

Abstract
The pyridine-based multimerization selective HIV-1 integrase (IN) inhibitors (MINIs) are a distinct subclass of allosteric IN inhibitors. MINIs potently inhibit HIV-1 replication during virion maturation by inducing hyper- or aberrant IN multimerization but are largely ineffective during the early steps of viral replication. Here, we investigated the mechanism for the evolution of a triple IN substitution (T124N/V165I/T174I) that emerges in cell culture with a representative MINI, KF116. We show that HIV-1 NL4-3(IN T124N/V165I/T174I) confers marked (>2000-fold) resistance to KF116. Two IN substitutions (T124N/T174I) directly weaken inhibitor binding at the dimer interface of the catalytic core domain but at the same time markedly impair HIV-1 replication capacity. Unexpectedly, T124N/T174I IN substitutions inhibited proteolytic processing of HIV-1 polyproteins Gag and Gag-Pol, resulting in immature virions. Strikingly, the addition of the third IN substitution (V165I) restored polyprotein processing, virus particle maturation, and significant levels of replication capacity. These results reveal an unanticipated role of IN for polyprotein proteolytic processing during virion morphogenesis. The complex evolutionary pathway for the emergence of resistant viruses, which includes the need for the compensatory V165I IN substitution, highlights a relatively high genetic barrier exerted by MINI KF116. Additionally, we have solved the X-ray structure of the drug-resistant catalytic core domain protein, which provides means for rational development of second-generation MINIs.
Keywords: human immunodeficiency virus (HIV), integrase, protein aggregation, protein drug interaction, viral replication, HIV, allosteric inhibitors
Introduction
Allosteric HIV-12 integrase (IN) inhibitors (ALLINIs; also referred to as noncatalytic site integrase inhibitors (NCINIs) or LEDGINs) are a promising class of antiviral compounds that are currently undergoing clinical trials. The mechanism of action of these inhibitors has been actively investigated. Most efforts have focused on the archetypal quinoline-based ALLINIs, which exhibit a multimodal mechanism of action. They inhibit IN binding to its cognate cellular binding partner lens epithelium–derived growth factor (LEDGF)/p75 and induce hypermultimerization of inactive IN with similar potencies in vitro (1–6). In infected cells, quinoline-based compounds display antiviral activity during both the late and early steps of HIV-1 infection, but their full potency is attributable to inhibition of HIV-1 maturation (4, 7–10). They promote hypermultimerization of IN in virions (7–9) and impair its ability to bind the viral RNA genome (11). As a result, ALLINI-treated virions have ribonucleoprotein (RNP) complexes misplaced outside of the protective capsid core and are defective for subsequent reverse transcription in target cells (7, 8). Initial quinoline-based ALLINIs exhibited relatively low genetic barriers to resistance (1, 12). For example, the single A128T substitution in IN, which had no detectable effect on viral replication, conferred significant resistance to these inhibitors.
These findings have prompted structure–activity relationship-guided medicinal chemistry efforts to develop improved compounds, which has led to the discovery of novel ALLINIs containing a pyridine core (13, 14). Unlike the quinoline-based compounds, the pyridine-based inhibitors exhibit marked selectivity for promoting hypermultimerization of IN; however, they are substantially less effective for blocking IN-LEDGF/p75 binding in vitro (13). Furthermore, similar selectivity was seen in infected cells where the pyridine-based compounds potently inhibited virion maturation through inducing hypermultimerization of IN but were 3 orders of magnitude less active during the early steps of replication. Because of these distinct properties, pyridine-based compounds have been grouped into a separate subclass of ALLINIs that are termed multimerization selective IN inhibitors (MINIs) to delineate them from their multifunctional quinoline-based counterparts.
HIV-1 IN is composed of three domains including the N-terminal domain, catalytic core domain (CCD), and C-terminal domain (for a review, see Ref. 15). The CCD harbors an extensive dimerization interface (16), which is where LEDGF/p75 principally engages IN (17). ALLINIs and MINIs both bind within the LEDGF/p75-binding pocket of the CCD dimer (1, 3, 4, 6, 7, 12, 18, 19). Shared features of these inhibitor interactions include hydrogen bonding with one IN subunit where the carboxylic acid and t-butoxy groups common to both highly potent ALLINIs and MINIs hydrogen bond with backbone amides of IN residues Glu-170 and His-171 and the side chain of Thr-174. However, the two subclasses of inhibitors markedly differ in their interactions with the other IN subunit. The rigid and bulky quinoline core extends toward Ala-128, and the substitution of this amino acid with the larger polar Thr residue allows the virus to confer marked resistance to archetypal quinoline-based compounds (12). These shortcomings have been considered during the development of pyridine-based MINIs (13, 14). For example, KF116, a highly potent MINI, contains a compact pyridine central core, which is linked to a benzimidazole group through a rotatable bond (13). These changes allow KF116 to both more effectively bridge between two IN subunits as compared with their quinoline-based counterparts and avoid the steric effects generated by the A128T substitution. Consequently, KF116 remained active against HIV-1 NL4-3(IN A128T). Selection of drug resistance in the presence of increasing concentrations of KF116 led to the evolution of the triple mutant HIV-1 NL4-3(IN T124N/V165I/T174I), indicating that the pyridine-based compound exerted a greater extent of genetic pressure on the virus than earlier quinoline-based counterparts (13).
Here, we have investigated the mechanism for drug resistance to KF116. Our findings uncover a complex evolutionary pathway for the emergence of HIV-1 NL4-3(IN T124N/V165I/T174I), which includes the need for the compensatory V165I substitution in IN. In addition, we have uncovered an architectural role of IN in the context of Gag-Pol that affects viral protease (PR) activity and polyprotein protein processing during virion morphogenesis.
