

Abstract
Lectins play important roles in infections by pathogenic bacteria, for example, in host colonization, persistence, and biofilm formation. The Gram-negative entomopathogenic bacterium Photorhabdus luminescens symbiotically lives in insect-infecting Heterorhabditis nematodes and kills the insect host upon invasion by the nematode. The P. luminescens genome harbors the gene plu2096, coding for a novel lectin that we named PllA. We analyzed the binding properties of purified PllA with a glycan array and a binding assay in solution. Both assays revealed a strict specificity of PllA for α-galactoside–terminating glycoconjugates. The crystal structures of apo PllA and complexes with three different ligands revealed the molecular basis for the strict specificity of this lectin. Furthermore, we found that a 90° twist in subunit orientation leads to a peculiar quaternary structure compared with that of its ortholog LecA from Pseudomonas aeruginosa. We also investigated the utility of PllA as a probe for detecting α-galactosides. The α-Gal epitope is present on wild-type pig cells and is the main reason for hyperacute organ rejection in pig to primate xenotransplantation. We noted that PllA specifically recognizes this epitope on the glycan array and demonstrated that PllA can be used as a fluorescent probe to detect this epitope on primary porcine cells in vitro. In summary, our biochemical and structural analyses of the P. luminescens lectin PllA have disclosed the structural basis for PllA's high specificity for α-galactoside–containing ligands, and we show that PllA can be used to visualize the α-Gal epitope on porcine tissues.
Keywords: carbohydrate, carbohydrate-binding protein, glycobiology, lectin, protein structure, structural biology
Introduction
Photorhabdus luminescens is a Gram-negative γ-proteobacterium belonging to the Enterobacteriaceae family. In its complex life cycle it lives symbiotically in the intestine of Heterorhabditidae entomopathogenic nematodes and pathogenically in insect larvae upon nematode invasion. P. luminescens was first isolated in 1977 as a symbiont bacterium of Heterorhabditidae nematodes and classified initially as Xenorhabdus luminescens (1, 2) and later renamed P. luminescens (3). The genus Photorhabdus consists of the four species P. luminescens, P. temperata, P. heterorhabditis, and P. asymbiotica (4–6), with the latter species being pathogenic to humans (7). Both Xenorhabdus and Photorhabdus species enter a wide range of insect larvae via Steinernematidae and Heterorhabditidae nematodes, respectively (4, 8). Once the nematodes enter into the insect at their infective juvenile developmental stage, their bacterial symbionts are released into the insect's blood, and both bacteria and nematode are able to kill the larvae within 48 h (9).
The complete genome of P. luminescens subsp. laumondii TTO1 was published in 2003 by Duchaud et al. (10). Compared with other bacteria, it shows a high number of genes predicted as toxins, and some of them, such as Tca and Tcd, have been extensively studied (10–12). Often, bacterial carbohydrate-binding proteins, i.e. lectins, also act as toxins in addition to their function as adhesins to enable host colonization. A number of predicted lectins are present in the genome of P. luminescens, but only one example, the fucose-binding protein PLL (13), has been experimentally characterized. In Pseudomonas aeruginosa, a ubiquitous opportunistic pathogen with a high current medical need for new therapeutics (14), the two lectins LecA and LecB are both toxins and adhesins with roles in biofilm formation and persistent infection (15–17). LecB (18, 19)-type proteins are relatively common, and several LecB homologs have also been previously characterized in other bacterial species such as Ralstonia solanaceum (20), Burkholderia cenocepacia (21–23), and Chromobacterium violaceum (24). The P. luminescens genome harbors several genes of predicted lectins that are homologs of LecB (25). In contrast, homologs of LecA are comparably rare and are not present in the genomes of the former species. In P. luminescens, a single gene encoding for a LecA homolog (plu2096) was predicted by Duchaud et al. (10).
Here, we report the biochemical and structural characterization of the P. luminescens lectin PllA. We demonstrate that PllA is highly specific for α-galactoside–containing ligands. By determining several crystal structures of PllA in complex with ligands, we were able to rationalize PllA's strong preference for α-galactosides. We further demonstrate that PllA can serve as a detection tool for the specific visualization of the α-Gal epitope present on porcine tissue. This epitope is responsible for hyperacute rejection of pig to primate organ xenotransplants.
Results and discussion
Identification and production of PllA
Although P. aeruginosa LecB orthologs have been widely studied, orthologs of LecA have not been characterized in detail. We searched the publicly accessible NCBI database using protein blast and the protein sequence of LecA from strain PAO1 as a template. A moderate number of orthologs was retrieved from only a few different genera of Gram-negative bacteria. These were mainly entomopathogenic Photorhabdus and Xenorhabdus species, as well as human opportunistic pathogens from the Enterobacter spp. and a few other pathogenic bacterial species (see supplemental Fig. S1).
In P. luminescens, the gene plu2096 was previously proposed as the coding gene for a LecA-like protein (10) and later confirmed (26) to be a galactose-binding lectin. An alignment of the retrieved orthologs of LecA in each Photorhabdus and Xenorhabdus species shows a high degree of similarity within these entomopathogenic species (Fig. 1). Although the residues involved in metal and ligand binding in LecA are relatively conserved, distinct differences to P. aeruginosa LecA were observed; LecA contains an insert flanked by two cysteines that are absent in PllA, and the Photorhabdus and Xenorhabdus homologs contain an additional C-terminal tail of 5–13 amino acids, which is partially conserved (Fig. 1).
Figure 1.

Sequence alignment of LecA from P. aeruginosa with hypothetical LecA-like proteins from Photorhabdus and Xenorhabdus species (one single ortholog per organism selected based on highest identity to LecA). Strictly conserved amino acids are shaded black, and similarly conserved amino acids are shaded gray. Black dot, amino acid of LecA involved in Ca2+ binding; black triangles, amino acids of LecA involved in sugar binding; asterisks, amino acids of LecA involved in both Ca2+ and sugar binding. Amino acid numbering follows the LecA crystal structure where the N-terminal methionine is lacking. The depicted protein sequence of P. luminescens (PllA) is encoded by the plu2096 gene.
PllA has 37% sequence identity to LecA, and all amino acids whose side chains are involved in calcium ion binding are conserved except for one Asn (LecA) to Asp (PllA) variation. Amino acids involved in carbohydrate recognition are only partially conserved.
Recombinant production and purification of PllA
The plu2096 gene was amplified from genomic DNA of P. luminescens subsp. laumondii TTO1 and cloned into the pET22b(+) vector. Recombinant expression in Escherichia coli BL21(DE3) yielded a protein product at a 13-kDa apparent molecular mass by SDS-PAGE (Fig. 2A), which corresponds to its predicted size of 12.95 kDa. Because of the sequence homology to galactose-binding LecA, we subjected the cell lysate to galactosylated Sepharose (27), and PllA was retained on this affinity resin. Subsequent elution was achieved using galactose in the elution buffer. Purification yielded ∼6 mg of PllA/liter of bacterial culture, which was later improved to 19 mg/liter by purification on melibiose-coupled Sepharose (see below).
Figure 2.
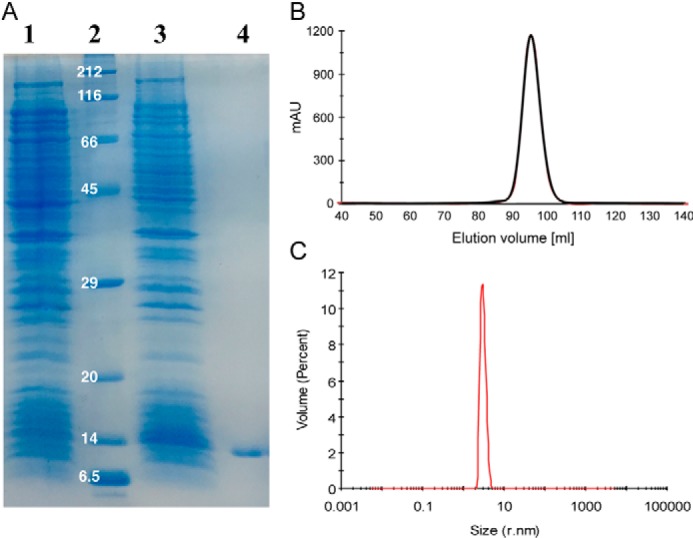
A, recombinant expression and affinity purification of PllA analyzed by SDS-PAGE (15%). E. coli whole-cell extracts of uninduced (lane 1) and IPTG-induced cultures (lane 3), and purified PllA (lane 4), molecular mass marker in kDa (lane 2) are shown. B, Sepharose size-exclusion chromatogram of PllA; C, DLS analysis of PllA.
Because lectins often oligomerize and LecA forms a tetramer (28), we analyzed PllA's multimeric state using size-exclusion chromatography (SEC).3 The protein's observed apparent molecular mass was 27 kDa, which suggested a PllA dimer (Figs. 2B and supplemental Fig. S2). When we used the more robust technique of dynamic light scattering (DLS), we observed a homogeneous sample with an apparent molecular mass of 52.1 kDa (Fig. 2C). The tetramer LecA was also studied by DLS under identical conditions, and its apparent molecular mass of 52.4 kDa (supplemental Fig. S3) matched the value for PllA and thus suggests the tetramerization of PllA in solution. The observed differences for PllA in SEC and DLS could result from weak interactions between PllA and the glycan-based Sepharose resin, altering the observed molecular mass to a smaller size.
Carbohydrate-binding specificity of PllA using the CFG glycan array
To assess the carbohydrate-binding specificity of PllA, the protein was fluorescently labeled using fluorescein isothiocyanate (FITC), and binding to a glycan array containing over 600 carbohydrate epitopes was performed at the Consortium for Functional Genomics (Fig. 3A). The LecA homolog PllA showed a strict specificity toward glycans with terminal α-galactosides, whereas β-galactosides and other carbohydrates only showed very weak or no binding. The highest apparent binding affinity was detected for the bivalent α-Gal-terminating N-glycan (Gal-α-1,3-Gal-β-1,4-GlcNAc)2Man3GlcNAc2 (glycan nos. 360 and 550) or its difucosylated derivative glycan no. 368 bearing blood group B antigens at the non-reducing end and the two monovalent disaccharides Gal-α-1,3-GalNAc (glycan no. 112) and Gal-α-1,4-GlcNAc (glycan no. 123).
Figure 3.

Carbohydrate specificity of PllA on a glycan array. A, profiling of the glycan-binding specificity of PllA on a glycan array. Glycans containing terminal α-galactoside are colored black; those with terminal β-galactoside are red, and oligosaccharides with neither of these terminal moieties are colored blue. Selected structures showing highest apparent binding are illustrated in CFG notation. B, comparison of glycan-binding specificity between PllA (this work) and LecA (PA-IL (32)) from P. aeruginosa based on normalized CFG glycan array data. Normalization was done by dividing RFU averages of the glycan of interest by the RFU average of the ligand with the highest apparent affinity on the respective array (i.e. glycan 550 for PllA and glycan 31 for LecA). Glycan structures are indicated and depicted with the different spacers of the array for LecA. For the PllA array, identical glycans generally contain β-Sp8 spacers except 105 (Sp0) and 116 (β-Sp0). Numbers on top of columns indicate glycan number of the respective arrays. A and B, error bars correspond to six replicates.
