

Abstract
Carrageenans are sulfated α-1,3-β-1,4-galactans found in the cell wall of some red algae that are practically valuable for their gelation and biomimetic properties but also serve as a potential carbon source for marine bacteria. Carbohydrate degradation has been studied extensively for terrestrial plant/bacterial systems, but sulfation is not present in these cases, meaning the marine enzymes used to degrade carrageenans must possess unique features to recognize these modifications. To gain insights into these features, we have focused on κ-carrageenases from two distant bacterial phyla, which belong to glycoside hydrolase family 16 and cleave the β-1,4 linkage of κ-carrageenan. We have solved the crystal structure of the catalytic module of ZgCgkA from Zobellia galactanivorans at 1.66 Å resolution and compared it with the only other structure available, that of PcCgkA from Pseudoalteromonas carrageenovora 9T (ATCC 43555T). We also describe the first substrate complex in the inactivated mutant form of PcCgkA at 1.7 Å resolution. The structural and biochemical comparison of these enzymes suggests key determinants that underlie the functional properties of this subfamily. In particular, we identified several arginine residues that interact with the polyanionic substrate, and confirmed the functional relevance of these amino acids using a targeted mutagenesis strategy. These results give new insight into the diversity of the κ-carrageenase subfamily. The phylogenetic analyses show the presence of several distinct clades of enzymes that relate to differences in modes of action or subtle differences within the same substrate specificity, matching the hybrid character of the κ-carrageenan polymer.
Keywords: crystal structure, enzyme kinetics, polysaccharide, processivity, site-directed mutagenesis, carbohydrate recognition, enzyme-substrate complex, kappa-carrageenan, sulfated polysaccharides
Introduction
Marine ecosystems house highly complex trophic networks. At the basis of these networks, photosynthetic organisms play a key role. As primary producers, they initiate and maintain the biogeochemical carbon cycle in the ocean and constitute the first link in the marine food chain. Among them are found macroalgae, which grow in the photic zone. Macroalgae are polyphyletic and belong to three distinct phyla, namely red, green, and brown algae. In addition to their role in climate regulation (1) and as shelter for many animal and microbial marine species (2), they are a major source of carbon for many communities. Particularly, they establish close interactions with marine bacteria living freely in the water column or entrapped in biofilm on the surface of macroalgae (3). There has been increasing interest in the study of the interactions between microbial communities and their associated host, especially in the case of terrestrial association in human gut (4).
Heterotrophic bacteria have developed sophisticated enzymatic tools to extract the carbon from plant cell walls. Notably, the ecological position of a bacterial strain in the microbial communities will influence the degradation strategy deployed. Some possess a wide set of enzymes that make them generalist organisms, like Bacteroidetes, others being more specialized in the degradation of certain families of polysaccharides (5). As an example of an integrative strategy, Bacteroidetes species have been shown to usually possess all of the genes involved in the degradation, recognition, and transport of a specific polysaccharide organized in clusters called polysaccharide utilization loci (6, 7).
However, the majority of studies are done on terrestrial bacteria, producing enzymes against land plant polysaccharides. In the case of marine heterotrophic bacteria, their arsenal of carbohydrate active enzymes is still largely unexplored (8). Indeed, they are adapted to the diversity of macroalgal polysaccharides, and many of these marine polysaccharides have no equivalent in land plants. One of their peculiarities is the abundance of sulfation motifs, which are absent in the land plant polysaccharides but also present in polysaccharides of the mammalian extracellular matrix (9).
One of the best-known families of algal sulfated polysaccharides is carrageenan. This family is composed of linear chains of α-1,3-β-1,4-galactans with an increasing number of sulfate groups per disaccharide, unit depending on the categories of carrageenans. Accordingly, the three main structures are called κ- (one sulfate group), ι- (two sulfate groups), and λ- (three sulfate groups) carrageenans (Fig. 1) (10). The carrageenases, which cleave the β-glycosidic linkages of carrageenans, belong to different families of glycoside hydrolases (GH),2 based on their amino acid sequences: GH16 for κ-carrageenases (11, 12), GH82 for ι-carrageenases (13, 14), and a new family of GH for λ-carrageenases (15, 16). In this study, we will focus on the family of κ-carrageenases.
Figure 1.
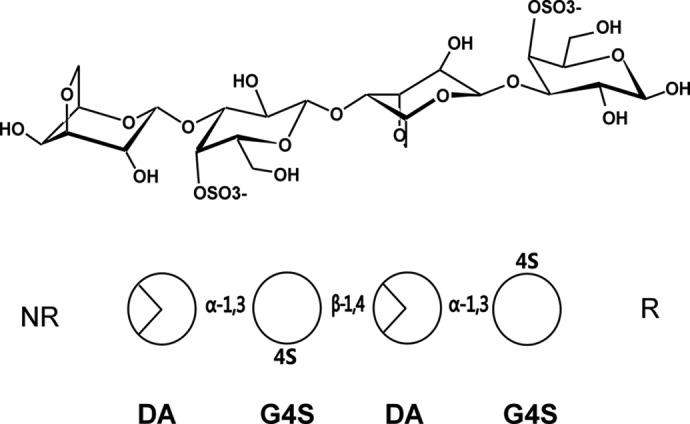
Schematic representations of the chemical structure of κ-neocarratetraose. At the top, the chemical representations of the units show the standard chair conformation. At the bottom, the simplified scheme follows the same order of monosaccharides and glycosidic linkages as above. NR, non-reducing end; R, reducing end.
During the past few years, many new κ-carrageenases have been identified in recently sequenced bacterial genomes (244 entries in UniProtKB), belonging mainly to the three phyla Bacteroidetes, Proteobacteria, and Planctomycetes. However, only seven have been cloned and biochemically studied, namely from Pseudoalteromonas carrageenovora 9T (ATCC 43555T) (17), Zobellia galactanivorans DsijT (11), Pseudoalteromonas porphyrae LL1 (18), Pseudoalteromonas tetraodonis JAM-K142 (19), Zobellia sp. ZM-2 (20), Shewanella sp. P1-14-1 (Kz7) (21), and Pseudoalteromonas sp. QY203 (22). Up to now, only one κ-carrageenase has been biochemically and structurally studied at 1.54 Å resolution, namely PcCgkAGH16 from the gammaproteobacterium P. carrageenovora 9T (12) that was isolated from the marine water column and algae off the Atlantic coast of Canada (23, 24).
We report here the second crystal structure of a catalytic module of a GH16 κ-carrageenase, ZgCgkAGH16 from Z. galactanivorans DsijT, a marine flavobacterium model for the bioconversion of algal polysaccharides that was isolated from the surface of the red macro-algae Delesseria sanguinea (15). In contrast to P. carrageenova, Z. galactanivorans grows perfectly well with κ-carrageenan as the sole carbon source (25). Both enzymes do not require any desulfation of natural κ-carrageenan substrate to be active. Despite the similarity of sequences between ZgCgkAGH16 and PcCgkAGH16 (40% identity, 50% similarity), several areas of divergence were observed, corresponding to the deletion or insertion of sequence stretches (Fig. 2A). The biochemical and structural comparison of both enzymes, along with a phylogenic analysis of κ-carrageenases, give a first insight into the diversity existing within this subfamily. This allowed us to propose some structural determinants that underlie the biochemical properties of these enzymes, active on polyanionic substrate. In addition, the mutagenesis and analysis of kinetic properties of PcCgkAGH16 point mutants allowed the proposal of potential targets of interest for enzymatic engineering of more efficient κ-carrageenases.
Figure 2.

Comparison of the catalytic modules of PcCgkAGH16 and ZgCgkAGH16. A, multiple-sequence alignment of the catalytic modules of both enzymes. The alignment was produced with MultAlin, and the figure was produced using ESPript version 3.x software. Secondary structure elements are symbolized with arrows for β-sheets, curls for α-helix, and T for turns; the “finger” regions surrounding the catalytic channel are boxed and numbered from F1 to F6; the sequence divergences that have structural consequences are boxed in green; the numbers 1 in green refer to the cysteine residues involved in the single disulfide bond of PcCgkAGH16; the black stars above the sequence highlight the amino acids that were mutated into alanine for PcCgkAGH16 in this study. B, schematic representation of the structural fold of ZgCgkAGH16. The color code is graduated from blue (N terminus) to red (C terminus) along the polypeptide chain. Catalytic residues Glu159-Asp161-Glu164 are shown as sticks and colored in red. C, schematic representation of the structural fold of PcCgkAGH16-E168D in complex with a κ-neocarratetraose, shown in stick representation (carbon atoms are white, oxygen atoms are red, and sulfur atoms are orange). The color code of the polypeptide chain is the same as in B. Catalytic residues Glu163-Asp165-E168D are shown as sticks and colored in red.
Results
Phylogenetic analysis of κ-carrageenases
A BLAST search with PcCgkAGH16 as query identified 76 putative κ-carrageenases in GenBankTM (identity ranging from 30 to 97%). All of these sequences have conserved several key residues for the recognition of κ-carrageenan (Arg196 and Arg260 in PcCgkAGH16). A phylogenetic tree was built (Fig. 3) based on the multiple alignment of the catalytic module sequences of these 76 κ-carrageenases and of three GH16 β-agarases chosen as an outgroup (supplemental Fig. S1).
Figure 3.

