files were searched against the NCBI Mus musculus FASTA database updated on April 29, 2015. A mass accuracy of ±5 ppm was used for precursor ions and 1.0 Da for product ions. Enzyme specificity was limited to trypsin, with at least one LysC or tryptic (Lys- or Arg-containing) terminus required per peptide and up to four miscleavages allowed. Cysteine carboxamidomethylation was specified as a static modification, and oxidation of methionine and succinylation on lysine residues were allowed as variable modifications. Reverse decoy databases were included for all searches to estimate false discovery rates and filtered using a 5% false discovery rate in the linear discriminant module of Core. Peptides were also manually filtered using a ±5-ppm mass error range and reagent-specific criteria. Results were filtered to include only peptides containing at least one succinyl-lysine residue. All quantitative results were generated using Progenesis version 4.1 (Waters Corp.) to extract the integrated peak area of the corresponding peptide assignments. Accuracy of quantitative data was ensured by manual review in Progenesis or in the ion chromatogram files.
Statistical analysis
Statistical analysis for all studies was performed using Prism version 6. Grubb's test was used to determine outliers. Outliers were removed from data. For studies with four conditions, a two-way ANOVA with multiple comparisons was performed. Bonferroni correction was used to correct for multiple comparisons. For studies with only WT and SIRT5KO comparisons, Student's t test was performed.
Author contributions
K. A. H., D. M. A., and M. D. H. conceptualized the work. A. S. M., H. G., and J. W. L. were responsible for methodology. K. A. H., D. M. A., L. M., J. L., and H. G. conducted the investigation. K. A. H. and M. D. H. wrote the original draft of the manuscript. All authors wrote, reviewed, and edited the final manuscript. J. W. L. and M. D. H. supervised the work. M. D. H. administered the project and acquired funding.
Supplementary Material
Acknowledgments
We thank Kelsey Fisher-Wellman and also Howard Rockman and other members of the dissertation committee of K. A. H. for feedback on this work. The Duke University Pharmacological Sciences Training Program was supported by Training Grant 5T32GM007105-40 from the NIGMS, the National Institutes of Health.
This work was supported by American Heart Association Grants 12SDG8840004 and 12IRG9010008; NIA, National Institutes of Health, Grant R01AG045351; and a pilot grant from the Duke O'Brien Center for Kidney Research supported by NIDDK, National Institutes of Health, Grant P30DK096493. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table 1 and Figs. 1 and 2.
- BCAA
- branched-chain amino acid
- TCA
- tricarboxylic acid
- OXPHOS
- oxygen phosphorylation
- PDH
- pyruvate dehydrogenase
- SDH
- succinate dehydrogenase
- HMGCS2
- 3-hydroxy-3-methlyglutaryl-CoA synthase 2
- TAC
- transverse aortic constriction
- EDD
- end-diastolic diameter
- ESD
- end-systolic diameter
- LV
- left ventricle
- PVA
- pressure-volume area
- IPA
- Ingenuity Pathway Analysis
- qPCR
- quantitative PCR
- ANOVA
- analysis of variance
- PV
- pressure-volume.