Results
We previously selected for KF116 resistance by repeatedly passaging HIV-1 in cell culture in the presence of increasing concentrations of the inhibitor over time (13). To examine the mechanism and evolutionary pathway for emergence of drug resistance, we studied the following mutant viruses: (i) HIV-1(IN T124N/V165I/T174I), which was the main viral species selected after 10 passages at the final concentration of ∼25 μm KF116; (ii) HIV-1(IN T124N/T174I), which was only a minor phenotype under the same conditions; and (iii) HIV-1(IN T124N), which was detected after five passages at a lower (∼10 μm) concentration of KF116 but disappeared with increasing concentrations of the inhibitor. Wild-type (WT) and IN mutant virions were produced by transfection of HEK293T cells with proviral plasmid DNA carrying the reporter gene for firefly luciferase (Luc), and levels of virus released into the cell supernatant were quantified by p24 ELISA. Each mutant yielded WT levels of virus particles under these conditions, suggesting that these IN substitutions did not affect virus particle production or release from cells (data not shown). Infectivity assays showed similar levels of Luc expression for WT and the single T124N IN mutant viruses, whereas the double T124N/T174I IN mutant virus was inactive (Fig. 1, A and B). Addition of the third V165I IN substitution allowed HIV-1 NL4-3(IN T124N/V165I/T174I) to exhibit ∼17% of WT levels of infectivity, which was ∼100-fold more infectious than HIV-1 NL4-3(IN T124N/T174I) (Fig. 1, A and B).
Figure 1.

Infectivity of drug-resistant HIV-1 NL4-3 variants. HEK293T cells were infected with the indicated luciferase reporter-HIV-1 (HIV-Luc). A, relative infectivity for the indicated amounts of the input viruses is shown with respect to 100 ng of WT HIV-Luc. Error bars indicate S.D. from three independent experiments. B, relative infectivity values for 100 ng of viruses.
We next assayed KF116 inhibitory profiles for the mutant viruses. The single and triple mutant viruses exhibited ∼300- and ∼2,000-fold resistance, respectively, whereas the double mutant virus was insufficiently infectious to measure KF116 potency. The triple mutant virus exhibited similarly high levels of cross-resistance with respect to quinoline-based inhibitors BI224436 and BI-D (Table 1).
Table 1.
Antiviral activities of IN inhibitors with respect to WT and drug resistant virus
All EC50 values were obtained under the same conditions
Previous data have shown that IN deletion constructs as well as several IN missense mutant viruses display alterations in HIV-1 particle morphology (7, 10, 20, 21). Therefore, we explored whether the KF116 resistance mutants could also affect HIV-1 particle morphology. WT and IN mutant virions produced from transfected HEK293T cells were processed for visualization by transmission electron microscopy (TEM). Under these conditions, the RNP in WT virions is often observed as a central electron density (ED), congruent with a conical capsid (CA) core (for a review, see Ref. 22). If sliced orthogonally, such particles would present a central, often circular ED as the ability to discern a distinct CA shell is obscured due to the plane of the microsection. To make this distinction, we have demarcated two types of particles with central ED: those with a discernible conical CA core and those where this distinction could not be made. In repeat experiments, ∼36% of WT virions harbored a conical core with central ED, whereas ∼47% harbored central ED without a discernible core structure (Fig. 2A; data from two independent experiments are quantified in Fig. 2E). Taken together, ∼83% of virions displayed central ED, which is fully consistent with our prior reports where both types of particles with central ED were lumped together (7, 10). Minor WT phenotypes included eccentric particles with the RNP visibly separate from a relatively ED-free (translucent) core structure as well immature particles with a characteristic outer ring ED. Of note, previously analyzed IN mutations accentuate the eccentric phenotype at the expense of particles with central ED (7, 10, 20, 21).
Figure 2.

Analysis of virion morphologies with TEM. The virions were prepared by transfecting full-length proviral clones into HEK293T cells. A–D, representative TEM images of WT HIV-1 NL4-3 (A), HIV-1 NL4-3(IN T124N) (B), HIV-1 NL4-3(IN T124N/T174I) (C), and HIV-1 NL4-3(IN T124N/V165I/T174I) (D) virions. Magnification, 20,000× (scale bar, 100 nm). E, quantification of virion morphologies from A–D from two independent experiments (average ± S.D. (error bars); number of counted virions from each sample in each experiment ranged from 99 to 639; brackets demarcate statistically significant comparisons using Student's t test analysis, two-tailed). Closed arrows indicate central ED with conical core, triangles indicate central ED without conical core, diamonds indicate immature virions, and open arrows indicate eccentric ED with translucent core.
The single T124N substitution in IN did not significantly alter the particle morphology frequencies observed for WT HIV-1 (Fig. 2, B and E). Unexpectedly, the addition of T174I to HIV-1 containing the T124N substitution yielded primarily immature particles (Fig. 2C). Strikingly, this phenotype reverted to virions displaying central ED by the addition of the V165I substitution (Fig. 2D). Although the total frequency of HIV-1 NL4-3(IN T124N/V165I/T174I) particles with central ED was similar to that of WT and HIV-1 NL4-3(IN T124N) virions, HIV-1 NL4-3(IN T124N/V165I/T174I) noticeably lacked the subclass with conical CA cores (Fig. 2E).
These findings prompted us to examine proteolytic processing of WT and IN mutant viral Gag and Gag-Pol precursor proteins (Fig. 3). Viral lysates separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were immunoblotted with antibodies against viral CA, matrix (MA), nucleocapsid (NC) (Fig. 3A), reverse transcriptase (RT) (Fig. 3B), IN (Fig. 3C), and PR (Fig. 3D), and the relative levels of these proteins in WT and mutant virions were quantitated (Fig. 3E). For each virus, a parallel control was analyzed in the presence of the PR inhibitor saquinavir (SQV), which afforded ready detection of Gag and Gag-Pol precursor proteins (Fig. 3, A–C). Consistent with the TEM analysis (Fig. 2), CA, MA, NC, RT, IN, and PR were similarly detected in the WT and T124N viral lysates. In sharp contrast, proteolytic processing was markedly impaired for the double mutant virus. Products corresponding to partial proteolytic cleavage products such as p40 (MA + CA + SP1) were detected in increased abundance as compared with WT, but the levels of fully processed CA, MA, NC, RT, IN, and PR were markedly reduced (Fig. 3, A–E). Consistent with the results of viral infectivity and TEM, the addition of the third V165I substitution significantly restored proteolytic processing activity (Fig. 3).
Figure 3.