Phylogenetic tree of the currently sequenced κ-carrageenases. The multiple alignments were done with 37 sequences of catalytic modules of κ-carrageenases. Clades A–D are defined according to the bootstrap values. Structural common features are identified for these clades and are represented in the circles. Purple, presence of finger F2 and absence of finger F4; blue, presence of finger F4 and absence of finger F2; black, presence of both fingers F2 and F4, subgroup I being the closer in sequence similarity for F2 and F4 motifs and subgroup II showing the higher divergence. Blue arrows identify the six κ-carrageenases from Algibacter sp. SK-16.
This phylogenetic tree can be divided into at least 10 major clades, based on the highest values of bootstrap of deepest nodes. Four major clades can be delineated. Clade A, which contains the κ-carrageenase from P. carrageenovora, and clade D correlate well with bacterial phyla from Proteobacteria, with a bootstrap value of 100% for both. Clade B is composed of two subgroups of sequences belonging to Bacteroidetes and Planctomycetes, with a bootstrap value of 98%. Clade C contains sequences from Bacteroidetes only, including ZgCgkAGH16, with a bootstrap value of 100%.
It is noteworthy that some bacterial strains contain several κ-carrageenase sequences that spread out throughout the whole phylogenetic tree, as exemplified by Algibacter sp. SK-16, which displays six sequences (highlighted by arrows in Fig. 3; specific alignment provided in supplemental Fig. S2) or Flammeovirga sp. OC4 with three sequences.
Interestingly, when looking at the specificities of the two clades A and C, we can notice that the presence or absence of sequence stretches in the alignment correlates with the phylogenetic clustering. Specific alignment of both PcCgkAGH16 and ZgCgkAGH16 allowed the definition of these sequence stretches more precisely as “fingers,” numbered from F1 to F6. They are boxed on the sequence alignment of Fig. 2A. Their delimitation is based on analysis of secondary structure elements. These fingers are generally constituted by a succession of a β-strand-[tight turn,loop or helix]-β-strand structural elements and were identified as regions containing a critical divergence in the amino acid sequences of both κ-carrageenases studied here (green boxes in Fig. 2A).
All of the enzymes belonging to clade A possess a “finger F2-type” motif, which is absent in clade C, and all of the enzymes from clade C possess a “finger F4-type” motif, absent in clade A enzymes. Conversely, clade B and D enzymes possess both types of motifs, clade D containing the most conserved sequences for both fingers and clade B showing modifications of the nature and of the length of “typical” sequences.
The remaining sequences of the phylogenetic tree possess bootstrap values that are too low to constitute a monophyletic clade and to allow the establishment of clear correlations between the sequences and structural features.
Another interesting feature observed in the phylogenetic tree of κ-carrageenases is that some enzymes possess a modular architecture (e.g. Z. galactanivorans), whereas others consist of catalytic modules only (e.g. Z. uliginosa, P. carrageenovora). Notably, the modular nature is not correlated with the clades, except for the clade B Planctomycetes sequences, which are all only composed of a catalytic module.
Biochemical characterization of ZgCgkAGH16 and ZgCgkAGH16-CBM16-PorSS and comparison with PcCgkAGH16
The κ-carrageenase from Z. galactanivorans, ZgCgkA, is a modular enzyme displaying an N-terminal GH16 catalytic module, a carbohydrate-binding module of family 16 (CBM16), and a C-terminal Type-IX secretion module (PorSS) (11, 25). Two forms of the enzyme were cloned from genomic DNA, the catalytic module alone (ZgCgkAGH16) and the entire form of the enzyme (ZgCgkAGH16-CBM16-PorSS). PcCgkA is composed of a GH16 catalytic domain and an Ig-like module of unknown function. Only the catalytic module (PcCgkAGH16) was previously cloned (17) and has been used in this study. The three proteins were successfully expressed in soluble form and purified to homogeneity.
The two forms of ZgCgkA, with and without CBM16, were studied in parallel to the recombinant enzyme PcCgkAGH16. At first, we determined the optimal conditions for pH, buffer, and temperature for both forms of ZgCgkA. Efficiency was highest at pH 6 in MES buffer for both constructions, and analysis by dynamic light scattering revealed that both forms are stable up to 40 °C (data not shown). The comparison of the kinetic parameters of the three enzymes on diluted solutions of κ-carrageenan shows that ZgCgkAGH16-CBM16-PorSS and ZgCgkAGH16 are more efficient than PcCgkAGH16 by 3 and 5 times, respectively (Table 1). The absence of CBM in the construct of ZgCgkAGH16 causes a 40% increase of efficiency in solution, compared with the entire enzyme.
Table 1.
Kinetic parameters of the different recombinant and point-mutant enzymes in the presence of liquid substrate
Calculations have been done from at least three replicates and treated with Hyper software. Kinetic parameters of the catalytic domains of PcCgkA and ZgCgkA are indicated in bold. ND, not determined.
| kcat | Km | kcat/Km | Percentage versus WT | |
|---|---|---|---|---|
| s−1 | m | m−1 s−1 | % | |
| PcCgkAGH16 (WT) | 135.84 | 6.83 × 10−7 | 1.99 × 108 | 100 |
| ZgCgkAGH16-CBM16-PorSS | 660.91 | 1.04 × 10−6 | 6.35 × 108 | 319 |
| ZgCgkAGH16 | 914.65 | 8.22 × 10−7 | 1.11 × 109 | 560 |
| PcCgkAGH16-R92A | 183.20 | 8.99 × 10−7 | 2.04 × 108 | 102 |
| PcCgkAGH16-R151A | 478.79 | 1.21 × 10−6 | 3.97 × 108 | 199 |
| PcCgkAGH16-Q171A | 120.17 | 3.85 × 10−7 | 3.12 × 108 | 157 |
| PcCgkAGH16-R196A | 6.13 | 1.15 × 10−6 | 5.31 × 106 | 3 |
| PcCgkAGH16-R260A | ND | ND | ND | <1 |
| PcCgkAGH16-W266A | 27.36 | 2.63 × 10−6 | 1.04 × 107 | 5 |
Degradation kinetics on a longer time course (5 h) gave a similar result in solution (Fig. 4A). The concentration of product released after 30 min of incubation was increased by 26 and 143 times for ZgCgkAGH16-CBM16-PorSS andZgCgkAGH16, respectively, as compared with PcCgkAGH16. Thus, ZgCgkAGH16 is 5.5 times more efficient in solution than ZgCgkAGH16-CBM16-PorSS. However on microgels (Fig. 4B), differences of released products between the three enzymes are far less important than reactions in solution. Under these conditions, subtle differences can be observed; ZgCgkAGH16-CBM16-PorSSreleases 2 times more product than PcCgkAGH16 and 1.5 times more than ZgCgkAGH16 after 90 min of incubation.
Figure 4.

Kinetics of degradation of κ-carrageenan. A and B, reducing sugar analysis in solution (A) and microgels (B) of κ-carrageenan for PcCgkAGH16 (white circles), ZgCgkAGH16 (light gray squares), and ZgCgkAGH16-CBM16-PorSS (dark gray triangles). Error bars, S.D. of triplicate determinations. C and D, HPAEC analyses of degradation products of ZgCgkAGH16 in solution (C) and in microgel (D). E and F, HPAEC analyses of degradation products of ZgCgkAGH16-CBM16-PorSS in solution (E) and in microgel (F). For C, D, E, and F, different curves correspond to kinetics at t0 = 0, t1 = 15 min, t2 = 45 min, t3 = 24 h, t4 = 1 week. The asterisks in C–F designate peaks corresponding to noncharacterized reaction products.
HPAEC analyses of reaction end products produced by the recombinant enzymes confirm the previously reported difference of product patterns observed for the native enzymes (15). Namely, ZgCgkAGH16 mainly produces oligosaccharides with degree of polymerization of 4 and 6 (DP4 and DP6) (supplemental Fig. S3), whereas PcCgkAGH16 produces oligosaccharides with DP2 and DP4 (data not shown). Moreover, for both constructs of Z. galactanivorans κ-carrageenase, the profile of degradation products is different in solution and in microgels, with a DP4/DP6 ratio always higher in microgels compared with that in solution (Table 2). But the presence of the CBM modifies the values of this ratio. In solution (Fig. 4, C–E), the ratio becomes >1 more quickly for ZgCgkAGH16-CBM16-PorSS than for ZgCgkAGH16 (after 45 and 90 min, respectively), and on microgels (Fig. 4, D–F), it reaches the highest levels with ZgCgkAGH16-CBM16-PorSS after 1 week of extensive digestion. This means that the construction with CBM (ZgCgkAGH16-CBM16-PorSS) favors the release of DP4, mainly in microgels but also in solution, to a higher extent than the construction without CBM (ZgCgkAGH16).
Table 2.