References
- 1. Hirschey M. D., and Zhao Y. (2015) Metabolic regulation by lysine malonylation, succinylation and glutarylation. Mol. Cell. Proteomics 14, 2308–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson K. A., Green M. F., Huynh F. K., Wagner G. R., and Hirschey M. D. (2014) SnapShot: mammalian sirtuins. Cell 159, 956–956.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., et al. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., Zwaans B. M. M., Tishkoff D., Ho L., Lombard D., He T. C., et al. (2011) The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell Protomics 10.1074/mcp.M111.012658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., et al. (2014) Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19, 605–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson K. A., Huynh F. K., Fisher-Wellman K., Stuart J. D., Peterson B. S., Douros J. D., Wagner G. R., Thompson J. W., Madsen A. S., Green M. F., Sivley R. M., Ilkayeva O. R., Stevens R. D., Backos D. S., Capra J. A., et al. (2017) SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 25, 838–855.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M. M., Skinner M. E., Lombard D. B., and Zhao Y. (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell. 50, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rardin M. J., He W., Nishida Y., Newman J. C., Carrico C., Danielson S. R., Guo A., Gut P., Sahu A. K., Li B., Uppala R., Fitch M., Riiff T., Zhu L., Zhou J., et al. (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boylston J. A., Sun J., Chen Y., Gucek M., Sack M. N., and Murphy E. (2015) Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 88, 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y., Bharathi S. S., Rardin M. J., Lu J., Maringer K. V., Sims-Lucas S., Prochownik E. V., Gibson B. W., and Goetzman E. S. (2017) Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 292, 10239–10249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishida Y., Rardin M. J., Carrico C., He W., Sahu A. K., Gut P., Najjar R., Fitch M., Hellerstein M., Gibson B. W., and Verdin E. (2015) SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell. 59, 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakamura Y., Ogura M., Ogura K., Tanaka D., and Inagaki N. (2012) SIRT5 deacetylates and activates urate oxidase in liver mitochondria of mice. FEBS Lett. 586, 4076–4081 [DOI] [PubMed] [Google Scholar]
- 13. Polletta L., Vernucci E., Carnevale I., Arcangeli T., Rotili D., Palmerio S., Steegborn C., Nowak T., Schutkowski M., Pellegrini L., Sansone L., Villanova L., Runci A., Pucci B., Morgante E., et al. (2015) SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 11, 253–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sadhukhan S., Liu X., Ryu D., Nelson O. D., Stupinski J. A., Li Z., Chen W., Zhang S., Weiss R. S., Locasale J. W., Auwerx J., and Lin H. (2016) Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. U.S.A. 113, 4320–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu J., Sadhukhan S., Noriega L. G., Moullan N., He B., Weiss R. S., Lin H., Schoonjans K., and Auwerx J. (2013) Metabolic characterization of a Sirt5 deficient mouse model. Sci. Rep. 3, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abel E. D., and Doenst T. (2011) Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 90, 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dorn G. W., 2nd (2007) The fuzzy logic of physiological cardiac hypertrophy. Hypertension 49, 962–970 [DOI] [PubMed] [Google Scholar]
- 18. Frey N., and Olson E. N. (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79 [DOI] [PubMed] [Google Scholar]
- 19. Rockman H. A., Ross R. S., Harris A. N., Knowlton K. U., Steinhelper M. E., Field L. J., Ross J. Jr., and Chien K. R. (1991) Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 88, 8277–8281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ruwhof C., and van der Laarse A. (2000) Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc. Res. 47, 23–37 [DOI] [PubMed] [Google Scholar]
- 21. van Oort R. J., Respress J. L., Li N., Reynolds C., De Almeida A. C., Skapura D. G., De Windt L. J., and Wehrens X. H. T. (2010) Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension 55, 932–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lamba S., and Abraham W. T. (2000) Alterations in adrenergic receptor signaling in heart failure. Heart Fail. Rev. 5, 7–16 [DOI] [PubMed] [Google Scholar]
- 23. Erickson J. R. (2014) Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engelhardt S., Böhm M., Erdmann E., and Lohse M. J. (1996) Analysis of β-adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: progressive reduction of β1-adrenergic receptor mRNA in heart failure. J. Am. Coll. Cardiol. 27, 146–154 [DOI] [PubMed] [Google Scholar]