Immunoblot analysis of Gag and Gag-Pol proteolytic processing products of WT HIV-1 NL4-3 and IN mutant virions. Virions were prepared by transfecting full-length proviral clones in HEK293T cells in the presence and absence of SQV. After normalizing with Gag p24 ELISA, the virions were subjected to SDS-PAGE. Gag polyclonal antibody was used to detect Gag, and its proteolytic processing products CA, MA, and NC (A). RT (B), IN (C), and PR (D) were detected using respective antibodies. Positions of mass standards are indicated to the left. The blots are representative of results obtained in two independent experiments. The band intensity for WT HIV-1 NL4-3 was considered 100%, and relative intensities for corresponding bands in mutant viruses were determined. E, quantitation of band intensity of two independent experiments is displayed with S.E. indicated by error bars.
To better understand the contributions of individual substitutions, we also analyzed mutant viruses containing single V165I or T174I IN substitutions (Fig. 4). The V165I IN virus displayed WT levels of infectivity and polyprotein proteolytic processing activity (Fig. 4, A–C). In contrast, the T174I IN substitution substantially reduced viral infectivity to ∼37% of the WT level (Fig. 4A). Furthermore, immunoblot analyses of HIV-1 NL4-3(IN T174I) particles revealed decreased amounts (by ∼25–60%) of fully processed CA, MA, and NC and substantially increased amounts (by ∼600%) of the partial proteolysis product p40 (MA + CA + SP1) as compared with WT virions (Fig. 4, B and C). These results suggest that the T174I substitution adversely affects the polyprotein proteolytic processing, contributing to the phenotype we observed in the context of the double T124N/T174I mutant virus (Figs. 1 and 2).
Figure 4.

Virology-based assays of V165I and T174I IN mutant viruses. A, infectivity of WT and single mutant viruses containing either V165I or T174I IN substitutions. Error bars indicate S.D. from three independent experiments. B, immunoblotting analysis of HIV-1 polyprotein processing products using Gag polyclonal antibodies. Positions of mass standards are indicated to the left. The blots are representative results obtained in two independent experiments. C, quantitation of partly processed intermediate p40 observed in B. The band intensity for WT HIV-1 NL4-3 was considered 100%, and relative intensities for corresponding bands in mutant viruses were determined. Error bars indicate S.D. from two independent experiments.
To elucidate the biochemical basis for KF116 resistance, we analyzed the effects of the amino acid substitutions in the context of recombinant IN. WT or IN mutant proteins fused to His6 affinity tags to facilitate protein purification were expressed in Escherichia coli. Somewhat surprisingly, the single T124N substitution reduced the level of IN expression to an extent that we were unable to obtain sufficient quantities of recombinant protein for our assays. In contrast, T124N/T174I and T124N/V165I/T174I INs were expressed in E. coli at WT levels and were successfully purified for biochemical characterization.
Size-exclusion chromatography (SEC) was utilized to characterize the potential effects of the amino acid substitutions on IN multimerization. Under these conditions, WT IN migrated as two forms: a tetramer, which was the main species, and a less abundant monomer (Fig. 5A). Both the double (T124N/T174I) and triple (T124N/V165I/T174I) amino acid substitutions altered IN multimerization with more pronounced effects seen with the double mutant protein (Fig. 5A). The double mutant IN exhibited similar amounts of three forms: tetramer, dimer, and monomer. Although the triple mutant also exhibited these three forms, the T124N/V165I/T174I IN tetramer was more prevalent than the dimeric and monomeric species. A homogenous time-resolved fluorescence (HTRF)-based assay for IN strand transfer activity was used to measure WT and mutant IN strand transfer activity. Both double and triple mutant INs exhibited reduced catalytic activities with the triple mutant being slightly more active (∼27% of WT level) than the double mutant (∼21% activity) (Fig. 5B). Binding to LEDGF/p75 was assessed by nickel-nitrilotriacetic (NTA) acid pulldown using His6-tagged WT and mutant INs. Both sets of mutations significantly diminished LEDGF/p75 binding (Fig. 5, C and D).
Figure 5.

Biochemical analysis of recombinant INs. A, SEC profiles for WT, T124N/T174I, and T124N/V165I/T174I INs. The peaks corresponding to monomeric (M), dimeric (D), and tetrameric (T) forms are indicated. Shown are representative chromatograms of two independent experiments. B, catalytic activities of WT and mutant INs. The error bars indicate the S.D. of three independent experiments. C, SDS-PAGE image showing LEDGF/p75 pulldown by WT and mutant INs. Lane 1, molecular weight markers. The gel was loaded with LEDGF/p75 (lane 2), WT IN (lane 3), T124N/T174I IN (lane 4), and T124N/V165I/T174I IN (lane 5). LEDGF/p75 pulldown without IN (lane 6; negative control), WT IN (lane 7), T124N/T174I IN (lane 8), and T124N/V165I/T174I IN (lane 9) is also shown. The gel is a representative of two independent experiments. D, quantitative analysis of the experimental results shown in C. The catalytic activity and LEDGF/p75 binding values for WT IN were considered 100%, and relative values for mutant INs were determined. The error bars indicate the S.E. from two independent experiments.
We next analyzed whether these substitutions affected KF116-induced hypermultimerization or aggregation of IN (Fig. 6A). WT or mutant IN was incubated with scalar levels of KF116 or vehicle control (dimethyl sulfoxide), and the reactions were fractionated by centrifugation. Analysis of supernatant and pellet fractions following SDS-PAGE afforded qualitative assessment of IN oligomerization (Fig. 6B). Expectedly, WT IN aggregated in the presence of KF116 in a dose-dependent manner (Fig. 6). In sharp contrast, both double and triple mutant INs exhibited marked resistance to the activity of the inhibitor as neither detectably pelleted even at the highest (20 μm) concentration of KF116 tested (Fig. 6).
Figure 6.

KF116-induced aggregation of IN. A, the indicated concentrations of KF116 were added to WT or mutant INs. Following centrifugation, supernatant (S) and pellet (P) fractions were analyzed by SDS-PAGE and visualized by Coomassie Blue staining. Mass standards are shown to the left. The gels are representative of two independent experiments. B, quantitative analysis of the results in A. Soluble IN fractions for DMSO controls and each indicated inhibitor concentration were determined by supernatant/(supernatant + precipitate). The error bars indicate the S.E. from two independent experiments.