Area ratio of chromatographic peaks corresponding to DP4 and DP6 in Fig. 4
ΔM/S corresponds to the ratio of DP4/DP6 obtained in microgels versus solutions. Maximum values for ZgCgkAGH16 and ZgCgkAGH16-CBM16-PorSS are underlined.
|
ZgCgkAGH16 |
ZgCgkAGH16-CBM16-PorSS |
|||||
|---|---|---|---|---|---|---|
| DP4/DP6 |
ΔM/S | DP4/DP6 |
ΔM/S | |||
| Solution | Microgel | Solution | Microgel | |||
| 15 min | 0.68 | 1.53 | 2.24 | 0.90 | 1.50 | 1.66 |
| 45 min | 0.85 | 1.79 | 2.12 | 1.04 | 1.90 | 1.83 |
| 90 min | 1.19 | 2.26 | 1.89 | 1.13 | 2.33 | 2.06 |
| 24 h | 1.37 | 2.68 | 1.95 | 1.49 | 4.17 | 2.80 |
| 1 week | 1.54 | 2.90 | 1.88 | 2.25 | 11.12 | 4.94 |
Crystal structure of the catalytic module of Z. galactanivorans κ-carrageenase (ZgCgkAGH16)
ZgCgkAGH16 was crystallized, and the three-dimensional structure was determined at 1.66 Å resolution (Fig. 2B) by molecular replacement, using the native PcCgkAGH16 structure as a model (PDB code 1DYP) (12). The asymmetric unit contains four copies of the protein, covering the same sequence from Gln30 to Ser307. The RMSD values, calculated for C-α atoms, between the four copies are low, ranging from 0.21 to 0.31 Å, and throughout the following, we will thus refer only to molecule A, except if otherwise specified. B factors of the overall chain (C-α and lateral chains) have been calculated for the different fingers defined upward as the extensions of β-strands and/or loops that flank the catalytic tunnel (Table 3). All further data statistics are summarized in Table 4.
Table 3.
Delimitation of structural “fingers” of the two catalytic modules of κ-carrageenases from Z. galactanivorans (5OCR) and P. carrageenovora, mutant E168D in complex with κ-neocarratetraose (5OCQ) and also the apoenzyme (1DYP), and associated values of B factor
B factor values that vary significantly from values of the full sequence are underlined.
| Finger | ZgCgkAGH16 | B factor | PcCgkAGH16-E168D | B factor | PcCgkAGH16 | B factor |
|---|---|---|---|---|---|---|
| Full sequence | 26.0 | Full sequence | 12.6 | Full sequence | 10.3 | |
| F1 | Trp62–Trp76 | 30.8 | Trp57–Trp69 | 6.2 | Trp57–Trp69 | 8.3 |
| F2 | Arg92–Tyr106 | 11.3 | Arg92–Tyr106 | 12.4 | ||
| F3 | Asp145–Val156 | 24.9 | Thr148–Gln160 | 22.4 | Thr148–Gln160 | 12.8 |
| F4 | Phe168–Asp177 | 32.3 | ||||
| F5 | Lys190–Lys201 | 28.5 | Val187–Gly198 | 25.3 | Val187–Gly198 | 14.6 |
| F6 | Lys264–Ala276 | 49.6 | Ala261–Pro273 | 11.2 | Ala261–Pro273 | 9.1 |
Table 4.
Data collection and refinement statistics for PcCgkAGH16-E168D mutant and ZgCgkAGH16 crystals
| PcCgkAGH16-E168D | ZgCgkAGH16 | |
|---|---|---|
| Data collection | ||
| ESRF beamline | ID14-1 | ID29 |
| Wavelength ( Å) | 0.99 | 0.97 |
| Space group | P212121 | P1 |
| Unit cell parameters | ||
| a, b, c (Å) | 62.63, 67.39, 159.44 | 40.96, 83.04, 85.71 |
| α, β, γ (degrees) | 90.00, 90.00, 90.00 | 72.60, 88.30, 89.50 |
| Vm | 2.69 | 2.22 |
| Solvent (%) | 54.4 | 44.7 |
| Resolution rangea | 79.0–1.710 (1.76–1.70) | 81.7–1.61 (1.70–1.61) |
| No. of observations | 268,064 | 261640 |
| No. of unique reflections | 74,710 | 132,172 |
| Rmerge (%) | 7.1 (15.5) | 0.052 (0.54) |
| Mean I/s(I) | 14.5 (6.2) | 11.97 (1.7) |
| Redundancy | 3.6 (3.4) | 1.98 |
| Completeness (%) | 99.6 (99.8) | 93.9 (91.0) |
| Molecular replacement (native κ-carrageenase) | ||
| Rfactor (%) | 37.0 | 58.2 |
| Correlation | 60.0 | 38.8 |
| Molecules/asymmetric unit | 2 | 4 |
| Refinement | ||
| Resolution range | 79.06 − 1.70 | 81.74 − 1.66 |
| Resolution | 1.70 | 1.66 |
| No. of unique reflections | 70358 | 114681 |
| Rfactor (%) | 14.6 | 16.9 |
| Rfree (%) | 17.7 | 20.5 |
| No. of protein atoms per molecule A; B; C; D (mean B factor in Å2) | 2252; 2311 (12.6) (9.8) | 2411; 2399; 2395; 2437 (26.0) (29.5) (30.6) (27.3) |
| No. of solvent atoms (mean B factor (solvent) in Å2) | 585 (24.40) | 793 (35.70) |
| Mean B factor (ligand) in molecule A;B in Å2 | 16.70; 11.30 | |
| RMSD | ||
| Bond length (Å) | 0.012 | 0.019 |
| Torsion angle (degrees) | 1.3 | 2.0 |
| Mean overall B factor (Å2) | 12.84 | 28.90 |
| Ramachandran plot (%) | ||
| Most favored | 97.06 | 95.5 |
| Additional allowed | 2.76 | 4.21 |
| Outliers | 0.18 | 0.29 |
| PDB code | 5OCQ | 5OCR |
a Values in parenthesis are for the high-resolution shell.
Crystal structure of the inactivated catalytic module of the κ-carrageenase from P. carrageenovora (PcCgkAGH16-E168D) in complex with a κ-neocarratetraose
To identify the key residues involved in the interaction with the sulfated substrate, we designed an inactive mutant of the κ-carrageenase from P. carrageenovora (PcCgkAGH16-E168D) to trap a stable enzyme-substrate complex. As a member of the GH16 family, PcCgkA possesses the characteristic catalytic triad EXDXXE responsible for the double displacement mechanism that results in the cleavage of the β-1,4 linkage with retention of the anomeric configuration (26–28). Glu163 is the nucleophile residue that binds the d-galactose 4-sulfate (G4S) unit at C1 to form the glycosyl–enzyme intermediate, and Glu168 is the acid/base catalyst. Asp165 is also involved in the mechanism by accelerating the deglycosylation step (12). Site-directed mutation of the catalytic Glu168 to aspartate resulted in a significant loss of enzymatic activity, although not abolished totally (data not shown). This allowed the co-crystallization of the recombinant enzyme in the presence of a mixture of κ-carrageenan oligosaccharides, purified from the digestion of κ-carrageenan polymer with PcCgkAGH16. The crystals obtained allowed us to solve the three-dimensional structure at 1.7 Å resolution (Fig. 2C) by molecular replacement again using the native PcCgkAGH16 as a model. The asymmetric unit contains two copies of the protein, covering residues from Met28 to Val297. The Fo − Fc difference density for the complex crystal that we obtained unambiguously showed a tetrasaccharide spanning the active-site cleft in subsites from −4 to −1 (subsite numbering according to Davies et al. (29)), and the oligosaccharide could readily be built into the density (Fig. 5A). 19 residues have been identified as involved in substrate binding (Fig. 5B). Bonding distances are listed in Table 5. 3,6-Anhydro-d-galactose (DA) moieties in subsites −4 and −2 interact with 3 and 5 lateral chains of the catalytic tunnel residues, respectively. Comparatively, G4S moieties in subsites −3 and −1 show the highest number of interactions with the enzyme, 12 and 13, respectively. Remarkably, Arg260 is involved in five interactions in −1 subsite, and of the 19 interacting residues, the three arginines are responsible for 12 of the 32 total interactions between the enzyme and its substrate. It can also be emphasized that the two sulfate functions of the substrate are particularly implicated in the network of hydrogen bonding, as underlined in Fig. 5B.
Figure 5.

Structural representations of the substrate–enzyme interactions in PcCgkAGH16-E168D. A, Fourier difference map (Fo − Fc) displaying the electron density (blue mesh) in the active site at a level of 3.5 σ that indicates the presence of the DP4 oligosaccharide (stick representation with the same color code as in Fig. 2). The catalytic sub-binding sites are numbered from −4 to −1. The polypeptide chain is represented as a purple schematic. B, schematic representation of the enzyme-substrate complex (produced with ChemDraw). All residues that interact with the substrate molecule are displayed. Possible hydrogen bonds are represented in dashes, and those established with sulfate groups are underlined in yellow, as well as the labels of the corresponding residues; water molecules are drawn as spheres; hydrophobic interactions are symbolized with arcs; the mutation E168D is preceded by an asterisk; the three arginine residues that were mutated into alanines are identified by a star; the three catalytic residues are labeled in red, and red waves symbolize the hydrogen bonds that are subsequently formed during the catalytic cleavage of the glycosidic linkage. C, schematic representation of PcCgkAGH16-E168D, showing the positions of the amino acids mutated into alanine, with respect to the catalytic sub-binding sites. The positions of residues Trp266 and Gln171 are on either side of the subsite +1. D, superposition of PcCgkAGH16 (PDB code 1DYP) in gray and PcCgkAGH16-E168D in purple, in complex with a neocarratetraose in light gray (DA-G4S-DA-G4S) (PDB code 5OCQ).
Table 5.