The highest apparent binding was observed for bivalent Gal-α-1,3-Gal-β-1,4-GlcNAc terminating glycans (glycans no. 360, 368, and 550). This glycan structure, called the α-Gal epitope (29), is a ubiquitous constituent of glycans in non-primate mammals and new world monkeys. The nematode Parelaphostrongylus tenuis also contains N-glycans decorated with this epitope (30). This epitope is mainly responsible for hyperacute rejection of porcine organ transplants in humans during xenotransplantation (29, 31). Interestingly, the corresponding monovalent glycans (no. 105 and 115) showed a 4–5-fold lower binding signal (Table 1), indicating that PllA binds carbohydrates multivalently as known for its ortholog LecA. Interestingly, when the same monovalent epitopes were presented on a shorter spacer (Sp0) with one mannose between spacer and epitope (no. 516 and 517), binding was reduced further.
Table 1.
Comparison between high binding glycans and structurally related glycans on the array, i.e. comparison of linkagetypes and multivalent-monovalent effect
Note: Spacers vary on the glycan array.
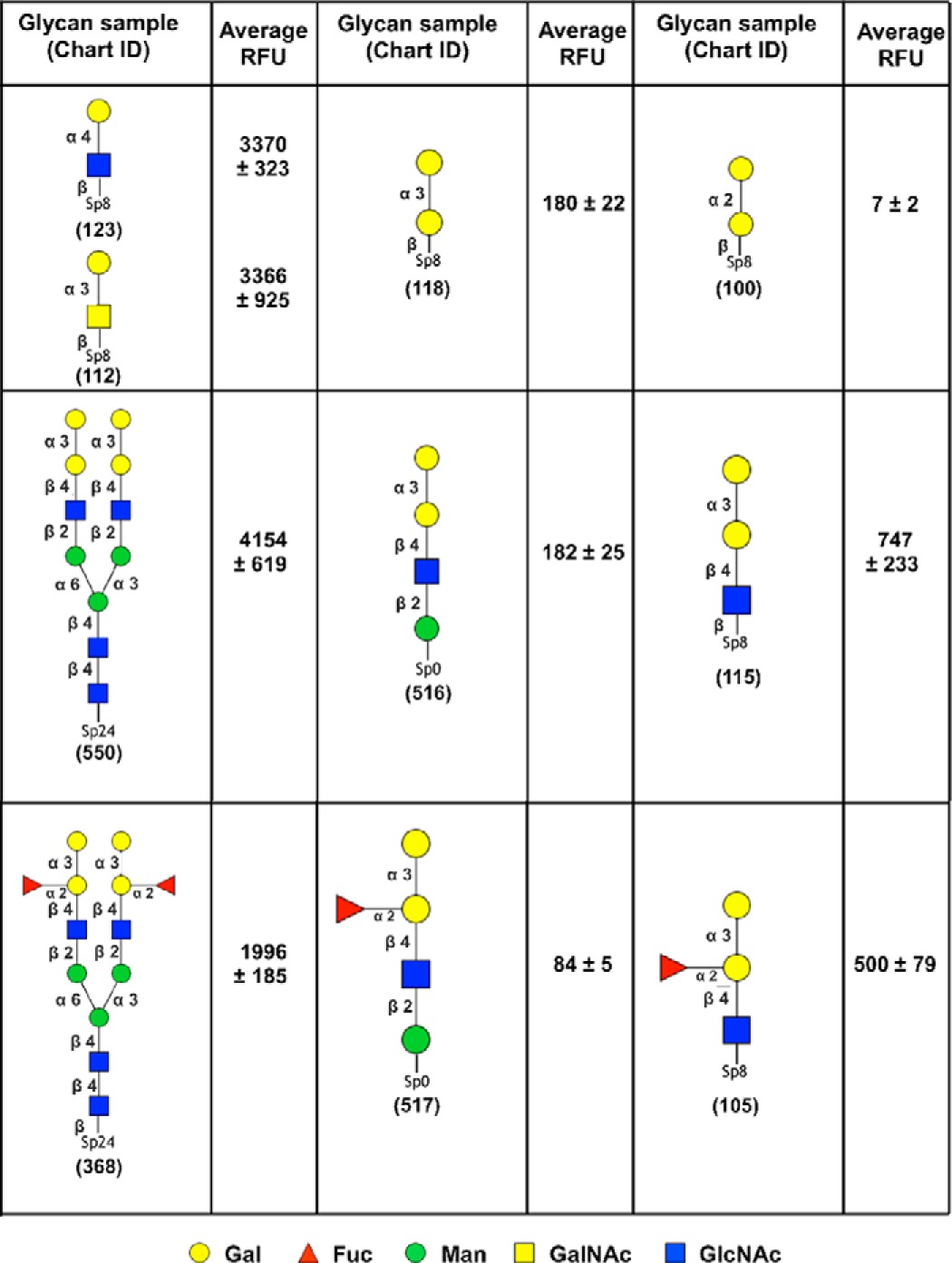
Apart from the divalent ligands, only the two disaccharides, Gal-α-1,3-GalNAc and Gal-α-1,4-GlcNAc, showed a high binding signal among the monovalent series (Fig. 3A). These two ligands are monovalently displayed and may reveal the intrinsic specificity of PllA, because other monovalent ligands showed only weak or no binding to PllA on this glycan array. Interestingly, these two ligands displayed much stronger binding than analogous Gal-α-1,3-Gal and Gal-α-1,2-Gal, suggesting an important role of the acetamide moiety in the penultimate residue for binding to PllA (Table 1). Among the monovalent ligands on the array, the observed linkage specificity of PllA was broad for glycans containing terminal Gal-α-1,3 and Gal-α-1,4 linkages. The single present Gal-α-1,6-linked ligand (Gal-α-1,6-Glc) was moderately bound, whereas the single Gal-α-1,2-linked ligand (Gal-α-1,2-Gal) was not recognized by PllA (Table 1).
To compare the carbohydrate specificity of PllA with its previously characterized ortholog LecA (32), glycan array binding data of monovalent glycan ligands for both lectins was normalized and plotted (Fig. 3B). Notably, LecA showed the best binding to Gal-α-1,4-Gal-β-1,4-GlcNAc, which is only weakly recognized by PllA. This glycan is part of the glycosphingolipid Gb3, which when bound by LecA triggers membrane bending, a process that was proposed as an entry pathway for P. aeruginosa invasion into the host cell (33). In contrast, PllA showed high apparent binding to the epitopes Gal-α-1,3-GalNAc and Gal-α-1,4-GlcNAc, whereas LecA only shows moderate apparent binding as observed for a number of other α-galactosides on the glycan array (Fig. 3B). In summary, LecA is a rather promiscuous receptor for a variety of monovalent galactosides. In addition to PllA's binding to the bivalent N-glycan structures described above, PllA was rather specific for Gal-α-1,3-GalNAc and Gal-α-1,4-GlcNAc, one or both of which may be the natural ligand of PllA.
Development of a competitive binding assay for PllA
To rapidly assess and quantify the binding specificity of PllA, we developed a competitive binding assay for PllA by utilizing fluorescence polarization, which is based on our previous work (19, 21, 27, 34) for four different lectins. Four different FITC-labeled d-galactosides (27) were titrated with increasing amounts of PllA (Fig. 4, A–C). All three β-linked galactosides (2–4) did not show binding up to 300 μm PllA. In contrast, titration of α-galactoside 1 resulted in binding with a Kd of 62.7 μm as observed by the increase in fluorescence polarization. These data confirmed the strict α-galactoside specificity of PllA in solution that was observed on the glycan array.
Figure 4.

Establishing a carbohydrate-binding assay for PllA in solution. A, structure of fluorescent ligands 1–4 based on d-galactose. B, titration of fluorescent ligands 1–4 with PllA. C, dissociation constant for 1 was obtained from a four-parameter fitting procedure to the dose-dependent increase in fluorescence polarization (Kd, 62.7 ± 3.8 μm). D, competitive inhibition of the binding of 1 to PllA with methyl α-d-galactoside (12, IC50 = 0.52 ± 0.07 mm) and raffinose (25, IC50 = 0.11 ± 0.01 mm). One representative titration experiment of triplicates on one plate is shown. Dissociation constant and standard deviations given were obtained from at least three independent replicates of triplicates on three plates each.
The latter system was then used for a competitive binding assay, where fluorescent α-galactoside 1 was competitively displaced from PllA using methyl α-d-galactoside (12), and a set of available d-galactose derivatives (Fig. 4D and Table 2). Interestingly, the obtained IC50 for 12 was 0.52 mm, which is ∼10-fold weaker than the determined affinity of the fluorescent analog 1. Among the monosaccharides tested, free d-galactose (9), showed inhibition of PllA with a reduced affinity (IC50 = 1.57 mm) compared with glycoside 12, probably due to the partial presence of the non-binding β-anomer. Methyl α-glycosides of d-glucose (5), l-fucose (6), d-mannose (7), or methyl β-d-arabinoside (8) did not show detectable binding to PllA. Replacement of the 2-hydroxy group of galactose with a free amine in d-galactosamine (10) led to a 2-fold increase in the binding affinity (IC50 = 0.86 mm). Acetamide substitution in the same position, i.e. N-acetylgalactosamine (11), resulted in a complete loss of binding. Next, we tested α-galactosides with different aglycons of varying size, e.g. methyl, allyl, 4-nitrophenyl, 4-methylumbelliferyl, and X-Gal (12–14, 16–18). These modifications only showed a small effect on the binding affinity. O-Alkylation of ring hydroxyl groups is a requirement for ligand binding of some lectins like the tectonins (35, 36). For PllA, O-methylation of the 3-hydroxyl group resulted in a complete loss of affinity (14 → 15). A similar loss of affinity was observed for O-methylation of the P. aeruginosa LecB or B. cenocepacia BC2L-A ligands fucose and mannose (21).
Table 2.
Evaluation of natural and synthetic inhibitors of PllA using the competitive binding assay
Averages and standard deviations were obtained from three independent experiments. n.i.: no inhibition observed up to 10 mm. SP: spacer –(CH2)3NH–CO(CH2)5NH–.