To examine whether the observed resistance to aggregation was due to the inability of KF116 to bind the mutated INs, we conducted surface plasmon resonance (SPR) experiments (Fig. 7). Full-length IN is not amenable to these assays due to poor solubility of the protein. We accordingly used IN mutant varieties of the CCD that additionally contained the solubilizing F185K substitution (16, 23). KF116 bound the CCD with a Kd of ∼55 nm, which is consistent with its antiviral IC50 value of ∼24 nm. In sharp contrast, the same concentration range (25–800 nm) of KF116 failed to yield detectable binding to CCD(T124N/T174I) or CCD(T124N/V165I/T174I). Instead, binding was only detected after increasing the inhibitor concentrations to 25–200 μm, suggesting Kd values of >200 μm (Fig. 7).
Figure 7.

A sample of sensorgrams used for SPR analysis of KF116 binding. SPR kinetics for KF116 binding to CCD (A), CCD(T124N/T174I) (B), and CCD(T124N/V165I/T174I) (C) are shown. All analyzed CCD constructs contained the additional F185K solubilizing substitution. D, calculated Kd values and -fold changes for the tested interactions.
To gain structural insight into how the T124N/V165I/T174I IN substitutions confer resistance to KF116, we determined the X-ray crystal structure of CCD(T124N/V165I/T174I/F185K), which contained the solubilizing F185K substitution (Fig. 8). CCD(T124N/V165I/T174I/F185K) crystals that were soaked in the presence of 1 mm KF116 did not contain any additional electron density, indicating a lack of inhibitor binding even at this relatively high drug concentration. To better understand how these drug resistance substitutions affect inhibitor binding, we superimposed the structure of CCD(T124N/V165I/T174I/F185K) with the previously determined CCD(F185K) structure bound to KF116 (13). Comparative analysis revealed that the hydrogen bonds formed between Thr-174 and the carboxylic acid and t-butoxy moieties of KF116 are abolished by the substitution to Ile. Thr-124 is within hydrogen bonding distance from the chlorine halogen of KF116, whereas Asn-124 is shifted away from the inhibitor (Fig. 8). Therefore, both the T124N and T174I substitutions are expected to markedly weaken inhibitor binding. In complete contrast, Val-165 is significantly distanced from the inhibitor-binding site, and its substitution with Ile is unlikely to impact KF116 binding to IN through a direct drug contact.
Figure 8.
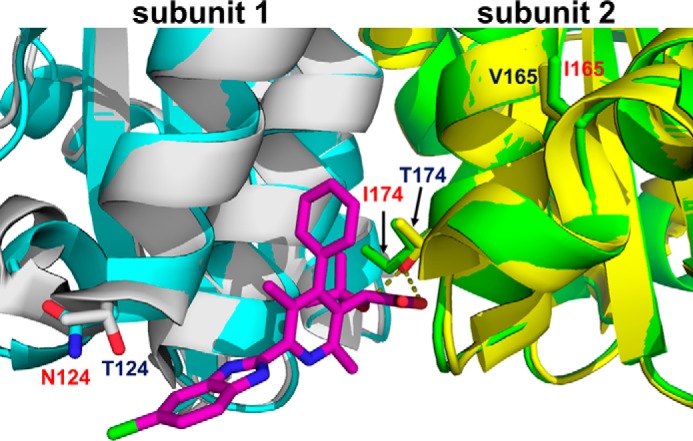
The X-ray crystal structure of the drug-resistant mutant (T124N/V165I/T174I) CCD dimer (cyan and green) overlaid with the structure of CCD dimer (gray and yellow) bound to KF116 (magenta). Side chains for drug resistance substitutions (in red) and their corresponding WT residues are shown. Both CCD constructs contained the additional F185K solubilizing substitution.
Discussion
In these studies, we have dissected an unexpectedly complex mechanism for the evolution of drug resistance to the pyridine-based MINI KF116. A key consequence of our work has revealed an unanticipated role of IN in regulating polyprotein proteolytic processing during virion morphogenesis.
Our virology, biochemistry, and structural biology experiments collectively provide a plausible explanation for the stepwise emergence of the drug-resistant HIV-1 NL4-3(IN T124N/V165I/T174I) phenotype. Scalar increases of KF116 concentration in infected cells initially led to the single T124N IN substitution, which was detected after five passages in the presence of ∼10 μm KF116 (13). Consistent with these earlier findings, our results reveal an IC50 value of ∼8 μm for KF116 against HIV-1 NL4-3(IN T124N). To withstand higher (∼25 μm) KF116 concentrations, which were achieved after 10 rounds of serial passaging, the virus was forced to acquire additional substitutions. Increased levels of resistance were attained when the original T124N substitution was coupled with a T174I change. Substitutions of these two residues, which are both located in the inhibitor-binding pocket, led to >4000-fold reduction in KF116 binding affinity (Fig. 7). However, the virus containing the double T124N/T174I IN substitutions was effectively non-infectious (Fig. 1). Therefore, the virus acquired the final V165I substitution, which is positioned outside the inhibitor-binding pocket, to allow the triple mutant virus to partly regain replication capacity (Fig. 1).
All three substitutions (T124N/T174I/V165I) are located at the IN dimer interface and affect the oligomeric state of purified recombinant IN (Fig. 5A). Therefore, it is not surprising that these changes substantially compromise IN catalytic activities in infected cells, which require a delicate assembly of higher-order IN oligomers into fully functional intasome nucleoprotein complexes for replication (24, 25). In addition, the three IN residues Thr-124, Val-165, and Thr-174 are located within or near the LEDGF/p75-binding pocket (17), and their substitutions markedly compromise interactions with the cellular cofactor (Fig. 5, C and D). Most notably, IN Thr-174 and LEDGF/p75 Asp-366 side chains are positioned within hydrogen bonding distance (17), and the T174I substitution could substantially weaken the interaction between these two proteins. Even with greatly diminished binding to LEDGF/p75, the triple (T124N/V165I/T174I) mutant virus exhibited ∼17% of WT levels of infectivity (Fig. 1), which is in line with previous studies (26–28) demonstrating ∼10-fold reductions in single-round HIV-1 infectivity in LEDGF/p75 knockout cells.