Close contacts (<3.6 Å) between κ-neocarratetraose and amino acid residues or water molecules in the crystal structure of PcCgkAGH16-E168D
| Oligosaccharide |
Interacting atoms | Distance | |
|---|---|---|---|
| Residues | Atoms | ||
| Å | |||
| DA −4 | O6 | Gln102 NE2 | 3.1 |
| O5/O2 | Arg151 NH1 | 2.6/3.2 | |
| Hydrophobic | Trp95 | ||
| G4S−3 | O4S1O | Trp95 NE2/H2O344 | 2.9/2.8 |
| Gln100 NE2/H2O344 | 3.5/2.8 | ||
| Gln270 OE1/H2O116/H2O344 | 2.7/2.71/2.8 | ||
| Cys268 OC1/H2O116/H2O344 | 2.9/2.71/2.8 | ||
| O4S3O | Trp194 NE1 | 3.0 | |
| Arg151 NH1/NH2 | 2.9/2.9 | ||
| O3 | Arg151 NH1 | 3.0 | |
| O4S2O | Asn269 OD1 | 3.2 | |
| O2 | Glu62 OE1/H2O343/H2O449 | 2.6/2.6/2.8 | |
| Asn63 OC1/H2O478 | 3.5/3.1 | ||
| O6 | Arg196 NH1/H2O322 | 2.9/3.0 | |
| Asn269 OC1 | 2.6 | ||
| DA−2 | O6 | Glu62 OE1/H2Ot343 | 2.6/2.8 |
| Hydrophobic clamp | Tyr64 OH/H2O14/Tyr146 OH | 2.8/2.8 | |
| O5 | Arg196 NH1/H2O322 | 2.9/3.0 | |
| O1 | Tyr161 OH | 3.5 | |
| G4S−1 | O2 | Arg196 NH2 | 3.0 |
| Glu163 OE1 | 2.9 | ||
| O4S3O | Arg196 NH2/H2O322/H2O95 | 2.9/3.0/2.9 | |
| O4S2O | Arg260 NH1 | 2.9 | |
| O4S1O | Arg260 NH2/H2O77/H2O109 | 3.0/2.8/2.8 | |
| O4 | Arg260 NH2/H2O331 | 2.5/2.4 | |
| O5/O6 | Arg260 NH2/H2O331 | 2.5/2.5–2.5 | |
| O6 | Arg260 NH2/H2O77 | 3.0/2.9 | |
| Asp168 OD1 | 2.6 | ||
| O5 | Asp168 OD1/H2O373 | 2.3/2.5 | |
| O1 | Asp168 OD1/H2O373 | 2.3/2.6 | |
| His183 NE2 | 3.1 | ||
| Hydrophobic | Trp144 | ||
By comparison with the available structure of the enzyme without substrate (PDB code 1DYP), we can notice that the general fold of the enzyme displays no significant difference (RMSD = 0.48 and 0.46 Å for molecules A and B, respectively). However, some side chains show translational movements, between 1.2 and 1.6 Å, and others show marked rotations, between 90 and 180° (Fig. 5D). The residues affected by these movements are mainly located in finger F2 above subsite −4 of the catalytic channel for the translational movements, namely Trp95, Gln100, and Gln102 (straight arrows in Fig. 5D), and in fingers F5-F6, which constitute the closed part of the tunnel between the −2 and −1 subsites, for the rotational movements (curved arrows). In particular, Arg196 and Asn269 adopt a “closed” conformation in the presence of the substrate, with their functional groups pointing into the channel, with an overall displacement of 3.6 Å when compared with the structure without substrate. Interestingly, Arg151 adopts two alternative conformations when the substrate is present as compared with the apo-structure of the enzyme, one pointing toward the −4 subsite and the second pointing toward the −3 subsite (Fig. 5C). The B factor of this arginine (20.0 Å2) is also higher than the overall mean B factor.
Structural comparison of ZgCgkAGH16 and PcCgkAGH16-E168D
As a member of the GH16 family, ZgCgkAGH16 adopts a β-jelly roll fold. Similar to PcCgkAGH16-E168D, the catalytic channel of ZgCgkAGH16 is partially closed, forming a tunnel, constituted by the junction of fingers F5 and F6 (Fig. 6A) through Arg199 and Asn272, the equivalent of Arg196 and Asn269 in PcCgkAGH16-E168D (supplemental Fig. S4). Despite the conservation of the overall fold, four main structural differences can be emphasized when superposing their structures (Fig. 6B).
Figure 6.

Structural comparison of the catalytic modules of PcCgkAGH16-E168D and ZgCgkAGH16 and their interactions with κ-neocarratetraose. A, schematic representation of the overall structure with the six identified fingers numbered from F1 to F6; the catalytic triad is identified with red letters; the substrate is symbolized by the orange arrow oriented from its non-reducing (NR) to its reducing (R) end; the specific fingers F2 and F4 are identified with a purple and a blue ring, respectively. B, ribbon representation of the two superposed structures with the oligosaccharide in orange, the catalytic triad in red, and the specific structural features of both enzymes in purple for PcCgkAGH16-E168D and blue for ZgCgkAGH16. C, ZgCgkAGH16 surface representation with its specific structural features in blue (top) superimposed with the oligosaccharide trapped in PcCgkAGH16-E168D in orange and PcCgkAGH16-E168D surface representation with its specific structural features in purple (bottom) and the oligosaccharide in orange. The tilt of the positive substrate binding sites by 45° is highlighted in orange. (+) and (−) indicate the regions of the respective subsites. D, schematic representation of the interactions between the catalytic subsites of ZgCgkAGH16 (blue) and PcCgkAGH16-E168D (purple) with a DP8 κ-carrageenan; the red dashed arrow symbolizes the cleavage site; white arrows symbolize the proposed direction of enzymatic processivity.
At first, substantial differences concern the extremity of the catalytic channel containing the negative subsites (−4 and −3 subsites). In PcCgkAGH16-E168D, the top finger F2 establishes a direct link to the substrate molecule through three amino acids, Trp95, Gln100, and Gln102. Residues of this finger also take part, together with finger F1, in binding a dense network of 34 water molecules that wraps around the oligosaccharide bound in the catalytic channel from subsite −4 to +2 (supplemental Fig. S5). Finger F2 is absent in ZgCgkAGH16, as well as Cys98 and Cys268, which form the disulfide bond between F2 and F6 in PcCgkAGH16-E168D. The dense water molecule network, described above, is also far from being as extended as that in ZgCgkAGH16, being confined to only seven water molecules located between subsites −1 and +2. It can also be noticed that the key basic residue Arg151 in PcCgkAGH16-E168D, located on finger F3, which adopts two alternative conformations, is replaced by an aromatic residue (Tyr148) in ZgCgkAGH16.
The second main difference between both enzymes is the presence of an additional finger, F4, at the extremity of the tunnel forming positive subsites for ZgCgkAGH16, in the place of a key residue, Lys172, identified in PcCgkAGH16-E168D (12). This finger F4 is composed of the loop Phe168–Asp177 and contains a combination of aromatic and charged residues (Fig. 2A). Notably, Trp170 forms a hydrophobic platform that extends the end of the binding cleft (supplemental Fig. S6). The distance of 2.8 Å between Asp169 in finger F4 and Lys201 in finger F5 is compatible with a hydrogen bond. Altogether, these features partially obstruct the base level of the tunnel exit, where the positive subsites are located in PcCgkAGH16-E168D, forcing the substrate into an orientation at 45° with respect to the substrate chain orientation on the side of negative subsites (Fig. 6C).
A third difference is the presence of a helix (α3 in Fig. 2A) on the very top of ZgCgkAGH16, in the prolongation of finger F6, just above the positive binding subsites of the catalytic cleft (Fig. 6B). Notably, two lysine residues, Lys284 and Lys288, contribute to a basic patch, together with Lys264, located just between this helix and finger F4. Altogether, finger F4, the top helix, and the charged side chains, create a unique basic environment located at the positive subsites of ZgCgkAGH16 (supplemental Fig. S6).
A fourth major difference between the two catalytic modules is the increased values of B factors of fingers F1 and F6 in ZgCgkAGH16 compared with those of PcCgkAGH16-E168D (Table 3 and supplemental Fig. S4). For finger F1, the relative value of the B factor with respect to the overall value changes by 49% in PcCgkAGH16-E168D (81% in PcCgkAGH16) and up to 119% in ZgCgkAGH16. This is associated with an extension of finger F1 by two residues in ZgCgkAGH16, resulting in a spatial displacement with respect to the position in PcCgkAGH16-E168D. Notably, Asn70 is oriented toward the −3 subsite with a rotation of 45° in contrast to the equivalent Asn63 in PcCgkAGH16-E168D, which points in the opposite direction (supplemental Fig. S4). Concerning finger F6, the relative B factor value changes by 89% in PcCgkAGH16-E168D (88% in PcCgkAGH16) and by 190% in ZgCgkAGH16 with respect to the overall B value. This increased mobility is especially marked for residue Asn272, which is equivalent to Asn269 of PcCgkAGH16-E168D that was previously identified for its implication in forming the tunnel above the catalytic site. We note that the side chain of this residue in ZgCgkAGH16 is in the “open” position, as compared with what is observed in the apo-structure of PcCgkAGH16 (PDB code 1DYP), and that the associated Arg199 (the equivalent of Arg196 in PcCgkAGH16) adopts two alternative conformations in the crystal structure of ZgCgkAGH16, one in the open and one in the closed position.
Site-directed mutagenesis of PcCgkAGH16
The structure of PcCgkAGH16-E168D in complex with a κ-neocarratetraose allowed the identification of 19 amino acids directly involved in the interaction with the substrate (listed in Table 5) in addition to some other residues more distant and involved in the network of water molecules. Six of these residues have been successfully mutated individually into alanine (Fig. 5C). The recombinant mutated proteins were purified and biochemically characterized in solution on κ-carrageenan substrate. Details of the determination of kinetic parameters are provided in supplemental Table S1.