- 25. Karamanlidis G., Lee C. F., Garcia-Menendez L., Kolwicz S. C. Jr., Suthammarak W., Gong G., Sedensky M. M., Morgan P. G., Wang W., and Tian R. (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin A. S., Abraham D. M., Hershberger K. A., Bhatt D. P., Mao L., Cui H., Liu J., Liu X., Muehlbauer M. J., Grimsrud P. A., Locasale J. W., Payne R. M., and Hirschey M. D. (2017) Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich's ataxia cardiomyopathy model. JCI Insight 10.1172/jci.insight.93885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horton J. L., Martin O. J., Lai L., Riley N. M., Richards A. L., Vega R. B., Leone T. C., Pagliarini D. J., Muoio D. M., Bedi K. C. Jr., Margulies K. B., Coon J. J., and Kelly D. P. (2016) Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 10.1172/jci.insight.84897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doenst T., Nguyen T. D., and Abel E. D. (2013) Cardiac metabolism in heart failure: implications beyond ATP production. Circ. Res. 113, 709–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xia J., Sinelnikov I. V., Han B., and Wishart D. S. (2015) MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia J., and Wishart D. S. (2016) Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinformatics 55, 14.10.1–14.10.91 [DOI] [PubMed] [Google Scholar]
- 31. Akki A., Smith K., and Seymour A.-M. L. (2008) Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol. Cell Biochem. 311, 215–224 [DOI] [PubMed] [Google Scholar]
- 32. Riehle C., and Abel E. D. (2012) PGC-1 proteins and heart failure. Trends Cardiovasc. Med. 22, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sankaralingam S., and Lopaschuk G. D. (2015) Cardiac energy metabolic alterations in pressure overload-induced left and right heart failure (2013 Grover Conference Series). Pulm. Circ. 5, 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rardin M. J., Wiley S. E., Naviaux R. K., Murphy A. N., and Dixon J. E. (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 389, 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krebs H. A., Williamson D. H., Bates M. W., and Page M. A. (1971) The role of ketone bodies in caloric homeostasis. Adv. Enzyme Regul. 10.1016/S0065-2571(71)80055-9 [DOI] [Google Scholar]
- 36. Chhoy P., Anderson K. A., Hershberger K. A., Huynh F. K., Martin A. S., Peterson E. M. B. S., Starzenski L. A., Backos D. S., Fritz K. S., and Hirschey M. D. (2016) Deacetylation by SIRT3 relieves inhibition of mitochondrial protein function. In Sirtuins (Houtkooper R. H., ed) pp. 105–138, Springer, New York [Google Scholar]
- 37. Fritz I. B. (1961) Factors influencing the rates of longchain fatty acid oxidation and synthesis in mammalian systems. Physiol. Rev. 41, 52–129 [DOI] [PubMed] [Google Scholar]
- 38. Wagner G. R., Bhatt D. P., O'Connell T. M., Thompson J. W., Dubois L. G., Backos D. S., Yang H., Mitchell G. A., Ilkayeva O. R., Stevens R. D., Grimsrud P. A., and Hirschey M. D. (2017) A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab. 25, 823–837.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das A. M., and Illsinger S. (2011) Inborn errors of metabolism and the heart in Procedings of the World Medical Conference, Prague 2011, pp. 135–141, WSEAS, Czech Republic [Google Scholar]
- 40. Hershberger K. A., Martin A. S., and Hirschey M. D. (2017) Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 13, 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee C. F., Chavez J. D., Garcia-Menendez L., Choi Y., Roe N. D., Chiao Y. A., Edgar J. S., Goo Y. A., Goodlett D. R., Bruce J. E., and Tian R. (2016) Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134, 883–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allard M. F., Schönekess B. O., Henning S. L., English D. R., and Lopaschuk G. D. (1994) Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 267, H742–H750 [DOI] [PubMed] [Google Scholar]
- 43. Kolwicz S. C. Jr., and Tian R. (2011) Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 90, 194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shermer M. (2014) How the survivor bias distorts reality. Sci. Am. 311, 94. [PubMed] [Google Scholar]
- 45. Zhang W. B., and Pincus Z. (2016) Predicting all-cause mortality from basic physiology in the Framingham Heart Study. Aging Cell 15, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sundaresan N. R., Bindu S., Pillai V. B., Samant S., Pan Y., Huang J.-Y., Gupta M., Nagalingam R. S., Wolfgeher D., Verdin E., and Gupta M. P. (2015) SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol. Cell. Biol. 36, 678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoo B., Lemaire A., Mangmool S., Wolf M. J., Curcio A., Mao L., and Rockman H. A. (2009) β1-Adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 297, H1377–H1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu X., Ser Z., and Locasale J. W. (2014) Development and quantitative evaluation of a high-resolution metabolomics technology. Anal. Chem. 86, 2175–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X.-M., Polakiewicz R. D., and Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 50. Stokes M. P., Farnsworth C. L., Gu H., Jia X., Worsfold C. R., Yang V., Ren J. M., Lee K. A., and Silva J. C. (2015) Complementary PTM profiling of drug response in human gastric carcinoma by immunoaffinity and IMAC methods with total proteome analysis. Proteomes 3, 160–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 52. Olsen J. V., de Godoy L. M. F., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., and Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 53. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 54. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Villén J., Beausoleil S. A., Gerber S. A., and Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.