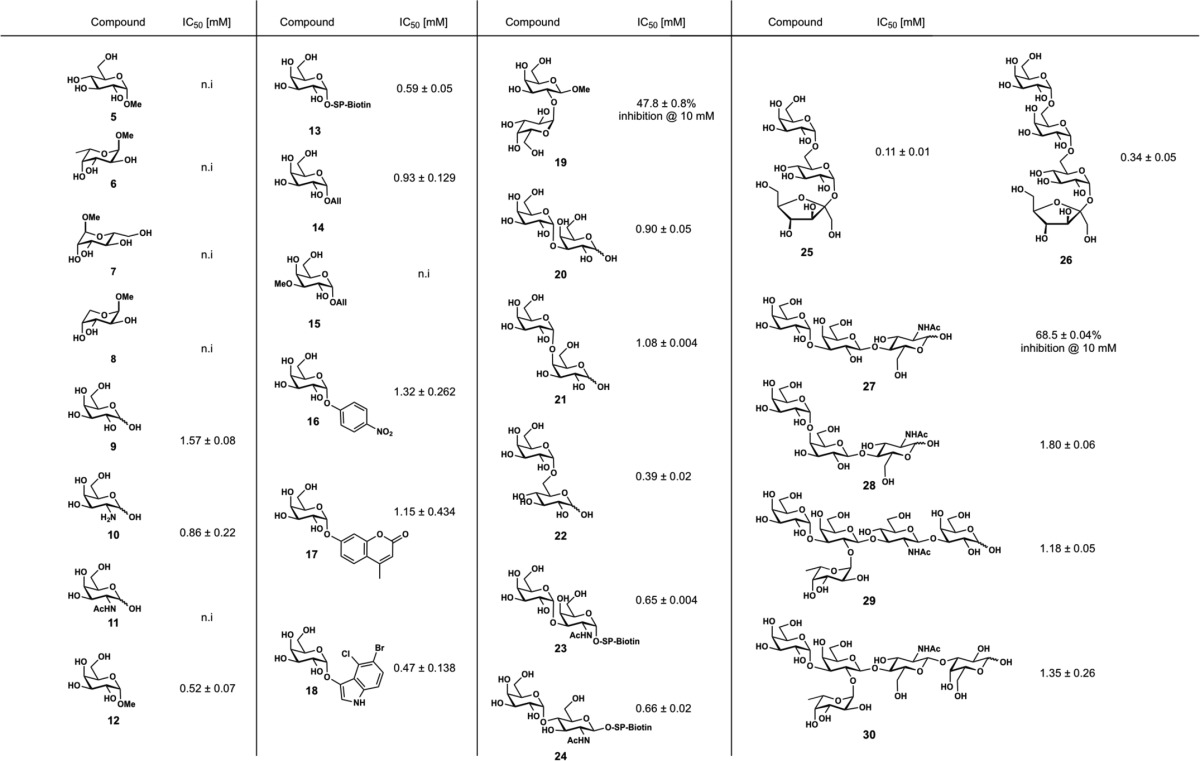
We also tested a set of oligosaccharides containing α-galactosyl residues for competitive binding to PllA. Gal-α-1,3-Gal (20, IC50 = 0.90 mm) and Gal-α-1,4-Gal (21, IC50 = 1.08 mm) showed a 2-fold lower binding affinity to PllA than melibiose (Gal-α-1,6-Glc, 22, IC50 = 0.39 mm), whereas Gal-α-1,2-Gal (19) was only weakly active and resulted in ∼50% inhibition at 10 mm. A comparable binding specificity for Gal-α-1,6-Glc has been reported for LecA from P. aeruginosa (37). The plant trisaccharide raffinose (25) contains a terminal melibiose motif and showed the highest binding to PllA among all tested compounds with an IC50 of 0.11 mm. This ubiquitous plant galactoside has also shown an inhibitory effect on P. aeruginosa biofilms and inhibits LecA in a similar affinity range (Kd = 32 μm) (37, 38). The tetrasaccharide stachyose (26) is another plant derivative of raffinose with an additional 1,6-linked α-galactoside moiety. For PllA an IC50 of 0.34 mm was observed, indicating that longer oligosaccharides do not improve the binding affinity to PllA, which is different for LecA. Bivalent oligosaccharides containing the α-Gal antigen Gal-α-1,3-Gal-β-1,4-GlcNAc were identified as the apparent best ligands of the glycan array (Fig. 3A). The corresponding monovalent trisaccharide 27 was tested in our competitive binding assay and showed only ∼70% inhibition of PllA at 10 mm. This weak binding is in good agreement with the glycan array data where the monovalent α-Gal epitope had reduced binding to PllA compared with its bivalent structure (see Table 1).
Because Gal-α-1,3-GalNAc and Gal-α-1,4-GlcNAc were identified as the monovalent ligands with the highest apparent affinity on the glycan array, we tested the corresponding biotinylated disaccharides 23 and 24, respectively. Both soluble glycans differ only in spacer identity and for 23 also in the anomeric configuration from those glycans used for the production of the glycan array. Surprisingly, they were as active as the comparably biotinylated α-galactosyl monosaccharide 13, displaying IC50 values from 0.59 to 0.66 mm.
The human blood group antigen P1 (28) (39) was also moderately recognized by PllA on the glycan array (ligand 121, supplemental Table S1) and in the competitive binding assay soluble monovalent 28 showed a moderate binding affinity (IC50 = 1.80 mm) to this lectin. Because PllA was shown to bind to α-Gal residues but not to GalNAc residues (see above) and it recognized the blood group B-terminating ligand 368 on the glycan array (Fig. 3A), we also tested soluble blood group B antigens 29 and 30. These oligosaccharides were inhibitors of PllA with moderate potency (IC50 of 1.18 and 1.35 mm, respectively). This specificity of PllA is in contrast to LecA, which binds to B and A antigens (27). Thus, PllA could be used as a new reagent for rapid identification of blood group B serotypes such as the currently used GS-IB4 isolectin from Griffonia simplicifolia (40).
PllA, a lectin with a unique tetrameric structure
Apo-PllA crystallizes in space group P3221 and crystals diffracted to 1.7 Å. Data collection and refinement statistics for all presented PllA structures can be found in Table 3. The core of PllA consists of two four-stranded anti-parallel β-sheets (Fig. 5). We did not observe the canonical Ca2+ ion found in other C-type lectins at the sugar-binding site, which may be a result of the crystallization buffer that contained a high concentration of citrate known to chelate calcium ions.
Table 3.
Data collection and refinement statistics
Statistics for the highest-resolution shell are shown in parentheses. r.m.s. is root mean square.
| PllA, apo, PDB 5OFZ | PllA, Me-α-Gal (12), PDB 5ODU | PllA, raffinose (25), PDB 5OFX | PllA, fluorescent ligand 1, PDB 5OFI | |
|---|---|---|---|---|
| Resolution range | 46.05–1.75 (1.81–1.75) | 47.2–1.56 (1.62–1.56) | 44.09–1.75 (1.81–1.75) | 43.76–2.0 (2.07–2.0) |
| Space group | P 32 2 1 | P 1 21 1 | P 1 | P 21 21 21 |
| Unit cell | 92.1 92.1 164.2 90 90 120 | 62.9 103.3 76.1 90 93.0 90 | 59.0 63.0 75.9 101.1 112.8 94.4 | 48.6 134.3 153.0 90 90 90 |
| Total reflections | 725,065 (34,414) | 597,157 (60,933) | 539,432 (54,563) | 126,530 (12,830) |
| Unique reflections | 81,583 (7832) | 133,175 (13,311) | 89,118 (9042) | 65,944 (6644) |
| Multiplicity | 8.9 (4.4) | 4.5 (4.6) | 6.1 (6.0) | 1.9 (1.9) |
| Completeness (%) | 99.59 (96.57) | 96.18 (95.63) | 90.73 (92.05) | 95.91 (97.62) |
| Mean I/σ(I) | 26.62 (3.32) | 11.19 (1.22) | 9.08 (2.02) | 8.23 (2.54) |
| Wilson B-factor | 23.67 | 14.93 | 13.78 | 23.65 |
| R-merge | 0.04649 (0.3966) | 0.09883 (1.261) | 0.1569 (0.7841) | 0.05735 (0.2516) |
| R-meas | 0.04921 (0.451) | 0.1121 (1.427) | 0.1719 (0.8578) | 0.08111 (0.3558) |
| R-pim | 0.01592 (0.2106) | 0.05232 (0.6619) | 0.06949 (0.345) | 0.05735 (0.2516) |
| CC1/2 | 0.999 (0.886) | 0.998 (0.358) | 0.994 (0.78) | 0.996 (0.907) |
| CC* | 1 (0.969) | 1 (0.726) | 0.999 (0.936) | 0.999 (0.975) |
| Reflections used in refinement | 81,572 (7830) | 132,517 (13,152) | 89,095 (9038) | 65,903 (6639) |
| Reflections used for R-free | 4183 (452) | 6491 (608) | 4458 (428) | 3335 (326) |
| R-work | 0.2006 (0.2858) | 0.2050 (0.3530) | 0.1849 (0.2669) | 0.1864 (0.2446) |
| R-free | 0.2202 (0.3105) | 0.2334 (0.3668) | 0.2133 (0.2986) | 0.2216 (0.3016) |
| CC(work) | 0.943 (0.876) | 0.960 (0.626) | 0.954 (0.867) | 0.957 (0.905) |
| CC(free) | 0.937 (0.823) | 0.958 (0.620) | 0.940 (0.834) | 0.939 (0.820) |
| No. of non-hydrogen atoms | 4138 | 8639 | 8749 | 8306 |
| Macromolecules | 3608 | 7264 | 7264 | 7264 |
| Ligands | 112 | 246 | 180 | |
| Solvent | 530 | 1263 | 1239 | 862 |
| Protein residues | 480 | 968 | 968 | 968 |
| r.m.s. (bonds) | 0.009 | 0.003 | 0.004 | 0.004 |
| r.m.s. (angles) | 1.00 | 0.58 | 0.69 | 0.61 |
| Ramachandran favored (%) | 97.46 | 98.00 | 97.69 | 97.37 |
| Ramachandran allowed (%) | 2.54 | 2.00 | 2.31 | 2.63 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | 0 |
| Rotamer outliers (%) | 0.26 | 0.76 | 0.51 | 0.25 |
| Clashscore | 3.35 | 3.92 | 2.18 | 1.85 |
| Average B-factor | 30.15 | 22.83 | 19.00 | 29.55 |
| Macromolecules | 28.81 | 20.95 | 17.24 | 28.84 |
| Ligands | 27.65 | 21.05 | 29.75 | |
| Solvent | 39.27 | 33.22 | 28.88 | 35.49 |
| No. of TLS groups | 28 | 50 | 76 | 46 |
Figure 5.

A, sequence of PllA. Secondary structure elements are shown above the sequence (blue arrows, β-strands; red barrel, α-helix). Residues responsible for sugar binding are highlighted with magenta stars, Ca2+-binding residues with cyan circles, and amino acids coordinating both as yellow triangles. Tail residues unique to PllA and its close homologs are highlighted with a blue box. B, schematic representation of a PllA apo monomer. C, fold diagram for the structure shown in B.
The asymmetric unit contained two PllA dimers, which form tetramers with symmetry mates in accordance with DLS data. The C-terminal five-residue extension (Fig. 1) of the four protomers are engaged in well-defined interactions leading to a 90° twist in the tetramer (Fig. 6A). Of the tail residues (YSPLK), Tyr-118 packs hydrophobically against Pro-120, and Ser-119 forms two hydrogen bonds with the tetramer partner (Ser-119 side-chain hydroxyl with side-chain amino group of Lys-82, Ser-119 carbonyl with the main chain of Thr-83). Residue Leu-121 is inserted into a tailored hydrophobic pocket of the tetramer partner, which is composed of residues Leu-22, Ile-28, Ala-60, Ile-68, Phe-73, Ile-75, Val-79, Val-84, and Leu-90 (Fig. 6B). When we compared the structure of PllA with the structure of the well-studied protein LecA, we found the structures to be very similar (Cα r.m.s.d. of 0.67 over 78 atoms, supplemental Fig. S11). The main differences are found in the region between β3 and β7 (Fig. 5C), which has a profound impact on carbohydrate binding (see below).