Depending on the position of the mutated amino acid, the effect on the catalytic efficiency is very different (Table 1). Indeed, R196A, R260A, and W266A cause a dramatic loss of catalytic efficiency, between 95 and 99%, as compared with the wild-type enzyme.
For Arg196 and Arg260, the loss of activity can be explained by their key role in binding to the charged sulfate groups of G4S in the −1 subsite. The importance of both of these residues and especially Arg260 in substrate binding was already detected by Michel et al. (12), based on a structural model of a docked substrate molecule in the catalytic active site of the native κ-carrageenase. It is noteworthy that these basic residues are strictly conserved in all of the sequences of κ-carrageenases, with a few exceptions, where Arg196 is replaced by a lysine, and thus must play an important role in the specificity of recognition of the substrate in this GH16 subfamily. In the case of our enzyme-substrate complex PcCgkAGH16-E168D, we show that the importance of Arg196 is connected to its involvement in shaping the tunnel above the catalytic active site and its concerted interaction with Asn269 in the binding of the substrate in subsites −1 and −2 (Table 5 and Fig. 5B). We also confirm the crucial importance of Arg260 in stabilizing the substrate in subsite −1, notably through the interactions with the sulfate group, which is the hallmark of κ-neocarrabiose (Table 5). As a consequence, the remaining activity of the protein with Arg260 mutated to alanine was <1% that of the wild-type enzyme, and kinetic parameters were not determined. By contrast, Trp266 is positioned in the catalytic groove, where the positive substrate-binding sites must be, and consequently does not interact with the substrate molecule in the crystal structure of this substrate-enzyme complex. Nevertheless, the drastic loss of activity when mutated to alanine clearly indicates that it plays an important role in the interaction with the saccharide units that bind to the positive sub-binding sites.
Surprisingly, R151A and Q171A have an opposite effect, causing an increase of efficiency by 1.4–1.8 times, respectively (Table 1). Arg151 is located between subsites −4 and −3 and establishes strong interactions with the substrate at the entrance of the catalytic groove, through a salt bridge and direct hydrogen bonds. On the other hand, Gln171 is located in the putative +1 sub-binding site, where it could establish hydrogen bonds with the leaving group G4S. This residue is highly conserved in κ-carrageenases, so it may be linked with the thermodynamic equilibrium of enzymatic reaction. When mutated into alanine, the interaction with the leaving group is strongly decreased, allowing a more rapid product release into the medium and thus accelerating the enzymatic turnover.
R92A is the only mutated residue not directly in contact with the substrate but involved in binding water molecules at the entrance of the channel (supplemental Fig. S5). The absence of any effect on catalytic efficiency when mutating Arg92 shows that this arginine does not seem to play a major role in substrate binding or mode of action, at least on soluble substrates.
Discussion
Although family GH16 κ-carrageenases are frequently identified in macroalgae-associated bacteria (3, 30), only seven enzymes have actually been produced (11, 17–22), and only one has been structurally characterized to date (12). In polyspecific GH families, such as the GH16 family, the subfamilies based on phylogenetic analyses usually correspond to distinct substrate specificities (11, 29, 31). Within a specific subfamily, the large diversity of sequences and the phylogenetic distribution in clades is rather related to differences in modes of action or subtle differences within the same substrate specificity (32). It is important to mention that carrageenans occur as hybrid polymers within the cell walls of red algae, containing numerous variant motifs beyond the frequent repeating units κ- and ι-carrabioses (e.g. the precursor units μ- and η-carrabiose and the desulfated units β- (33) and α-carrabiose (34, 35)). These complex structures are further modified by the addition of methyl or pyruvate groups. In this context, one can expect significant differences between κ-carrageenases, and the results presented here are the first example of exploring this biochemical diversity.
The first obvious difference between PcCgkA and ZgCgkA is the modular architecture of these κ-carrageenases. Even if both are secreted enzymes, only ZgCgkA possesses a CBM (11, 36). The presence of CBMs appended to catalytic GH modules is in general related to the capacity of the adjacent enzyme to tackle recalcitrant, complex substrates in the solid form, as encountered in the context of the plant cell wall (37–39). Four main roles are attributed to CBMs: 1) to allow proximity between the catalytic domain and its substrate; 2) to target the catalytic module to specific parts of the cell wall; 3) to help disrupt the substrate organization; and 4) to anchor the enzyme to the bacterial cell wall (40, 41). The isolated CBM16 module of ZgCgkA was cloned and expressed as a recombinant protein in an independent study and was indeed shown to bind κ-carrageenan in ELISA tests,3 indicating that this secreted enzyme most probably degrades semi-crystalline κ-carrageenans within the algal cell wall.
Moreover, contrary to PcCgkA, DP6 is not hydrolyzed by ZgCgkA, which confirms what Potin et al. (15) suggested, that is to say DP8 is the minimal size of oligosaccharide that can be degraded by ZgCgkA. According to the displacement scheme proposed by Lemoine et al. (42) for PcCgkA, DP6 is exclusively produced by random processing, whereas DP4 is produced by random and processive modes of action. Similarly, we propose to use the DP4/DP6 ratio as a marker of processivity for ZgCgkA. In this study, comparison of behavior of recombinant ZgCgkA with and without CBM, and also on microgels compared with κ-carrageenan solutions, suggests that ZgCgkA is an endo-processive enzyme, mainly on microgel, and that its appended CBM16 contributes to the processivity, even on soluble substrate, probably by favoring and maintaining the proximity between the catalytic module and substrate. The difference in processivity also explains the difference in catalytic efficiency observed with and without CBM16, due to the fact that the selective advantage of processivity on insoluble substrate is in balance with the loss of catalytic efficiency on soluble substrate (43). This phenomenon has already been observed for processive endoglucanases, where deletion of the CBM associated with the catalytic module leads to an increased activity on soluble substrate (44).
Dissection of the molecular details of protein–carbohydrate interactions within the catalytic active site is crucial to understand the enzymatic efficiency, the mode of action, and the substrate specificity of a given enzyme class. From our structural and mutagenesis study, we can establish several key parameters in the κ-carrageenase subfamily.
Importance of arginine residues
First emphasized in the 1970s, with the study of carboxypeptidase A (45), the positive charge and basic character of arginines allow them to establish salt bridges and/or multiple hydrogen bonds with negatively charged substrates like phosphate groups (46) or sulfated polysaccharides, as observed in the case of chondroitin lyase (47).
Importance of enzymatic flexibility and substrate binding in catalytic efficiency
Loss of finger F2 and the stabilizing disulfide bond between F2 and F6, as well as the associated water network, result in a more flexible F6 finger that correlates well with the 5-fold increase of enzymatic efficiency of ZgCgkAGH16 in solution compared with PcCgkAGH16. Interestingly, the mobility of fingers also seems to be inversed between both enzymes, because finger F6 at the top of the groove in ZgCgkAGH16 has 2 times higher B factors than overall, whereas fingers F3 and F5 of PcCgkAGH16 display 2 times higher than average values (Table 3). Moreover, the units at the reducing end of the oligosaccharide might be more tightly bound in ZgCgkAGH16 (at least four positive sub-binding sites constituted by fingers F4-F5-F6 than in PcCgkAGH16 (two positive sub-binding sites: fingers F5–F6), and less tightly bound at the non-reducing end, with only three negative sub-binding sites (fingers F1–F3) instead of four (fingers F1-F2-F3). These findings, along with the experiments on microgels, lead us to assume that ZgCgkAGH16 is an endoprocessive enzyme with at least seven sub-binding sites, moving from the reducing end toward the non-reducing end of a κ-carrageenan chain, movement opposite to that proposed for PcCgkAGH16 (Fig. 6D) (12).
Requirements for processivity on charged substrates
Processivity necessitates a compromise between affinity and mobility, which can be fulfilled by a combination of basic and mobile residues, such as arginines and lysines, associated with aromatic residues (Arg151/Trp95 for PcCgkAGH16; Lys201/Trp170 for ZgCgkAGH16). The alternative conformations of the side chain of Arg151 observed in presence of the ligand, together with the relative high B factor of this residue lead us to assume that it might assist the establishment of a first contact to the substrate, “grabbing” a sulfate group, but also assist in sliding the substrate further, after a catalytic cleavage. We can further assume that the mutation of Arg151 into an alanine induces a weakening of the substrate binding at the non-reducing end of the polysaccharide chain, possibly hindering the processivity and thus leading to an increase of the catalytic efficiency on a soluble substrate. The effect of this point mutation can thus be compared with what is observed for the processive cellobiohydrolase of T. reesei, where the deletion of one of the active-site loops leads to an increased efficiency of the enzyme on amorphous cellulose (48).
Importance of subsites associated with the leaving group
A balance is needed between the necessity of maintaining the stability of the enzyme-substrate complex before cleavage and facilitating the release of the product. In PcCgkAGH16, Gln171 is possibly involved in a process defined as “product inhibition” by binding the product tightly to the enzyme, a feature that is frequently observed in processive enzymes active on crystalline substrates, such as cellulases or chitinases (43, 49). We can assume a similar function for Asn70 in ZgCgkAGH16. Conversely, Trp266 might facilitate the sliding of the released product, thus leading to a loss of enzymatic efficiency when mutated into alanine. Indeed, hydrophobic amino acids and tryptophans in particular are well known for their role in facilitating the sliding of oligosaccharide substrates in glycoside hydrolases (50). In ZgCgkAGH16, an equivalent residue could be Phe146.