The unexpected finding of our study is that the double IN substitutions impaired proteolytic processing of Gag and Gag-Pol polyproteins and resulted in immature virus particles, whereas the additional V165I substitution restored substantial levels of proteolytic processing activity and virion maturation. Prior to proteolytic processing in immature virions, IN is part of the Gag-Pol polyprotein, and substitutions at the IN dimer interface that have been shown to alter the multimeric state of purified recombinant IN could also potentially affect Gag-Pol dimerization in virions, which in turn is critical for PR activity (29). In WT virions, properly dimerized PR has to first excise itself from Gag-Pol, which can then cleave the multiple cognate sites within Gag and Gag-Pol to form fully matured infectious virions. The release of mature PR is significantly reduced by the T124N/T174I IN substitutions (Fig. 3, D and E). Residual amounts of active PR seem to be responsible for some partial cleavage products but are insufficient to fully process Gag and Gag-Pol even after 48 h post-transfection. It is striking that the additional V165I IN substitution allowed the virus to regain substantial PR activity, suggesting that Gag-Pol architecture and related PR activity are intimately linked with IN structure.
The immature virion morphology observed for the double mutant virus (Fig. 2) is clearly distinct from the eccentric core phenotype seen with MINI- and ALLINI-treated WT viruses (7, 8, 10, 13). These inhibitors do not affect proteolytic processing of Gag and Gag-Pol and instead yield eccentric, non-infectious virions where the RNPs are mislocalized outside of the translucent capsid core. The inhibitors induce hyper- or aberrant multimerization of free IN after its proteolytic cleavage and release from Gag-Pol, which in turn impairs its ability to bind and encapsulate the viral RNA genome within the protective capsid core (11).
At present, it is unclear why HIV-1 NL4-3(IN T124N/V165I/T174I) virions lack conical core structures as assessed by TEM. HIV-1 CA cores can assume multiple shapes (30–32), so it is possible that HIV-1 NL4-3(IN T124N/V165I/T174I) cores are smaller in size than WT cores and hence not distinct from the ED. This could account for the partial (∼17%) replication capacity of HIV-1 NL4-3(IN T124N/V165I/T174I) as compared with WT HIV-1 NL4-3. Additional work will be needed to fully characterize the biochemical basis for the lack of discernible conical core structures in HIV-1 NL4-3(IN T124N/V165I/T174I) particles.
Numerous amino acid changes in the IN coding sequence that have been shown to yield non-infectious virions have been broadly grouped into class II mutant viruses to delineate them from class I mutations that selectively affect IN catalytic activity during integration (33). The morphologies of several class II IN substitutions have been examined by TEM, which showed some to have eccentric virions with mislocalized RNPs (7, 8, 10, 20, 21). However, a more detailed analysis of a larger number of class II IN mutant viruses is warranted to comprehensively characterize how various IN substitutions affect virus particle maturation. For example, might additional IN substitutions, similar to the T124N/T174I substitutions detailed herein, alter Gag-Pol proteolytic processing? We envisage that some class II IN mutations would also directly or indirectly impair the ability of IN to bind the viral RNA genome (11).
In addition to virion morphogenesis, the structure of IN seems to influence virus particle release from cells as IN deletion mutant constructs are reportedly defective for particle production (20, 34). Interestingly, particle production was restored when PR was inactivated in the context of IN deletion mutant viruses or when a stop codon was placed after the PR-encoding sequence to preclude both RT and IN from Gag-Pol (34). Collectively, our findings together with these published studies argue for a potential architectural interplay between IN and PR within Gag-Pol prior to PR excision. At the same time, it is important to point out the following important differences between prior analyses of IN deletion mutant constructs and the double mutant T124N/T174I IN virus. Unlike the deletion mutant viral studies, HIV-1 NL4-3(IN T124N/T174I) yielded WT levels of virions in cell supernatants. Furthermore, in common with other described class II IN viruses, TEM analysis of the IN deletion mutant virus revealed the predominance of eccentric cores with mislocalized RNPs (7, 20), whereas the double mutant virus exhibited almost exclusively immature virions.
In conclusion, the present studies reveal an architectural role of IN during Gag-Pol proteolytic processing, which in turn forces HIV-1 to undertake a complex evolutionary pathway to confer resistance to MINI KF116. These findings together with the crystal structure of the drug-resistant HIV-1 IN CCD(T124N/V165I/T174I) provide a means for developing second-generation ALLINIs.
Experimental procedures
Compounds
Syntheses of KF116 and BI-D were performed as described previously (13, 26). BI224436 was synthesized at Haoyuan Chemexpress Co., Ltd. (Shanghai, China). SQV (catalog number 4658) was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program.
Mutant virus generation
Site-directed mutagenesis was performed using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies) following the manufacturer's protocol in a subclone that contained the IN region of the HIV-1 pol gene. The resulting PCR product was digested, gel-purified, ligated into pNL4-3 (full-length or Luc-containing single-round derivative), and transformed into Stbl2 (Invitrogen) cells. The entire viral genome was sequenced by The Ohio State University's Plant-Microbe Genomics Facility to confirm that only the desired substitutions were introduced.
Cells, transfections, and infections
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented to contain 10% fetal bovine serum (Invitrogen), 1% penicillin and streptomycin (Gibco), and 0.1% Normocin (Invitrogen) at 37 °C and 5% CO2. Viral stocks were produced as described previously (13).