Figure 6.

Overall structure of PllA and comparison to LecA. A, schematic representation of the PllA tetramer. Two parallel dimers (yellow/magenta and green/cyan) form tail-to-tail dimers with a 90° twist. B, detailed view of the PllA tail-to-tail interface. We observe two hydrogen bonds between the side chains of tail Ser-119 (cyan) and Lys-82 (yellow) and the C terminus of the tail and the backbone nitrogen of Ala-74 (yellow). In addition, tail residue Leu-121 is inserted into a hydrophobic pocket of its binding partner. C, LecA tetramer is planar, formed by tail-to-tail dimerization of two parallel dimers (yellow/magenta and green/cyan). D, much shorter tail of LecA provides several stabilizing hydrogen bonds (dashed lines), but the interactions are not sufficient to cause a twist of the two dimers relative to each other.
The sequence alignment of PllA with LecA from P. aeruginosa and the LecA homologs from Photorhabdus and Xenorhabdus species showed that PllA and LecA homologs from the latter two species possess an extension at their C termini. This extension led to the surprising result that the dimerization of dimers is twisted by 90° in PllA. In contrast, LecA does not possess the five C-terminal residues. As a result, the LecA tetramer is planar and formed by the tail-to-tail arrangement of two dimers (Fig. 6, C and D). We have no indication as to the biological significance of this arrangement, but we believe that the interactions between the tails of the tetramer partners will lead to a significant stabilization of the tetrameric assembly.
Structural basis of α-galactoside specificity of PllA
To understand the α-galactoside specificity of PllA, we determined the crystal structures of several PllA–carbohydrate complexes in the presence of 3 mm calcium chloride in the crystallization buffer (Fig. 7): PllA in complex with the monosaccharide methyl α-d-galactoside (12), the trisaccharide raffinose (25), and an α-d-galactoside linked to fluorescein (1). Complex crystals of PllA with 12 were obtained by co-crystallization. The resulting crystals belonged to space group P21 and were diffracted to 1.56 Å. The overall structure of PllA does not change upon complex formation (Cα r.m.s.d. of 0.12 Å), and we observed unambiguous electron density for Ca2+ and the ligand in each of the eight protomers in the asymmetric unit. The canonical Ca2+ ion at the sugar-binding site is coordinated by the side chains of Asp-96, Thr-100, Asp-103, and Asn-104 as well as the main-chain carbonyl oxygen atoms of Tyr-38 and Thr-100. The ligand is not involved in crystal contacts and oriented in the same way in each protomer. It sits in a shallow binding pocket and is engaged in a total of 10 hydrogen bonds (protein and the Ca2+ ion): the anomeric oxygen (O1) and galactoside C2 hydroxyl with the side chain of Glu-44; sugar C3 hydroxyl with the Ca2+ ion, the side chain of Asp-103, and the main chain of Tyr-38; sugar C4 hydroxyl with the Ca2+ ion, the side chain of Asp-96, and the main chain of Tyr-38; sugar ring O5 with the side chain of Gln-57; and sugar C6 hydroxyl with the side chain of Gln-57. From this complex structure, it is clear that PllA is only able to bind to α-galactosides; the side chains of the two amino acids Val-43 and Asn-55 that are absent in LecA form a barrier and any β-linkage would result in a clash with the PllA surface (Fig. 8). The structure also allowed us to rationalize why d-galactosamine (10) is a better binder than d-galactose (9), and why N-acetylgalactosamine (11) shows no detectable binding. By swapping the C2 hydroxyl group for an amino group, the sugar can now engage in an additional hydrogen bond with the side chain of Asp-103 (see supplemental Fig. S8). Acetylation of the amino group leads to a clash with the protein and thus abolishes binding.
Figure 7.

PllA–carbohydrate complex structures. A and B, PllA bound to methyl α-d-galactoside (12). This interaction is stabilized through 10 hydrogen bonds (dashed lines). Eight of them are between the ligand and the protein, and two are provided by the Ca2+ ion. C and D, PllA bound to raffinose (25). In addition to the hydrogen bonds observed in A, the glucose moiety forms a hydrogen bond with the side chain of Gln-57, whereas terminal fructofuranoside forms two hydrogen bonds with the side chain of Glu-44, which results in the ligand adopting a horseshoe shape. E and F, PllA bound to fluorescent tracer 1. No interactions with the protein are observed beyond the carbohydrate moiety. The fluorescein can only be observed as the result of fortuitous crystal contacts in half of the monomers in the asymmetric unit. PllA is shown as a yellow schematic/surface representation, ligand as gray sticks, oxygen atoms in red, nitrogen atoms in blue, sulfur atoms in yellow, and Ca2+ ions as green spheres. Difference electron density (Fo − Fc) contoured to 3σ with phases calculated from a model that was refined in the absence of metal ions is shown as gray isomesh (B, D, and F).
Figure 8.
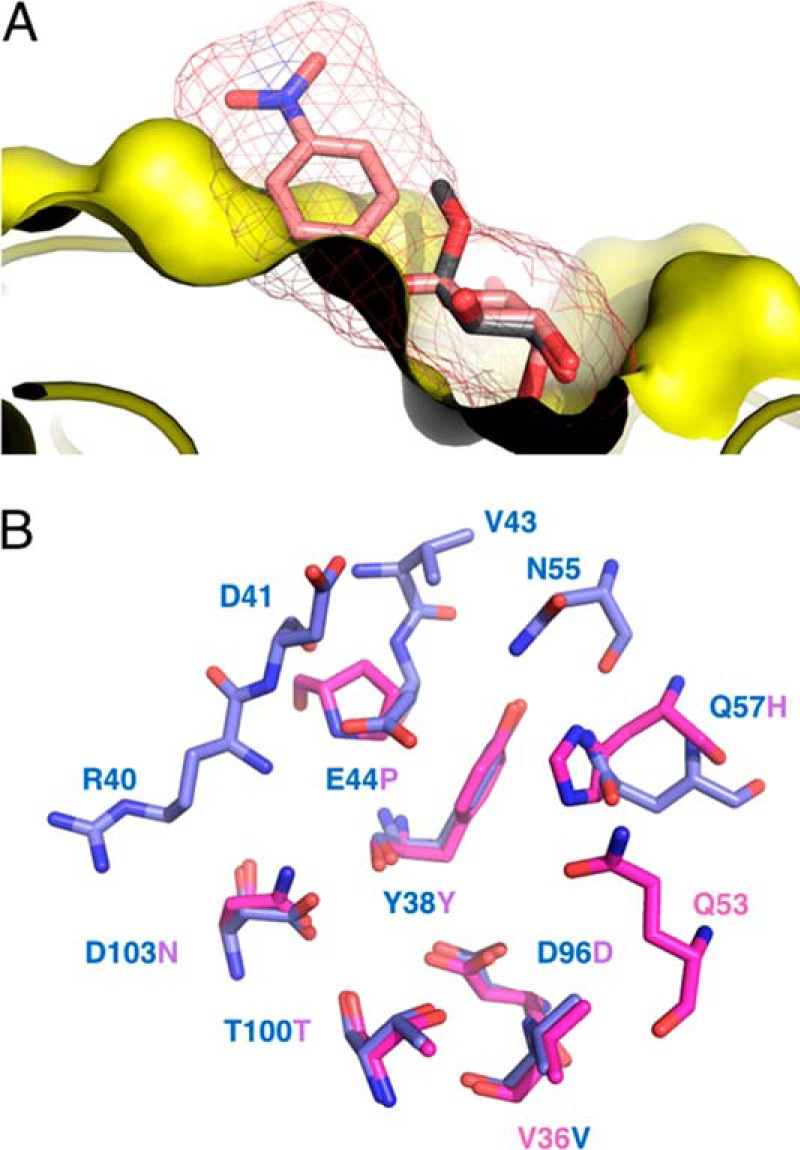
Rationalizing PllA α-galactoside specificity. A, representation of the PllA (yellow)-binding pocket with methyl α-d-galactoside (12, gray sticks). 4-Nitrophenyl β-d-galactoside (salmon sticks and isomesh, taken from PDB 3ZYF) was superposed onto α-d-galactoside. Because of the restricted ligand-binding site of PllA only α-substituted ligands, leading away from the surface, can be accommodated, whereas β-substituted ligands clash. B, superposition of the binding site amino acid residues of PllA (blue) with LecA (magenta), oxygen atoms, red; nitrogen atoms, blue. Residue numbers correspond to PllA.
Complex crystals of PllA with raffinose were also obtained by co-crystallization. The resulting crystals belonged to space group P1 and diffracted to 1.75 Å. As expected, the overall structure of PllA does not change upon complex formation (Cα r.m.s.d. of 0.12 Å). There are eight protomers in the asymmetric unit, and we observed unambiguous electron density for raffinose in all of them. Although some of the raffinose molecules are involved in crystal contacts, the orientation and shape of the trisaccharide is virtually identical in each protomer. The orientation of the galactose moiety does not differ between the monosaccharide and raffinose structures, and the same hydrogen bonds are formed. The α-1,6-linkage leads glucose away from the PllA surface, but the C4 hydroxyl forms one hydrogen bond with the side chain of Gln-57. Interestingly, the final fructose moiety is pointing back toward the PllA surface, giving the raffinose an overall horseshoe shape. The fructose C3 and C4 hydroxyls are engaged in a hydrogen bond with the side chain of Glu-44. Glu-44 is also involved in hydrogen bond formation with the galactose C2 hydroxyl, thus linking the two ends of the raffinose horseshoe resulting in an additional intraligand hydrogen bond between fructose C6 hydroxyl with galactose C2 hydroxyl.
Because we used fluorescent probe 1 in our competitive binding assays, we wanted to understand how the probe binds to PllA. Complex crystals of PllA with 1 were also obtained by co-crystallization. The resulting crystals belonged to space group P212121 and diffracted to 2.0 Å. The overall structure of the eight protomers in the asymmetric unit did not differ significantly from the apo structure (Cα r.m.s.d. of 0.15 Å), and we observed unambiguous density for 1 in four of the protomers (Fig. 7, E and F, and supplemental Fig. S10). The interactions of the galactose include all of those observed in the other two structures. Through fortuitous crystal packing, we were able to obtain good electron density for the fluorophore and were able to fit it. The ordered nature of the fluorophore is the result of π-stacking between the tricyclic ring systems of two molecules of 1 bound to symmetry mates.
The strict specificity toward α-galactosides is unique for PllA when compared with LecA. From the crystal structure of PllA with 12, it becomes clear that β-galactosides cannot be recognized without a steric clash with the protein surface of PllA (Fig. 8A). In contrast, LecA opens a shallow cleft close to its anomeric center that allows the accommodation of large β-linked aglycons, such as in 4-nitrophenyl β-d-galactoside. From a superposition of the binding site amino acid residues of PllA with LecA, it can be deduced that the additional amino acids Val-43 and Asn-55 present in PllA are responsible for preventing PllA from binding β-galactosides (Fig. 8B).