To summarize, our data indicate that despite sharing a global common ancestor, PcCgkAGH16 and ZgCgkAGH16 have evolved unique structural features, which shape distinct tunnel topologies and probably result in opposite directions of processivity. Interestingly, from the point of view of industrial applications, the enzyme from Z. galactanivorans is 5 times more efficient in solution. The two GH16 enzymes described here are representatives of different clades of κ-carrageenases (clades A and C, respectively; Fig. 3). Whereas clade C enzymes clearly originate from marine Bacteroidetes and clades A and D from Proteobacteria, we cannot currently determine whether the common ancestor of clade B κ-carrageenases belonged to the Bacteroidetes or Planctomycetes phylum. The exact taxonomic nature of the common ancestor of the entire κ-carrageenase subfamily is even more difficult to establish, considering the low bootstrap values of the deep nodes of the remaining sequence clusters. Remarkably, some marine bacteria have multigenic families of κ-carrageenases, such as, for example, Algibacter sp. SK-16 that possesses three clade B sequences, one clade C sequence, and two other sequences (including the current deepest sequence of the κ-carrageenase subfamily). This diversity of enzymes active on κ-carrageenan within one organism most likely results from various evolutionary events, such as horizontal gene transfers or sequence duplications followed by diverging evolution. The presence of this multigenic family might also give an adaptive advantage to this species to face the chemical complexity of carrageenans, perhaps also indicating that this organism potentially adopts a strategy even different from that of Z. galactanivorans and P. carragenovora for the degradation of this abundant marine polysaccharide.
Experimental procedures
Bioinformatics analysis
Homologous sequences have been extracted with the BLASTp suite from the NCBI server (51) (https://blast.ncbi.nlm.nih.gov) from the non-redundant protein sequence database, manually curated before being aligned with the MAFFT program available at http://www.ebi.ac.uk/Tools/msa/mafft/ and edited with BioEdit free software (52) available at http://www.mbio.ncsu.edu/BioEdit/bioedit.html to eliminate all of the insertion areas before calculation of the phylogenetic tree with MEGA software (http://www.megasoftware.net/).4 Bootstrap values inferior to 50% do not appear on the tree.
Figures of multiple alignment were done using the ESPript version 3.0 server (53) available at http://espript.ibcp.fr/ESPript/ESPript/index.php from the MAFFT alignment, edited with BioEdit when necessary to fit the structural data.4
Cloning of the two forms of κ-carrageenases of Z. galactanivorans in E. coli
Two different forms of the gene cgkA (zobellia_236) encoding the κ-carrageenase from Z. galactanivorans have been cloned. Briefly, primers (sequences provided in supplemental Table S2) were designed to amplify by PCR, from Z. galactanivorans genomic DNA, the coding regions corresponding to the complete sequence of the κ-carrageenase without the signal peptide sequence in the N-terminal position (ZgCgkAGH16-CBM16-PorSS) and to the catalytic module alone (ZgCgkAGH16). After digestion with the restriction enzymes BamHI and PstI, the purified PCR products were ligated using T4 DNA ligase into the expression vector pFO4 predigested by a compatible couple of restriction enzymes. The recombinant proteins encoded in the plasmids pZG237 and pZG238 correspond to the peptide sequences Gln30–Lys306 (ZgCgkAGH16) and Gln30–Glu546 (ZgCgkAGH16-CBM16-PorSS), respectively, with an added N-terminal hexahistidine tag. The plasmids were transformed into the E. coli BL21 (DE3) strain for protein expression.
Mutagenesis of κ-carrageenase from P. carrageenovora
The plasmid pCGK that was obtained by cloning the native κ-carrageenase gene into the C-terminal His tag encoding expression plasmid pET20b (Invitrogen) was subsequently used as a template for mutagenesis using the QuikChange mutagenesis kit (Stratagene). The primers used to amplify the different mutants are summarized in supplemental Table S2. The plasmids obtained were used to transform the E. coli C43 (DE3) strain for protein expression.
Expression and purification of the recombinant enzymes
A single colony containing the desired plasmid (pCGK or pZG) was used to inoculate LB medium (supplemented with 100 μg/ml ampicillin). This preculture was incubated at 37 °C and was then used to inoculate ZIP5052-autoinducing medium for cell growth at 17 °C. The culture was stopped after 2 days when A600 had reached 15 and pelleted (30 min). If needed, the pellet was frozen and stored at −20 °C before use. The bacterial pellet was lysed by means of a French press after resuspension in buffer A (Tris-HCl (10 mm, pH 7.2, for P. carrageenovora enzymes or 50 mm, pH 7.5, for Z. galactanivorans enzymes), 500 mm NaCl, 40 mm imidazole), with an anti-protease mixture (Complete EDTA-free, Roche Applied Science), DNase (0.1 mg/ml), and the addition of lysozyme (0.2 mg/ml). The supernatant after lysis containing the soluble proteins was separated from the pellet by centrifugation (20,000 × g for 30 min at 4 °C) and then filtered before the two-step purification by nickel affinity chromatography (equilibrated with buffer A) and size exclusion chromatography on a Sephadex column (Amersham Biosciences). Elution on immobilized metal ion affinity chromatography was done with a gradient of buffer B, which differed from buffer A only in the concentration of imidazole, which was 250 mm for P. carrageenovora enzymes and 500 mm for Z. galactanivorans enzymes. Size exclusion chromatography was done with a flow rate at 1 ml/min in buffer C (50 mm Tris-HCl, pH 7.2, 150 mm NaCl for P. carrageenovora enzymes and 10 mm Tris-HCl, pH 7.2, 250 mm NaCl for Z. galactanivorans enzymes). The different fractions were analyzed by SDS-PAGE. Those containing the protein purified to homogeneity were pooled and concentrated by ultrafiltration on a styrene acrylonitrile membrane (10- or 30-kDa cutoff) (Millipore) to the desired concentration for biochemistry analysis and to 6.9 and 5 mg/ml for crystallogenesis of PcCgkAGH16-E168D and of ZgCgkAGH16, respectively.
Crystallization of PcCgkAGH16-E168D and ZgCgkAGH16, structure determination, and refinement
In the first step, crystallization conditions were screened for using the commercial kits PACT and JCSG+ (Qiagen), dispensed by a robot (200 nl of protein solution mixed with 100 nl of reservoir solution). For both proteins, initial crystallization conditions identified from these screens were then manually optimized. Single crystals of suitable size were obtained for PcCgkAGH16-E168D in complex with substrate as follows. 200 μl of enzyme at 6.9 mg/ml were supplemented with 2 mg of a purified mix of κ-carrabiose, tetraose, and hexaose (30:60:10, w/w/w), obtained from the digestion of κ-carrageenan by PcCgkAGH16. Of this solution, 2 μl were mixed with 2 μl of reservoir solution containing 1.0 m sodium citrate and 100 mm cacodylate at pH 6.5, in hanging drops equilibrated against 500 μl of reservoir solution at 19 °C. Single crystals of ZgCgkAGH16 were obtained from 2 μl of the enzyme solution (5.0 mg/ml) that were added to 1 μl of reservoir solution containing 28–29% of PEG 3350, 100 mm MES buffer, pH 6.5, and 0.3 m NaNO3, in hanging drops equilibrated against 250 μl of reservoir solution at 19 °C. Before flash-freezing in a nitrogen stream at 100 K, single crystals were quickly soaked in their respective crystallization solution supplemented with 20% glycerol. Diffraction data for PcCgkAGH16-E168D complex crystals were collected on beamline ID14-1, and data for ZgCgkAGH16 were collected on beamline ID29 (ESRF, Grenoble, France). X-ray diffraction data were integrated using Mosflm and scaled with SCALA (54). Both structures of ZgCgkAGH16 and PcCgkAGH16-E168D in complex with κ-carratetraose were determined by molecular replacement with MolRep (55) using chain A of the PcCgkAGH16 native structure (PDB code 1DYP) as a starting model. The structure of ZgCgkAGH16 was then manually corrected and built using COOT (56). For both structures, the initial structural models were refined with the program REFMAC5 (57), alternating with cycles of further manual rebuilding using COOT. A subset of 5% randomly selected reflections was excluded from computational refinement to calculate the Rfree factors throughout the refinement. The addition of the ligand sugar units for the complex structure was performed manually using COOT. Water molecules were added automatically with REFMAC-ARP/wARP and visually verified. All data collection and refinement statistics are presented in Table 1. The structure obtained for the ligand tetrasaccharide has also been checked with the software Privateer in CCP4 (58).
Enzymatic activity assays on κ-carrageenan by reducing sugar analysis
The processive character of an enzyme can only be clearly observed on a non-soluble substrate (42). However, the determination of kinetic parameters implies a total availability of the substrate to fulfill Michaelis–Menten conditions (59). Thus, we decided to do the biochemical experiments in diluted solutions of κ-carrageenan to have an estimate of the apparent efficiency under similar conditions for all studied enzymes and mutants, although not entirely reflecting their behavior in “natural” conditions (i.e. on solid algal cell walls).