For viral infectivity experiments, HIV-Luc virions for WT HIV-1 NL4-3, HIV-1 NL4-3(IN T124N), HIV-1 NL4-3(IN T124N/T174I), and HIV-1 NL4-3(IN T124N/V165I/T174I) were generated by transfecting HEK293T with pNL4-3.Luc.Env− and pCG-VSV-G using X-tremeGene DNA transfection reagent (Sigma-Aldrich). Twenty-four hours post-transfection, the culture supernatant was replaced with fresh complete medium after washing once with complete medium. Forty-eight hours post-transfection, the virus-containing supernatant was collected and filtered through 0.45-μm filters. Virus concentration was determined by HIV-1 Gag p24 ELISA (Zeptometrix) following the manufacturer's protocol. HEK293T cells (2 × 105) were infected with 10–100 ng of p24 of WT or IN mutant virus in the presence of 8 μg/ml Polybrene (Sigma). Two hours post-infection, the culture supernatant was removed, and the cells were washed once with fresh medium, and then fresh complete medium was added. Cells were cultured for 48 h, and then cell extracts were prepared using 5× reporter lysis buffer (Promega). Luciferase activity was determined using a commercially available kit (Promega).
Virus production and TEM
HEK293T cells grown in two 15-cm dishes (30 × 106 cells/dish) were transfected with 30 μg of plasmids of full-length proviral clones encoding WT HIV-1 NL4-3, HIV-1 NL4-3(IN T124N), HIV-1 NL4-3(IN T124N/T174I), or HIV-1 NL4-3(IN T124N/V165I/T174I) using PolyJet in vitro DNA transfection reagent (SignaGen Laboratories). After 5 h, medium was aspirated, and fresh medium was added. After 48 h from the start of transfection, viruses were harvested from the medium by filtration through a 0.45-μm filter and pelleted by ultracentrifugation using a Beckman SW32-Ti rotor at 26,000 rpm for 2 h at 4 °C. Following centrifugation, medium was removed, and the virus pellets were fixed in a solution of 1.25% formaldehyde, 2.5% glutaraldehyde, and 0.03% picric acid in 0.1 m sodium cacodylate (pH 7.4) for at least 1 h at room temperature. Virus pellets were dehydrated in a graduated ethanol series and embedded in Epon resin. Thin sections (70 nm) were stained with uranyl acetate and observed under a JEOL 1200EX transmission electron microscope equipped with an Advanced Microscopy Techniques 2k charge-coupled device camera at the Harvard Medical School EM core facility. Images were captured at 20,000× magnification, and virion morphological phenotype was distinguished and recorded by eye. ImageJ Cell Counter plugins were utilized to mark the particles that had been counted.
Virion protein processing
HEK293T cells (5 × 106 cells/175-cm2 cell culture flask in 35 ml of complete medium) were transfected with plasmids of full-length proviral clones encoding WT HIV-1 NL4-3, HIV-1 NL4-3(IN T124N), HIV-1 NL4-3(IN T124N/T174I), or HIV-1 NL4-3(IN T124N/V165I/T174I) and cultured for 24 h as described above. Next, half of the cell samples were treated with 10 μm SQV for 1 h. The culture supernatant was replaced with fresh complete medium, and SQV was added to the those previously treated, and the cells were then cultured for an additional 24 h to allow for the production of HIV-1 viral particles in the presence and absence of protease inhibitor. The samples were normalized by p24 and prepared for immunoblotting as described previously (11). Quantitation was performed using ImageJ software. The intensity of each analyzed protein band was considered 100% for the WT virus, and the relative values for the corresponding protein in mutant viruses were determined.
Immunoblotting
Standard Western blotting procedures were used with the following antibodies: HIV-1 Gag (Abcam ab63917), which allowed detection of Gag as well as its proteolytic processing products including CA, MA, and NC; HIV-1 PR (Thermo Fisher 1696), HIV-1 RT (National Institutes of Health mAb 21), and HIV-1 IN (Abcam ab66645).
Antiviral assays
Antiviral activities of the inhibitors were determined as reported (7, 13). The EC50 values and standard deviations were determined from three independent experiments. Luciferase activity was monitored using a commercially available kit (Promega) and normalized with the BCA kit (Thermo Fischer). Excel software was used to calculate the average luciferase readings and corresponding standard deviations for each concentration point of the inhibitors. These values were then compared with WT and converted to a percentage. Origin 8 software (OriginLab, Inc.) was used to build dose-response curves and calculate EC50 values.
Recombinant proteins
Site-directed mutagenesis was used to introduce T124N, T124N/T171I, and T124N/V165I/T171I substitutions in the background of WT full-length IN or the CCD containing the F185K solubilizing mutation. The His6-tagged proteins were expressed in E. coli BL21 (DE3) and purified as described previously (6).
IN catalytic activity
The HTRF-based assay (3, 35) was used to compare strand transfer activities of WT, T124N/T174I, and T124N/V165I/T174I INs. Briefly, 400 nm IN was incubated with 50 nm Cy5-labeled viral donor DNA and 10 nm biotinylated target acceptor DNA. Detection was based on europium-streptavidin, and the HTRF signal was recorded using a PerkinElmer Life Sciences Enspire multimode plate reader.
Inhibitor-induced IN aggregation
Serial dilutions of KF116 were added to 5 μm IN in 20 μl of reaction buffer containing 20 mm HEPES (pH 7.5), 1 m NaCl, and 5 mm β-mercaptoethanol (BME). The reactions were incubated overnight at 4 °C and centrifuged at 10,000 × g for 10 min. All of the supernatant was collected and diluted with 100 μl of 1.2× NuPAGE lithium dodecyl sulfate sample buffer (Novex). The remaining pellet was resuspended with 120 μl of 1× NuPAGE lithium dodecyl sulfate sample buffer to match the overall volume of the corresponding supernatant fraction. Supernatant and pellet fractions were then analyzed by SDS-PAGE, and IN was visualized by Coomassie staining. IN bands in individual fractions were quantified by ImageJ software.
IN-LEDGF/p75 binding assay
Nickel-Sepharose 6 Fast Flow beads (GE Healthcare) were used to assay the interaction of recombinant His6-tagged IN with tag-free LEDGF/p75. IN (1 μm) was added to equimolar LEDGF/p75 in binding buffer containing 50 mm HEPES (pH 7.1), 300 mm NaCl, 2 mm MgCl2, 20 mm imidazole, 2 mm BME, and 0.2% (v/v) Nonidet P-40. The reactions were incubated for 30 min at room temperature and then added to pre-equilibrated nickel beads. The reaction mixture was rotated for 1 h at room temperature, and unbound fractions were removed by repeated washing in the binding buffer. The pulldown fractions were then analyzed by SDS-PAGE.