Application of PllA for the detection of the α-Gal epitope
PllA showed the highest apparent binding on the glycan array to a biantennary N-glycan structure carrying the α-Gal epitope on its antenna. This antigen (Gal-α-1,3-Gal-β-1,4-GlcNAc) is a ubiquitous epitope in non-primate mammals and new world monkeys. This carbohydrate structure is the major factor of hyperacute rejection of xenotransplanted organs in humans (29, 41, 42). In pigs, genetic engineering resulted in animals lacking the corresponding galactosyltransferase thus reducing the risk of severe immune responses (31, 43, 44). By using modern techniques such as CRISPR/Cas to engineer animals or animal tissue lacking the α-Gal epitope, quality controls for the complete suppression of the biosynthetic machineries are of crucial importance. Currently, the isolectin GS-IB4 purified from the plant G. simplicifolia is used as a tool to identify a wide range of α-galactoside epitopes, among which is the α-Gal epitope (45). Because of the high selectivity of PllA, this bacterial lectin could be an alternative to the currently used GS-IB4.
The crystal structure of GS-IB4 in complex with the terminal disaccharide Gal-α-1,3-Gal (20) as a methyl glycoside shows extensive interactions between the terminal galactose residue and the protein but no contacts with the reducing-end galactose moiety (46). To compare the recognition features of both proteins to this epitope, we have docked the methyl glycoside of 20 into the carbohydrate-binding site of PllA (supplemental Fig. S9). In this computed structure, the terminal saccharide moiety forms extensive contacts with the lectin receptor. In contrast to GS-IB4, the reducing-end galactose established two more hydrogen bonds with Glu-44 and Asn-55 of PllA, which may serve as an explanation for the high specificity of PllA for the Gal-α-1,3-Gal epitope.
We thus tested the suitability of PllA to detect the α-Gal epitope in wild-type primary pig kidney cells and cells derived from the corresponding α-1,3-galactosyltransferase knock-out (GTKO) animals (Fig. 9). Both PllA and the current standard lectin GS-IB4 visualized the α-Gal antigen in wild-type porcine cells similarly. Because the α-Gal antigen is also present on glycolipids in red blood cells (41), we performed hemagglutination experiments with red blood cells (RBC) from wild-type and GTKO pigs. PllA agglutinated wild-type porcine red blood cells but was unable to agglutinate RBCs from the GTKO pig (Fig. 10, A and B). In addition, we could further show that this agglutination was galactose-dependent and could be inhibited by the presence of raffinose (Fig. 10C).
Figure 9.

Staining of primary porcine kidney cells from wild-type pigs (WT) and GGTA1 KO (GTKO) animals unable to produce the α-Gal antigen. Fluorescein-tagged PllA or GS-IB4 were used as probes and detected the α-Gal antigen in WT cells. Lectin concentration: PllA, 50 μg/ml; GS-IB4, 500 μg/ml, 400× magnification.
Figure 10.

Hemagglutination of porcine red blood cells by PllA. A, wild-type pig RBCs. B, GTKO pig RBCs. C, inhibition of PllA-mediated agglutination of wild-type pig RBCs with raffinose.
Conclusion
The opportunistic pathogen P. aeruginosa utilizes the two soluble lectins LecA and LecB for infection of the host and biofilm formation. Although numerous LecB-like proteins have been characterized, LecA orthologs are scarce. Here, we show that various orthologs of LecA are present in the insect pathogenic bacteria from the Photorhabdus and Xenorhabdus species as well as in the human gut bacterium and pathogen Enterobacter spp. A high degree of similarity was observed among those orthologs with LecA having a sequence insert and lacking an otherwise conserved C-terminal tail.
The gene plu2096 from the entomopathogenic bacterium P. luminescens was cloned and recombinantly produced in high production yields. It encodes the galactose-binding lectin PllA with 37% identity to LecA. The carbohydrate-binding specificity of PllA was assessed on a glycan array containing over 600 different carbohydrate epitopes. Interestingly, PllA showed very strict specificity toward α-galactosides with high apparent binding to the α-Gal epitope as well as to Gal-α-1,4-GlcNAc and Gal-α-1,3-GalNAc.
To date, the biological role of the Gal-α-1,4-GlcNAc epitope remains unclear, and natural sources have not been identified despite the fact that anti-Gal-α-1,4-GlcNAc antibodies are present in human serum (47). In contrast, Gal-α-1,3-GalNAc epitopes are present in nematodes and have, for example, been described in glycolipids from the worms Ascaris suum and Caenorhabditis elegans (48). Furthermore, Gal-α-1,3-GalNAc is a ubiquitous epitope present on glycoproteins of the nematode Hemonchus contortus, and vaccination of lambs with glycoproteins of the sheep parasite H. contortus specifically elicited anti-Gal-α-1,3-GalNAc IgG antibodies (49). In addition, α-linked galactosyl residues have been identified in C. elegans N-glycans attached to mannose residues (50, 51) or attached to core fucose residues (52). Importantly, the nematodes C. elegans and H. contortus are both phylogenetically closely related to Heterorhabditis, whereas A. suum is more distantly related (53). It is thus reasonable to speculate that the Gal-α-1,3-GalNAc epitope recognized by the bacterial lectin PllA is also present in the nematode Heterorhabditis and plays a role in bacterial attachment or symbiosis of Photorhabdus species with their native nematode hosts. Moreover, this epitope has been described as one terminal constituent of glycosphingolipids of the insect Calliphora vicina pupae (54) and members of the order of diptera, i.e. flies, are generally susceptible to infection with Heterorhabditis and P. luminescens. Therefore, it is possible that Gal-α-1,3-GalNAc is one natural ligand bound by PllA both in the nematode symbiont and in infected insects.
Based on the carbohydrate specificity of PllA as determined by the glycan array, PllA was tested in a porcine cell culture staining experiment for the detection of the α-Gal epitope, the prime reason for hyperacute organ rejection in xenobiotic transplants. PllA proved to be a suitable detection tool and specifically detected the α-Gal epitope in porcine tissue and on red blood cells. This fact qualifies recombinantly produced PllA for the efficacy assessment of methods to genetically manipulate cells, such as CRISPR/Cas, for the production of alternative animal cells, tissue, or organisms lacking the α-Gal epitope as donors for xenotransplantation.
We are currently analyzing the biological role of PllA in P. luminescens for its life cycle in nematodes and insects. Furthermore, it will be of interest to analyze the role of PllA orthologs in the human pathogens P. asymbiotica and the Enterobacter spp., a group of bacteria that are part of the normal human gut flora with pathogenic potential.
Materials and methods
Chemicals
Methyl α-l-fucoside (6), methyl α-d-mannoside (7), and d-galactose (9) were purchased from Dextra Laboratories (Reading, UK); d-galactosamine (10), methyl α-d-galactoside (12), p-nitrophenyl α-d-galactoside (16), 4-methylumbelliferyl α-d-galactoside (17), 5-bromo-4-chloro-3-indolyl α-d-galactoside (18), isopropyl β-d-1-thiogalactoside (IPTG) were from Carbosynth Ltd. (UK); methyl β-d-arabinoside (8) was from Tokyo Chemical Industry (Japan); methyl α-d-glucoside (5), N-acetyl-d-galactosamine (11), and stachyose (26) were from Sigma (Germany); melibiose (22) was from MP Biomedicals Llc. (France); raffinose (25) was from Th. Geyer Laboratories (Germany); Gal-α-1,3-Gal (20), Gal-α-1,4-Gal (21), Xeno antigen (27), P1 antigen (28), and blood group B antigens (29, 30) were from Elicityl OligoTech (France); Gal-α-SP-biotin (13), Gal-α-1,3-GalNAc-α-SP-biotin (23), and Gal-α-1,4-GlcNAc-β-SP-biotin (24) were from Lectinity (Russia); and Gal-α-1,2-Gal-β-1-OMe (19) was from Carbohydrate Synthesis (Oxford, UK). Fluorescent ligands 1–4 were synthesized as described (27).
Allyl α-d-galactopyranoside (14) was synthesized from galactose in a Fischer-type glycosylation with allyl alcohol in presence of Amberlite IR120/H+ (supplemental Scheme 1). The title compound was obtained by recrystallization. 1H NMR (400 MHz, MeOH-d4) δ 5.98 (dddd, J = 17.2, 10.4, 6.1, 5.2 Hz, 1H, CH2CHCH2O-), 5.33 (dq, J = 17.2, 1.7 Hz, 1H, CH2CHCH2O-), 5.17 (dq, J = 10.4, 1.4 Hz, 1H, CH2CHCH2O-), 4.87 (d, J = 3.0 Hz, 1H, H1), 4.23 (ddt, J = 13.0, 5.3, 1.5 Hz, 1H, CH2CHCH2O-), 4.04 (ddt, J = 13.0, 6.1, 1.4 Hz, 1H, CH2CHCH2O-), 3.93–3.89 (m, 1H, H4), 3.85–3.78 (m, 1H, H5), 3.78–3.76 (m, 2H, H2, H3), 3.73–3.69 (m, 2H, H6). 13C NMR (101 MHz, MeOH-d4) δ 135.65 (CH2CHCH2O-), 117.48 (CH2CHCH2O-), 99.46 (C1), 72.37 (C5), 71.51 (C2/3), 71.16 (C4), 70.21 (C2/3), 69.39 (CH2CHCH2O-), 62.78 (C6). Transcripts of NMR spectra are shown in supplemental Figs. 4 and 5).
For allyl 3-O-methyl-α-d-galactopyranoside (15), a microwave vial was filled with allyl galactoside 14 (57 mg, 0.26 mmol) and dibutyltinoxide (71 mg, 0.29 mmol), and the reagents were dried in vacuo. Dry PhMe/MeCN (5:1, 660 μl) was added, and the suspension in the sealed tube was exposed to microwave irradiation for 20 min at 150 °C. The clear solution was allowed to cool to 50 °C, and to the resulting suspension was added MeI (405 μl, 6.5 mmol) dropwise. The reaction was stirred for 48 h at 50 °C, after removal of the volatiles in vacuo and purification of the crude product by MPLC, the title compound was obtained as a pure product (40 mg, 65%). 1H NMR (400 MHz, MeOH-d4) δ 6.08–5.89 (m, 1H, CH2CHCH2O-), 5.34 (dd, J = 17.2, 1.8 Hz, 1H, CH2CHCH2O-), 5.17 (dd, J = 10.4, 1.6 Hz, 1H, CH2CHCH2O-), 4.85 (d, J = 4.0 Hz, 1H, H1), 4.23 (ddt, J = 13.0, 5.3, 1.6 Hz, 1H, 1H, CH2CHCH2O-), 4.13 (dd, J = 3.3, 1.2 Hz, 1H, H4), 4.04 (ddt, J = 13.0, 6.1, 1.4 Hz, 1H, CH2CHCH2O-), 3.85 (dd, J = 10.1, 3.9 Hz, 1H, H2), 3.82–3.77 (m, 1H, H5), 3.76–3.63 (m, 2H, H6), 3.46 (s, 3H, CH3), 3.42 (dd, J = 10.1, 3.2 Hz, 1H, H3). 13C NMR (101 MHz, MeOH-d4) δ 135.65 (CH2CHCH2O-), 117.50 (CH2CHCH2O-), 99.37 (C1), 81.15 (C3), 72.35 (C5), 69.38 (CH2CHCH2O-), 69.22 (C2), 66.91 (C4), 62.74 (C6), 57.24 (CH3). Transcripts of NMR spectra are shown in supplemental Figs. 6 and 7).