A stock solution of 0.5% (w/v) κ-carrageenan (Danisco) was prepared in 50 mm MOPS, pH 7, 150 mm NaCl for PcCgkAGH16 and its mutants, in Teorell buffer (60) to determine the best range of pH for both construction of ZgCgkAGH16 and then in 50 mm MES, pH 6, 300 mm NaCl for the determination of kinetic constants. Aliquots (14 μl) of enzyme at the appropriate concentration were incubated in triplicate in the presence of κ-carrageenan solution (126 μl) at a final concentration of 0.125% (w/v) at 40 °C. The amount of reducing sugars released was assayed using a ferricyanide method adapted from that of Kidby and Davidson (61). The ferricyanide reagent consisted of 300 mg of potassium hexacyanoferrate III and of 28 g of hydrated Na2CO3 dissolved in 1 liter of distilled water, to which 1 ml of 1 m aqueous NaOH was added. Aliquots (20 μl) of the incubation medium were mixed with 180 μl of ferricyanide reagent, and absorbance was recorded at 420 nm. Reducing sugar concentrations were calibrated using glucose as a standard.
The initial reaction rates of κ-carrageenan hydrolysis were measured at least in triplicate by the ferricyanide reagent method for 10 κ-carrageenan concentrations (1.25, 1.05, 0.85, 0.65, 0.45, 0.30, 0.25, 0.20, 0.15, and 0.13 g/liter). The Michaelis constant (Km) and the reaction rate at infinite substrate concentration (Vm) were calculated with the software Hyper, with the hyperbolic regression model applying weighting. Note that for ZgCgkAGH16, parameters were calculated without data at 0.13 g/liter and for PcCgkAGH16-R196A and PcCgkAGH16-W266A without data at 1.25 and 1.05 g/liter. For PcCgkAGH16-R260A, the concentration of enzyme needed to obtain comparable kinetic curves at 1.25 g/liter of substrate was 100 times higher than for PcCgkAGH16, thus preventing us from calculating kinetic parameters, due to the fact that we were no longer in conditions consistent with the Michaelis–Menten assumption of [S] ≫ [E].
For long time course kinetics, digestions were conducted at 30 °C, in a final volume of 250 μl of 0.2% (w/v) κ-carrageenan solutions, in 10 mm Tris buffer, pH 7.2, 150 mm NaCl. A 3.5 nm concentration of each enzyme was dissolved in the κ-carrageenan solution, or 14 nm in microgels, obtained from the previous solution at 0.2% κ-carrageenan supplemented with 60 mm KCl. Aliquots (20 μl) of the incubation medium were pipetted after 15, 30, 45, 60, 90, 180, and 300 min and mixed with 180 μl of 2.5× ferricyanide reagent and treated the same way as above. Experiments were done in triplicate, and the S.D. was drawn for each spot on the graphics.
HPAEC analysis
Analyses of degradation products generated by the different enzymes were conducted by HPAEC on an ICS-5000 system (Dionex). 20 μl of samples were injected (AS50 Automatic Autosampler, Dionex) onto an AS11 analytical anion-exchange column (4 × 250 mm, IonPac®, Dionex) equipped with an AG11 precolumn (4 × 50 mm, IonPac®, Dionex). Elution was performed at a flow rate of 0.5 ml/min (gradient pump SP-5, Dionex) by NaOH multistep gradients from 8 to 280 mm (40 min). Oligosaccharides were detected in conductivity mode (analytical DC-5000 detector, Dionex) with a suppressor (AERS 500, 4 mm, Dionex) delivering a current from 50 to 300 mA. Acquisition of chromatograms was realized by Chromeleon software version 6.8 (Dionex).
Author contributions
M. M. M., P. N. C., and M. C. designed the study and wrote the paper. R. L., T. Bernard, and M. M.-M. performed the molecular biology experiments. A. J., M. J., T. Bernard, and M. C. performed crystallizations, and T. Bernard, C. L., M. C., and M. M.-M. performed the crystal structure determinations. M. M.-M. performed the biochemical characterization of the recombinant proteins. M. J. and A. P. provided technical assistance for chromatography and HPAEC analysis of the samples. G. M. and T. Barbeyron performed the phylogenetic analysis. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are indebted to local contacts for support during data collection at beamlines ID14-1 and ID29, ESRF (European Synchrotron, Grenoble, France).
This work was supported by ANRT and Olmix group Ph.D. Grant CIFRE-684/2014 (to M. M.-M.). This work was also supported by the French National Research Agency with regard to the investment expenditure program IDEALG (Grant Agreement ANR-10-BTBR-04) and supported by the “Region Bretagne.” The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2 and Figs. S1–S6.
The atomic coordinates and structure factors (codes 5OCQ and 5OCR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. A. Salméan, A. Guillouzo., D. Duffieux, M. Jam, M. Matard-Mann, R. Larocque, H. L. Pedersen, G. Michel, M. Czjzek., W. G. T. Willats, and C. Hervé, manuscript in preparation.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- GH
- glycoside hydrolase(s)
- CBM
- carbohydrate-binding module
- PDB
- Protein Data Bank
- DP
- degree of polymerization
- DA
- 3,6-anhydro-d-galactose
- G4S
- d-galactose 4-sulfate
- HPAEC
- high-performance anion-exchange chromatography
- RMSD
- root mean square deviation.
References
- 1. O'Dowd C. D., Jimenez J. L., Bahreini R., Flagan R. C., Seinfeld J. H., Hämeri K., Pirjola L., Kulmala M., Jennings S. G., and Hoffmann T. (2002) Marine aerosol formation from biogenic iodine emissions. Nature 417, 632–636 [DOI] [PubMed] [Google Scholar]
- 2. Duggins D. O., Simenstad C. A., and Estes J. A. (1989) Magnification of secondary production by kelp detritus in coastal marine ecosystems. Science 245, 170–173 [DOI] [PubMed] [Google Scholar]
- 3. Martin M., Portetelle D., Michel G., and Vandenbol M. (2014) Microorganisms living on macroalgae: diversity, interactions, and biotechnological applications. Appl. Microbiol. Biotechnol. 98, 2917–2935 [DOI] [PubMed] [Google Scholar]
- 4. Larsbrink J., Rogers T. E., Hemsworth G. R., McKee L. S., Tauzin A. S., Spadiut O., Klinter S., Pudlo N. A., Urs K., Koropatkin N. M., Creagh A. L., Haynes C. A., Kelly A. G., Cederholm S. N., Davies G. J., Martens E. C., and Brumer H. (2014) A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grondin J. M., Tamura K., Déjean G., Abbott D. W., and Brumer H. (2017) Polysaccharide utilization loci: fuelling microbial communities. J. Bacteriol. 199, e00860–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bjursell M. K., Martens E. C., and Gordon J. I. (2006) Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J. Biol. Chem. 281, 36269–36279 [DOI] [PubMed] [Google Scholar]
- 7. Martens E. C., Chiang H. C., and Gordon J. I. (2008) Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michel G., and Czjzek M. (2013) Polysaccharide-degrading enzymes from marine bacteria. in Marine Enzymes for Biocatalysis: Sources, Biocatalytic Characteristics and Bioprocesses of Marine Enzymes (Trincone A., ed) pp. 429–464, Elsevier, Amsterdam [Google Scholar]
- 9. Senni K., Pereira J., Gueniche F., Delbarre-Ladrat C., Sinquin C., Ratiskol J., Godeau G., Fischer A. M., Helley D., and Colliec-Jouault S. (2011) Marine polysaccharides: a source of bioactive molecules for cell therapy and tissue engineering. Mar. Drugs 9, 1664–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knutsen S. H., Myslabodski D. E., Larsen B., and Usov A. I. (1994) A modified system of nomenclature for red algal galactans. Botanica Marina 37, 163–170 [Google Scholar]
- 11. Barbeyron T., Gerard A., Potin P., Henrissat B., and Kloareg B. (1998) The κ-carrageenase of the marine bacterium Cytophaga drobachiensis: structural and phylogenetic relationships within family-16 glycoside hydrolases. Mol. Biol. Evol. 15, 528–537 [DOI] [PubMed] [Google Scholar]
- 12. Michel G., Chantalat L., Duee E., Barbeyron T., Henrissat B., Kloareg B., and Dideberg O. (2001) The κ-carrageenase of P. carrageenovora features a tunnel-shaped active site: a novel insight in the evolution of Clan-B glycoside hydrolases. Structure 9, 513–525 [DOI] [PubMed] [Google Scholar]
- 13. Michel G., Helbert W., Kahn R., Dideberg O., and Kloareg B. (2003) The structural bases of the processive degradation of ι-carrageenan, a main cell wall polysaccharide of red algae. J. Mol. Biol. 334, 421–433 [DOI] [PubMed] [Google Scholar]
- 14. Rebuffet E., Barbeyron T., Jeudy A., Jam M., Czjzek M., and Michel G. (2010) Identification of catalytic residues and mechanistic analysis of family GH82 ι-carrageenases. Biochemistry 49, 7590–7599 [DOI] [PubMed] [Google Scholar]
- 15. Potin P., Sanseau A., Le Gall Y., Rochas C., and Kloareg B. (1991) Purification and characterization of a new κ-carrageenase from a marine Cytophaga-like bacterium. Eur. J. Biochem. 201, 241–247 [DOI] [PubMed] [Google Scholar]
- 16. Guibet M., Colin S., Barbeyron T., Genicot S., Kloareg B., Michel G., and Helbert W. (2007) Degradation of λ-carrageenan by Pseudoalteromonas carrageenovora λ-carrageenase: a new family of glycoside hydrolases unrelated to κ- and ι-carrageenases. Biochem. J. 404, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barbeyron T., Henrissat B., and Kloareg B. (1994) The gene encoding the κ-carrageenase of Alteromonas carrageenovora is related to β-1,3–1,4-glucanases. Gene 139, 105–109 [DOI] [PubMed] [Google Scholar]