Size-exclusion chromatography
The oligomeric state of WT, T124N/T174I, and T124N/V165I/T174I INs was analyzed using a Superdex 200 10/300 GL column (GE Healthcare) and elution buffer containing 20 mm HEPES (pH 6.8), 1 m NaCl, 10 mm MgSO4, 0.2 mm EDTA, 10% glycerol, and 5 mm BME. The loading concentration of each IN was 15 μm, and the absorbance was recorded at 280 nm. The column was calibrated with a gel filtration standard (Bio-Rad) containing a mixture of the following proteins: thyroglobulin (bovine), 670,000 Da; γ-globulin (bovine), 158,000 Da; ovalbumin (chicken), 44,000 Da; and myoglobin (horse), 17,000 Da.
X-ray crystallography
IN CCD(T124N/V165I/T174I/F185K) was crystallized by hanging-drop vapor diffusion at 4 °C. Protein at 5 mg/ml was diluted 1:1 in 0.2 m ammonium sulfate, 0.1 m Bis-Tris (pH 5.5), and 25% PEG 3,350. Data collected on a Rigaku Micromax-003 at 100 K yielded a diffraction limit of 1.76 Å with space group P1. Data were integrated and scaled using HKL3000 (36) and Scalepack (37). Phaser (38) in the PHENIX suite (39) was used to run molecular replacement using Protein Data Bank code 1ITG as a search model (16). Phenix.refine (40) was used for data refinement, and manual refinement was done in Coot (41). The structure had a final Rwork/Rfree of 15.9/19.2. The coordinates are deposited in the Protein Data Bank under accession code 5JL4, and the refinement statistics are given in supplemental Table S1.
Surface plasmon resonance
SPR experiments were performed on a Biacore T100 as described (35). Briefly, His6-tagged CCD(F185K) and mutant domains were captured onto an NTA chip to about 4,500 response units. The indicated concentrations of KF116 were then flowed over the chip, and the sensorgrams were recorded. Origin 8 software was used to calculate the Kd values.
Author contributions
A. C. H., A. V. J., P. C. K., and M. J. K. performed the experiments. R. C. L. and J. R. F. generated and provided reagents. M. K. and A. N. E. conceived and guided the studies. A. C. H. and M. K. wrote the manuscript with contributions from all other authors.
Supplementary Material
Acknowledgments
We are grateful to Jared Lindenberger, Lei Feng, Stephanie Rebensburg, Nikoloz Shkriabai, and Jason Anderson for helpful advice and critical reading of the manuscript.
This work was supported by National Institutes of Health Grants R01 AI110310 and R01 AI062520 (to M. K.), R01 AI039394 (to A. N. E.), U54 GM103368 (to A. N. E. and M. K.), and KL2 TR001068 (to R. C. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1.
The atomic coordinates and structure factors (code 5JL4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HIV-1
- human immunodeficiency virus, type 1
- ALLINI
- allosteric HIV-1 integrase inhibitor
- BME
- β-mercaptoethanol
- CA
- capsid
- CCD
- catalytic core domain
- ED
- electron density
- HTRF
- homogenous time-resolved fluorescence
- IN
- integrase
- LEDGF
- lens epithelium-derived growth factor
- Luc
- luciferase
- MA
- matrix
- MINI
- multimerization selective integrase inhibitor
- NC
- nucleocapsid
- NTA
- nitrilotriacetic acid
- PR
- protease
- RNP
- ribonucleoprotein
- RT
- reverse transcriptase
- SEC
- size-exclusion chromatography
- SPR
- surface plasmon resonance
- SQV
- saquinavir
- TEM
- transmission electron microscopy
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Christ F., Voet A., Marchand A., Nicolet S., Desimmie B. A., Marchand D., Bardiot D., Van der Veken N. J., Van Remoortel B., Strelkov S. V., De Maeyer M., Chaltin P., and Debyser Z. (2010) Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6, 442–448 [DOI] [PubMed] [Google Scholar]
- 2. Fader L. D., Malenfant E., Parisien M., Carson R., Bilodeau F., Landry S., Pesant M., Brochu C., Morin S., Chabot C., Halmos T., Bousquet Y., Bailey M. D., Kawai S. H., Coulombe R., et al. (2014) Discovery of BI 224436, a noncatalytic site integrase inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 5, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsiang M., Jones G. S., Niedziela-Majka A., Kan E., Lansdon E. B., Huang W., Hung M., Samuel D., Novikov N., Xu Y., Mitchell M., Guo H., Babaoglu K., Liu X., Geleziunas R., et al. (2012) New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 287, 21189–21203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le Rouzic E., Bonnard D., Chasset S., Bruneau J. M., Chevreuil F., Le Strat F., Nguyen J., Beauvoir R., Amadori C., Brias J., Vomscheid S., Eiler S., Lévy N., Delelis O., Deprez E., et al. (2013) Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 10, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Bel N., van der Velden Y., Bonnard D., Le Rouzic E., Das A. T., Benarous R., and Berkhout B. (2014) The allosteric HIV-1 integrase inhibitor BI-D affects virion maturation but does not influence packaging of a functional RNA genome. PLoS One 9, e103552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kessl J. J., Jena N., Koh Y., Taskent-Sezgin H., Slaughter A., Feng L., de Silva S., Wu L., Le Grice S. F., Engelman A., Fuchs J. R., and Kvaratskhelia M. (2012) A multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 287, 16801–16811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jurado K. A., Wang H., Slaughter A., Feng L., Kessl J. J., Koh Y., Wang W., Ballandras-Colas A., Patel P. A., Fuchs J. R., Kvaratskhelia M., and Engelman A. (2013) Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U.S.A. 110, 8690–8695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Balakrishnan M., Yant S. R., Tsai L., O'Sullivan C., Bam R. A., Tsai A., Niedziela-Majka A., Stray K. M., Sakowicz R., and Cihlar T. (2013) Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One 8, e74163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Desimmie B. A., Schrijvers R., Demeulemeester J., Borrenberghs D., Weydert C., Thys W., Vets S., Van Remoortel B., Hofkens J., De Rijck J., Hendrix J., Bannert N., Gijsbers R., Christ F., and Debyser Z. (2013) LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fontana J., Jurado K. A., Cheng N., Ly N. L., Fuchs J. R., Gorelick R. J., Engelman A. N., and Steven A. C. (2015) Distribution and redistribution of HIV-1 nucleocapsid protein in immature, mature, and integrase-inhibited virions: a role for integrase in maturation. J. Virol. 