Bioinformatics
A BLAST search (blastp) was done using the amino acid sequences of LecA from P. aeruginosa as query (accession number Q05097). The search was carried out choosing non-redundant protein sequence database with exclusion of P. aeruginosa (taxid: 287). The best 100 matches were chosen for an alignment using the COBALT tool (55). The aligned sequences were clustered using the SECATOR algorithm (56), which relies on BIONJ (57) to build the phylogenetic tree. The best LecA-like sequences (with lowest E-value) from each Xenorhabdus and Photorhabdus species were aligned with LecA using COBALT, and the conserved sequence was colored using Color Align Conservation (58). The amino acid sequence of hypothetical LecA homologs (with lowest E-value in Xenorhabdus and Photorhabdus species) were WP_011146351.1 (P. luminescens), WP_046975865.1 (P. temperata), WP_065824676.1 (P. asymbiotica), WP_054480913.1 (P. heterorhabditis), WP_038256436.1 (Xenorhabdus bovienii), WP_013184196.1 (Xenorhabdus nematophila), WP_047963870.1 (Xenorhabdus khoisanae), WP_074019816.1 (Xenorhabdus thuongxuanensis), WP_038237499.1 (Xenorhabdus szentirmaii), and GenBankTM number SFO04414.1 (Xenorhabdus japonica), and SFJ01328.1 (Xenorhabdus mauleonii).
Cloning, expression, and purification of recombinant PllA
Genomic DNA was isolated from P. luminescens subsp. laumondii TTO1 using GenElute Bacterial Genomic DNA Kit (Sigma). The plu2096 gene sequence was amplified by PCR with Phusion polymerase (New England Biolabs, UK) and primers introducing NdeI (5′-GGAATTCCATATGTCTGATTGGTCAGGAAG-3′) and BamHI (5′-CGGGATCCTTATTTTAAAGGGGAGTATCGAG-3′) restriction sites. After digestion of the expression vector pET22b(+) (Novagen, Germany) and the PCR product with NdeI and BamHI (New England Biolabs, UK), ligation of the insert was performed with T4 DNA ligase (New England Biolabs, UK) resulting in plasmid pET22b-pllA. The sequence was confirmed by sequencing (GATC Biotech, Germany) with primers T7 promotor (5′-TAATACGACTCACTATATAGG-3′) and T7 terminator (5′-GCTAGTTATTGCTCAGCGG-3′).
For expression, pET22b-pllA was transformed into chemically competent E. coli BL21(DE3), and the expression strain was selected on LB agar supplemented with ampicillin (100 μg ml−1). 2 liters of LB supplemented with ampicillin (100 μg ml−1) were inoculated with a preculture and grown at 37 °C and 180 rpm to an A600 of 0.5–0.6. Expression was induced with addition of IPTG (0.5 mm final concentration), and bacteria were then further cultured for 6 h at 30 °C and 180 rpm. The cells were harvested by centrifugation (3000 × g, 10 min), and the pellet was washed with TBS/Ca (20 mm Tris, 137 mm NaCl, 2.6 mm KCl, pH 7.4, supplemented with 100 μm CaCl2). The cells were resuspended in 25 ml of TBS/Ca with PMSF (1 mm) and lysozyme (0.4 mg ml−1) and subsequently disrupted by five cycles in a microfluidics homogenizer (Microfluidics Corp.). Cell debris was removed by centrifugation (10,000 × g, 60 min), and the supernatant was loaded onto a column containing galactosylated (59) or later melibiose-coupled Sepharose CL-6B. The column was washed with TBS/Ca, and PllA was eluted by addition of 100 mm galactose or 100 mm raffinose to the buffer. The eluted fractions were extensively dialyzed against distilled water and then TBS/Ca buffer. The concentration was determined by UV absorbance at 280 nm using a calculated molar extinction coefficient of 19,480 m−1 cm−1. The yield of purified PllA was 6 mg (galactose-column) or 19 mg (melibiose-column) per liter of culture volume.
Gel filtration
A HiLoad 16/600 Superdex 200 pg (GE Healthcare) was equilibrated with TBS/Ca buffer (20 mm Tris, 137 mm NaCl, 2.6 mm KCl, pH 7.4, supplemented with 1 mm CaCl2) with a flow rate of 1 ml/min. A calibration curve for molecular size estimation was generated by loading 10 μm of mixture of standard proteins (lysozyme, DNase I, ovalbumin, and BSA). Thereafter, 10 μm PllA was loaded on the column and analyzed with the same flow rate.
Dynamic light scattering (DLS) measurements
DLS measurements were performed on a Zetasizer Nano-ZS (Malvern Instruments, UK). Stock solutions were filtered with a syringe filter before measurements. 50 μl of PllA or LecA (100 mm) in TBS/Ca (20 mm Tris, 137 mm NaCl, 2.6 mm KCl, pH 7.4, supplemented with 1 mm CaCl2) was measured at 25 °C.
Fluorescent labeling of PllA and glycan array analysis
PllA (700 μl, 58 μm in Na2CO3 buffer, pH 9.3) was incubated at room temperature under shaking (500 rpm) with fluorescein isothiocyanate (FITC, 33 μl, 3 mg ml−1, in sodium carbonate buffer, pH 9.3) for 1 h. Purification of the labeled protein was performed as described above for unlabeled PllA; the protein concentration was determined as described previously for LecB-PA14 (19) using an extinction coefficient of 19,480 m−1 cm−1 for PllA.
FITC-labeled PllA was tested on the Consortium for Functional Glycomics (CFG) mammalian glycan array (Core H) version 5.3 containing 600 printed glycans in replicates of 6. Standard procedures of Core H (details see http://www.functionalglycomics.org/glycomics/publicdata/selectedScreens.jsp)4 were run at 5 and 50 μg ml−1 protein based on the protocol by Blixt et al.(60). Raw data of the PllA binding experiments are available as supplemental Tables S1 and S2 as an XLS spreadsheet.
Direct binding of fluorescent ligands 1–4 to PllA
10 μl of a serial dilution of PllA in TBS/Ca (618–0.30 μm) was added in triplicate to a 384-well plate (Greiner Bio-One, Germany, catalog no. 781900). Then, 10 μl of fluorescent ligand 1–4 dissolved in TBS/Ca were added to PllA to a final concentration of 10 nm. After incubation for 1 h at room temperature, blank corrected fluorescence intensity was recorded using a PheraStar FS microplate reader (BMG Labtech GmbH, Germany) with excitation filters at 485 nm and emission filters at 535 nm, and fluorescence polarization was calculated. The data were analyzed using a four-parameter fit of the MARS Data Analysis Software (BMG Labtech GmbH, Germany). A minimum of three independent experiments on three plates was performed for each fluorescent ligand.
Competitive binding assay for PllA
10 μl of a serial dilution of each tested compounds in TBS/Ca (20 to 0.01 mm) were added in triplicate to a 384-well plate (Greiner Bio-One, Germany, catalog no. 781900). Afterward, 10 μl of PllA and 1 were added to each well at final concentrations of 55 μm and 10 nm, respectively. After incubation for 1 h at room temperature, fluorescence polarization was determined using a microplate reader as described above. The data were analyzed using a four-parameter fit of the MARS Data Analysis Software (BMG Labtech GmbH, Germany). A minimum of three independent experiments on three plates was performed for each compound.
X-ray crystallography
Crystals of apo-PIIA were obtained in 1.6 m sodium citrate tribasic dihydrate, pH 6.5. To solve the PIIA–ligand complex structures, PIIA was co-crystallized in the presence of 10 mm ligand and 3 mm calcium chloride. Optimized crystals of PIIA-12, PIIA-25, and PIIA-1 were grown under conditions of 0.2 m ammonium acetate, 0.1 m BisTris buffer, pH 5.5, and 25% PEG 3350; 0.2 m magnesium acetate and 20% PEG 3350; and 0.15 m dl-malic acid and 20% PEG 3350, respectively. Diffraction data for all proteins was collected from single crystals at 100 K. Data for apo-PIIA and PllA-12 were obtained at beamline ID23-2 (ESRF) at a wavelength of 0.873 Å, whereas data for the PIIA-25 and PIIA-1 were collected at beamline ID30-B (ESRF) at a wavelength of 0.967 Å. Data were processed using Xia2 (61) or XDS (62), and the structures were solved using PHASER (63) molecular replacement with LecA (PDB code 1L7L) as a search model. The models were manually rebuilt with COOT (64) and refined using PHENIX (65) and Refmac5 (66). The structures were validated using MolProbity, and all images were created using PyMOL (67).
Molecular modeling
Docking was performed using PLANTS version 1.1 (68). The calculation of charge and energy minimization of the protein and ligand was performed with Molecular Operating Environment (MOE) version 2014.09 (Chemical Computing Group Inc., Montreal, Quebec, Canada). Then, the standard docking procedure was used to dock d-galactosamine (10) and the methyl glycoside of 20 into the binding pocket of the apo-PllA crystal structure. The docking site was limited to a 13 Å radius sphere centered in the mass center (coordination: X = −8.624, Y = 15.131, and Z = 45.115) of the crystallized protein. Asp-103, Asp-96, Gln-57, and Glu-44 were set as flexible residues in the input file.
Generation of primary GTKO cells
Pigs lacking the GTKO were generated by disrupting the causative galactosyltransferase gene GGTA1, according to the procedure described in Klymiuk et al. (69). First, a bacterial artificial chromosome (BAC) containing the target region of the porcine genome, CH242-21F3, was modified by bacterial recombineering in a way that it contained a STOP box right after the START codon of GGTA1, resulting in the termination of protein translation as well as RNA transcription of the gene, and a floxed resistance cassette for neomycin selection. Then, this modified BAC was nucleofected into pig primary cells according to Richter et al. (70), and single-cell clones were generated under antibiotic selection and propagated to yield cells for DNA isolation and somatic cell nuclear transfer (SCNT). Single-cell clones were screened for homologous recombination by a quantitative PCR-based loss-of-wild-type allele approach, and cell clones that indicated a heterozygous modification of the GGTA1 allele were used for SCNT to generate heterozygous knock-out pigs. After birth, one of the animals was sacrificed, and primary cells were cultivated and nucleofected with a plasmid encoding Cre recombinase. Again, single-cell clones were generated and now screened for the removal of the neomycin selection cassette. Another round of SCNT was performed to generate heterozygous KO animals lacking the neomycin selection cassette. Pigs were then maintained and bred to achieve homozygous GTKO pigs after two generations. Primary cells from such GTKO animals were isolated according to the procedure described by Richter et al. (70), and these cells were used for evaluating the specificity and sensitivity of the PllA lectin.