- 18. Liu G. L., Li Y., Chi Z., and Chi Z. M. (2011) Purification and characterization of κ-carrageenase from the marine bacterium Pseudoalteromonas porphyrae for hydrolysis of κ-carrageenan. Process Biochem. 46, 265–271 [Google Scholar]
- 19. Kobayashi T., Uchimura K., Koide O., Deguchi S., and Horikoshi K. (2012) Genetic and biochemical characterization of the Pseudoalteromonas tetraodonis alkaline κ-carrageenase. Biosci. Biotechnol. Biochem. 76, 506–511 [DOI] [PubMed] [Google Scholar]
- 20. Liu Z., Li G., Mo Z., and Mou H. (2013) Molecular cloning, characterization, and heterologous expression of a new κ-carrageenase gene from marine bacterium Zobellia sp. ZM-2. Appl. Microbiol. Biotechnol. 97, 10057–10067 [DOI] [PubMed] [Google Scholar]
- 21. Wang L., Li S., Zhang S., Li J., Yu W., and Gong Q. (2015) A new κ-carrageenase CgkS from marine bacterium Shewanella sp. Kz7. J. Ocean Univ. China 14, 759–763 [Google Scholar]
- 22. Xu X., Li S., Yang X., Yu W., and Han F. (2015) Cloning and characterization of a new κ-carrageenase gene from marine bacterium Pseudoalteromonas sp. QY203. J. Ocean Univ. China 14, 1082–1086 [Google Scholar]
- 23. Yaphe W., and Baxter B. (1955) The enzymic hydrolysis of carrageenin. Appl. Microbiol. 3, 380–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weigl J., and Yaphe W. (1966) The enzymatic hydrolysis of carrageenan by Pseudomonas carrageenovora: purification of a κ-carrageenase. Can. J. Microbiol. 12, 939–947 [DOI] [PubMed] [Google Scholar]
- 25. Barbeyron T., Thomas F., Barbe V., Teeling H., Schenowitz C., Dossat C., Goesmann A., Leblanc C., Oliver Glöckner F., Czjzek M., Amann R., and Michel G. (2016) Habitat and taxon as driving forces of carbohydrate catabolism in marine heterotrophic bacteria: example of the model algae-associated bacterium Zobellia galactanivorans DsijT. Environ. Microbiol. 18, 4610–4627 [DOI] [PubMed] [Google Scholar]
- 26. Koshland D. E. (1953) Stereochistry and the mechanism od enzymatic reactions. Biol. Rev. Camb. Philos. Soc. 28, 416–436 [Google Scholar]
- 27. Zechel D. L., and Withers S. G. (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res. 33, 11–18 [DOI] [PubMed] [Google Scholar]
- 28. Planas A. (2000) Bacterial 1,3–1,4- l-glucanases: structure, function and protein engineering. Biochim. Biophys. Acta 1543, 361–382 [DOI] [PubMed] [Google Scholar]
- 29. Davies G. J., Wilson K. S., and Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin M., Barbeyron T., Martin R., Portetelle D., Michel G., and Vandenbol M. (2015) The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-polysaccharide-degrading bacteria. Front. Microbiol. 6, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aspeborg H., Coutinho P. M., Wang Y., Brumer H. 3rd, and Henrissat B. (2012) Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stam M. R., Danchin E. G. J., Rancurel C., Coutinho P. M., and Henrissat B. (2006) Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562 [DOI] [PubMed] [Google Scholar]
- 33. Knutser S. H., and Grasdalen H. (1987) Characterization of water-extractable polysaccharides from Norwegian Furcellaria lumbricalis (Huds.) Lamour (Gigartinales, Rhodophyceae) by IR and NMR spectroscopy. Bot. Mar. 30, 497–506 [Google Scholar]
- 34. Falshaw R., Furneaux R. H., Wong H., Liao M. C., Bacic A., and Chandrkrachang S. (1996) Structural analysis of carrageenans from Burmese and Thai samples of Catenella nipae Zanardini. Carbohydr. Res. 285, 81–98 [Google Scholar]
- 35. Usov A. I. (2011) Advances in Carbohydrate Chemistry and Biochemistry. in Polysaccharides of the Red Algae, 1st Ed., pp. 116–219, Elsevier, Amsterdam: [DOI] [PubMed] [Google Scholar]
- 36. Potin P., Richard C., Barbeyron T., Henrissat B., Gey C., Petillot Y., Forest E., Dideberg O., Rochas C., and Kloareg B. (1995) Processing and hydrolytic mechanism of the cgkA-encoded κ-carrageenase of Alteromonas carrageenovora. Eur. J. Biochem. 228, 971–975 [DOI] [PubMed] [Google Scholar]
- 37. Tomme P., Van Tilbeurgh H., Pettersson G., Van Damme J., Vandekerckhove J., Knowles J., Teeri T., and Claeyssens M. (1988) Studies of the cellulolytic system of Trichoderma reesei QM 9414. Eur. J. Biochem. 170, 575–581 [DOI] [PubMed] [Google Scholar]
- 38. Din N., Gilkes N. R., Tekant B., Miller R. C., Warren R. A. J., and Kilburn D. G. (1991) Non-hydrolytic disruption of cellulose fibres by the binding domain of a bacterial cellulase. Nat. Biotechnol. 9, 1096–1099 [Google Scholar]
- 39. Black G. W., Rixon J. E., Clarke J. H., Hazlewood G. P., Theodorou M. K., Morris P., and Gilbert H. J. (1996) Evidence that linker sequences and cellulose-binding domains enhance the activity of hemicellulases against complex substrates. Biochem. J. 319, 515–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilbert H. J., Knox J. P., and Boraston A. B. (2013) Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules. Curr. Opin. Struct. Biol. 23, 669–677 [DOI] [PubMed] [Google Scholar]
- 42. Lemoine M., Nyvall Collén P., and Helbert W. (2009) Physical state of κ-carrageenan modulates the mode of action of κ-carrageenase from Pseudoalteromonas carrageenovora. Biochem. J. 419, 545–553 [DOI] [PubMed] [Google Scholar]
- 43. Horn S. J., Sikorski P., Cederkvist J. B., Vaaje-Kolstad G., Sorlie M., Synstad B., Vriend G., Varum K. M., Eijsink V. G. H., Sørlie M., Synstad B., Vriend G., Vårum K. M., and Eijsink V. G. H. (2006) Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 103, 18089–18094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gilad R., Rabinovich L., Yaron S., Bayer E. A., Lamed R., Gilbert H. J., and Shoham Y. (2003) Cell, a noncellulosomal family 9 enzyme from Clostridium thermocellum, is a processive enduglucanase that degrades crystalline cellulose. J. Bacteriol. 185, 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riordan J. F., and Hayashida H. (1970) Chemical evidence for a functional carboxyl group in carboxypeptidase A. Biochem. Biophys. Res. Commun. 41, 122–127 [DOI] [PubMed] [Google Scholar]
- 46. Riordan J. F., McElvany K. D., and Borders C. L. (1977) Arginyl residues: anion recognition sites in enzymes. Science 195, 884–886 [DOI] [PubMed] [Google Scholar]
- 47. Huang W., Boju L., Tkalec L., Su H., Yang H. O., Gunay N. S., Linhardt R. J., Kim Y. S., Matte A., and Cygler M. (2001) Active site of chondroitin AC lyase revealed by the structure of enzyme-oligosaccharide complexes and mutagenesis. Biochemistry 40, 2359–2372 [DOI] [PubMed] [Google Scholar]
- 48. von Ossowski I., Ståhlberg J., Koivula A., Piens K., Becker D., Boer H., Harle R., Harris M., Divne C., Mahdi S., Zhao Y., Driguez H., Claeyssens M., Sinnott M. L., and Teeri T. T. (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A: a comparison with Phanerochaete chrysosporium Cel7D. J. Mol. Biol. 333, 817–829 [DOI] [PubMed] [Google Scholar]
- 49. Watanabe T., Ariga Y., Sato U., Toratani T., Hashimoto M., Nikaidou N., Kezuka Y., Nonaka T., and Sugiyama J. (2003) Aromatic residues within the substrate-binding cleft of Bacillus circulans chitinase A1 are essential for hydrolysis of crystalline chitin. Biochem. J. 376, 237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Divne C., Ståhlberg J., Teeri T. T., and Jones T. A. (1998) High-resolution crystal structures reveal how a cellulose chain is bound in the 50 Å long tunnel of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 275, 309–325 [DOI] [PubMed] [Google Scholar]
- 51. NCBI Resource Coordinators (2017) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 45, D12–D17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hall T. (2011) BioEdit: an important software for molecular biology. GERF Bull. Biosci. 2, 60–61 [Google Scholar]
- 53. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 30, 1022–1025 [Google Scholar]
- 56. Emsley P., and Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 57. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 58. Agirre J., Iglesias-Fernández J., Rovira C., Davies G. J., Wilson K. S., and Cowtan K. D. (2015) Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 [DOI] [PubMed] [Google Scholar]
- 59. Michaelis L., Menten M. L., Johnson K. A., and Goody R. S. (2011) The original Michaelis constant: translation of the 1913 Michaelis–Menten paper. Biochemistry 50, 8264–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Östling S, and Virtama P. (1946) A modified preparation of the universal buffer described by Teorell and Stenhagen. Acta Physiol. Scand. 11, 289–293 [Google Scholar]
- 61. Kidby D. K., and Davidson D. J. (1973) A convenient ferricyanide estimation of reducing sugars in the nanomole range. Anal. Biochem. 55, 321–325 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.