89, 9765–9780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kessl J. J., Kutluay S. B., Townsend D., Rebensburg S., Slaughter A., Larue R. C., Shkriabai N., Bakouche N., Fuchs J. R., Bieniasz P. D., and Kvaratskhelia M. (2016) HIV-1 integrase binds the viral RNA genome and is essential during virion morphogenesis. Cell 166, 1257.e12-1268.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng L., Sharma A., Slaughter A., Jena N., Koh Y., Shkriabai N., Larue R. C., Patel P. A., Mitsuya H., Kessl J. J., Engelman A., Fuchs J. R., and Kvaratskhelia M. (2013) The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 288, 15813–15820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma A., Slaughter A., Jena N., Feng L., Kessl J. J., Fadel H. J., Malani N., Male F., Wu L., Poeschla E., Bushman F. D., Fuchs J. R., and Kvaratskhelia M. (2014) A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 10, e1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fader L. D., Carson R., Morin S., Bilodeau F., Chabot C., Halmos T., Bailey M. D., Kawai S. H., Coulombe R., Laplante S., Mekhssian K., Jakalian A., Garneau M., Duan J., Mason S. W., et al. (2014) Minimizing the contribution of enterohepatic recirculation to clearance in rat for the NCINI class of inhibitors of HIV. ACS Med. Chem. Lett. 5, 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grawenhoff J., and Engelman A. N. (2017) Retroviral integrase protein and intasome nucleoprotein complex structures. World J. Biol. Chem. 8, 32–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dyda F., Hickman A. B., Jenkins T. M., Engelman A., Craigie R., and Davies D. R. (1994) Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266, 1981–1986 [DOI] [PubMed] [Google Scholar]
- 17. Cherepanov P., Ambrosio A. L., Rahman S., Ellenberger T., and Engelman A. (2005) Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U.S.A. 102, 17308–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta K., Brady T., Dyer B. M., Malani N., Hwang Y., Male F., Nolte R. T., Wang L., Velthuisen E., Jeffrey J., Van Duyne G. D., and Bushman F. D. (2014) Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains. J. Biol. Chem. 289, 20477–20488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta K., Turkki V., Sherrill-Mix S., Hwang Y., Eilers G., Taylor L., McDanal C., Wang P., Temelkoff D., Nolte R. T., Velthuisen E., Jeffrey J., Van Duyne G. D., and Bushman F. D. (2016) Structural basis for inhibitor-induced aggregation of HIV integrase. PLoS Biol. 14, e1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelman A., Englund G., Orenstein J. M., Martin M. A., and Craigie R. (1995) Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 69, 2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson B. C., Métifiot M., Ferris A., Pommier Y., and Hughes S. H. (2013) A homology model of HIV-1 integrase and analysis of mutations designed to test the model. J. Mol. Biol. 425, 2133–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sundquist W. I., and Kräusslich H. G. (2012) HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2, a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jenkins T. M., Hickman A. B., Dyda F., Ghirlando R., Davies D. R., and Craigie R. (1995) Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc. Natl. Acad. Sci. U.S.A. 92, 6057–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Passos D. O., Li M., Yang R., Rebensburg S. V., Ghirlando R., Jeon Y., Shkriabai N., Kvaratskhelia M., Craigie R., and Lyumkis D. (2017) Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 355, 89–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ballandras-Colas A., Maskell D. P., Serrao E., Locke J., Swuec P., Jónsson S. R., Kotecha A., Cook N. J., Pye V. E., Taylor I. A., Andrésdóttir V., Engelman A. N., Costa A., and Cherepanov P. (2017) A supramolecular assembly mediates lentiviral DNA integration. Science 355, 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang H., Jurado K. A., Wu X., Shun M. C., Li X., Ferris A. L., Smith S. J., Patel P. A., Fuchs J. R., Cherepanov P., Kvaratskhelia M., Hughes S. H., and Engelman A. (2012) HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 40, 11518–11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shun M. C., Raghavendra N. K., Vandegraaff N., Daigle J. E., Hughes S., Kellam P., Cherepanov P., and Engelman A. (2007) LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 21, 1767–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fadel H. J., Morrison J. H., Saenz D. T., Fuchs J. R., Kvaratskhelia M., Ekker S. C., and Poeschla E. M. (2014) TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J. Virol. 88, 9704–9717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wondrak E. M., and Louis J. M. (1996) Influence of flanking sequences on the dimer stability of human immunodeficiency virus type 1 protease. Biochemistry 35, 12957–12962 [DOI] [PubMed] [Google Scholar]
- 30. Welker R., Hohenberg H., Tessmer U., Huckhagel C., and Kräusslich H. G. (2000) Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 74, 1168–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Accola M. A., Ohagen A., and Göttlinger H. G. (2000) Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6(gag). J. Virol. 74, 6198–6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao G., Perilla J. R., Yufenyuy E. L., Meng X., Chen B., Ning J., Ahn J., Gronenborn A. M., Schulten K., Aiken C., and Zhang P. (2013) Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engelman A. (1999) In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 52, 411–426 [DOI] [PubMed] [Google Scholar]
- 34. Bukovsky A., and Göttlinger H. (1996) Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. J. Virol. 70, 6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Slaughter A., Jurado K. A., Deng N., Feng L., Kessl J. J., Shkriabai N., Larue R. C., Fadel H. J., Patel P. A., Jena N., Fuchs J. R., Poeschla E., Levy R. M., Engelman A., and Kvaratskhelia M. (2014) The mechanism of H171T resistance reveals the importance of Nδ-protonated His171 for the binding of allosteric inhibitor BI-D to HIV-1 integrase. Retrovirology 11, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 37. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 38. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.