Lectin staining of porcine cells
For lectin staining, 1 × 104 cells were seeded in 6-channel slides (IBIDI, Martinsried, Germany), coated with collagen type 1 (Serva Electrophoresis, Heidelberg, Germany), and cultivated under conventional conditions (70). When reaching a confluence of 80–100%, cells were stained for 15 min with 5 μg/ml Hoechst 33342 and subsequently with FITC-labeled isolectin B4 (GS-IB4, Sigma, 500 μg/ml) or FITC-labeled PllA (50 μg/ml) for 1 h at room temperature. After washing with PBS, cells were visualized in a fluorescence microscope (Axiovert 200, Zeiss).
Hemagglutination of porcine red blood cells
Hemagglutination was done in analogy to a previously published protocol (34). Lithium-heparinized pig blood was centrifuged at 1000 × g for 5 min. Plasma was removed, and the pRBCs were washed with 45 ml of PBS three times. A 10% pRBC solution was prepared by diluting 1 ml of pRBCs with 9 ml of PBS (A600 = 7). Then, 50 μl of PBS was added to each well of a 96-well plate. Thereafter, 50 μl of PIIA (2.5 mg/ml) were added to the first well and mixed, and 50 μl of this mixture was transferred to the second well. Serial dilution of PIIA was continued until 23 dilutions were obtained. 50 μl of 10% pRBCs from WT or GTKO pig were added to each well, and the plate was incubated for 2 h at room temperature. Inhibition of PllA-mediated WT pRBC agglutination was then tested with raffinose. A serial dilution of raffinose (20 mm) was mixed with the lowest lectin concentration showing agglutination (2.4 μg/ml). After incubation for 30 min at room temperature, pRBCs were added. The plate was incubated for 2 h at room temperature.
Author contributions
A. T. conceived and coordinated the study. G. B., J. K., and A. T. wrote the paper. G. B. and A. T. designed, performed, and analyzed the experiments shown in Figs. 1–4 and Tables 1 and 2. G. B. performed docking experiments. D. H. performed chemical synthesis of compounds. S. W. designed and performed cloning of PllA expression vector with assistance of G. B., A. S., and J. K. designed, performed, and analyzed the experiments shown in Figs. 5–8 and Table 3 with assistance of G. B., E. M. J., N. K., and E. W. designed, performed, and analyzed the experiments shown in Figs. 9 and 10. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Prof. Rolf Müller (HIPS Saarbrücken) for providing the P. luminescens strain used in this study and to the Consortium for Functional Glycomics (Core H, Protein–Glycan Interaction Resource of the Consortium for Functional Glycomics and National Institutes of Health Supporting Grant R24 GM098791) for the glycan array analysis of PllA. We acknowledge use of the ESRF synchrotron (beamlines ID23-2 and ID30-B). GTKO pigs were produced with funding from Deutsche Forschungsgemeinschaft Grant TRR 127 “Biology of xenogeneic cell, tissue, and organ transplantation–from bench to bedside.”
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S11, Tables S1–S2 and Scheme S1.
The atomic coordinates and structure factors (codes 5OFZ, 5ODU, 5OFX, and 5OFI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- SEC
- size-exclusion chromatography
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DLS
- dynamic light scattering
- GTKO
- α-1,3-galactosyltransferase knock-out
- SCNT
- somatic cell nuclear transfer
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- BAC
- bacterial artificial chromosome.
References
- 1. Poinar G. O., Thomas G. M., and Hess R. (1977) Characteristics of the specific bacterium associated with Heterorhabditis bacteriophora (Heterorhabditidae: Rhabditida). Nematologica 23, 97–102 [Google Scholar]
- 2. Thomas G. M., and Poinar J. R. (1979) Xenorhabdus gen. nov., a genus of entomopathogenic, nematophilic bacteria of the family Enterobacteriaceae. Int. J. Syst. Evol. Microbiol. 29, 352–360 [Google Scholar]
- 3. Boemare N., Akhurst R., and Mourant R. (1993) DNA relatedness between Xenorhabdus spp. (Enterobacteriaceae), symbiotic bacteria of entomopathogenic nematodes, and a proposal to transfer Xenorhabdus luminescens to a new genus, Photorhabdus gen. nov. Int. J. Syst. Evol. Microbiol. 43, 249–255 [Google Scholar]
- 4. Forst S., and Nealson K. (1996) Molecular biology of the symbiotic-pathogenic bacteria Xenorhabdus spp., and Photorhabdus spp. Microbiol. Rev. 60, 21–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischer-Le Saux M., Viallard V., Brunel B., Normand P., and Boemare N. E. (1999) Polyphasic classification of the genus Photorhabdus and proposal of new taxa: P. luminescens subsp. luminescens subsp. nov., P. luminescens subsp. akhurstii subsp. nov., P. luminescens subsp. laumondii subsp. nov., P. temperata sp. nov., P. temperata subsp. temperata subsp. nov., and P. asymbiotica sp. nov. Int. J. Syst. Bacteriol. 49, 1645–1656 [DOI] [PubMed] [Google Scholar]
- 6. Ferreira T., van Reenen C. A., Endo A., Tailliez P., Pagès S., Spröer C., Malan A. P., and Dicks L. M. (2014) Photorhabdus heterorhabditis sp. nov., a symbiont of the entomopathogenic nematode Heterorhabditis zealandica. Int. J. Syst. Evol. Microbiol. 64, 1540–1545 [DOI] [PubMed] [Google Scholar]
- 7. Hapeshi A., and Waterfield N. R. (2017) Photorhabdus asymbiotica as an insect and human pathogen. Curr. Top. Microbiol. Immunol. 402, 159–177 [DOI] [PubMed] [Google Scholar]
- 8. Akhurst R., and Boemare N. (1990) in Entomopathogenic Nematodes in Biological Control (Gaugler R., and Kaya H. K., eds) pp. 75–90, CRC Press Inc., Boca Raton, FL [Google Scholar]
- 9. Akhurst R., and Dunphy G. (1993) in Parasites and Pathogens of Insects (Beckage N. E., Thompson S. A., and Federici B. A., eds) Vol. 2, pp. 1–23, Academic Press, New York [Google Scholar]
- 10. Duchaud E., Rusniok C., Frangeul L., Buchrieser C., Givaudan A., Taourit S., Bocs S., Boursaux-Eude C., Chandler M., Charles J.-F., Dassa E., Derose R., Derzelle S., Freyssinet G., Gaudriault S., et al. (2003) The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat. Biotechnol. 21, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 11. Bowen D., Rocheleau T. A., Blackburn M., Andreev O., Golubeva E., Bhartia R., and ffrench-Constant R. H. (1998) Insecticidal toxins from the bacterium Photorhabdus luminescens. Science 280, 2129–2132 [DOI] [PubMed] [Google Scholar]
- 12. Guo L., Fatig R. O. 3rd., Orr G. L., Schafer B. W., Strickland J. A., Sukhapinda K., Woodsworth A. T., and Petell J. K. (1999) Photorhabdus luminescens W-14 insecticidal activity consists of at least two similar but distinct proteins. Purification and characterization of toxin A and toxin B. J. Biol. Chem. 274, 9836–9842 [DOI] [PubMed] [Google Scholar]
- 13. Kumar A., Sýkorová P., Demo G., Dobes̆ P., Hyrs̆l P., and Wimmerová M. (2016) A novel fucose-binding lectin from Photorhabdus luminescens (PLL) with an unusual heptabladed β-propeller tetrameric structure. J. Biol. Chem. 291, 25032–25049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagner S., Sommer R., Hinsberger S., Lu C., Hartmann R. W., Empting M., and Titz A. (2016) Novel strategies for the treatment of Pseudomonas aeruginosa infections. J. Med. Chem. 59, 5929–5969 [DOI] [PubMed] [Google Scholar]
- 15. Diggle S. P., Stacey R. E., Dodd C., Cámara M., Williams P., and Winzer K. (2006) The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ. Microbiol. 8, 1095–1104 [DOI] [PubMed] [Google Scholar]
- 16. Tielker D., Hacker S., Loris R., Strathmann M., Wingender J., Wilhelm S., Rosenau F., and Jaeger K.-E. (2005) Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology 151, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 17. Garber N. (1997) Specific Adherence Mechanisms in Microbiology and Immunology. Nova Acta Leopoldina 1997, 75 [Google Scholar]
- 18. Mitchell E., Houles C., Sudakevitz D., Wimmerova M., Gautier C., Pérez S., Wu A. M., Gilboa-Garber N., and Imberty A. (2002) Structural basis for oligosaccharide-mediated adhesion of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Nat. Struct. Biol. 9, 918–921 [DOI] [PubMed] [Google Scholar]
- 19. Sommer R., Wagner S., Varrot A., Nycholat C. M., Khaledi A., Häussler S., Paulson J. C., Imberty A., and Titz A. (2016) The virulence factor LecB varies in clinical isolates: consequences for ligand binding and drug discovery. Chem. Sci. 7, 4990–5001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sudakevitz D., Kostlánová N., Blatman-Jan G., Mitchell E. P., Lerrer B., Wimmerová M., Katcoff D. J., Imberty A., and Gilboa-Garber N. (2004) A new Ralstonia solanacearum high-affinity mannose-binding lectin RS-IIL structurally resembling the Pseudomonas aeruginosa fucose-specific lectin PA-IIL. Mol. Microbiol. 52, 691–700 [DOI] [PubMed] [Google Scholar]
- 21. Beshr G., Sommer R., Hauck D., Siebert D. C. B., Hofmann A., Imberty A., and Titz A. (2016) Development of a competitive binding assay for the Burkholderia cenocepacia lectin BC2L-A and structure activity relationship of natural and synthetic inhibitors. Med. Chem. Commun. 7, 519–530 [Google Scholar]
- 22. Lameignere E., Shiao T. C., Roy R., Wimmerova M., Dubreuil F., Varrot A., and Imberty A. (2010) Structural basis of the affinity for oligomannosides and analogs displayed by BC2L-A, a Burkholderia cenocepacia soluble lectin. Glycobiology 20, 87–98 [DOI] [PubMed] [Google Scholar]
- 23. Marchetti R., Malinovska L., Lameignère E., Adamova L., de Castro C., Cioci G., Stanetty C., Kosma P., Molinaro A., Wimmerova M., Imberty A., and Silipo A. (2012) Burkholderia cenocepacia lectin A binding to heptoses from the bacterial lipopolysaccharide. Glycobiology 22, 1387–1398 [DOI] [PubMed] [Google Scholar]
- 24. Zinger-Yosovich K., Sudakevitz D., Imberty A., Garber N. C., and Gilboa-Garber N. (2006) Production and properties of the native Chromobacterium violaceum fucose-binding lectin (CV-IIL) compared to homologous lectins of Pseudomonas aeruginosa (PA-IIL) and Ralstonia solanacearum (RS-IIL). Microbiology 152, 457–463 [DOI] [PubMed] [Google Scholar]
- 25. Imberty A., Wimmerová M., Mitchell E. P., and Gilboa-Garber N. (2004) Structures of the lectins from Pseudomonas aeruginosa: insight into the molecular basis for host glycan recognition. Microbes Infect. 6, 221–228 [DOI] [PubMed] [Google Scholar]
- 26. McMahon K. (2009) Structural and Functional Characterisation of Lectins from the PA-IL Superfamily. Ph.D. thesis, Dublin City University, Dublin, Ireland [Google Scholar]
- 27. Joachim I., Rikker S., Hauck D., Ponader D., Boden S., Sommer R., Hartmann L., and Titz A. (2016) Development and optimization of a competitive binding assay for the galactophilic low affinity lectin LecA from Pseudomonas aeruginosa. Org. Biomol. Chem. 14, 7933–7948 [DOI] [PubMed] [Google Scholar]
- 28. Cioci G., Mitchell E. P., Gautier C., Wimmerová M., Sudakevitz D., Pérez S., Gilboa-Garber N., and Imberty A. (2003) Structural basis of calcium and galactose recognition by the lectin PA-IL of Pseudomonas aeruginosa. FEBS Lett. 555, 297–301 [DOI] [PubMed] [Google Scholar]
- 29. Galili U. (2005) The α-Gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol. Cell Biol. 83, 674–686 [DOI] [PubMed] [Google Scholar]
- 30. Duffy M. S., Morris H. R., Dell A., Appleton J. A., and Haslam S. M. (2006) Protein glycosylation in Parelaphostrongylus tenuis–first description of the Galα1–3Gal sequence in a nematode. Glycobiology 16, 854–862 [DOI] [PubMed] [Google Scholar]
- 31. Ekser B., and Cooper D. K. (2010) Overcoming the barriers to xenotransplantation: prospects for the future. Expert Rev. Clin. Immunol. 6, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blanchard B., Nurisso A., Hollville E., Tétaud C., Wiels J., Pokorná M., Wimmerová M., Varrot A., and Imberty A. (2008) Structural basis of the preferential binding for globo-series glycosphingolipids displayed by Pseudomonas aeruginosa lectin I. J. Mol. Biol. 383, 837–853 [DOI] [PubMed] [Google Scholar]
- 33. Eierhoff T., Bastian B., Thuenauer R., Madl J., Audfray A., Aigal S., Juillot S., Rydell G. E., Müller S., de Bentzmann S., Imberty A., Fleck C., and Römer W. (2014) A lipid zipper triggers bacterial invasion. Proc. Natl. Acad. Sci. U.S.A. 111, 12895–12900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hauck D., Joachim I., Frommeyer B., Varrot A., Philipp B., Möller H. M., Imberty A., Exner T. E., and Titz A. (2013) Discovery of two classes of potent glycomimetic inhibitors of Pseudomonas aeruginosa LecB with distinct binding modes. ACS Chem. Biol. 8, 1775–1784 [DOI] [PubMed] [Google Scholar]
- 35. Wohlschlager T., Butschi A., Grassi P., Sutov G., Gauss R., Hauck D., Schmieder S. S., Knobel M., Titz A., Dell A., Haslam S. M., Hengartner M. O., Aebi M., and Künzler M. (2014) Methylated glycans as conserved targets of animal and fungal innate defense. Proc. Natl. Acad. Sci. U.S.A. 111, E2787–E2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sommer R., Hauck D., Varrot A., Imberty A., Künzler M., and Titz A. (2016) O-Alkylated heavy atom carbohydrate probes for protein X-ray crystallography: Studies towards the synthesis of methyl 2-O-methyl-l-selenofucopyranoside. Beilstein J. Org. Chem. 12, 2828–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen C. P., Song S. C., Gilboa-Garber N., Chang K. S., and Wu A. M. (1998) Studies on the binding site of the galactose-specific agglutinin PA-IL from Pseudomonas aeruginosa. Glycobiology 8, 7–16 [DOI] [PubMed] [Google Scholar]
- 38. Kim H.-S., Cha E., Kim Y., Jeon Y. H., Olson B. H., Byun Y., and Park H.-D. (2016) Raffinose, a plant galactoside, inhibits Pseudomonas aeruginosa biofilm formation via binding to LecA and decreasing cellular cyclic diguanylate levels. Sci. Rep. 6, 25318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landsteiner K., and Levine P. (1927) Further observations on individual differences of human blood. Exp. Biol. Med. 24, 941–942 [Google Scholar]
- 40. Khan F., Khan R. H., Sherwani A., Mohmood S., and Azfer M. A. (2002) Lectins as markers for blood grouping. Med. Sci. Monit. 8, RA293–RA300 [PubMed] [Google Scholar]
- 41. Macher B. A., and Galili U. (2008) The Galα1,3Gaβ1,4GlcNAc-R (α-Gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim. Biophys. Acta 1780, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li S., Waer M., and Billiau A. D. (2009) Xenotransplantation: role of natural immunity. Transpl. Immunol. 21, 70–74 [DOI] [PubMed] [Google Scholar]
- 43. Yamada K., Yazawa K., Shimizu A., Iwanaga T., Hisashi Y., Nuhn M., O'Malley P., Nobori S., Vagefi P. A., Patience C., Fishman J., Cooper D. K., Hawley R. J., Greenstein J., Schuurman H.-J., et al. (2005) Marked prolongation of porcine renal xenograft survival in baboons through the use of α1,3-galactosyltransferase gene-knockout donors and the cotransplantation of vascularized thymic tissue. Nat. Med. 11, 32–34 [DOI] [PubMed] [Google Scholar]
- 44. Klymiuk N., Aigner B., Brem G., and Wolf E. (2010) Genetic modification of pigs as organ donors for xenotransplantation. Mol. Reprod. Dev. 77, 209–221 [DOI] [PubMed] [Google Scholar]
- 45. Puga Yung G. L., Li Y., Borsig L., Millard A.-L., Karpova M. B., Zhou D., and Seebach J. D. (2012) Complete absence of the αGal xenoantigen and isoglobotrihexosylceramide in α1,3galactosyltransferase knock-out pigs. Xenotransplantation 19, 196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tempel W., Tschampel S., and Woods R. J. (2002) The xenograft antigen bound to Griffonia simplicifolia lectin 1-B(4). X-ray crystal structure of the complex and molecular dynamics characterization of the binding site. J. Biol. Chem. 277, 6615–6621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Obukhova P., Rieben R., and Bovin N. (2007) Normal human serum contains high levels of anti-Gal α1–4GlcNAc antibodies. Xenotransplantation 14, 627–635 [DOI] [PubMed] [Google Scholar]
- 48. Gerdt S., Dennis R. D., Borgonie G., Schnabel R., and Geyer R. (1999) Isolation, characterization and immunolocalization of phosphorylcholine-substituted glycolipids in developmental stages of Caenorhabditis elegans. Eur. J. Biochem. 266, 952–963 [DOI] [PubMed] [Google Scholar]
- 49. van Stijn C. M., van den Broek M., Vervelde L., Alvarez R. A., Cummings R. D., Tefsen B., and van Die I. (2010) Vaccination-induced IgG response to Galα1–3GalNAc glycan epitopes in lambs protected against Haemonchus contortus challenge infection. Int. J. Parasitol. 40, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yan S., Jin C., Wilson I. B., and Paschinger K. (2015) Comparisons of Caenorhabditis fucosyltransferase mutants reveal a multiplicity of isomeric N-glycan structures. J. Proteome Res. 14, 5291–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yan S., Brecker L., Jin C., Titz A., Dragosits M., Karlsson N. G., Jantsch V., Wilson I. B., and Paschinger K. (2015) Bisecting galactose as a feature of N-glycans of wild-type and mutant Caenorhabditis elegans. Mol. Cell. Proteomics 14, 2111–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yan S., Bleuler-Martinez S., Plaza D. F., Künzler M., Aebi M., Joachim A., Razzazi-Fazeli E., Jantsch V., Geyer R., Wilson I. B., and Paschinger K. (2012) Galactosylated fucose epitopes in nematodes: increased expression in a Caenorhabditis mutant associated with altered lectin sensitivity and occurrence in parasitic species. J. Biol. Chem. 287, 28276–28290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meldal B. H., Debenham N. J., De Ley P., De Ley I. T., Vanfleteren J. R., Vierstraete A. R., Bert W., Borgonie G., Moens T., Tyler P. A., Austen M. C., Blaxter M. L., Rogers A. D., and Lambshead P. J. (2007) An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol. Phylogenet. Evol. 42, 622–636 [DOI] [PubMed] [Google Scholar]
- 54. Dennis R. D., Geyer R., Egge H., Menges H., Stirm S., and Wiegandt H. (1985) Glycosphingolipids in insects. Chemical structures of ceramide monosaccharide, disaccharide, and trisaccharide from pupae of Calliphora vicina (Insecta: Diptera). Eur. J. Biochem. 146, 51–58 [DOI] [PubMed] [Google Scholar]
- 55. Papadopoulos J. S., and Agarwala R. (2007) COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079 [DOI] [PubMed] [Google Scholar]
- 56. Wicker N., Perrin G. R., Thierry J. C., and Poch O. (2001) Secator: a program for inferring protein subfamilies from phylogenetic trees. Mol. Biol. Evol. 18, 1435–1441 [DOI] [PubMed] [Google Scholar]
- 57. Gascuel O. (1997) BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 [DOI] [PubMed] [Google Scholar]
- 58. Stothard P. (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28, 1102. [DOI] [PubMed] [Google Scholar]
- 59. Fornstedt N., and Porath J. (1975) Characterization studies on a new lectin found in seeds of Vicia ervilia. FEBS Lett. 57, 187–191 [DOI] [PubMed] [Google Scholar]
- 60. Blixt O., Head S., Mondala T., Scanlan C., Huflejt M. E., Alvarez R., Bryan M. C., Fazio F., Calarese D., Stevens J., Razi N., Stevens D. J., Skehel J. J., van Die I., Burton D. R., Wilson I. A., Cummings R., Bovin N., Wong C.-H., and Paulson J. C. (2004) Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 17033–17038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 [Google Scholar]
- 62. Kabsch W. (2010) XDS. Acta Crystallogr D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol. Crystallogr. 67, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schrödinger L. (2015) The PyMOL Molecular Graphics System, Version 1.8, New York [Google Scholar]
- 68. Korb O., Stützle T., and Exner T. E. (2006) PLANTS: application of ant colony optimization to structure-based drug design. Lecture Notes in Computer Science 4150, 247–258 [Google Scholar]
- 69. Klymiuk N., Mundhenk L., Kraehe K., Wuensch A., Plog S., Emrich D., Langenmayer M. C., Stehr M., Holzinger A., Kröner C., Richter A., Kessler B., Kurome M., Eddicks M., Nagashima H., et al. (2012) Sequential targeting of CFTR by BAC vectors generates a novel pig model of cystic fibrosis. J. Mol. Med. 90, 597–608 [DOI] [PubMed] [Google Scholar]
- 70. Richter A., Kurome M., Kessler B., Zakhartchenko V., Klymiuk N., Nagashima H., Wolf E., and Wuensch A. (2012) Potential of primary kidney cells for somatic cell nuclear transfer mediated transgenesis in pig. BMC Biotechnol. 12, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.